
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA design strategy for single-molecule magnet materials with fullerene confinement-induced unpaired f-electrons†
Xiao-Kun
Zhao
 ab,
Jing
Zhao
a,
Shi-Ru
Wei
b,
Yun-Ze
Qiu
b,
Yang
He
b,
Han-Shi
Hu
*b and
Jun
Li
*abc
ab,
Jing
Zhao
a,
Shi-Ru
Wei
b,
Yun-Ze
Qiu
b,
Yang
He
b,
Han-Shi
Hu
*b and
Jun
Li
*abc
aDepartment of Chemistry, Guangdong Provincial Key Laboratory of Catalytic Chemistry, Southern University of Science and Technology, Shenzhen 518055, China
bDepartment of Chemistry, Engineering Research Center of Advanced Rare-Earth Materials of Ministry of Education, Tsinghua University, Beijing 100084, China. E-mail: hshu@mail.tsinghua.edu.cn; junli@mail.tsinghua.edu.cn
cFundamental Science Center of Rare Earths, Ganjiang Innovation Academy, Chinese Academy of Sciences, Ganzhou 341000, China
First published on 28th May 2025
Abstract
Endohedral metallofullerenes (EMFs) are promising platforms for single-molecule magnets (SMMs) due to their internal cavities, which enable effective coupling of magnetically anisotropic metal ions through direct covalent bonding. However, the practical application of EMF-SMMs remains challenging, particularly in the robust assembly of the cage structures. In this study, we propose a strategy for designing two-dimensional (2D) diactinide EMF-SMM materials (M2@C60-2D, M = U, Th) by doping thorium and uranium into a 2D quasi-hexagonal-phase fullerene (qHPC60) monolayer. Ab initio molecular dynamics (AIMD) simulations and density functional theory (DFT) calculations confirm the thermal and thermodynamic stability of these materials, suggesting their synthetic feasibility. Further investigations show that fullerene confinement tends to eliminate the traditional Lewis-type electron-pair bonds in neutral M2 dimers and induces multiple single-electron M–M bonding in M2@C60, thus facilitating the enhanced magnetic properties of 2D EMF monolayers. These findings provide valuable insights for designing space-confined metal diatomic systems for magnetic applications.
Introduction
Single-molecule magnets (SMMs) have attracted significant research interest since the discovery of {Mn12} clusters, owing to their ability to store and process quantum information at the molecular level.1–3 By utilizing fullerene cages with confined internal nanospace, endohedral metallofullerenes (EMFs) have emerged as promising SMM candidates.4–7 Dimetallofullerenes (di-EMFs), in particular, exhibit high SMM blocking temperatures and large magnetic anisotropy barriers, making them suitable for applications in quantum information processing and information storage.8–11In di-EMFs of C60, direct single-electron metal–metal (M–M) bonds can be formed, which enable strong magnetic coupling between the two metal ions.12,13 Recently, Long et al. reported record SMMs in mixed-valence dilanthanide complexes (CpiPr5)2Ln2I3 (Ln = Gd, Tb, Dy, Ho), featuring a single-electron Ln–Ln (d–d)σ bond.14,15 These findings demonstrated that coupling two magnetic metal ions via direct bonding is an effective strategy for designing high-performance SMMs.16,17 However, while the 4f orbitals in lanthanides are radially too contracted to form effective chemical bonds, leading to extremely weak direct 4f–4f coupling, the more radially extended and chemically accessible 5f orbitals in actinides make actinide-based EMFs intriguing for exploring unique bonding motifs to achieve stronger magnetic coupling.18,19
In 2007, Wu et al. proposed six-fold ferromagnetic U–U bonding inside a U2@C60 cage.20 Gagliardi and colleagues later suggested that these U–U bonds might be artifacts of confinement within the small C60 cavity.21 Similarly, the Straka group described the ferromagnetic U–U bonds in U2@C80 as “unwilling bonding”.22 These theoretical insights spurred experimental investigations into diactinide EMFs. In 2018, Zhang et al. provided the first experimental evidence for actinide–actinide bonding in the diuranium endofullerene U2@C80.19 Shortly thereafter, dithorium endofullerene Th2@C80 was also prepared.23 Notably, M–M bonding in diactinide EMFs is significantly influenced by the size of the carbon cages, resulting in unpaired single-electron M–M bonds in the C60 cage, which further enhance the magnetism of the diactinide EMFs.24
The cage assembly of SMMs is crucial for their practical application in information storage. Current approaches include attaching SMMs to conducting surfaces to form 2D molecular assemblies25–27 and incorporating SMMs into metal–organic frameworks (MOFs) to create 3D molecular structures.28–30 Recently, the successful synthesis of a two-dimensional quasi-hexagonal-phase fullerene (2D qHPC60) material has demonstrated distinct electronic structures, showing great potential for applications in physical devices and chemical catalysis.31–34 Given the single-electron M–M bonds in diactinide EMFs, encapsulating two actinide atoms within a 2D qHPC60 monolayer provides a promising approach for developing EMF-SMM materials.
In this work, we have performed a theoretical study on a 2D diactinide EMF monolayer with diactinide doped in the 2D qHPC60 monolayer. Our theoretical analyses confirm that these 2D EMF monolayers are thermodynamically stable, indicating their feasibility for experimental synthesis under suitable conditions. Quantum-chemical studies with DFT and CASSCF calculations reveal that fullerene confinement induces single-electron M–M bonding in the encapsulated M2 dimer, contributing to the magnetism of the 2D fullerene monolayer. It is found that U2@C60-2D exhibits an interfullerene antiferromagnetic (AFM) singlet ground state, attributed to the U2–C60⋯C60–U2 super-superexchange interactions along the interfullerene [2 + 2] cycloaddition bonds. In contrast, Th2@C60-2D has an interfullerene ferromagnetic (FM) ground state. Our findings present a novel strategy for designing space-confined EMF-SMM materials that could provide insights for designing high-density information storage, quantum computing, and molecular spintronic devices.
