
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic catalysis between In single atoms and In nanoparticles for highly selective electrocatalytic CO2 reduction to formate with high current densities†
Yuxin Chena,
Junyoung Choib,
Fangkui Liangc,
Xinyi Tan*d,
Yudi Chene,
Jiahui Yanga,
Song Hong a,
Xin Zhangc,
Alex W. Robertsonf,
Yousung Jung*bg and
Zhenyu Sun*a
a,
Xin Zhangc,
Alex W. Robertsonf,
Yousung Jung*bg and
Zhenyu Sun*a
aState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: sunzy@mail.buct.edu.cn
bDepartment of Chemical and Biological Engineering, Seoul National University, 1 Kwanak-ro, Seoul 08826, South Korea. E-mail: yousung.jung@snu.ac.kr
cCollege of Mathematics and Physics, Beijing University of Chemical Technology, Beijing 100029, P. R. China
dSchool of Materials Science and Engineering, Beijing Institute of Technology, Beijing Key Laboratory of Environmental Science and Engineering, Beijing 100081, P. R. China. E-mail: xinyitan@bit.edu.cn
eBeijing Academy of Science and Technology, Beijing 100089, P. R. China
fDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
gInstitute of Engineering Research, Institute of Chemical Processes, Seoul National University, 1 Kwanak-ro, Seoul 08826, South Korea
First published on 14th May 2025
Abstract
The practical realization of the electrocatalytic reduction of CO2 to formate is limited by the lack of suitable highly active and selective electrocatalysts, particularly candidates compatible with operation at high current densities. Herein, we report a dual-active site electrocatalyst consisting of In single atoms and In nanoparticles supported on N, S-codoped porous carbon (In–NSC/NPs), which enables a remarkable formate faradaic efficiency (FE) of 92% with a large absolute partial current density for formate of up to 1.1 A cm−2. By using membrane electrode assembly cells, a formate FE exceeding 90% and an energy conversion efficiency of over 44% are attainable within a wide cell voltage range of 2.4–3 V. The maximum formate generation rate reaches 10.5 mmol cm−2 h−1 at a cell voltage of 2.9 V. By coupling with anodic glycerol oxidation, the formate yield rate in a full electrolytic cell is significantly improved to 23.2 mmol cm−2 h−1 while using the same reaction conditions as the standard anodic oxygen evolution reaction. A combination of control experiments and in situ characterization methods reveals that In nanoparticles facilitate the generation of the *OCHO and the subsequent hydrogenation step to generate formate while the In single atoms boost H2O dissociation. The generated *H migrates to the surface of the In nanoparticles, increasing the proton concentration and promoting the hydrogenation reaction.
Introduction
The sustained consumption of fossil fuels has led to a substantial increase of CO2 in the atmosphere, resulting in severe and continuing climate change. The electrocatalytic CO2 reduction (ECR) uses water and renewably generated electricity to convert CO2 into high-value chemicals and fuels under mild conditions (room temperature, atmospheric pressure), presenting a closed carbon cycle that holds significant promise for reducing greenhouse gas emissions.1–3 The reaction process entails a multiple electron/proton transfer that can produce a rich variety of C1 (containing one carbon atom) and C2+ (containing two or more carbon atoms) hydrocarbons and oxygenates.1 Among the produced compounds, the formation of formate is considered to be one of the most economically feasible routes. As a high-energy density molecule, formate has been applied in hydrogen storage and in proton exchange membrane fuel cells.4,5 The design and development of efficient, selective, and stable electrocatalysts for formate formation is highly desirable for its use in these applications.In a pioneering work, Pb, Hg, Tl, In, Sn, Cd, and Bi metals were reported to be able to catalyze the ECR reaction to produce formate.6,7 In particular, In has attracted attention due to its decent performance, low toxicity, and rich earth reserves. Extensive efforts have been devoted to enhancing the performance of ECR to formate of In-based catalysts through defect engineering,8 bimetallic alloying,9 catalyst morphology design,10 and heterostructure creation.11 However, the concurrent competitive hydrogen evolution reaction (HER) and tendency to form multiple carbon-containing intermediates during the ECR still significantly restrict the highly selective production of formate, especially under a wide potential range and high current densities. Further development of high-performance indium-based catalysts is therefore required.
It is generally considered that the ECR to formate needs to go through a two-step electron–proton pair transfer process, (1). CO2 + e− + H+ → *OCHO, (2). *OCHO + e− + H+ → HCOOH.12,13 Constructing dual or multi-active site electrocatalysts that can simultaneously satisfy the activation of CO2 and provide sufficient protons to participate in the reaction, especially in alkaline environments may markedly boost ECR performance.14,15 In recent years, due to their high atomic utilization and impressive catalytic performance, single atom catalysts have been widely studied in their application to ECR. The low metal loading and the single configuration of active sites are the challenges facing the further development of single atom catalysts.16–20 One approach to improve single atom catalyst performance is to introduce secondary active sites during their synthesis, coupling the new sites with the single atom sites (SAs) so as to leverage the synergistic effect between the multiple active sites to promote the reaction. However, this often requires additional additives and complicates the catalyst preparation process. It is well known that metal nanoparticles (NPs) are a product that is difficult to avoid during the preparation of single atom catalysts, and they are often associated with adverse activities. However, some recent studies have shown that the synergistic effect produced when SAs coexist with NPs can actually promote the reaction.21–23 For example, Zhao et al. found that when Ni SAs and Ni NPs coexist, the electronic interaction between Ni SAs and Ni NPs could promote ECR to produce CO.24 Shao et al.25 constructed a catalyst in which Cu SAs and Cu NPs coexisted and found a tandem catalytic effect between the two sites. At present, there are relatively few reports in this field. How to further develop the synergistic catalytic effect between SAs and NPs is a topic worth further study.
