
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOriented design and engineering of advanced metal–organic frameworks for light hydrocarbon separations
Hujun
Zhang†
b,
Jie
Tang†
a,
Chunze
Yu†
b,
Muyu
Zhang
a,
Jiaqi
Wang
a and
Jingui
Duan
 *ab
*ab
aState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: duanjingui@njtech.edu.cn
bState Key Laboratory of Chemistry and Utilization of Carbon-Based Energy Resources, College of Chemistry, Xinjiang University, Urumqi, 830017, China
First published on 2nd June 2025
Abstract
Light olefins, such as ethylene (C2H4) and propylene (C3H6), are essential feedstocks for the production of chemical products. However, the current purification strategy of distillation is energy-intensive and results in high carbon emissions. Adsorptive separation, the selective capture of gas from mixtures by porous materials, is considered a promising alternative or transitional technology. Metal–organic frameworks (MOFs), a kind of porous material with highly tunable nature, have emerged as an innovative chemistry in the past two decades, offering solutions for separating these small gases. This review highlights recent advances in the design and engineering of advanced MOFs, with a focus on precise control over their pore structure and functionality for the adsorption-based purification of C2H4 and C3H6 from the corresponding hydrocarbons with the same carbon number. The importance of rational design in achieving specific functionalities, such as functional sites and molecular sieving in rigid MOFs and local/global dynamics in soft MOFs, is underscored, with examples demonstrating enhanced performance in selective adsorption separation. Additionally, methods and examples of large-scale synthesis of MOFs are briefly described. The goal is to present the state-of-the-art chemistry and applications of MOFs and to offer an outlook towards discovering and designing further new materials.
 Hujun Zhang | Hujun Zhang obtained his master's degree from Xinjiang University in 2022. Now, he is a PhD candidate at Xinjiang University, under the supervision of Prof. Jingui Duan. His current research interests include the design and synthesis of porous coordination polymers and their applications in gas separation. |
 Jie Tang | Jie Tang graduated from the Anhui University of Science and Technology of China with a BS Degree in Chemical Engineering and Technology. She is currently a PhD student under the supervision of Prof. Jingui Duan at Nanjing Tech University. Her research focuses on the design and synthesis of porous coordination polymers for gas separation. |
 Chunze Yu | Chunze Yu graduated from Yili Normal University with a B.S. degree in 2024. In the same year, he joined Xinjiang University as a master’s student under the supervision of Prof. Jingui Duan. His main interests are in the field of porous coordination polymers for efficient light hydrocarbon separation. |
 Jingui Duan | Jingui Duan obtained his PhD from the Coordination Chemistry Institute of Nanjing University in 2011. He was subsequently awarded a JSPS fellowship at Kyoto University, under the supervision of Professor S. Kitagawa, from 2011 to 2014. In 2015, he joined Nanjing Tech University. He was promoted to a full professor in 2018, and subsequently moved to Xinjiang University in 2024. His current research interests focus on the design and synthesis of porous coordination polymers and membranes for gas separations. |
1. Introduction
The global production and demand for light olefins, especially polymer-grade ethylene (C2H4) and propylene (C3H6), exceed that of all other commodity organic compounds. The majority of these products are widely used in chemical manufacturing, such as plastics, synthetic rubber, and synthetic fibers.1 Additionally, they are essential raw materials for agriculture and fine chemicals. Currently, C2H4 and C3H6 are isolated from the cracking units through the strategies of energy-intensive catalytic hydrogenation and cryogenic distillation methods, which account for nearly 3% of all separation energy.2,3 Furthermore, the high pressure and low temperature bring significant risks during the separation process. Therefore, there is a strong need to develop energy-efficient separation technologies, such as non-thermally driven processes, for downstream processing of cracked gas.4–6Adsorption separation is often considered as an alternative technology due to its ability to separate gas molecules based on their chemical nature or size, rather than their boiling points. This non-thermal process can be carried out under mild conditions, such as at room temperature, resulting in significantly reduced energy inputs. However, the main challenge of this technology lies in the construction or selection of optimal porous materials that can achieve the desired uptakes and separation factors. This is particularly difficult due to the extremely similar molecular properties of light hydrocarbons, especially when dealing with mixtures containing the same number of carbon atoms (Table 1). This similarity in properties, both physical and chemical, makes the efficient separation of these molecules a highly challenging task.
| Molecular dimensions (Å3) | Kinetic diameter (Å) | Polarizability (×10–24 cm−3) | Boiling point (K) | |
|---|---|---|---|---|
| C2H2 | 3.32 × 3.34 × 5.70 | 3.3 | 3.33–3.93 | 188.4 |
| C2H4 | 3.28 × 4.18 × 4.82 | 4.16 | 4.25 | 169.45 |
| C2H6 | 3.81 × 4.08 × 5.70 | 4.44 | 4.43–4.47 | 184.6 |
| C3H4 | 4.16 × 4.01 × 6.51 | 4.2 | 5.55 | 249.95 |
| C3H6 | 4.65 × 4.16 × 6.44 | 4.6 | 6.26 | 225.45 |
| C3H8 | 4.20 × 4.60 × 6.80 | 4.3//5.12 | 6.29–6.37 | 231.05 |
Porous materials, such as zeolites and activated carbons, have long been studied for their potential in separating light hydrocarbons.7–10 For example, silver ZK-5 zeolite, known for its unique pore structure and surface chemistry, has shown promise in separating C2H4 and C2H6. However, the strong interaction between the host and guest molecules makes it difficult to desorb them at higher temperatures (>200 °C), limiting their practical applications.11 Activated carbons, on the other hand, have an irregular pore system with meso- or macropores, leading to co-adsorption and making it challenging to obtain pure C2H4. In summary, these traditional porous materials, lacking structural diversity and design flexibility, are not suitable for meeting the demands of industrial hydrocarbon separations.
Metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), are a new class of crystalline porous materials, which are built from the coordination assembly of organic linkers and inorganic nodes in two or three dimensions.12–21 MOFs are emerging porous materials due to their charming diversity, exceptional porosity, functional pore surface and tunable nature (pore size, shape and distribution).22–29 Over the past two decades, there have been numerous reports on MOFs, with many designs specifically targeting the separation of light hydrocarbon mixtures (Scheme 1). These designs have led to significant improvements in key properties, such as capacity and selectivity (Table 2). Typically, as a kind of rigid framework with open Cu sites, HKUST-1 was investigated as the first MOF for the separation of C2H4/C2H6 mixtures as early as 2002.30 After modifying the isolated open metal sites to a linear configuration, Fe2(dobdc) (dobdc: 2,5-dioxido-1,4-benzenedicarboxylate) demonstrated a sharply promoted separation performance of C2H4/C2H6 and C3H6/C3H8 at 318 K, as the linear Fe atoms exhibited a strong affinity towards C2H4 and C3H6.31 Since the prediction of structural dynamics by Prof. Kitagawa in 1998,75 active research shifted to design and synthesis of soft MOFs for such separation, given the structural sensitivity of these structures to the tiny difference in the light hydrocarbons.76 With a rational pyridyl ring (also called a confined-rotational shutter) in the confined nanospace, NTU-88 creates a maximum aperture of 4.4 Å, allowing dedicated propyne C3H4 (4.4 Å) adsorption from a C3H6 (5.4 Å) containing mixture under ambient conditions.69 Moreover, the dynamic nature of the molecular pockets in JNU-3a is pivotal for achieving molecular sieving of C3H6 and C3H8.65 In this regard, MOFs have shown significant advantages over traditional materials in the separation of light hydrocarbons. Consequently, the present review outlines recent advances in MOF chemistry in the separation of light hydrocarbons, with a particular focus on the oriented design strategy, including functional sites in rigid MOFs, molecular sieving in rigid MOFs, and local dynamics and global flexibility in soft MOFs. The discussion will incorporate computational modelling and gas-loaded crystallographic structure to reveal the host–guest interaction and emerging functions in the most promising and innovative achievements. In addition, the strategies for large-scale MOF synthesis were also analysed. Separations of methane and carbon dioxide from light hydrocarbons are not included.