Results and discussion
Geometric structure of 2D diactinide EMF monolayers: M2@C60-2D (M = Th, U)
The experimentally synthesized 2D qHPC60 monolayer features each C60 molecule surrounded by six neighbouring cages, forming a hexagonal lattice with a space group of Pmna. Among these neighbours, four are linked by interfullerene C–C single bonds and two are connected through interfullerene [2 + 2] cycloaddition bonds. The interfullerene bonds reduce the local symmetry of C60 from Ih to C2h in the 2D qHPC60 monolayer. Therefore, the 20 hexagonal rings of C60 can be categorized into six distinct types (positions 1 to 6 in Scheme 1). Both experimental and theoretical studies have revealed that encapsulated diactinide dimers in EMFs tend to be sandwiched between two hexagonal rings.19–21,23 We initially explored the encapsulation sites of actinide dimers within distorted C2h C60 cages by constructing six different diuranium EMF models based on U2@C66H20. In these models, the U2 dimer was placed at the centroids of six distinct hexagonal rings (positions 1 to 6). Geometry optimizations identified position 1 as the most favourable (Fig. S1†), where the U2 dimer is encapsulated while preserving the C2h symmetry.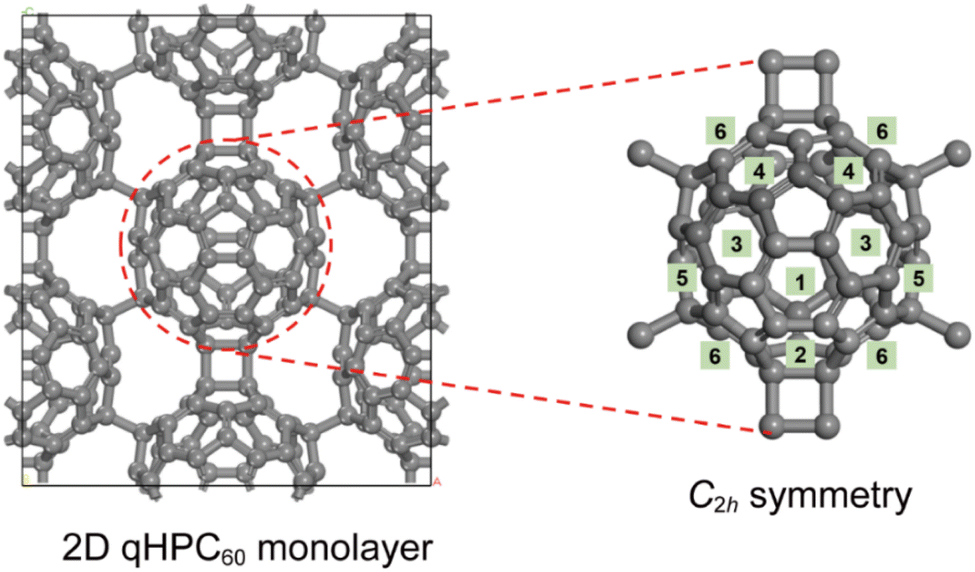 | ||
| Scheme 1 The geometric structure of the 2D qHPC60 monolayer and its constituent C60 unit with local C2h symmetry. The numbers indicate the six distinct types of hexagonal rings found within the C2h-symmetric C60 unit. | ||
We then constructed the 2D diactinide EMF monolayers with M2 dimers located at position 1. The calculated geometric structure shows that both U2@C60-2D and Th2@C60-2D monolayers share the same structure and symmetry as the 2D qHPC60 monolayer (Fig. 1). The lattice and structural parameters are compared to those of the 2D qHPC60 monolayer (Table 1). The lattice lengths of the a-axis and b-axis are 15.90 and 9.24 Å for the U2@C60-2D monolayer, and 15.80 and 9.20 Å for the Th2@C60-2D monolayer, respectively, showing slight elongation along the b-axis and shrinkage along the a-axis when compared to the 2D qHPC60 monolayer. Furthermore, the bond lengths of interfullerene C–C single bonds and [2 + 2] cycloaddition bonds remain virtually unchanged in both the pristine 2D fullerene monolayer and M2@C60-2D. The calculated M–M bond distances are 2.70 Å for U–U and 2.86 Å for Th–Th, which are comparable to the sum of the Pyykkö covalent double-bond radii of 2.68 Å for U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) U and of 2.86 Å for ThTh.35 Notably, the space-confined Th–Th distance is notably shorter than that reported for thorium systems such as the tri-thorium cluster (3.99 Å)36 and the dithorium complex (4.01 Å).37 These relatively short M–M bond lengths in our system indicate strong bonding interactions between two endohedral actinide atoms under the structure confinement of the fullerene cage. In addition, the M–C contacts range from 2.46 to 2.48 Å for U–C bonds and from 2.48 to 2.50 Å for Th–C bonds, suggesting strong bonding interactions between the encapsulated M2 dimers and the adjacent carbon atoms of the C60 cages.