Herein, we report an In based electrocatalyst with the coexistence of In SAs and NPs (In–NSC/NPs). A combination of control experiments and in situ characterizations reveals that the coexistence of In SAs and NPs in a suitable ratio could maximize the synergistic catalytic effect to promote ECR to produce formate. In NPs dominate the hydrogenation process of CO2, while In SAs boost H2O dissociation to increase the proton concentration on the surface of In NPs and therefore promote the hydrogenation reaction. Notably, a formate faradaic efficiency (FE) of up to 92% has been achieved on the as-obtained In–NSC/NPs with a high current density of 1.2 A cm−2 at −1.32 V versus reversible hydrogen electrode (vs. RHE). A formate FE exceeding 90% was attained over a wide cell voltage range from 2.4 to 3 V with a formate yield rate reaching 10.5 mmol cm−2 h−1 at 2.9 V using a membrane electrode assembly (MEA) system. More interestingly, by coupling with the glycerol oxidation reaction (GOR) that has a much more negative thermodynamic oxidation potential (0.003 V vs. RHE) in place of the kinetically sluggish oxygen evolution reaction (OER) occurring at a more positive thermodynamic potential (1.23 V vs. RHE), the formate generation rate of the full electrolytic cell was further improved to 23.2 mmol cm−2 h−1 at 2.9 V.
Results and discussion
The In–NSC/NPs was easily obtained via a simple wet-chemical reduction process without using any stabilizing agents and toxic solvents, as illustrated in Fig. S1.† The NSC support was derived from cheap and abundant commercially purchased broad bean shells through dehydration, graphitization, and demineralization. X-ray diffraction (XRD) measurements verified the formation of crystalline In (PDF#85-1409) in In–NSC/NPs (Fig. S2†) with a corresponding content of ∼2.9 wt% based on inductively coupled plasma optical emission spectrometry (ICP-OES) results (Table S1†). Scanning electron microscopy (SEM) observations revealed that In–NSC/NPs is composed of tightly packed flakes decorated with particles (Fig. S3†). High-resolution transmission electron microscopy (HRTEM) showed that a number of crystalline In NPs were uniformly distributed on NSC sheets (Fig. S4†). Lattice fringes with a d spacing value of ∼0.27 and 0.15 nm correspond to the (101) and (103) facets of In, respectively, consistent with the XRD results (Fig. S2†). Scanning transmission electron microscopy (STEM) combined with energy dispersive spectroscopy (EDS) analysis confirmed In species coexist in the form of NPs and SAs, which indicates that there was an interaction between In species with the NSC, causing it to be partially anchored on the support (Fig. 1).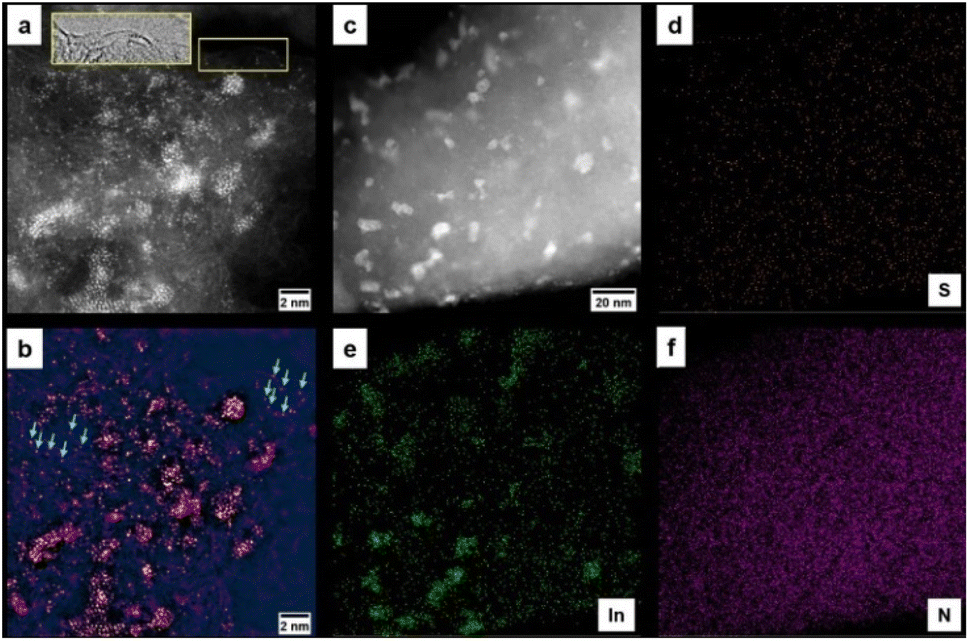 | ||