 | ||
| Scheme 1 The representative MOFs for the separation of light hydrocarbons.6,30–74 | ||
| Typical MOFs | Structural properties | C2/C3 separation | Adsorption uptake (mmol g−1) | Selectivity | T/P | Ref. |
|---|---|---|---|---|---|---|
| HKUST-1 | 3D-channel with Cu sites | C2H6/C2H4 | 5.0/5.9 mol kg−1 | — | 295 K 0.8 bar | 30 |
| [ZIF(2-Cim2)] | SOD network | C3H6/C3H8 | 160/155 mg g−1 | — | 303 K 0.8 bar | 45 |
| ZIF-7 | SOD network | C2H6/C2H4 | 2.24/2.2 | 1.75 | 298 K 1 bar | 38 |
| M-MOF-3 | Hexagonal network | C2H2/C2H4 | 6.56/1.35 | 25.5 | 195 K 1 bar | 59 |
| MAF-49 | 3D framework with narrow 1D zigzag channels | C2H6/C2H4 | 1.73/1.7 | 9 | 316 K 1 bar | 49 |
| SIFSIXs-2-Cu-i | Cubic topology | C2H2/C2H4 | 4.02/2.19 | 44.54 | 298 K 1 bar | 34 |
| KAUST-7 | 3D framework with square-shaped channels | C3H6/C3H8 | 1.4/0.04 | — | 318 K 1 bar | 32 |
| Y-abtc | Ftw network and cage-like pores | C3H8/C3H6 | 2.0/0.07 | — | 318 K 1 bar | 6 |
| UTSA-200 | Doubly interpenetrated nets | C2H2/C2H4 | 3.65/0.63 | 6320 | 198 K 1 bar | 44 |
| UTSA-280 | 1D Ca–C4O4 chain with pentagonal bipyramidal structure | C2H4/C2H6 | 2.5/0.098 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
298 K 1 bar | 70 |
| Fe2(O2)(dobdc) | Iron–peroxo sites | C3H8/C3H6 | 3.32/2.53 | 4.4 | 298 K 1 bar | 46 |
| JNU-3 | 1D diffusion channel with dynamic molecular pockets | C3H6/C3H8 | 58.6/42.5 cm3 g−1 | 513 | 303 K 1 bar | 65 |
| ZnAtzPO4 | 2D pillared framework with pocket-bottleneck channels | C2H4/C2H6 | 1.1/0/3 | 31 | 273 K 1 bar | 35 |
| CuTiF6-TPPY | Pillared semi-cage 1D channels | C2H2/C2H6/C2H4 | 3.62/2.82/2.42 | 1.50 C2H2/C2H4 | 298 K 1 bar | 67 |
| 1.17 C2H6/C2H4 | ||||||
| NTU-85 | Square-apertured H2O channels | C3H6/C3H8 | 0.45/0.003 | 1570 | 298 K 1 bar | 37 |
| NTU-88 | Sql layer and rhombic pores | C3H4/C3H6 | 86.0/2.0 cm3 g−1 |
— | 298 K 1 bar | 69 |
| FDMOF-2 | 3D framework | C3H8/C3H6 | 5.04/4.15 | 2.18 | 298 K 1 bar | 73 |
| CdIF-13 | SOD network | C3H8/C3H6 | 2.56/2.32 | 2.04 | 288 K 1 bar | 43 |
| MOF-808Bzz | Octahedral morphologies | C2H2/C2H6/C2H4 | 2.98/2.20/1.43 | 3.15 C2H2/C2H4 | 298 K 1 bar | 53 |
| 1.90 C2H6/C2H4 | ||||||
| 1.92 C2H6/C2H4 | ||||||
| ZU-609 | 3D networks with large 1D channels | C3H6/C3H8 | 2.0 | >10 | 298 K 1 bar | 33 |
| HAF-1 | 3D framework with channels and molecular pockets | C3H8/C3H6 | 101.61 cm3 cm−3/— | 1.67 × 107 | 298 K 1 bar | 55 |
| X-dia-1-Ni0.89Co0.11 | Flexible diamondoid networks | C2H4/C2H6 | 4.96/0.54 | — | 273 K 1 bar | 56 |
| TYUT-17 | Spindle-like cages | C2H6/C2H4 | 67.4/61.3 cm3 g−1 | — | 298 K 1 bar | 66 |
| NTU101-NH2 | H-bond-tuned interpenetrated pcu framework | C2H6/C2H4 | 40.1/15.2 cm3 g−1 | — | 328 K 0.5 bar | 62 |
| NKMOF-1-Ni | 3D framework with 1D channels and dual gas-binding sites | C3H4/CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2/C3H6 CCH2/C3H6 |
3.5/2.1/— | 1271.6(C3H4) | 298 K 1 bar | 52 |
| JNU-9-CH3 | 3D framework with cubane SBUs | C3H4/CH2CCH2/C3H8/C3H6 |
3.6/3.4/2.95/2.9 | 1.5 (C3H8/C3H6) | 298 K 1 bar | 60 |
| 2.1 (C3H4/C3H6) | ||||||
| 1.3 (propadiene/C3H6) |
2. Design strategies
In the context of the continuous advancement of sophisticated MOFs for the separation of gases, a range of design strategies have been put forward. These can be broadly categorized into two groups based on their principles of gas molecular adsorption separation: (1) functional sites, molecular sieving and diffusion strategies using rigid MOFs, and (2) framework dynamics strategies in soft MOFs (Fig. 1). And, we compare the advantages and limitations of each strategy in order to provide clear suggestions and guidance for the synthesis of future MOFs (Table 3). Furthermore, the incorporation of host–guest interactions has been comprehensively examined in relation to these design strategies. | ||
| Fig. 1 Oriented design and engineering strategies of advanced MOFs. | ||
| Design strategy | Advantages | Limitations |
|---|---|---|
| Open metal sites | High selectivity | Synthesis and scale-up challenges |
| Highly adjustable | Stability issues | |
| High adsorption capacity | Vulnerability to impurity gases | |
| Free organic sites | Highly selective | Diffusion resistance |
| Highly adjustable | Difficulty and cost of synthesis | |
| Good stability to avoid toxicity of metal sites | Vulnerable to other impurity gases | |
| Low regenerative energy consumption | Selectivity may be low | |
| Multiple sites | Synergistically enhanced selectivity | Synthesis complexity increase |
| Broadening the applicable separation system | Risk of site-to-site interference | |
| Balancing adsorption strength and regeneration energy | Challenging kinetic balance | |
| Improve stability | ||
| Molecular sieving | Highly selective separation | Aperture regulation is difficult |
| Gentle operating conditions | Low pressure adsorption capacity | |
| High stability (not dependent on chemisorption) | Limited separation of similar-sized molecules | |
| Low energy consumption for regeneration | Diffusion kinetic limitations | |
| Suitable for kinetic separations | ||
| Diffusion controlled | Highly dynamic selectivity | Highly sensitive to an orifice structure. |
| Low energy consumption for regeneration | Low pressure separation efficiency | |
| High stability | Limited separation of similar sized molecules | |
| Gentle operating conditions | May be affected by the ‘blocking effect’ | |
| Global softness of the framework | High selectivity and dynamic adaptability | Structural stability issues |
| Low energy consumption and efficient regeneration | High material preparation requirements | |
| Outfield response characteristics | Representation challenge | |
| Local softness of the framework | Highly selective | Complex interaction mechanisms |
| Fast adsorption kinetics | Difficult to synthesize | |
| Good stability | Industrial scale-up challenges | |
| Precise molecular recognition | ||
| Low energy consumption | ||
| Softness of the confined moiety in the framework | Highly selective | Synthesis is complex |
| Energy saving potential | Difficulty in industrial scale-up | |
| Regeneration difficulties |
2.1 Oriented design strategies in rigid MOFs
Given the finding that HKUST-1 can separate C2H4/C2H6,30 the configuration of open metal sites has been investigated. This has been achieved by altering the transition metal ions in M-MOF-74 (M = Co, Mn, and Mg), resulting in varying abilities of the rod-shaped and high-density open metal sites to interact with C3H6 through π-complexation.22 It is worth noting that Co sites exhibit the largest difference in binding energies between C3H6 and C3H8. As pressure increases, the open Co sites become increasingly occupied by C3H6, leading to a strong suppression of C3H8 adsorption and a significant increase in C3H6/C3H8 selectivity (Fig. 2a). Following the alteration of the redox-active Fe(II) within the same MOF platform, the material exhibits olefin adsorption selectivity that surpassed that of paraffin.31 The neutron diffraction data confirmed that the unsaturated hydrocarbons such as C2H2, C2H4 and C3H6 exhibited side-on binding modes, with Fe–C distances of 2.42(2) to 2.60(2) Å. A comparison with the shorter distance of 2.020(5) to 2.078(4) Å observed in the diamagnetic complex [Fe(C2H4)4]2− indicated that the Fe centres within Fe-MOF-74 maintain a high spin electron configuration when binding these unsaturated gases. The interactions of both C2H6 and C3H8 with Fe in Fe-MOF-74 are even weaker, as evidenced by the longer Fe–C distance of ∼3 Å (Fig. 2b).