U and of 2.86 Å for ThTh.35 Notably, the space-confined Th–Th distance is notably shorter than that reported for thorium systems such as the tri-thorium cluster (3.99 Å)36 and the dithorium complex (4.01 Å).37 These relatively short M–M bond lengths in our system indicate strong bonding interactions between two endohedral actinide atoms under the structure confinement of the fullerene cage. In addition, the M–C contacts range from 2.46 to 2.48 Å for U–C bonds and from 2.48 to 2.50 Å for Th–C bonds, suggesting strong bonding interactions between the encapsulated M2 dimers and the adjacent carbon atoms of the C60 cages.
 | ||
Fig. 1 Top view and side view of the 2D diactinide EMF monolayer. (a) U2@C60-2D monolayer, and (b) Th2@C60-2D monolayer. C1 and  represent the C atoms of the interfullerene [2 + 2] cycloaddition bond. C2 and represent the C atoms of the interfullerene [2 + 2] cycloaddition bond. C2 and  represent the C atoms of the interfullerene C–C single bond. represent the C atoms of the interfullerene C–C single bond. | ||
| 2D qHPC60 | U2@C60-2D | Th2@C60-2D | |
|---|---|---|---|
| Lattice parameters | |||
| a (Å) | 15.92 | 15.90 | 15.80 |
| b (Å) | 9.16 | 9.24 | 9.20 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Interfullerene bond | |||
| C–C single bonds (Å) | 1.61 | 1.59 | 1.60 |
| [2 + 2] cycloaddition bonds (Å) | 1.61 | 1.62 | 1.61 |
| M–M bond (Å) | 2.70 | 2.86 | |
| M–C bond (Å) | 2.46–2.48 | 2.48–2.50 | |
Stability analysis
The phonon spectrum that describes lattice vibration in crystals is relevant to its stability with respect to structural phase transitions or other distortions. Our 2D diactinide EMF monolayers do not show imaginary phonon modes despite the numerical noise, indicating dynamic stability of these monolayers (Fig. S2 and S3†). To investigate the thermal stability, we conducted temperature-accelerated AIMD simulations at various high temperatures, each for 10 ps, to enhance the atomic mobility and explore a larger configurational space in a shorter simulation time. The temperatures used are T = 1400, 1800, 2200, and 2600 K for U2@C60-2D and T = 600, 1000, 1400, and 1800 K for Th2@C60-2D. The AIMD simulations reveal that U2@C60-2D exhibits greater thermal stability. The hexagonal framework of U2@C60-2D remains intact up to 2200 K (Fig. S4†), whereas Th2@C60-2D is stable only up to 1400 K (Fig. S5†). The 2D fullerene framework breaks down into single C60 molecules at 2600 K for U2@C60-2D and at 1800 K for Th2@C60-2D. These results indicate that encapsulating the U2 dimer enhances the stability of the 2D fullerene monolayer, while Th2 dimer doping has little influence on stability.33The stability of these fullerene monolayers was further elucidated through crystal orbital Hamilton population (COHP) analysis, which reconstructs orbital-resolved wavefunctions via projecting delocalized plane-wave basis functions into localized atomic-like basis sets.38 This analysis aims at describing the interaction between two orbitals centred at neighbouring atoms by considering the product of their corresponding Hamiltonian matrix element and the density-of-states matrix, thus measuring the bonding contribution and bonding strength between neighbouring atoms upon integrating the COHP below the Fermi level. The integrated projected COHP (IpCOHP) of interfullerene bonds in the 2D qHPC60 monolayer is about −7.90 eV (Table S1†). In contrast, the IpCOHP values of the [2 + 2] cycloaddition bonds and C–C single bonds in U2@C60-2D are up to −8.46 and −8.49 eV, respectively, compared to −7.82 and −8.03 eV in Th2@C60-2D. Consequently, the U2@C60-2D monolayer is more stable than the pristine 2D qHPC60 monolayer. Furthermore, to quantitatively evaluate changes in the 2D fullerene framework during AIMD simulations, we examined the bond length variations of interfullerene bonds  (four-membered ring bond) and
(four-membered ring bond) and  (single bond). The bond lengths of
(single bond). The bond lengths of 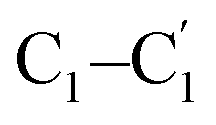 and
and  in U2@C60-2D fluctuate around the equilibrium bond length in simulations at temperatures below 2200 K (Fig. S8†). However, at 2600 K, these bond lengths experience significant changes after 4 ps of simulation, indicating the collapse of the 2D hexagonal structure. Similarly, the Th2@C60-2D monolayer undergoes fission after 5 ps of simulations at 1800 K (Fig. S9†). Additionally, the U–U and Th–Th bonds oscillate around their equilibrium bond length even after the 2D EMF monolayers break down into individual EMF cages.
in U2@C60-2D fluctuate around the equilibrium bond length in simulations at temperatures below 2200 K (Fig. S8†). However, at 2600 K, these bond lengths experience significant changes after 4 ps of simulation, indicating the collapse of the 2D hexagonal structure. Similarly, the Th2@C60-2D monolayer undergoes fission after 5 ps of simulations at 1800 K (Fig. S9†). Additionally, the U–U and Th–Th bonds oscillate around their equilibrium bond length even after the 2D EMF monolayers break down into individual EMF cages.
To verify the thermodynamic feasibility of synthesizing these 2D diactinide EMF monolayers, we computed the Gibbs free energies from the reactants (2D qHPC60 monolayer and bulk uranium or thorium metal). The proposed reaction is shown in eqn (1):
| 2M (bulk) + 2D qHPC60 → M2@C60-2D | (1) |
The Gibbs free energy of the reactants, corrected for entropy and zero-point energies, is provided in Table S2.† The Gibbs free energy (ΔG300 K) at 300 K was calculated using the formula ΔG300 K = G(M2@C60-2D)300 K − 2G(Mbulk)300 K − G(2D qHPC60)300 K. The calculated ΔG300 K is negative (−3.02 kcal mol−1) for the formation of the U2@C60-2D monolayer, but positive (+14.8 kcal mol−1) for the Th2@C60-2D monolayer, indicating the experimental feasibility of the U2@C60-2D monolayer under ambient conditions. Despite the endergonic feature, the possibility of experimental preparation of the Th2@C60-2D monolayer cannot be excluded because it remains metastable.