| Fig. 1 STEM analysis of In–NSC/NPs. (a) High-magnification high-angle annular dark field STEM (HAADF-STEM) with bright dots indicating In atoms and nanoparticles on carbon. The insert shows a bright field STEM (BF-STEM) image from the region indicated with yellow, showing the carbon support with In atoms decorating the edge. (b) False-color lookup table of the image in (a) following application of a bandpass filter, revealing that some In species exists in the form of single atoms (e.g., blue arrows) (c) low magnification HAADF-STEM image showing well-dispersed In on the carbon support. (d–f) EDS maps of the area in (c) of S, In, and N, respectively. | ||
The surface composition and chemical states of In–NSC/NPs was further probed by X-ray photoelectron spectroscopy (XPS). The wide-survey XPS spectrum of In–NSC/NPs clearly indicated the presence of C, O, N, S and In (Fig. 2a). No other impurities were observed. The contents of N and S in In–NSC/NPs were estimated to be ∼1.8 and 0.6 at%, respectively (Table S1†). The In 3d5/2 XPS peak with a binding energy at 444.3 eV (Fig. 2b) was found located between In0 (443.8 eV) and In3+ (445.7 eV). This indicates that in In–NSC/NPs, In exists in both metallic In and its oxidized form.26 Fig. 2c shows the N 1s XPS spectrum of In–NSC/NPs, which could be deconvoluted into four peaks at 403.6, 401.4, and 399.4, and 397.8 eV, assigned to the graphitic N, pyrrolic N, In–N, and pyridinic N, respectively.27,28 The S 2p XPS spectrum validated the presence of C–S–C species (∼163.6 eV for S 2p3/2 and 164.9 eV for S 2p1/2) and oxidized S groups (C–SOx–C at 168.2 eV for S 2p3/2 and 169.8 eV for S 2p1/2).29,30
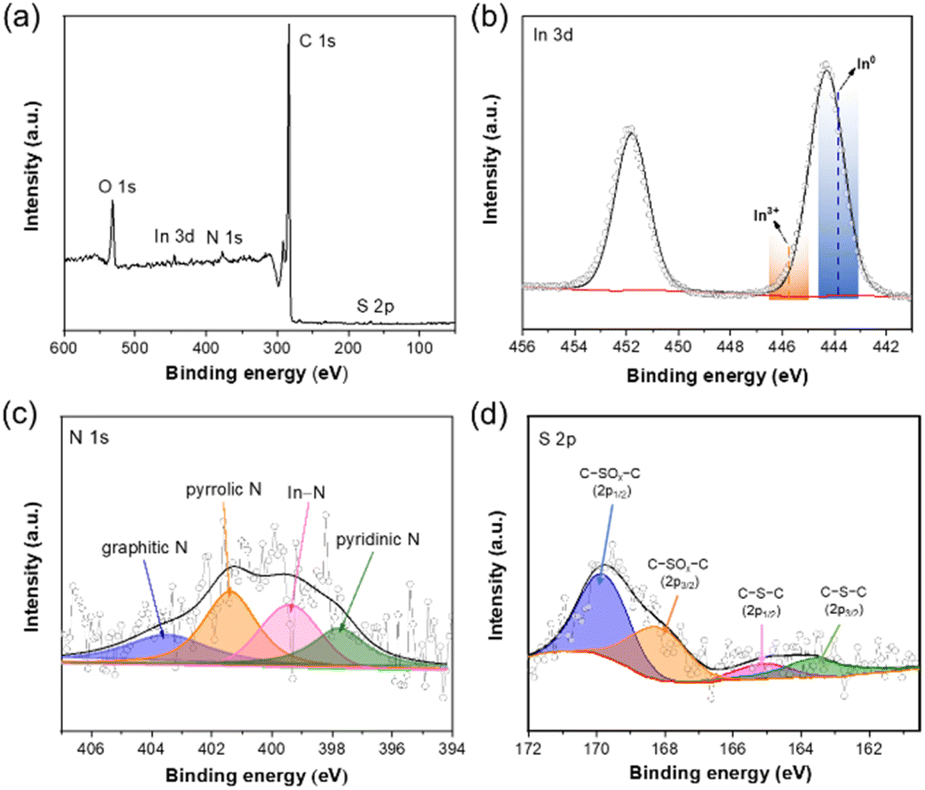 | ||
| Fig. 2 (a) The full-scale, (b) In 3d, (c) N 1s, and (d) S 2p XPS spectra of In–NSC/NPs. | ||
Fig. S5† depicts the Raman spectra of In–NSC/NPs with two characteristic peaks at 1330 and 1580 cm−1, attributed to the D and G bands of sp3-and sp2-coordinated carbon, respectively. The absolute peak intensity ratio of D band and G band (ID/IG) for In–NSC/NPs is 1.1. This indicates that the carbon skeleton of In–NSC/NPs contained defects, originating from the fact that incorporating S with a larger atomic radius broke the integrity of the hexagonal carbon honeycomb lattice and introduced defects.31
The specific surface area and pore structure of In–NSC/NPs were analyzed by N2 adsorption/desorption measurements. The Brunauer–Emmett–Teller (BET) specific surface areas of In–NSC/NPs were determined to be ∼348.9 m2 g−1 (Fig. S6a†). The pore distribution curves prove the formation of micropores and mesopores in In–NSC/NPs with an average pore size of 1.9 nm (Fig. S6b†). It is believed that the rich pore structure and a large specific surface area can promote mass transfer during the ECR reaction, especially the adsorption and diffusion of CO2 to the surface of active sites, facilitating the ECR.32 The room temperature CO2 adsorption experiment (Fig. S7†) showed In–NSC/NPs has a strong CO2 adsorption capacity (41.5 cm3 g−1 under one atmosphere). This enables a sufficiently high CO2 concentration near the active sites and promotes the formation of gas–solid–liquid interface to improve the reaction performance.