 | ||
| Fig. 2 (a) A snapshot from a GCMC simulation for C3H6 adsorption in Co-MOF-74, and the propylene molecule binds to the Co atom. (b) A portion of the solid-state structure of Fe2(dobdc)·2C2D4, and C2H4, and C3H6 coordination with Fe2(dobdc). Reprinted with permission from ref. 22. Copyright 2012 John Wiley & Sons, ref. 31. Copyright 2012 The American Association for the Advancement of Science, respectively. | ||
In the context of the larger channel aperture of 11–12 Å in the MOF-74-series, a microporous framework, Fe(pyrazine)Pt(CN)4, with a channel size of 4.0 Å and mixed open metal sites was designed and prepared for the separation of C3H6/C3H8. C3H6 was located between two pyrazine rings, forming π–π interactions. Notably, the distance between CC3H6 and the face-to-face packed open Pt sites is short, ranging from 2.8–3.2 Å. In contrast, the CC3H6⋯Ni distance becomes longer (3.5 Å) in FeNi-M’MOF. The finely tailored pore size of 4.0 Å facilitates the passage of C3H6 through the channel, but significantly hinders the movement of the larger C3H8 molecule.
Supramolecular interactions derived from organic sites and light hydrocarbons are visualized and understood in the case of NOTT-300.80 The results of neutron scattering, synchrotron X-ray and neutron diffraction, and computational modelling revealed that the free OH and benzene ring are simultaneously incorporated with C2H2 through stronger hydrogen-bonding (C⋯H = 2.96–3.64 Å), π⋯π stacking interactions (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C⋯C6 = 3.81 Å) and intermolecular dipole interactions (CI⋯HII = 3.24 Å, CII⋯HI = 3.12 Å). However, the reduced π-electron density of C2H4 compared to that of C2H2 enables a longer CC2H4⋯HO distance (4.62 Å), which suggests a weaker hydrogen bonding. Despite the π⋯π interactions between C2H4 and phenyl rings of the ligand, C2H4 was displaced towards the centre of the cavity and interacted majorly with C2H2 molecules (captured by OH), upon the introduction of an equimolar mixture of the two gases to the system (Fig. 3a).
C⋯C6 = 3.81 Å) and intermolecular dipole interactions (CI⋯HII = 3.24 Å, CII⋯HI = 3.12 Å). However, the reduced π-electron density of C2H4 compared to that of C2H2 enables a longer CC2H4⋯HO distance (4.62 Å), which suggests a weaker hydrogen bonding. Despite the π⋯π interactions between C2H4 and phenyl rings of the ligand, C2H4 was displaced towards the centre of the cavity and interacted majorly with C2H2 molecules (captured by OH), upon the introduction of an equimolar mixture of the two gases to the system (Fig. 3a).
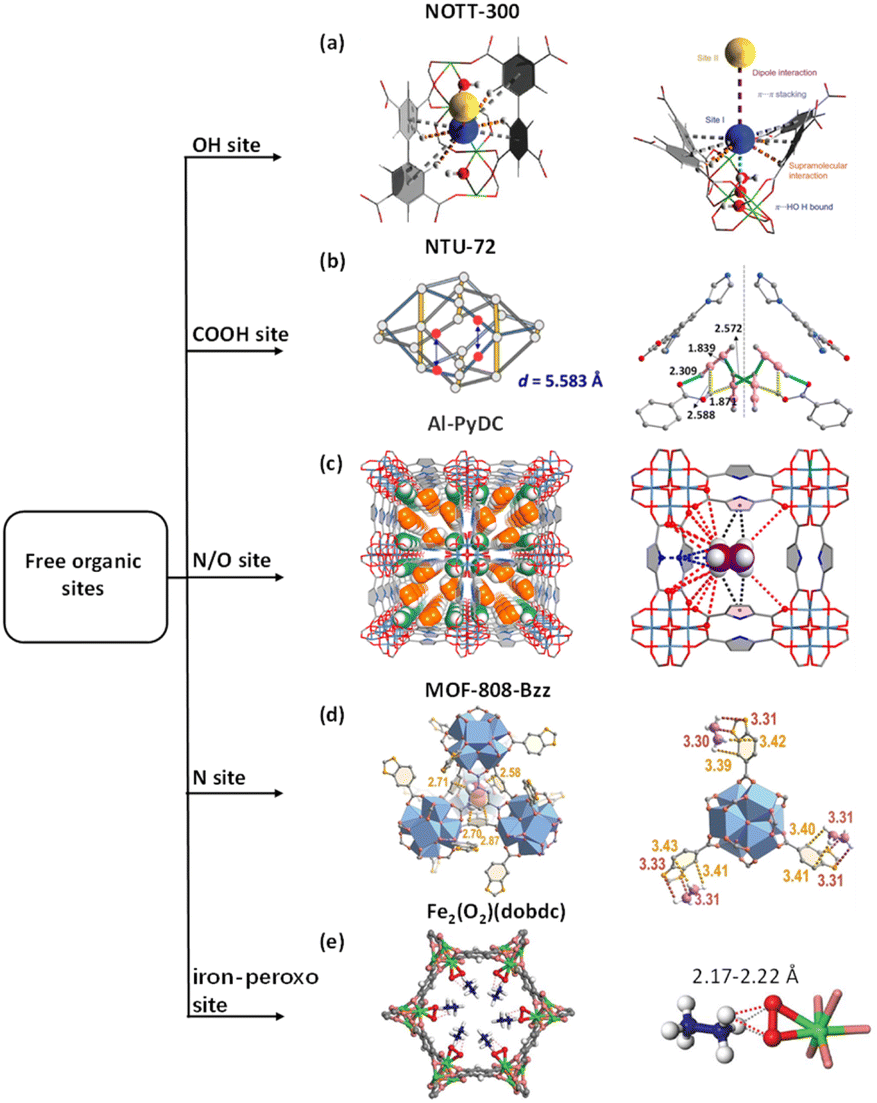 | ||
| Fig. 3 (a) Schematic representation of the action of combining C2H2 and C2H4 molecules. (b) The pincer distance of NTU-72 and structure of C2H2- loaded NTU-72. (c) The SCXRD structure of C2H2-loaded Al-PyDC and the C2H6 binding site in gas-loaded Al-PyDC. (d) C2H4 binding sites I and sites II in MOF-808-Bzz. (e) Structure of Fe2(O2) (dobdc)⊃C2D6. Reprinted with permission from ref. 80. Copyright 2015 Springer Nature Limited, ref. 81. Copyright 2023 The Royal Society of Chemistry, ref. 83. Copyright 2023 Springer Nature Limited, ref. 53. Copyright 2023 John Wiley & Sons, ref. 46. Copyright 2018 The American Association for the Advancement of Science, respectively. | ||
By changing the OH group into COOH, we recently found the unique ability for the adsorption of C2H2 from C2H4 containing mixtures by a MOF with crab-like carboxylic pincers. On a new platform of a 6-c topology network, pore size engineering of the carboxylic-functionalized MOFs was finely tuned by altering the MF62− pillars (M = Si, Ti and Zr).81 Following the increased distance of the M–F, the pincer distance increased gradually from 5.02 to 5.91 Å, yielding a fitted nanospace in NTU-72 for selective recognition of a C2H2 tetramer. The OCOOH interacts with terminal HC2H2 with a short distance of 2.309 Å, while the OHCOOH chelates to two C2H2 through O–H⋯CC2H2. The formation of a C2H2 tetramer with C2h symmetry is characterised by these two groups of gas molecules, which is distinctly different from the S4 symmetric C2H2 tetramer in SIFSIX-Cu-i.34 In contrast, two types of longer hydrogen bonds are observed between COOH⋯C2H4, reflecting the relative weak carboxylic–C2H4 interaction. Importantly, the captured C2H2 tetramer not only facilitates the direct harvesting of highly pure C2H4 at the adsorption step, but also benefits the collection of pure C2H2 during the desorption step (Fig. 3b).