Single-electron actinide–actinide bonds in 2D fullerene
DFT calculations reveal that the actinides of the encapsulated M2 dimers exhibit a ferromagnetic state (Table S3†), with calculated magnetic moments 2.75 μB for uranium (U) and 0.68 μB for thorium (Th) atoms. We used Bader charge analysis to assist the determination of the oxidation states of encapsulated actinides by comparing with reported EMFs, including U2@C80,19 U2C2@C80,39 Th2@C80,23 and ThC2@C82.40 Previous experimental and quantum-chemical calculation results indicate that U has a formal oxidation state of +4 in U2C2@C80 and +3 in U2@C80. The Bader charges for U4+ and U3+ in these EMFs are +2.00 and +1.54, respectively (Table S4†). In U2@C60-2D, the Bader charge of U is +1.45, which is close to +1.54 observed for U3+ in U2@C80, suggesting a +3 formal oxidation state for encapsulated U atoms. Similarly, the Bader charge for Th in Th2@C60-2D is +1.64, comparable to the +1.68 in Th2@C80, indicating a +3 formal oxidation state for Th atoms. Thus, the valence state of the M2@C60-2D monolayer can be described as a (M2)6+@(C60-2D)6− assignment.As depicted in Fig. 2(a), the U2@C60-2D monolayer has an indirect energy gap of 0.31 eV, which is significantly smaller than the 1.6 eV band gap of the 2D qHPC60 monolayer. This reduction is attributed to the charge transfer from the U2 dimer to the C60 cage. In the band structure, several red flat bands below the Fermi level are ascribed to the f-bonding orbitals of the U2 dimer, as confirmed by the projected density of states (pDOS). The valence band minimum (VBM) results from interactions between the U2 dimer and the C60 cage, with real-space wavefunctions indicating φ-type non-bonding orbitals of U2 interacting with the C60 cage.41
 | ||
| Fig. 2 (a) Band structure and projected density of states (pDOS) at the HSE06 level for the U2@C60-2D monolayer. The high-symmetry points in the first Brillouin zone of reciprocal space are denoted as G (0, 0, 0), X (0.5, 0, 0), S (0.5, 0.5, 0) and Y (0, 0.5, 0). The red curves represent contributions from uranium (U), while the blue curves indicate contributions from carbon (C) atoms. (b) Spin-up real-space wave functions for the bands at the G point illustrate the bonding patterns of the U2 dimer. Note that only the real space wave functions of a single EMF cage are shown due to the interfullerene AFM state of the U2@C60-2D monolayer. | ||
The f-bonding orbitals are further revealed by the real-space wavefunctions shown in Fig. 2(b). Specifically, VBM-5 and VBM-9 correspond to U(5f) δ-bonding orbitals, VBM-8 corresponds to U(5f) σ-bonding orbitals, and VBM-10 and VBM-13 correspond to U(5f) π-bonding orbitals. Therefore, there are five single-electron U–U bonds formed within EMF cages. The calculation of the U2@C68H20 molecular model further confirms the five single-electron U–U bonds, consistent with the solids model calculation results (Fig. S10 and S11†). Additionally, the calculated band structure and projected DOS reveal Th–Th bonding in the Th2@C60-2D monolayer. As shown in Fig. S12,† spin-up real-space wavefunctions show two nearly degenerate single-electron Th–Th π bonds in EMF cages. The orbital interaction diagram for the Th2@C68H20 molecular model confirms two degenerate Th–Th π bonds (40 au and 50 bu) in the encapsulated Th2 dimer as well (Fig. S13 and S14†).
Natural localized molecular orbitals (NLMOs) from one-electron reduced density matrix analysis provide insight into the nature of actinide–actinide bonding in diactinide EMF cages. Fig. 3 shows five single-electron NLMOs, comprising one σ bond, two π bonds, and two δ bonds, primarily contributed by U(5f) orbitals (with minor 6d character) within the U2@C68H20 cage. In contrast, the Th2@C68H20 cage forms only two π bonds, with Th(5f) and Th(6d) orbitals contributing almost equally. To verify the multiconfigurational character of single-electron actinide–actinide bonds, we performed complete-active-space self-consistent field (CASSCF) calculations. As shown in Fig. S15,† the U–U bonding can be interpreted as σ1π2δ2f1, with an effective bond order (EBO) of 1.90. The Th–Th bonding of the Th2@C68H20 molecule model consists of two single-electron π bonds with an EBO of 0.98 (Fig. S16†). As a result, the CASSCF calculations further confirm the single-electron M–M bonding formed in the 2D EMF monolayers. Additionally, relativistic effects significantly influence heavier elements and their compounds, particularly in the bonding patterns of diatomic molecules.42 The effect of spin–orbit coupling (SOC) was examined by comparing it with the scalar relativistic (SR) states to understand the nature of the U–U and Th–Th bonding in the diactinide EMFs. As shown in Fig. S17 and S18,† the SOC effect does not significantly alter the U–U and Th–Th bonding scheme compared to the SR results. Therefore, the SR results can accurately describe bonding motifs of U–U and Th–Th.