The ECR properties of In–NSC/NPs were investigated in a liquid-phase flow cell (Fig. S8 and S9†) with a gas diffusion electrode (GDE) using 1 M KOH solution as a catholyte. The gaseous and liquid products were detected by gas chromatography (GC) and 1H nuclear magnetic resonance (1H NMR) with DMSO as an internal standard (Fig. S10 and S11†), respectively. Carbon monoxide and hydrogen are the main gas products and formate is the only liquid product from In–NSC/NPs. Linear sweep voltammetry (LSV) tests illustrated a marked increase in current density under a CO2 atmosphere with respect to an Ar environment (Fig. 3a), indicative of the preferred occurrence of ECR over HER on In–NSC/NPs. Constant current electrolysis was conducted within a current density range from 0.2 to 1.2 A cm−2 (Fig. 3b). It is worth noting that In–NSC/NPs presented high formate selectivity throughout the entire electrochemical window. The formate FE values exceed 90% over a wide current density range (0.2–1.2 A cm−2). At 0.6 A cm−2, the formate FE approached 99% and only a small amount of H2 and CO were produced as by-products. More importantly, In–NSC/NPs still achieved a formate FE over 90% at a high current density of 1.2 A cm−2. The as-obtained In–NSC/NPs also surpasses many recently reported electrocatalysts for ECR to formate (Fig. 3c and Table S2†).
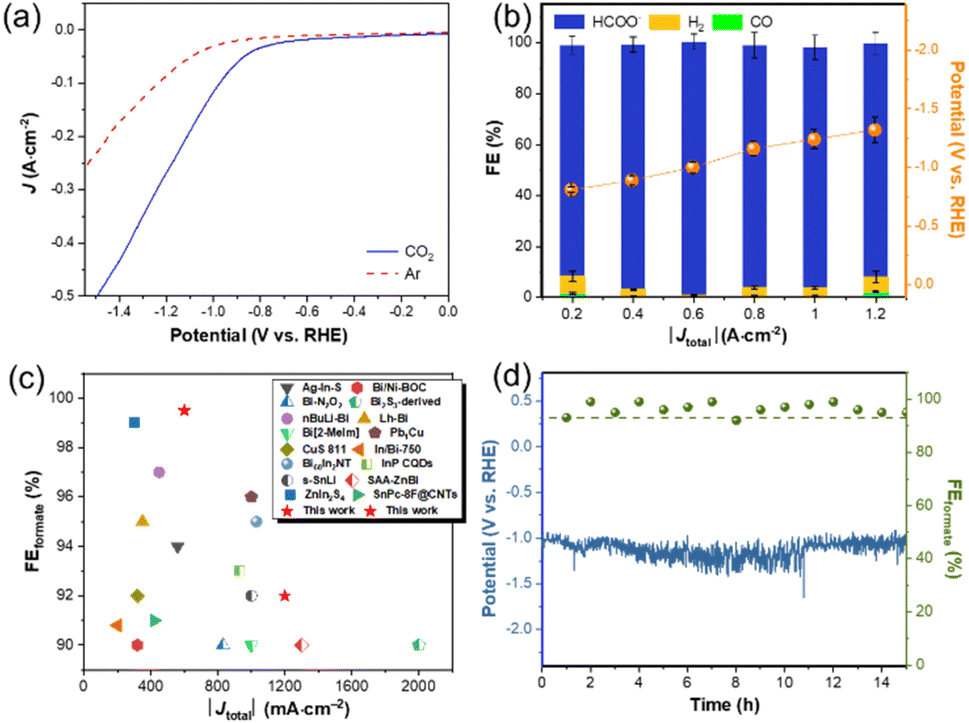 | ||
| Fig. 3 (a) LSV curves of In–NSC/NPs under a CO2 atmosphere at a scan rate of 20 mV s−1. (b) Constant current electrolysis tests of In–NSC/NPs. The data are represented as mean ± standard deviation of three independent measurements. (c) Comparison of In–NSC/NPs with recently reported electrocatalysts in terms of formate FEs at different current densities. (d) Potential and FE toward formate formation versus electrolysis time over In–NSC/NPs at 0.6 A cm−2. | ||
A stability test was conducted through chronopotentiometry at 0.6 A cm−2 for 15 h and the GDE was washed and refreshed after 11 h of continuous operation to address the issue of salt precipitation (the specific operation process is provided in the ESI†). Fig. 3d shows that the FE of formate retained over 90% on In-NSC/NPs during the electrolysis. Post characterization for the In–NSC/NPs after extended electrolysis by XRD, HRTEM, STEM, ICP-OES, and XPS (Fig. S12 and S13, and Table S1†) confirmed that its morphology, structure, and surface oxidation state were well preserved.