To promote the ability for purification of C2H4 from C2 ternary hydrocarbons, fine material design is highly required as the strength of the interactions between open metal sites and hydrocarbons typically increases in the order of C2H6 < C2H4 < C2H2. Based on the same 6-c platform, we replaced the functional group of COOH by –CH3 and CH3.48 The varied pore environment and organic sites endow NTU-73-CH3 with remarkable capability for the direct production of poly-grade C2H4 from ternary C2 hydrocarbons under ambient conditions. Comparably, the precursor framework of NTU-73-COOH cannot purify C2H4, while NTU-73-CF3 exhibits only negligible capability in this regard. This is primarily attributable to the modified binding sites in the NTU-73 series, which not only eliminate the channel obstruction caused by the formed C2H2 tetramer, but also enhance the interactions of host-C2H2/C2H6. These interaction changes were clearly observed by gas-loaded structures, and TJT-100 also demonstrates the ability for C2H4 purification from ternary (C2H2/C2H6/C2H4) mixtures by utilizing the abundant O sites.82
To further promote the host-C2H6 and host-C2H2 interactions, multiple supramolecular binding sites have been designed in the MOFs with suitable pore sizes. For example, Al-PyDC was assembled with ligands containing N and a large number of polar O sites, providing abundant supramolecular binding sites for C2H6, while the electronegative O and N sites form hydrogen bonds with C2H2, as confirmed by gas loaded structural analysis.83 This unique design endows Al-PyDC with the capacity to effectively purify C2H4 from mixtures containing C2H2 and C2H6 in one step. Furthermore, this MOF has been shown to be highly stable under harsh conditions (Fig. 3c).
By expanding the dimensions of the individual organic sites to nitrogen-containing heterocyclic ligands, including indole-5-carboxylic acid (ind), benzimidazole-5-carboxylic acid (bzz), and indazole-5-carboxylic acid (izo), the functionalized MOF-808 materials attain a uniform distribution and specific configuration of such extensive binding sites. MOF-808-Bzz has multiple strong supramolecular interactions, providing an excellent geometric configuration for C2H6, resulting in the highest C2H2/C2H4 (1/99, v/v) and C2H6/C2H4 (50:50, v/v) selectivity among these four materials (Fig. 3d).53
To achieve efficient C2H4 and C2H6 separation, an inverse interaction, namely the stronger host–C2H6 interaction, has been considered. Inspired from the structures of metalloenzymes, Fe2(O2)(dobdc) exhibited a preferential binding of C2H6 over C2H4.46 High-resolution neutron powder diffraction (NPD) measurements revealed that the peroxo site binds C2D6 through a C–D/O interaction, of which the D⋯O distance varies in a narrow range of 2.17 to 2.22 Å. In addition, the nonplanar C2D6 molecule matches better to the pore surface in Fe2(O2)(dobdc) than the planar C2D4 molecule, resulting in stronger hydrogen bonds with the Fe-peroxo site and stronger van der Waals interactions with the ligand surface (Fig. 3e).
In light of the terminal acidic H on C3H4, the basic organic site strategy has also been explored in the design of MOFs for the removal of trace C3H4 from C3H6. Taking the pore-partitioned MOF as a platform, aminophthalic acid (NH2-BDC) was selected to construct the framework.84 The amino sites, which are densely arranged, possess a distinctive capacity to recognise the acetylic and methyl hydrogen atoms of C3H4, resulting in the formation of short hydrogen bonds. Moreover, the customised nanospace induces C3H4⋯C3H4 molecules to aggregate together with short H⋯C distances. Although the NH2 group also interacts with C3H6, the interaction distance is longer, yielding a sharp adsorption difference (84.5 cm3 g−1) at about 1 kPa, 298 K. Furthermore, such an interaction difference was also finely illustrated by in situ infrared spectroscopy measurement.
To further promote the ability to selectively capture C2H2 with a terminal acidic H, the weakly basic hybrid anion MF62− (M = Si, Ti and Zr) was selected. As a typical example, SIFSIX-1-Cu [SIFSIX, hexafluorosilicate (SiF62−); 1, 4,4′-bipyridine)] was prepared.34 The prototypical primitive cubic network has previously been reported for high volumetric CH4 and CO2 uptake.86,87 Notably, the periodically arrayed SiF62− allowed preferential binding of C2H2 molecules through strong C–H⋯F H-bonding (2.017 Å) and van der Waals (vdW) interactions with the organic linkers. Constrained by the narrow pores, the four neighbouring adsorbed molecules assemble to form a gas cluster through multiple Hδ+···Cδ− dipole–dipole interactions, further increasing the energy for adsorption. Comparably, the calculated H-bond distances between C2H4 and SiF62− sites are longer than those between C2H2 and SiF62− sites. This unique interaction allows SIFSIX-1-Cu to show balanced performance between C2H2 uptake and C2H2/C2H4 selectivity (Fig. 4a). This idea has been further expanded in the family of cubic net of SIFSIX-1-Cu, SIFSIX-2-Cu (2: 4.4′-dipyridylacetylene), SIFSIX-2-Cu-i, SIFSIX-3-Cu (3: pyrazine), SIFSIX-3-Zn, SIFSIX-3-Ni, and TIFSIX-2-Cu-i (TIFSIX = TiF62−) by changing the length of the linkers, the node, and/or the framework interpenetration for separation of C2H4 from ternary (C2H2/C2H6/C2H4) or quaternary (CO2/C2H2/C2H6/C2H4) mixtures (Fig. 4a).88
 | ||
| Fig. 4 (a) Optimized C2H2 configuration and adsorption binding sites in SIFSIX-1-Cu. (b) View of the inner space in channel A, and view of the structure of a molecular trap with pure gas of NTU-67⊃C2H4. (c) Coordination environment of Ni3(pzdc)2(7Hade)2, binding configurations of C2H2, and the open Ni2+ strong polarization capacity and the calculated CC–H bond angle. (d) Coordination environment of Ca2+ ions and preferential adsorption sites for C2H2. (e) Densely arranged high-density strong binding sites and C3H6 adsorption sites based on theoretical calculations. Reprinted with permission from ref. 34. Copyright 2016 The American Association for the Advancement of Science, ref. 36. Copyright 2022 John Wiley & Sons, ref. 85. Copyright 2020 John Wiley & Sons, ref. 89. Copyright 2021 American Chemical Society, ref. 90. Copyright 2023 John Wiley & Sons, respectively. | ||
By changing pyridine to imidazole at the coordination sites of the linear ligand, a new MOF (NTU-67) with a trap-and-flow channel structure was prepared by crystal conversion. With the narrowest window aperture of 3.4 Å, the flow channel acted as a sieve channel for faster diffusion of the linear molecules of C2H2/CO2 (3.3 Å) and the butterfly-like molecule of C2H4 (4.2 Å). Meanwhile, the confined nanospace regulated by three SiF62− anions and six imidazoles in another channel works as a molecular trap for adsorbed molecules. Crystallographic study under pure gas revealed that the linear C2H2 interacts with SiF62− anions and imidazole carbon to form short hydrogen bonds in the range of 2.470–2.984 Å. In addition, the existing hydrogen bonds between the adsorbed C2H2 molecules enable the formation of gas clusters, which benefits the C2H2 uptake at very low pressure. For CO2, they are observed in the molecular trap around the SiF62− anions, forming a typical dipolar–dipolar (Si–F⋯CO: δ+–δ−···δ+ = δ−) interaction. However, relatively longer hydrogen bonds of F⋯H– CC2H4 (2.554–2.715 Å) were observed for C2H4. Notably, the competitive binding of the gases was validated by crystallographic study under mixed gas. Therefore, NTU-67 was able to directly harvest highly-pure C2H4 from the ternary (CO2/C2H2/C2H4) mixtures (Fig. 4b).36