 | ||
| Fig. 3 Natural localized molecular orbitals (NLMOs). (a) Five single-electron NLMOs of the U2 dimer in the U2@C68H20 molecular model, and (b) two single-electron NLMOs of the Th2 dimer in the Th2@C68H20 molecular model. The hydrogen atoms are omitted for clarity. (Isovalue = 0.04 a.u.) | ||
The role of fullerene confinement in single-electron bond formation
In addition to the ferromagnetic state of encapsulated M2 dimer observed in individual EMF cages in solids (Table S3†), cluster models also show a preference for high-spin states. Specifically, U2@C68H20 adopts a septet ground state, while Th2@C68H20 favours a triplet ground state (Table S5†), both exhibiting ferromagnetic single-electron bonds within the EMFs. To further investigate the mechanism behind the formation of these single-electron bonds, simplified U2@C60 and Th2@C60 models with D3d symmetry were used.20,24As illustrated in Fig. 4, the neutral Th2 molecule exhibits a quadruple bonding scenario consisting of (7sσg)2(6dπu)4(6dσg)1(6dδg)1.43 However, similar to the case of ligand-destabilized dx2−y2 antibonding orbitals in the [Re2Cl8]2− dianion of Cotton's multiple bonding model,44 the quadruple bonds in Th2 are disrupted when encapsulated inside the fullerene due to strong Th–fullerene interactions. Furthermore, the hybridization of the 6d and 5f orbitals in Th3+ is highly effective because of the small 6d–5f energy gap (Fig. S20†), allowing the 5fπ and 6dπ bonding orbitals to hybridize and form two more stable (f–d)π bonding orbitals (see details in Fig. S21†). However, the encapsulated Th26+ dimer contains only two electrons, resulting in the formation of two degenerate single-electron Th–Th (f–d)π bonds in the Th2@C60 molecule.
 | ||
| Fig. 4 Orbital interaction diagram for Th2@C60 molecules, illustrating the bonding scheme between neutral C60 and the Th2 dimer. The arrows represent the unpaired single-electron bonds of the Th2 dimer, and the dots represent the electron-pair bonds. The orbitals of the neutral Th2 dimer are shown in Fig. S19.† | ||
In the case of the U2@C60 molecule, strong U–fullerene interactions eliminate the (7sσg)2 and (6dπu)4 electron-pair bonds from the valence manifold of neutral U2, leaving only the 5f electrons available for U–U bonding.45 However, the U(5f) orbitals are radially too contracted to form effective Lewis-type electron-pair bonds at the relatively long U–U distance of 2.70 Å. Instead, single-electron bonds are energetically more favourable due to the exchange stabilization, leading to the formation of five single-electron U–U bonds in the U2@C60 molecule (Fig. S22 and S23†).
Additionally, the atomic radial distribution function (RDF) curves in Fig. S24† provide insights into the U–fullerene orbital interactions, revealing a stronger overlap between U(6d) and C(2p) orbitals compared to U(5f) and C(2p) orbitals. Further analysis using principal interacting orbital (PIO) and energy decomposition analysis with the extension of natural orbitals for chemical valence (EDA-NOCV) methods for the encapsulated U2 and C60 fragments show that the U(6d) orbitals play a more significant role in U–fullerene bonding interaction than U(5f) orbitals (Fig. S25 and S26†).
The single-electron M–M bonds within M2@C60 represent a balance between M–M bonding and M–fullerene interactions. The confinement effect of the fullerene is evident in the projected DOS of neutral C60, M2 and M2@C60 molecules (Fig. S27†). The strong electron-pair bonds in neutral M2 dimers, primarily contributed by 7s and 6d orbitals, are shifted to unoccupied states within M2@C60 due to the M2–fullerene orbital interaction with space-confinement. This shift results in the formation of single-electron M–M bonds, which are responsible for introducing magnetism into 2D diactinide EMF monolayers.
The COHP analysis provides valuable insights into the relationships between magnetic states and the bonding characteristics of encapsulated M2 dimers from the perspective of chemical bonding in solids.38 As shown in Fig. S28,† the non-spin-polarized COHP curves indicate that the Fermi levels in antiferromagnetic M2 dimers cross the bonding regions of the M–M COHP curves, leaving many bonding orbitals unoccupied. In contrast, for ferromagnetic M2 dimers, the spin-polarized COHP curves show that the spin-up states occupy lower energy levels, while the spin-down states shift upward. As a result, most of the spin-up bonding states are positioned below the Fermi levels, resulting in strengthened M–M bonding (0.24 eV for U–U and 0.44 eV for Th–Th). Moreover, the single-electron M–M bonding strength, calculated as IpCOHP(spin up) − IpCOHP(spin down), reveals bonding strengths of −3.28 eV for U–U and −2.31 eV for Th–Th.