A series of controlled experiments and characterizations were carried out to understand the underlying structure–performance relationship of In–NSC/NPs. First, we controlled the In loading to investigate the effect of the ratio of In NPs to SAs on catalytic performance. The comparison samples with lower and higher In loadings were marked as In–NSC/NPs-0.1 and In–NSC/NPs-0.3, respectively (here 0.1 and 0.3 correspond to the molar concentration of the In precursor, respectively. Detailed preparation information can be found in the SI). ICP-OES showed that the actual In contents of In–NSC/NPs-0.1 and In–NSC/NPs-0.3 were 1.6 and 4.7 wt%, respectively. XRD (Fig. S14†) showed that with the increase of In loading, the intensity of the diffraction peaks attributable to the metallic In increased significantly, indicating that the content of In NPs were increased. The In 3d XPS spectrum (Fig. S15†) showed that with the increase of In loading, the In 3d XPS signals gradually shifted to the negative, which indicates that the proportion of In NPs (In0) increased. LSV tests (Fig. 4a) showed that under the CO2 atmosphere In–NSC/NPs exhibited a higher current density response and a more positive reduction potential than In–NSC/NPs-0.1 and In–NSC/NPs-0.3, suggesting its more kinetically favorable nature for the ECR. Constant current electrolysis at 0.6 A cm−2 (Fig. S16†) showed that with the increased of In loadings, the FE of ECR to formate presented a volcano-shaped curve. Furthermore, Tafel plot (Fig. S17†) analysis revealed that In–NSC/NPs possessed a smaller Tafel slope (159 mV dec−1) than In–NSC/NPs-0.1 (185 mV dec−1) and In–NSC/NPs-0.3 (190 mV dec−1), suggesting faster reaction kinetics on In–NSC/NPs.33 The above experiments showed that the coexistence of In SAs and NPs in a suitable ratio was more conducive for the ECR to formate.
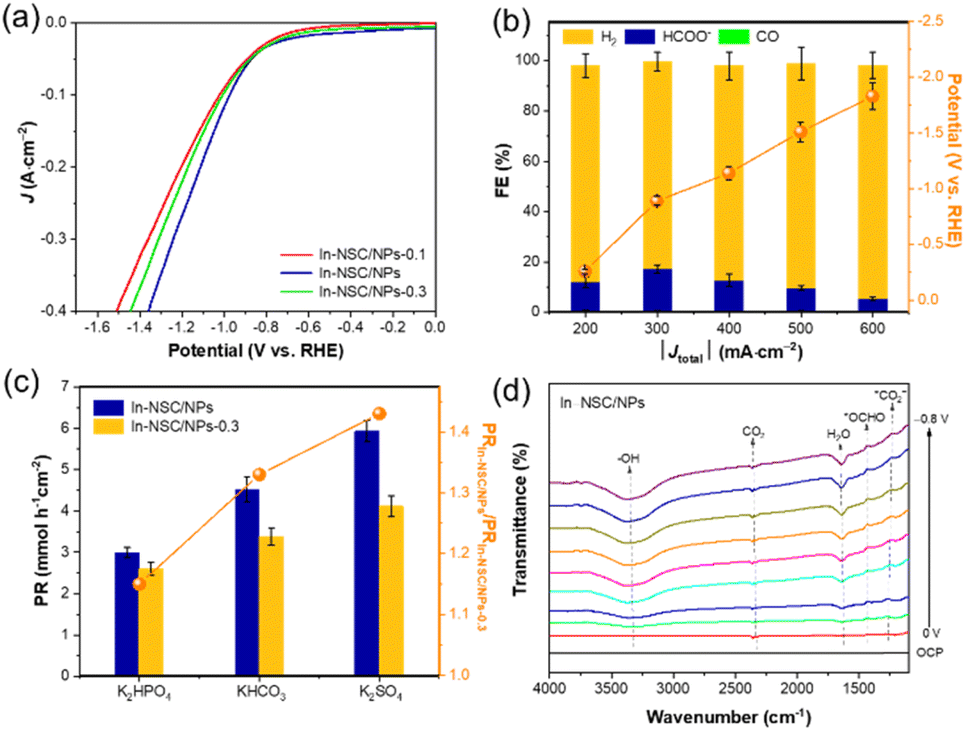 | ||
| Fig. 4 (a) LSV curves of In–NSC/NPs-0.1, In–NSC/NPs and In–NSC/NPs-0.3 under CO2 atmosphere at a scan rate of 20 mV s−1. (b) Constant current electrolysis of In–NSC under CO2 atmosphere. (c) PR and the ratio of PR over In–NSC/NPs and In–NSC/NPs-0.3 in different electrolytes under constant voltage electrolysis at −1 V (vs. RHE). (d) The In–NSC/NPs's ECR ATR-SEIRAS spectra obtained under applied potentials from 0 to −0.8 V (vs. RHE). The data in (b) and (c) are represented as mean ± standard deviation of three independent measurements. | ||
We then prepared the In–NSC single atom catalyst from In–NSC/NPs with a sufficient acid etching treatment, removing the In NPs, allowing us to investigate the catalytic performance when In SAs existed independently. The HRTEM images (Fig. S18†) showed that In NPs were completely removed after acid washing. The XRD pattern (Fig. S19†) also confirmed this conclusion, with the broad peak at 25°attributed to graphitic carbon. The In 3d XPS signals for In–NSC (Fig. S20†) were found at 445.4 eV (In 3d5/2) and 452.9 eV (In 3d3/2), indicate that In species exist in oxidation states (Inδ+, 0 < δ < 3) rather than In metallic states, unraveling their isolated states as single atoms. ICP-OES results showed that the actual content of In in In–NSC was 0.12 wt%. LSV tests illustrated a marked increase in current density under an Ar atmosphere with respect to a CO2 environment (Fig. S21†), which indicates that HER is more favorable than ECR on In–NSC. The constant current electrolysis experiment under CO2 atmosphere (Fig. 4b) also showed that within a wide range of applied current, H2 was the dominant product over In–NSC, accompanied by the generation of a small amount of formate and CO. This indicates that when In SAs existed independently, they were more conducive to the generation of H2 through HER and could not effectively catalyze ECR to formate. In order to exclude the catalytic ability of the NSC support itself, we further compared the H2 production capacity of NSC and In–NSC. LSV tests (Fig. S22†) showed that In–NSC achieved a higher current density than NSC under Ar atmosphere, which indicates that the In SAs were indeed conducive to the dissociation of H2O. A certain water dissociation ability is beneficial to improve the coverage of hydrogen on the surface of the catalytic active sites in alkaline environment and thus promote the reaction.21,22
In order to further highlight the importance of the water dissociation ability provided by In SAs in the system of In–NSC/NPs catalyzed ECR to produce formate, the pH effect of the electrolyte solutions was investigated. Since the three electrolytes K2HPO4, KHCO3 and K2SO4 have been found to provide a correspondingly increased local pH at the electrode/electrolyte interface,34 we selected these three solutions to study the effect of local pH on the In–NSC/NPs and In–NSC/NPs-0.3 that have moderate and high ratios of NPs with SAs, respectively. Fig. 4c shows that with the increased local pH, the product formation rate (PR) of formate both increased in In–NSC/NPs and In–NSC/NPs-0.3, which indicates that higher local pH is beneficial to ECR, which is consistent with previous reports.35 It is worth noting that the ratio of PR on In–NSC/NPs and In–NSC/NPs-0.3 gradually increased with the increase of local pH, which indicated that In–NSC/NPs was more reactive at high local pH. This experiment shows that the In SAs could enhance formate formation by accelerating water dissociation, which becomes more difficult at a higher pH.