To investigate the combined effect of open metal sites and electronegative sites in a single domain, a super-microporous MOF, Ni3(pzdc)2(7Hade)2 was designed and prepared. This structure features S-shaped one-dimensional ultramicroporous channels decorated with a high-density of open Ni sites and electronegative oxygen and nitrogen sites. First-principles density functional theory (DFT) calculations revealed that C2H2 interacts with the Ni site. The slight bending of the CC–H bond angle suggests that the unsaturated Ni2+ site can polarize the C2H2 molecule. In addition, the highly polar HC2H2 interacts with the basic or electronegative O and N sites on the pore surface through electrostatic interactions at very short distances of 3.10–3.60 Å. In contrast, π-complexation occurs between Ni and C2H4 with a longer distance of 4.00–4.40 Å, despite the vdW interaction between C2H4 and the pore surface being very similar in type and geometry to that observed for C2H2. Therefore, the difference in the synergistic effect significantly improves the adsorption capacity and selectivity of C2H2 from C2H4 containing mixtures (Fig. 4c).85
Another example is Ca-MOF, derived from a N, O-donor ligand 2,5-di(2H-tetrazol-5-yl) pyrophosphate.89 This material exhibits unique metal carboxylate-nitrogen heterocyclic oxide units, and a high density of open metal sites and organic functional sites that enable it to selectively adsorb and separate C2H2. This interaction of the open metal sites with the π-electron of the C2H2 molecules is a key feature, while the organic groups play a regulatory role in the pore structure and surface properties through organic sites. This group forms hydrogen bonds and other interactions with C2H2, thereby generating additional binding forces. Monte Carlo simulations revealed that C2H2 forms strong C–H⋯O/N bonds with benzene rings and interacts with Ca2+ sites through M⋯π interactions. In comparison, C2H4 lacks C–H⋯π interaction (Fig. 4d). Additionally, CuTiF6-TPPY (TPPY: 5,10,15,20-tetra(4-pyridyl)-21h,23h-porphyrin) with semi-cage-like 1D channels decorated with synergistic binding sites of TiF62− and TPPY exhibits a noticeable adsorption of C2H2 and C2H6 over C2H4.67 Therefore, efficient single-step C2H4 purification from a C2H2/C2H4/C2H6 mixture has been achieved based on this MOF. Similar to the structure of Fe(pyrazine)Pt(CN)4, a new MOF Co(pyz-NH2)[Ni(CN)4] (ZJU-75a, pyz-NH2 = 2-aminopyrazine) has been prepared for the separation of C3H6/C3H8.90 The open Ni site and the organic NH2 sites point into the narrow channel (4.4 Å), allowing multiple interactions with C3H6, including double π-complexation interactions (3.46 and 3.76 Å) with face-to-face packed Ni sites and multiple supramolecular interactions with N sites (C–H⋯NCN: 3.17–3.84 Å and C–H⋯NNH2: 2.79–3.24 Å). In contrast, only weak supramolecular interactions (C–H⋯NCN: 3.07–3.81 Å) and C–H⋯NNH2: 2.63–3.55 Å) were observed in host@C3H8, due to the lack of a double bond in C3H8 (Fig. 4e).
As a typical example, [Ca(C4O4)(H2O)] (UTSA-280), synthesized from calcium nitrate and squaric acid, has two parallel 1D open cylindrical channels with similar cross-sectional areas of about 14.4 Å2.70 Importantly, this value is larger than the minimum cross-sectional area of C2H4 (13.7 Å2), but smaller than that of C2H6 (15.5 Å2). Therefore, it exhibits exclusive C2H4 adsorption, but not of C2H6 at 298 K. This observation was further confirmed by the adsorption isotherms of the two gases at 273 and 195 K. Further diffraction experiments and calculations show that the C2H4 molecules adopt a head-to-head configuration inside the 1D channel, associated with weak C–H⋯O hydrogen bonding, π⋯π stacking and vdW interactions with the rings of the ligand or coordinated water molecules. However, C2H6, regardless of its orientation, is strongly restricted by the aperture of the channel (Fig. 5a).
 | ||
| Fig. 5 (a) Preferential binding site for C2H4 molecules, and size/shape sieving based on the minimum cross-sectional areas of C2H4 and C2H6 molecules. (b) Crystal structure of NbOFFIVE-1-Ni, and simulation of the maximum open structure of NbOFFIVE-1-Ni. (c) Sieving gate of ZU-609 and the molecular sizes of propylene and propane. Reprinted with permission from ref. 70. Copyright 2018 Springer Nature Limited, ref. 32. Copyright 2016 The American Association for the Advancement of Science, ref. 33. Copyright 2023 The American Association for the Advancement of Science, respectively. | ||
Moving to the target of C3H6/C3H8 mixtures, the two have <0.4 Å size difference, and design and construction of the sieving channel becomes more challenging. Selecting or tailoring the length of the ligands is not effective, as the required change is located in a wider range. Inspired by the sub-Å level size change of the hybrid ions, a chemically stable fluorinated MOF, KAUST-7, was prepared according to the reticular chemistry approach.32 Initially, by changing the SiF62− pillar in SIFSIX-3-Ni (adsorbs both C3H6 and C3H8 due to the free rotation of the pyrazine ligands) to a slightly bigger cation NbOF52−, the short proximal distance between adjacent F atoms provides a plausible window opening of 3.0–4.8 Å, associated with the restricted rotation and tilting of the pyrazine linker. Therefore, KAUST-7 displayed full exclusion of C3H8 from C3H6-containing mixtures. Sieving separation of C3H6/C3H8 has also been observed in Y-abtc (abtc = 3,3′,5,5′-azobenzenete tracarboxylates), which has cage-like pores connected through small windows of 4.72 Å (Fig. 5b).
Although the strategy of molecular sieving can provide complete separation of the mixtures, the narrow pore strongly restricts molecule diffusion, a long-standing issue of adsorption separation. ZU-609, a 2D network composed of an inorganic metal node and organic linkers (EDS2−, 1,2-ethanedisulfonate; dps, 4,4′-dipyridyl sulfide), exhibits a large 1D channel with a size of 7.5–11.1 Å.33 Meanwhile, such a channel was connected by a narrow neck (4.2 × 5.1 Å). This cross-sectional area falls just between the molecular dimensions of C3H6 and C3H8. In other words, by incorporating a molecular sieve gate and a fast diffusion channel in a single domain, ZU-609 exhibits precise exclusion of C3H8 from C3H6-containing mixtures, as well as fast C3H6 adsorption kinetics. Particularly, the diffusion coefficient of this MOF is 1–2 orders of magnitude higher than that of KAUST-7 and Co-gallate. The rapid diffusion and high sorption were finely supported by in situ PXRD analysis and dispersion corrected calculation (Fig. 5c).