Magnetism in 2D diactinide EMF monolayers
As the fullerene confinement can significantly affect the bonding between the diactinide metals, the magnetic properties of the solid are dictated by the competition of the fullerene–metal and metal–metal interactions. To further investigate the magnetic properties in these 2D diactinide EMF monolayers, we constructed a supercell model with three different interfullerene magnetic states: ferromagnetic (FM), antiferromagnetic (AFM), and antiferromagnetic/ferromagnetic (AF/FM) states (Fig. S29†). In these magnetic states, the electrons of M2 dimers are ferromagnetic and are represented by a single red arrow, indicating the orientation of the magnetic moment of the unpaired electrons in the encapsulated M2 dimers (displayed in Fig. 5). DFT calculation results, as shown in Table S6,† reveal that the U2@C60-2D monolayer has an interfullerene AFM singlet ground state, which is more stable than the ferromagnetic (FM) state by 36.4 meV per U2 dimer. In contrast, the ground state of the Th2@C60-2D monolayer is the interfullerene FM state. | (2) |
 | ||
| Fig. 5 (a) Magnetic coupling scheme of 2D diactinide EMF monolayers. Schematic presentation of the (b) AFM ground state of U2@C60-2D and (c) FM ground state of the Th2@C60-2D monolayer. (d) Schematic representation of super-superexchange (SSE) interactions in U2@C60-2D and Th2@C60-2D monolayers. The magnetic coupling along the interfullerene [2 + 2] cycloaddition bonds and C–C single bonds is indicated by the blue solid lines (J1) and green dashed lines (J2), respectively. | ||
In addition, the Heisenberg model (see eqn (2)) with FM, AFM, and AF/FM states was applied to calculate the exchange coupling constants along the interfullerene [2 + 2] cycloaddition bonds (J1, represented by the blue solid lines) in Fig. 5, and along interfullerene C–C single bonds (J2, represented by the green dashed lines). In eqn (2), J is the exchange coupling constant, H0 is the nonmagnetic Hamiltonian, and ![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) i and j are the spin momentum at sites i and j, respectively. The exchange coupling constant J1 is calculated to be −16.1 cm−1 and J2 = −0.3 cm−1, leading to an interfullerene AFM ground state within the U2@C60-2D monolayer.46 Since each U2 dimer has six unpaired electrons, a 1D Heisenberg antiferromagnetic chain with S = 3 on encapsulated U2 dimers forms in the U2@C60-2D monolayer due to the relatively small J2 exchange coupling (J2 ≪ J1). In the Th2@C60-2D monolayer, the calculated exchange coupling constants J1 (6.4 cm−1) and J2 (5.4 cm−1) are comparable, leading to an interfullerene ferromagnetic ground state.
i and j are the spin momentum at sites i and j, respectively. The exchange coupling constant J1 is calculated to be −16.1 cm−1 and J2 = −0.3 cm−1, leading to an interfullerene AFM ground state within the U2@C60-2D monolayer.46 Since each U2 dimer has six unpaired electrons, a 1D Heisenberg antiferromagnetic chain with S = 3 on encapsulated U2 dimers forms in the U2@C60-2D monolayer due to the relatively small J2 exchange coupling (J2 ≪ J1). In the Th2@C60-2D monolayer, the calculated exchange coupling constants J1 (6.4 cm−1) and J2 (5.4 cm−1) are comparable, leading to an interfullerene ferromagnetic ground state.
The interfullerene AFM ground state of the U2@C60-2D monolayer is further evaluated using EDA-NOCV for cluster models of the (C66H16)2, (Th2@C66H16)2, and (U2@C66H16)2 dimers, where the C60 cages are connected by [2 + 2] cycloaddition bonds. The orbital interactions constitute approximately 60% of the total interactions between two EMF fragments in molecular dimers, indicating a significant covalent bonding character among these 2D EMF monolayers (see Tables S7–S9† for details). The NOCV deformation densities between two EMF fragments are summarized in Fig. S30.† The third deformation orbital of the (U2@C66H16)2 dimer, highlighted in the red box, reveals that the 5f orbitals of the U2 dimer contribute about 5.0% to the orbital interactions between two U2@C66H16 fragments. In contrast, the EDA-NOCV analysis of the (Th2@C66H16)2 dimer is similar to the (C66H16)2 dimer, showing no f-orbital involvement.
The f-orbital involved interfullerene interaction in the (U2@C66H16)2 dimer suggests a possible pathway for the super-superexchange (SSE) interactions47–49 among encapsulated U2 dimers in different EMF cages, which results in the AFM ground state in the U2@C60-2D monolayer. As illustrated in Fig. 5(d), the unpaired f-orbital electrons (solid arrows) of the U2 dimer can hop to the unoccupied orbitals of the C60 cage (dashed arrows), and then interact with electrons of opposite spin in the adjacent C60 cage, thereby stabilizing the U2@C60-2D monolayer. These U2–C60⋯C60–U2 SSE interactions help explain the AFM ground state of the U2@C60-2D monolayer. In contrast, the Th2@C60-2D monolayer lacks f-orbital mediated interfullerene interactions, resulting in a ferromagnetic ground state.
Conclusions
In summary, we present a strategy for designing EMF-SMM materials by doping diactinide into a 2D qHPC60 monolayer. Through DFT calculations and AIMD simulations, we confirm the dynamic, thermal, and thermodynamic stability of these monolayers, demonstrating their feasibility in experimental preparation. The oxidation states of Th and U in the M2@C60-2D monolayer are assigned as +3, which corresponds to a (M2)6+@(C60-2D)6− charge state. This relative low oxidation states of Th+3 and U+3 are attributed to the low electronegativity of the C60 cage, which disfavours high oxidation states of actinides. Theoretical investigations, including DFT and multi-configurational ab initio CASSCF calculations on molecular models, reveal that fullerene confinement induces single-electron M–M bonds in diactinide EMFs, enhancing the magnetic properties of these 2D monolayers. Further supercell model calculations using periodic DFT reveal that the U2@C60-2D monolayer adopts an interfullerene AFM singlet ground state, driven by strong exchange coupling via U2–C60⋯C60–U2 super-superexchange interactions along the interfullerene [2 + 2] cycloaddition bonds. In contrast, the Th2@C60-2D monolayer displays an interfullerene FM ground state. This study expands our understanding of fullerene confinement and its role in promoting unique single-electron M–M bonding and magnetism, offering a novel strategy for designing EMF-SMM materials.Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
J. L. and H.-S. H. conceived and directed the research. X.-K. Z. performed all the calculations. X.-K. Z., J. Z., and S.-R. W. designed the study and analysed the data. Y.-Z. Q. assisted with the CASSCF calculations. Y. H. contributed to the discussion of magnetic calculations. X.-K. Z., H.-S. H. and J. L. co-wrote the paper. All the authors discussed the results and provided comments on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Project (No. 2022YFA1503900), the National Natural Science Foundation of China (Grant No. 22033005, 22222605, and 22076095), the NSFC Center for Single-Atom Catalysis, and the startup fund for the Fundamental Science Center of Rare Earths, Ganjiang Innovation Academy, Chinese Academy of Sciences. Computational resources were supported by the Taiyi and Qiming high-performance supercomputer cluster of the Center for Computational Science and Engineering of the Southern University of Science and Technology and the high-performance supercomputer cluster of the Department of Chemistry (CHEM-HPC) of the Southern University of Science and Technology. The Tsinghua National Laboratory for Information Science and Technology and the Tsinghua Xuetang Talents Program are also acknowledged for providing computational resources.References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 Search PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- M. Shiddiq, D. Komijani, Y. Duan, A. Gaita-Ariño, E. Coronado and S. Hill, Nature, 2016, 531, 348–351 CrossRef CAS PubMed.