In situ attenuated total reflection-surface-enhanced IR absorption spectroscopy (ATR-SEIRAS) spectra were collected to capture the evolution of reaction intermediates during ECR from 0 to −0.8 V (vs. RHE) over In–NSC/NPs and In–NSC to help us understand the reaction process. As shown in Fig. 4d, the peaks appearing at 3350 and 1650 cm−1 are ascribed to water molecules.36 We assign the peak at 2360 cm−1 to CO2 molecules, and the peak at 1250 cm−1 can be ascribed to the formation of *CO2−,37 which is the first-step adsorption of CO2 molecules during ECR. The peak at 1400 cm−1 is due to the asymmetric stretching vibration of *OCHO, which is a key intermediate generated from hydrogenation of *CO2−.37 Notably, the presence of *OCHO is particularly evident starting from 0 V (vs. RHE), confirming In–NSC/NPs could promote the formation of formate at a lower overpotential. The *OCHO peak intensity increased first and then stabilized, indicating that the more negative potential first helps to increase the rate of *OCHO formation and then to the continuous conversion in the subsequent hydrogenation step (*OCHO + e− + *H → HCOOH).36 By contrast, in ATR-SEIRAS of In–NSC (Fig. S23†), no peak corresponding to the ECR hydrogenation intermediate was observed with the increase in applied potential, indicating that In SAs could not effectively catalyze ECR to formate when they exist independently, which is consistent with the catalytic test results.
Density functional theory (DFT) calculations were employed to investigate the ECR on In–NSC/NPs at the molecular level. A configuration in which In–N3S and Inx dual active sites coexist according to prior literature23,38 was adopted for modeling of catalytic active sites of In–NSC/NPs. First, the Gibbs free energy of the reduction intermediate adsorbed on the catalyst surface in the ECR to HCOOH and ECR to CO reaction paths was calculated. As presented in Fig. 5a, in the ECR to HCOOH reaction path, the potential-determining step (PDS) for both In-NSC and In-NSC/NPs is the second step of proton/electron pair transfer (*OCHO + *H+ + e− → *HCOOH). It can be seen that with the introduction of NP sites, the thermodynamic energy barrier required for the PDS on In–NSC/NPs was greatly lowered to be only 0.218 eV. This indicates that the In NPs are conducive to the conversion of CO2 to HCOOH. In the ECR to CO reaction pathway, the PDS for both In–NSC and In–NSC/NPs is the first step of proton/electron pair transfer, both of which require a high reaction energy barrier. This implies that neither In–NSC nor In–NSC/NPs favors the ECR to CO, consistent with the above experimental results. The hydrogen evolution ability of In–NSC and In–NSC/NPs was further calculated, as shown in Fig. 5b. We first calculated the adsorption energy of *H+ on the NP and NSC site of In–NSC/NPs, which was 0.748 and 0.241 eV, respectively, indicating that NSC is more conducive to stabilizing *H+. Therefore, HER was assumed to occur on the NSC site of In–NSC/NPs. The limiting potential differences between ECR to HCOOH, ECR to CO and HER (represented as UL(HCOOH)–UL(CO) and UL(HCOOH)–UL(H2), respectively) were used as metrics to describe the ECR selectivity. Fig. S24† shows the markedly higher selectivity of In–NSC/NPs toward HCOOH over CO or HER than In–NSC.
 | ||
| Fig. 5 (a) The Gibbs free energy diagrams for CO2 to HCOOH and CO, and (b) HER on In-NSC and In-NSC/NPs. The balls in grey, blue, yellow, and brown represent carbon, nitrogen, sulfur, and indium, respectively. | ||
Through the above characterization, experimental analysis, and theoretical calculations, we proposed a possible synergistic catalytic mechanism between In SAs and NPs, as shown in the Fig. 6. Initially, In NPs adsorb and activate CO2 to generate *CO2− intermediate. At the same time, H2O dissociates on In SAs to generate *H, which diffuses to the surface of In NPs, increasing the proton concentration and combining with *CO2− to form *OCHO. As the concentration of *H and *OCHO is enriched, the second step of hydrogenation of *OCHO is promoted, resulting in the production of HCOOH, which diffuses into the reaction system to complete the catalytic cycle.