 | ||
| Fig. 6 (a) Crystal and pore structures of MAF-23-O, and breakthrough curves and adsorption kinetic curves using an equimolar C3H6/C3H8 mixture at 298 K and 1 atm. (b) Structure of DL-mal-MOF and MD-derived self-diffusion rates of C3H6 and C3H8. (c) Three-dimensional structure of ZnAtzPO4 and distribution of the aperture size of the bottleneck. Reprinted with permission from ref. 88. Copyright 2019 John Wiley & Sons, ref. 27. Copyright 2023 The American Chemical Society, ref. 35. Copyright 2020 Science, respectively. | ||
To further tune the synergistic effect of thermodynamic and kinetic adsorption performance of C3H6 from C3H8, a group of MOFs were synthesized using chiral ligands. With L-malic acid, the L-mal-MOF has a homogeneous one-dimensional pore structure measured to be 5.3 × 5.5 Å. In contrast, the racemic material DL-mal-MOF synthesized from a mixture of D/L-malic acid ligands exhibited periodic contraction–expansion pore structures, forming quasi-discrete pore structures. This difference was caused by the different orientations of the chiral group in the nanosapce. Similar to MAF-23-O, abundant oxygen atoms were found in these contraction–expansion pore structures. Adsorption isotherms showed that the DL-mal-MOF has a significantly higher equilibrium-kinetic combined selectivity of C3H6/C3H8, which was further illustrated by energy fluctuations and corresponding binding energy during the diffusion of the C3H6 and C3H8 molecules in such quasi-discrete pore structures.27 This result suggests that the narrow-neck channel, along with the O-rich environment, may make the MOF a C3H6/C3H8 separator (Fig. 6b). Utilizing a similar concept, a phosphate anion (PO43−) functionalized MOF ZnAtzPO4 with a narrow (4.94 Å)-neck (3.82 Å) channel, has been reported for C2H4/C2H6 separation (Fig. 6c).35
2.2 Oriented design strategies in soft MOFs
Soft MOFs, a kind of crystalline porous solid with reversible phase transformation, have been considered as the most interesting discovery in materials chemistry in the last two decades. The combination of diverse structural nature and an external stimulus (such as gas adsorption, temperature, pressure, light, and electric field) creates emergent softness of the frameworks; particularly, some of them can show fully different structural responses to the gases, even if they have very similar properties. This unique characteristic thereafter sheds light on the development of adsorption based light hydrocarbon separation technology with significant vitality. According to the modified parts of the framework, they can be broadly divided into three types: global softness, local softness and confined softness. However, the design and construction of the desired softness for the accurate detection of light hydrocarbons, as well as the thorough understanding of the softness involved, remains a critical challenge.2.2.1.1 Rapid change. Typically, an ultramicroporous MOF UTSA-300 ([Zn(dps)2(SiF6)], dps = 4,4′-dipyridylsulfide) with 2D channels of about 3.3 Å was reported. Interestingly, the fully activated framework transformed into a closed pore phase with 0D nanospaces. This is due to the conformational changes of the dps ligands and the rotation of the SiF62− ions, leading to a shrinkage of the framework. However, this 0D nanospace starts to open under C2H2 at 273 K, but not for C2H4 and CO2. Neutron powder diffraction and modelling studies pointed out that two F atoms from the two adjacent SiF62− ions bind a C2H2 molecule (via short hydrogen bonds) in a head-on configuration, leading to a recovery of the pore system from 0D to 2D.91 The framework change allows UTSA-300a to selectively capture C2H2 from C2H4 or CO2 containing mixtures.
In contrast, NTU-65, the global soft framework demonstrated different structural changes, resulting in efficient C2H4 purification ability from C2H2/CO2/C2H4.92 The as-synthesized NTU-65, constructed from 1,4-di(1H-imidazole-1-yl)benzene, Cu2+ and SiF62−, exhibits a 3D framework with pcu topology. The two adjacent, rather than the two opposite, F atoms on SiF62− join the coordination of the framework, while other F atoms form hydrogen bonds with the ligands. In addition, NTU-65 has two types of nanochannels with opening sizes of 2.6 × 3.4 and 5.2 × 6.3 Å2. Therefore, the activated framework showed sharply different PXRD and parameters of the unit cell, indicating the global framework change. Interestingly, this activated framework showed a temperature-dependent response for C2H2, CO2, and C2H4 at 195 K, but only for C2H2 at 298 K. On further temperature optimization to 263 K, C2H2 and CO2 were adsorbed, resulting in a one-step purification of C2H4 (Fig. 7a).
 | ||
| Fig. 7 (a) The crystal structure of NTU-65 via the c axis and benzene ring rotation of NTU-65. (b) The crystal structure of X-dia-1-Ni0.89Co0.11 and C2H4 and C2H6 binding sites at X-dia-1-Ni0.89Co0.11. (c) The crystal structure of NTU-88 via an axis and open phase of NTU-88o under C3H4 at 100 kPa. Reprinted with permission from ref. 92. Copyright 2020 John Wiley & Sons, ref. 56. Copyright 2024 American Chemical Society, ref. 69. Copyright 2023 John Wiley & Sons, respectively. | ||
In addition, sudden pore opening of X-dia-1-Ni occurs on C2H6, but not on C2H4, affording direct harvesting of C2H4 in the adsorption step.56 The pressure of C2H6 triggered sudden opening can be finely tuned through partial Co2+ doping, due to the better shape-fitting between the host and C2H6 under increased pressure. Further modelling calculation results confirmed that the open phase of X-dia-1-Ni exhibited strong C2H6 interaction through multiple C–H⋯O bonding with distances of 2.40 and 3.61 Å, whereas only two C–H⋯O bonds with distances of 3.75 and 3.83 Å were observed between the host and C2H4 (Fig. 7b).
On the base of an NTU-65 precursor with Cu–F (from SiF62−) and Cu–N (from ligand) coordination bonds, the same group developed a new approach to fine regulate the gradient gate-opening in a series of MOFs (NTU-65-FeZr, NTU-65-FeTi, NTU-65-CoZr and NTU-65-CoTi) via node substitution.51 Due to the systemically altered strength of the coordination bond, the sole structural response toward C3H4 in NTU-65-FeZr gradually evolves into a sequential response to C3H4 (1.6 kPa), C3H6 (19.4 kPa), and C3H8 (57.2 kPa) in NTU-65-CoTi at 273 K, as the incorporation of multiple nodes determines the energy barrier of the global softness of the framework. This unique phenomenon allows NTU-65-CoTi to show sieve separation of C3H4/C3H6/C3H8 in one step.
Notably, co-adsorption is a kind of common phenomenon once the inherent large pore has been opened by the molecule with strong host–guest interactions, which then leads to issues with selectivity and separation efficiency. To tackle this problem, we designed a rotational shutter (a rotated pyridyl ring) in NTU-88, a soft MOF composed of 4,4′-dipyridylnitride and NiCl2.69 The 2D coordination framework connected by halogen bonds exhibits a zig-zag channel with a rhomboid pore of 5.3 × 8.8 Å. But, the structure of the fully activated framework exhibits significant changes, particularly the sharply tailored open size of the channel (1.6 Å). However, this framework demonstrated quick C3H4 uptake, but no C3H6 uptake. Structural models under typical pressure and modelling calculations showed that a rotation of the pyridine rings in L was observed in the open phase of this MOF, creating an open pore with a maximum size of 4.4 Å, very close to the molecular size of C3H4 (4.4 Å), but smaller than that of C3H6 (5.4 Å). Thus, this is a rare soft example that can comprehensively suppress co-adsorption (Fig. 7c).
2.2.1.2 Stepwise change. It is worth noting that the adsorption isotherms of these corresponding MOFs show a clear step in a slightly wider pressure range. However, the guest- or stimuli-triggered phase transition in soft MOFs passes through a closed phase, an intermediate phase and then an open phase. Such stepwise phase transitions represent a novel and responsive platform for molecular recognition, even for light hydrocarbons. As an early example, the MIL-53-series demonstrates stepwise adsorption isotherms of CO2 and alkanes.93,94 A similar stepwise N2 adsorption isotherm was also found in a flexible framework Co(BDP).95,96 However, the stepwise structural change dominated by coordination bonds usually requires high energy input (high gas pressure), which could not distinguish the small difference in the light hydrocarbons. Inspired by the stepwise isotherms of CO2 in a partially interpenetrated MOF, MFM-202, an interpenetrated dia framework, showed a stepwise C2H2 uptake at 195 K.97 However, the unique function of such a stepwise change was not investigated in detail.