- Z. Hu, Y. Wang, A. Ullah, G. M. Gutiérrez-Finol, A. Bedoya-Pinto, P. Gargiani, D. Shi, S. Yang, Z. Shi, A. Gaita-Ariño and E. Coronado, Chem, 2023, 9, 3613–3622 CAS.
- F. Liu, D. S. Krylov, L. Spree, S. M. Avdoshenko, N. A. Samoylova, M. Rosenkranz, A. Kostanyan, T. Greber, A. U. B. Wolter, B. Buchner and A. A. Popov, Nat. Commun., 2017, 8, 16098 CrossRef CAS PubMed.
- F. Liu, L. Spree, D. S. Krylov, G. Velkos, S. M. Avdoshenko and A. A. Popov, Acc. Chem. Res., 2019, 52, 2981–2993 Search PubMed.
- R. Westerström, J. Dreiser, C. Piamonteze, M. Muntwiler, S. Weyeneth, H. Brune, S. Rusponi, F. Nolting, A. Popov, S. Yang, L. Dunsch and T. Greber, J. Am. Chem. Soc., 2012, 134, 9840–9843 CrossRef PubMed.
- W. Fu, J. Zhang, T. Fuhrer, H. Champion, K. Furukawa, T. Kato, J. E. Mahaney, B. G. Burke, K. A. Williams, K. Walker, C. Dixon, J. Ge, C. Shu, K. Harich and H. C. Dorn, J. Am. Chem. Soc., 2011, 133, 9741–9750 CrossRef CAS PubMed.
- G. Velkos, D. S. Krylov, K. Kirkpatrick, X. Liu, L. Spree, A. U. B. Wolter, B. Büchner, H. C. Dorn and A. A. Popov, Chem. Commun., 2018, 54, 2902–2905 RSC.
- Z. Hu, B.-W. Dong, Z. Liu, J.-J. Liu, J. Su, C. Yu, J. Xiong, D.-E. Shi, Y. Wang, B.-W. Wang, A. Ardavan, Z. Shi, S.-D. Jiang and S. Gao, J. Am. Chem. Soc., 2018, 140, 1123–1130 CrossRef CAS PubMed.
- F. Liu, G. Velkos, D. S. Krylov, L. Spree, M. Zalibera, R. Ray, N. A. Samoylova, C.-H. Chen, M. Rosenkranz, S. Schiemenz, F. Ziegs, K. Nenkov, A. Kostanyan, T. Greber, A. U. B. Wolter, M. Richter, B. Büchner, S. M. Avdoshenko and A. A. Popov, Nat. Commun., 2019, 10, 571 CrossRef CAS PubMed.
- Z. Zhu and J. Tang, Chem. Soc. Rev., 2022, 51, 9469–9481 RSC.
- Z. Hu and S. Yang, Chem. Soc. Rev., 2024, 53, 2863–2897 Search PubMed.
- C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E.-S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- H. Kwon, K. R. McClain, J. G. C. Kragskow, J. K. Staab, M. Ozerov, K. R. Meihaus, B. G. Harvey, E. S. Choi, N. F. Chilton and J. R. Long, J. Am. Chem. Soc., 2024, 146, 18714–18721 CrossRef CAS PubMed.
- Y.-C. Chen and M.-L. Tong, Chem. Sci., 2022, 13, 8716–8726 RSC.
- J. Xin, Z. Hu, Y.-R. Yao, A. Ullah, X. Han, W. Xiang, H. Jin, Z. Jiang and S. Yang, J. Am. Chem. Soc., 2024, 146, 17600–17605 CrossRef CAS PubMed.
- X. Zhang, W. Li, L. Feng, X. Chen, A. Hansen, S. Grimme, S. Fortier, D. C. Sergentu, T. J. Duignan, J. Autschbach, S. Wang, Y. Wang, G. Velkos, A. A. Popov, N. Aghdassi, S. Duhm, X. Li, J. Li, L. Echegoyen, W. H. E. Schwarz and N. Chen, Nat. Commun., 2018, 9, 2753 CrossRef PubMed.
- X. Zhang, Y. Wang, R. Morales-Martínez, J. Zhong, C. de Graaf, A. Rodríguez-Fortea, J. M. Poblet, L. Echegoyen, L. Feng and N. Chen, J. Am. Chem. Soc., 2018, 140, 3907–3915 CrossRef CAS PubMed.
- X. Wu and X. Lu, J. Am. Chem. Soc., 2007, 129, 2171–2177 CrossRef CAS PubMed.
- I. Infante, L. Gagliardi and G. E. Scuseria, J. Am. Chem. Soc., 2008, 130, 7459–7465 CrossRef CAS PubMed.