 | ||
| Fig. 6 Possible ECR to formate mechanism diagram showing the synergy between In NPs and SAs. | ||
In order to eliminate solution resistance, improve energy utilization efficiency, and overcome the damage to the GDE structure caused by electrolyte flooding, a zero-gap membrane-electrode assembly (MEA) was used to further study the performance of In–NSC/NPs. The reaction device is shown in Fig. S25 and S26.† Moistened CO2 was introduced into the cathode side, and 50 mL of 1 M KOH was circulated as the anode electrolyte. The reaction results (Fig. 7a) showed that within the cell voltage range of 2.4–3 V, the formate FEs were maintained above 90%, indicating In–NSC/NPs could effectively catalyze ECR to formate in a wide potential window, and at 2.9 V, the formate FE reached a maximum value of 98.7%, with a current density of 570 mA cm−2. In addition, several sets of ECR application indicators were evaluated: Energy conversion efficiency (ECE) refers to the percentage of electrical energy stored in the target product to the total electrical energy input (Fig. S27†). The result shows that the ECE was maintained above 44% in the cell voltage range of 2.4–3 V, which indicates that electrical energy was effectively converted into formate production in a wide potential range. Single-pass carbon conversion efficiency (SPCE) refers to the proportion of reactant CO2 actually converted into target products. The result showed that under a CO2 flow rate of 40 mL min−1, the SPCE could reach a maximum of 23.2% under 2.9 V, correspondingly the PR of formate was 10.5 mmol cm−2 h−1 (Fig. 7b). Compared to a recently reported similar system, the In–NSC/NPs maintained a leading performance (Table S3†).
 | ||
| Fig. 7 (a) FEs of products and the total current densities at different applied potentials. The data are represented as mean ± standard deviation of three independent measurements. (b) SPCEs and PRs at different applied potentials. (c) LSV curves of ECR//GOR and ECR//OER systems. (d) Comparison of the PR of ECR//OER and ECR//GOR systems. | ||
At present, the anode half reaction of ECR coupling is an OER, in which the OER has slow kinetics and the added value of the product O2 is low. In order to further improve the energy efficiency and increase the enrichment rate of formate, replacing OER with the thermodynamically more favorable GOR is an attractive strategy (Fig. S28†). NiCo2O4 was synthesized as an efficient GOR catalyst to match In–NSC/NPs for construction of a dual-electrode MEA reactor for the simultaneous production of formate on the cathode and anode sides. The LSV curves of ECR//GOR compared to ECR//OER showed that in the potential range of 1.6–2.5 V (Fig. 7c), the ECR//GOR system has a higher onset current and a lower cell voltage than the ECR//OER system. When the total current density was 110 mA cm−2, the cell voltage of ECR//GOR system was reduced by 251 mV compared to ECR//OER. In addition, when electrolyzed under constant current (Fig. S29†), the cell voltage decreased significantly before and after the addition of 0.2 M glycerol to the anolyte, which further proves the thermodynamic advantage of GOR over OER. Impressively, the formate generation rate increased by 2.1 times compared to the ECR//OER system, reaching 23.2 mmol cm−2 h−1 under 2.9 V, showing a high potential for application of electrochemical conversion of CO2 and glycerol to synthesize formate.
Conclusions
In summary, we have reported a catalyst concept where In NPs and SAs coexist to promote ECR to product formate. In this catalytic system, In NPs dominate the hydrogenation process of CO2, while In SAs promote the dissociation of H2O to provide sufficient protons that migrate to the surface of In NPs to accelerate the reaction. Specifically, in the flow cell, In–NSC/NPs achieved almost 100% formate FE under 0.6 A cm−2 constant current electrolysis. Even at a current density of 1.2 A cm−2, the formate FE still remained at 92% and the reduction potential was as low as −1.32 V (vs. RHE). Particularly, when using the MEA, in the cell voltage range of 2.4–3 V, In–NSC/NPs maintained a formate FE above 90%. The SPCE reached a maximum of 23.2% at 2.9 V and the formate FE approached 98.7%. The partial current density was 563 mA cm−2, the ECE was 48.7%, and the formate generation rate was up to 10.5 mmol cm−2 h−1. By coupling GOR at the anode, the current density and formate generation rate of the ECR//GOR system were further improved compared with the ECR//OER system at 2.9 V, reaching 636 mA cm−2 and 23.2 mmol cm−2 h−1, respectively. Our study provided a high performance ECR electrocatalyst, and by coupling GOR at the anode, the generation rate of formate was greatly improved in an integrated MEA reactor.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Y. C. wrote the draft. Y. C. and J. Y. prepared samples and conducted electrocatalytic tests. J. C. and Y. J. performed DFT calculations. F. L. Y. C. and X. Z. conducted characterizations including XRD, XPS, and SEM, etc. S. H. and A. W. R. made STEM measurements. X. T. supported the project and revised the manuscript. Z. S. supervised the project and co-wrote and revised the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors of this manuscript have no conflicts of interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFC2105900), National Natural Science Foundation of China (22372007 and 21972010), and IITP (RS-2021-II211343) of the Korean government.Notes and references
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. R. Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 Search PubMed.
- L. Zhang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 Search PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 Search PubMed.
- Y. Hori, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Y. Zhang, S. Liu, N. Ji, L. Wei, Q. Liang, J. Li, Z. Tian, J. Su and Q. Chen, J. Mater. Chem. A, 2024, 12, 7528–7535 Search PubMed.