As an early reported soft MOFs, ELM-11 adopts a 2D square-grid coordination framework. The trans-axial positions of the metal centres were occupied by two BF4− anions. Interestingly, the author found S-shaped adsorption isotherms of CO2, N2, O2, Ar, and CH4, caused by stepwise layer expansion. Utilizing this characteristic, ELM-11 was found to have a stepwise C2H2 uptake and trace C2H4 adsorption, resulting in efficient separation performance, when the partial pressure of C2H2 changed from 50 kPa to 10 kPa.98
To develop the function of the MOFs that involves stepwise opening, Zn2(bpdc)2(bpee), (bpdc = 4,4′-biphenyldicarboxylate; bpee = 1,2-bipyriylethylene) was investigated.71 The framework showed emerging stepwise adsorption isotherms of short alkanes at 298 K, as well as C2 hydrocarbons. Raman spectra of the C2H6 loaded sample revealed that the methyl group of C2H6 interacts with the uncoordinated COCOO on the bpdc ligand, causing a decrease in the dihedral angle (Δϕ = −2.0°) between the two rings of the bpdc ligand. Comparably, the dihedral angle increases (Δϕ = 1.5°) when C2H4 was adsorbed, reflecting a competitive alternative binding site with similar binding strength to that of the H-bonding. In other words, a higher pressure of C2H4 (π orbital plays a minimal role) is required to open the gate of such a framework. Furthermore, the chain length of short alkanes is crucial for the gate opening of the pressure dependence (C2 < C3 < C4). Therefore, tuning the H-bond strength is shown to be a strategy for optimizing the MOFs with the desired gate-opening pressure (Fig. 8a).
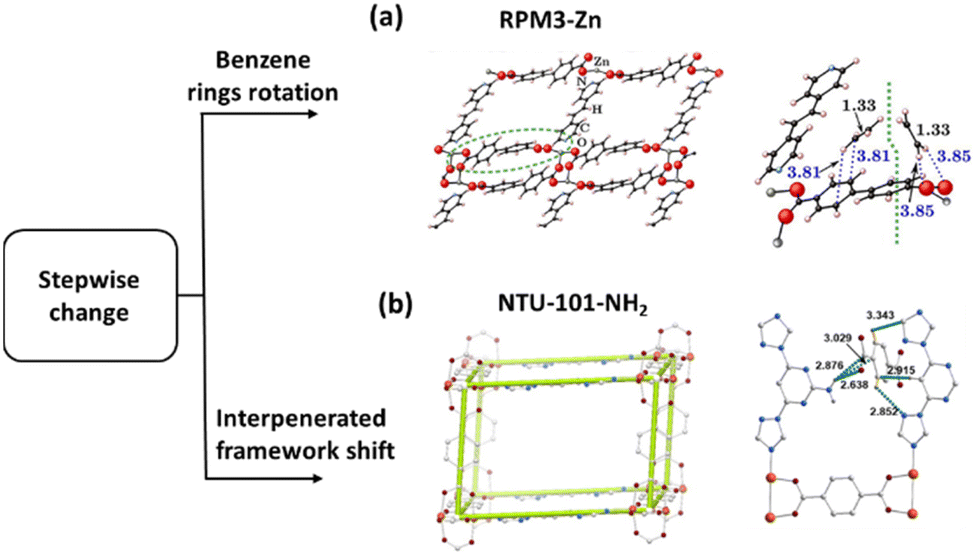 | ||
| Fig. 8 (a) The lateral perspective of the RPM3-Zn structure and C2H2, C2H4 and C2H6 binding sites at Zn2(bpdc)2(bpee). (b) The crystal structure of NTU-101-NH2. Reprinted with permission from ref. 71. Copyright 2012 American Chemical Society, ref. 62. Copyright 2025 John Wiley & Sons, respectively. | ||
Inspired by this finding and also by the dynamics of the interpenetrated MOF, we recently presented an approach to control the temperature-dependent dynamics in hydrogen-bonded interpenetrated frameworks. NTU-101-NH2, a single H-bond linked interpenetrated porous framework, exhibited stepwise structural dynamics in response to CO2.62 Importantly, this MOF showed gas and pressure dominated dynamics towards C2H6 (37 kPa, 328 K) and showed an inverse ability for C2H6/C2H4 separation at an elevated temperature of 328 K. This was due to the shift of the dynamics towards C2H6 (37 kPa, 328 K) and C2H4 (53 kPa, 328 K). This is because the displacement of the interpenetrating frameworks here requires a relatively weak stimulus, allowing the adsorption to be optimised in a higher temperature range. However, NTU-101, the precursor framework linked by three H-bonds, shows structural dynamics at very low temperatures. Therefore, hydrogen-bonded frameworks were expected to pave the way for the design of soft families capable of challenging separations at higher temperatures (Fig. 8b).
2.2.1.3 Gradual change. Sudden pore opening and stepwise pore opening both result from overcoming the clearly defined host–guest interactions, but the change is not always a sudden transition from a closed phase to an open phase. During the adsorption process, the pores or structure of MOF materials can be gradually adjusted to match the size and shape of the adsorbate molecules, resulting in a progressively enhanced host–guest interaction. This characteristic is different from the structural response of MOFs with a sudden global change and gradual global change.
Yet an earlier framework of CPL-1, one of the attractive adsorbents, has bridging pillar ligands and 2D porous sheets. The formed small channel with a pore size of 4.0 × 6.0 Å2 features abundant O atoms in a one-to-one fashion.99 Interestingly, the structure was shown to change gradually under increased pressure of C2H2, as the gradually enhanced host–C2H2 interaction promotes the changes in the structure.100 The same group then investigated the separation potential of CPL-1 on C2H4 and C2H6.101 The flexibility of the framework allowed stepped adsorption isotherms, particularly for C2H4 in the pressure range of 200 to 400 kPa, at 273 K; however, the C2H6 uptake is very low in the measured region. Shifting the target to the C3 hydrocarbons, CPL-1 showed a gradually changing thermo-responsive gate-opening behavior towards C3H6 in the temperature range of 273 to 288 K, but no structural response was found under C3H8.102
Due to the global framework change, the temperature assistant framework response usually occurred at relatively low temperature. To overcome this issue, a microporous MOF, [Zn3OH)2(btca)2] (JNU-1, H2btca = benzotriazole-5-carboxylic acid), with gas-induced fit was synthesized. The activated JNU-1 exhibits a 1D microporous channel of 8 Å diameter with an accessible open zinc site.103 A self-adaption framework change was observed upon increasing C2H2 loading, namely a gradually closed form. Gas-loading crystal analysis and modelling calculations showed a side-on configuration of the trapped C2H2 interacting with two adjacent open Zn sites. Furthermore, one C2H2 molecule interacts with other two additional molecules bound to Zn sites via diploe interactions. The gradual increase in adsorption leads to enhanced host–guest interactions, which in turn trigger framework shrinkage. This phenomenon is distinctly different from the general observation of gradual pore enlargement of the framework associated with increased pressure of the adsorbed gas (Fig. 9a).
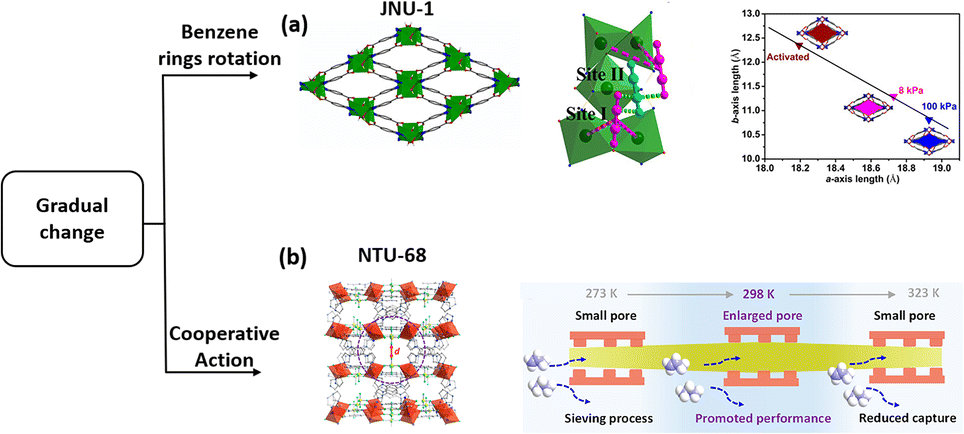 | ||
| Fig. 9 The structure of JNU-1 (a) and illustration of the accelerated separation process in NTU-68a (b). Reprinted with permission from ref. 103. Copyright 2019 John Wiley & Sons, ref. 40. Copyright 2023 American Chemical Society, respectively. | ||
For the gradual change in the framework, we have recently found a new system that exhibits a subtle expansion and contraction of the channel neck. NTU-68 was prepared via crystal conversion of NTU-65 in boiling water.40 Crystal analysis (from 200 to 340 K) revealed that the distance of the channel neck defined by F–F gradually expanded and then contracted to around 4 Å (turning point: 328 K), which falls between the centre dimensions of C3H6 (3.44 Å) and C3H8 (4.02 Å). Therefore, an unprecedented phenomenon of thermodynamically dominant C3H6 uptake, but kinetically regulated C3H8 adsorption behaviour, was observed. The calculated energy profile reveals that the maximum energy for passing through the cell is 42.8 kJ mol−1. However, C3H8 has to overcome two strong interactions, corresponding to 46.3 and 63.8 kJ mol−1, respectively, as C3H8 diffusion requires a configuration change. This temperature-dependent gradual change allows an enhancement in separation efficiency as the temperature increases from 273 to 298 K (Fig. 9b).