- C. Foroutan-Nejad, J. Vicha, R. Marek, M. Patzschke and M. Straka, Phys. Chem. Chem. Phys., 2015, 17, 24182–24192 RSC.
- J. Zhuang, R. Morales-Martinez, J. Zhang, Y. Wang, Y. R. Yao, C. Pei, A. Rodriguez-Fortea, S. Wang, L. Echegoyen, C. de Graaf, J. M. Poblet and N. Chen, Nat. Commun., 2021, 12, 2372 CrossRef CAS PubMed.
- X. Ge, X. Dai, H. Zhou, Z. Yang and R. Zhou, Inorg. Chem., 2020, 59, 3606–3618 CrossRef CAS PubMed.
- C. F. Hermanns, M. Bernien, A. Krüger, C. Schmidt, S. T. Waßerroth, G. Ahmadi, B. W. Heinrich, M. Schneider, P. W. Brouwer, K. J. Franke, E. Weschke and W. Kuch, Phys. Rev. Lett., 2013, 111, 167203 CrossRef PubMed.
- R. Westerström, A.-C. Uldry, R. Stania, J. Dreiser, C. Piamonteze, M. Muntwiler, F. Matsui, S. Rusponi, H. Brune, S. Yang, A. Popov, B. Büchner, B. Delley and T. Greber, Phys. Rev. Lett., 2015, 114, 87201 CrossRef PubMed.
- T. Greber, A. P. Seitsonen, A. Hemmi, J. Dreiser, R. Stania, F. Matsui, M. Muntwiler, A. A. Popov and R. Westerström, Phys. Rev. Mater., 2019, 3, 14409 CrossRef CAS.
- Y. Feng, T. Wang, Y. Li, J. Li, J. Wu, B. Wu, L. Jiang and C. Wang, J. Am. Chem. Soc., 2015, 137, 15055–15060 CrossRef CAS PubMed.
- D. Aulakh, J. B. Pyser, X. Zhang, A. A. Yakovenko, K. R. Dunbar and M. Wriedt, J. Am. Chem. Soc., 2015, 137, 9254–9257 CrossRef CAS PubMed.
- H. Meng, C. Zhao, M. Nie, C. Wang and T. Wang, J. Phys. Chem. C, 2019, 123, 6265–6269 CrossRef CAS.
- L. Hou, X. Cui, B. Guan, S. Wang, R. Li, Y. Liu, D. Zhu and J. Zheng, Nature, 2022, 606, 507–510 CrossRef CAS PubMed.
- B. Peng, J. Am. Chem. Soc., 2022, 144, 19921–19931 CrossRef CAS PubMed.
- X.-K. Zhao, Y.-Y. Zhang, J. Zhao, H.-S. Hu and J. Li, Inorg. Chem., 2024, 63, 11572–11582 CrossRef CAS PubMed.
- H. Suo, M. Li, G. Yang, Y. Du, X. Zhao, X. Ren and S. Li, Phys. Rev. B, 2024, 110, 125405 Search PubMed.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- J. T. Boronski, J. A. Seed, D. Hunger, A. W. Woodward, J. Van Slageren, A. J. Wooles, L. S. Natrajan, N. Kaltsoyannis and S. T. Liddle, Nature, 2021, 598, 72–75 CrossRef CAS PubMed.
- W. Sheng, F. Xie, T. Rajeshkumar, Y. Zhao, Y. Jiang, W. Chen, S. Ye, L. Maron and C. Zhu, Nat. Synth., 2025 DOI:10.1038/s44160-025-00789-5.
- S. Steinberg and R. Dronskowski, Crystals, 2018, 8, 225 CrossRef.
- J. Zhuang, L. Abella, D. C. Sergentu, Y. R. Yao, M. Jin, W. Yang, X. Zhang, X. Li, D. Zhang, Y. Zhao, X. Li, S. Wang, L. Echegoyen, J. Autschbach and N. Chen, J. Am. Chem. Soc., 2019, 141, 20249–20260 CrossRef CAS PubMed.
- Y. Shen, X. Yu, Q. Meng, Y. R. Yao, J. Autschbach and N. Chen, Chem. Sci., 2022, 13, 12980–12986 RSC.
- J. Burkhardt and W.-L. Li, Inorg. Chem., 2024, 63, 18313–18322 CrossRef CAS PubMed.
- Y.-L. Wang, H.-S. Hu, W.-L. Li, F. Wei and J. Li, J. Am. Chem. Soc., 2016, 138, 1126–1129 CrossRef CAS PubMed.
- B. O. Roos, P.-Å. Malmqvist and L. Gagliardi, J. Am. Chem. Soc., 2006, 128, 17000–17006 CrossRef CAS PubMed.
- F. A. Cotton, Inorg. Chem., 1965, 4, 334–336 CrossRef CAS.
- L. Gagliardi and B. O. Roos, Nature, 2005, 433, 848–851 CrossRef CAS PubMed.
- X. K. Zhao, C. S. Cao, J. C. Liu, J. B. Lu, J. Li and H. S. Hu, Chem. Sci., 2022, 13, 8518–8525 RSC.
- H.-J. Koo, D. Dai and M.-H. Whangbo, Inorg. Chem., 2005, 44, 4359–4365 CrossRef CAS PubMed.
- I. P. Muthuselvam, R. Sankar, V. N. Singh, G. N. Rao, W.-L. Lee, G.-Y. Guo and F.-C. Chou, Inorg. Chem., 2015, 54, 4303–4309 CrossRef CAS PubMed.
- S. N. Sofronova and I. I. Nazarenko, Phys. Status Solidi B, 2019, 256, 1900060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details. See DOI: https://doi.org/10.1039/d5sc01607j |
| This journal is © The Royal Society of Chemistry 2025 |