- D. Yang, Q. Zhu, X. Sun, C. Chen, W. Guo, G. Yang and B. Han, Angew. Chem., Int. Ed., 2019, 59, 2354–2359 CrossRef PubMed.
- J. Zhou, L. Li, H. Ren, H. Wang, Y. Li, K. Liu, L. Huang, X. Yang, Z. Hao, Y. Zhang, Z. Wang, X. Wang, J. Ding, Y. Ji, L. Wang and H. Liang, Inorg. Chem. Front., 2024, 11, 1703–1709 Search PubMed.
- Z. Li, D. He, X. Yan, S. Dai, S. Younan, Z. Ke, X. Pan, X. Xiao, H. Wu and J. Gu, Angew. Chem., Int. Ed., 2020, 59, 18572–18577 Search PubMed.
- X. Zhao, M. Huang, B. Deng, K. Li, F. Li and F. Dong, Chem. Eng. J., 2022, 437, 135114 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- F. A.-P. A. A. Peterson, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 Search PubMed.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 Search PubMed.
- M. Wang, S. Liu, B. Chen, M. Huang and C. Peng, Chem. Eng. J., 2023, 451, 139056 Search PubMed.
- J. Fu, K. Liu, H. Li, J. Hu and M. Liu, Environ. Chem. Lett., 2021, 20, 243–262 CrossRef.
- S. G. Han, D. D. Ma and Q. L. Zhu, Small Methods, 2021, 5, 2100102 Search PubMed.
- X.-L. Lu, X. Rong, C. Zhang and T.-B. Lu, J. Mater. Chem. A, 2020, 8, 10695–10708 Search PubMed.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- W. Sun, S. Liu, H. Sun, H. Hu, J. Li, L. Wei, Z. Tian, Q. Chen, J. Su and L. Chen, Adv. Energy Mater., 2025, 2500283 Search PubMed.
- D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 Search PubMed.
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Nat. Comm., 2023, 14, 4615 Search PubMed.
- X. Wang, X. Sang, C. L. Dong, S. Yao, L. Shuai, J. Lu, B. Yang, Z. Li, L. Lei, M. Qiu, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2021, 60, 11959–11965 Search PubMed.
- M. Li, Y. Hu, W. Fang, S. Xin, Y. Wu, Y. Cao, W. Cui, Z. Li and H. Zhao, Chem. Eng. J., 2024, 480, 148014 CrossRef CAS.
- Q. Zhao, Y. Wang, M. Li, S. Zhu, T. Li, J. Yang, T. Lin, E. P. Delmo, Y. Wang, J. Jang, M. Gu and M. Shao, SmartMat, 2022, 3, 183–193 CrossRef CAS.
- B. Sun, X. Wang, Z. Li, H. Liu, W. Jiang, K. Song, Z. Wang, P. Wang, Y. Liu, Z. Zheng, Y. Dai, B. B. Huang and H. F. Cheng, Chem Catal., 2024, 4, 100862 CrossRef CAS.
- J. Bi, P. Li, J. Liu, Y. Wang, X. Song, X. Kang, X. Sun, Q. Zhu and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202307612 Search PubMed.
- X. Yang, J. Cheng, H. Lv, X. Yang, L. Ding, Y. Xu, K. Zhang, W. Sun and J. Zhou, Chem. Eng. J., 2022, 450, 137950 CrossRef CAS.
- F. Pan, B. Li, E. Sarnello, S. Hwang, Y. Gang, X. Feng, X. Xiang, N. M. Adli, T. Li, D. Su, G. Wu, G. Wang and Y. Li, Nano Energy, 2020, 68, 104384 Search PubMed.
- G. Xu, J. Han, B. Ding, P. Nie, J. Pan, H. Dou, H. Li and X. Zhang, Green Chem., 2015, 17, 1668–1674 Search PubMed.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2014, 54, 2131–2136 Search PubMed.
- W. Li, C. Yu, X. Tan, Y. Ren, Y. Zhang, S. Cui, Y. Yang and J. Qiu, ACS Catal., 2024, 14, 8050–8061 Search PubMed.
- C. W. Lee, N. H. Cho, K. D. Yang and K. T. Nam, ChemElectroChem, 2017, 4, 2130–2136 Search PubMed.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 Search PubMed.
- J. Zhou, J. Lin, X. Huang, Y. Zhou, Y. Chen, J. Xia, H. Wang, Y. Xie, H. Yu, J. Lei, D. Wu, F. Liu, Q. Fu, Q. Zeng, C.-H. Hsu, C. Yang, L. Lu, T. Yu, Z. Shen, H. Lin, B. I. Yakobson, Q. Liu, K. Suenaga, G. Liu and Z. Liu, Nature, 2018, 556, 355–359 Search PubMed.
- C. Wang, Z. Lv, Y. Liu, R. Liu, C. Sun, J. Wang, L. Li, X. Liu, X. Feng, W. Yang and B. Wang, Angew. Chem., Int. Ed., 2024, 63, e202404015 Search PubMed.
- Y. Li, E. P. Delmo, G. Hou, X. Cui, M. Zhao, Z. Tian, Y. Zhang and M. Shao, Angew. Chem., Int. Ed., 2023, 62, e202404015 Search PubMed.
- E. Zhang, L. Tao, J. An, J. Zhang, L. Meng, X. Zheng, Y. Wang, N. Li, S. Du, J. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202117347 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01580d |
| This journal is © The Royal Society of Chemistry 2025 |