A coordination framework was synthesised and prepared from a butterfly-type ligand comprising isophthalic acid and phenothiazine-5,5-dioxide (OPTz) moieties (OPTz-IPA).72 After activation, the framework features a nanocage connected by eight nanochannels. Interestingly, one OPTz moiety and one isophthalic unit adopt a face-to-face configuration, forming a gate of 3 Å inside the channel. Therefore, the thermal flipping of the OPTz units provides a gate function that enables different kinetic responses towards C2H4 and C2H6, as well as O2/Ar. It is worth noting that the ability for preferred adsorption is strongly temperature-dependent, of which the Tmax is as low as 270 K for the adsorption separation of C2H4 and C2H6. The function of such thermal local-dynamics was extended into the separation of water isotopologues, which have extremely similar physicochemical properties (Fig. 10a).105
 | ||
| Fig. 10 (a) Simulation of the structure of activated Cu(OPTz)-loaded C2H4 and the diffusion structure between the interlayer and intralayer. (b) Crystal structure of JNU-3a with molecular pockets and schematic diagram of molecular pockets. (c) The structure of ZU-33 and bonding sites for loaded C3H4 and C3H4(PD). Reprinted with permission from ref. 72. Copyright 2019 American Association for the Advancement of Science, ref. 106. Copyright 2024 Springer Nature Limited, ref. 107. Copyright 2022 Springer Nature Limited, respectively. | ||
Recently, a 3D porous MOF, named JNU-3, was prepared by using a branched ligand with carboxylate, triazole and pyridine coordination sites. JNU-3 possesses a 1D channel with dimensions of approximately 4.5 A × 5.3 Å. The yielded cross-sectional area (23.85 Å2) is slightly larger than that of the minimum cross-sections of C3H6 (19.34 Å2) and C3H8 (18.17 Å2). In addition, a molecular pocket with an aperture size of 3.7 Å is lined up on both sides of the 1D channel. Given the larger kinetic parameters of C3H6 and C3H8, it appears improbable that these pockets can accommodate them. However, the tilting and rotation of the “guard” moiety of aromatic rings on the ligand can enlarge the aperture (4.4 for C3H6 and 4.7 Å for C3H8) of the pocket, facilitating the adsorption of C3H6 and C3H8. Consequently, the unique structure can be regarded as an orthogonal-array dynamic molecular sieve, allowing both fast adsorption–desorption kinetics and large capacity. The presence of a gas-responsive pocket has been demonstrated to facilitate the purification of C2H4 from C2–C4 mixtures in one-step (Fig. 10b).106 In addition, this strategy has been expanded in HAF-1, which features channels and shrinkage throats, the latter being defined as narrower channels that connect the main channels and a molecular pocket.55
As a molecule separator, GeFSIX-14-Cu-i (ZU-33) can also exhibit an adaptable response to guest molecules.107 The coordination of 4,4′-azopyridine with a GeF62− anion provides ZU-33 with local rotational flexibility, which plays a crucial role in the recognition of C2H2, C3H4, and C3H4 (PD), and avoids the competitive adsorption of olefins and paraffins. The responsive properties to different stimuli endow this MOF with multiple regulations for challenging separations (Fig. 10c).
 | ||
| Fig. 11 Crystal structure of loaded C3H6 and C3H8 samples: (a) NTU-85-WNT⊃C3H6 and (b) NTU-85-WNT⊃C3H8 and (c, d) detailed H-bonding interactions of C3H6 in one-dimensional nanochannels. Reprinted with permission from ref. 37. Copyright 2023 American Chemical Society. | ||
3. Applications
In recent years, there has been significant research interest in the field of adsorption separation of light hydrocarbons using MOFs. According to the demand for highly pure olefins, the separation process can be broadly classified into olefin-selective adsorption and alkane-selective adsorption.108–112 To obtain pure olefins from an olefin-selective adsorption process, further desorption–adsorption cycles via inert gas sweeping or a vacuum pump are required. In contrast, alkane-selective adsorption has been shown to produce pure olefins directly, a process that is notably more energy efficient. This section will discuss recent research advances in the purification of C2H4 and C3H6 from light hydrocarbon mixtures, particularly those with the same carbon numbers, in recent years.2,111,113 Furthermore, we compared MOFs with framework dynamics induced by light hydrocarbons (Table 4), and the separation performance and the isosteric heat of adsorption (Qst) of MOFs used for light hydrocarbon separation (Table 5).| MOFs | Reasons for phase changing | P gas for phase changing (kPa) | Temperature (K) | Ref. |
|---|---|---|---|---|
| ZIF-7 | Configuration change in benzimidazole | C2H6: 0.0008 | 298 | 38 |
| C2H4: 0.0012 | ||||
| NTU-88 | Pyridine ring rotation | C3H4: 1.7 | 273 | 69 |
| C3H6: no change | ||||
| GeFSIX-dps-Zn | Pyridine ring rotation | C3H4: 0.13 | 298 | 114 |
| GeFSIX-dps-Cu | C3H4: 0.015 | |||
| NTU-65-CoTi | Benzene ring rotation | C3H4: 1.6 | 273 | 51 |
| C3H6: 19.4 | ||||
| C3H8: 57.2 | ||||
| Zn2(bpdc)2(bpee) | bpdc benzene ring rotation | C2H2: 0.2 | 298 | 71 |
| C2H4: 0.4 | ||||
| C2H6: 0.26 | ||||
| MFM-202 | Benzene ring rotation | C2H2: 2.7 | 195 | 97 |
| C2H4: 4.0 | ||||
| C2H6: no change | ||||
| NTU-101-NH2 | Shifting of the interpenetration framework | C2H6: 36 | 293 | 62 |
| C2H4: 57 | ||||
| X-dia-1-Ni | Pyridine ring and pyridine ring rotation | C2H6: 51.7 | 273 | 56 |
| ZU-13 | Pyridine ring rotation | C3H4: 0.05 | 298 | 115 |
| NTU-68 | Benzene ring rotation | Gradual change | 40 | |
| Cu(OPTz) | Phenothiazine-5,5-dioxide rotation | Gradual change | 72 | |
| TYUT-17 | 3-Methylisonicotinic rotation | C2H6: <0.1 | 298 | 66 |
| JNU-3 | Pyridine ring rotation | Gradual change | 65 | |
| ZU-33 | 4,4′-azopyridine ligand and GeF62− anion rotation | CH2CCH2: 0.002 |
298 | 107 |
| NTU-85 | Dynamic of water nanotubes | Gradual change | 298 | 37 |
| MOFs | Feed gas | Velocity of feed gas (mL min−1) and working temperature (K) | Q st (kJ mol−1) | Purity and productivity | Ref. |
|---|---|---|---|---|---|
| UTSA-280 | C2H4/C2H6 (1/1, v/v) | 2.0, 298 | C2H4: 34.1 | Highly pure C2H4, 1.86 mol kg−1 | 70 |
| ZU-901 | C2H6/C2H4 (1/1, v/v) | 1.7, 273 | C2H4: 24.85 | C2H4, 95%, — | 123 |
| Co(aip)(pyz)0.5 | C2H4/C2H6 (1:1, v/v) |
1.0, 298 | C2H4: 33.6 | C2H4 > 97%, 19.1 L kg−1 | 117 |
| Fe2(O2)(dobdc) | C2H6/C2H4 (1/1, v/v) | 5.0, 298 | C2H6: 66.8 | C2H4, 99.99%, 0.79 mmol g−1 | 46 |
| Ni-MOF 2 | C2H6/C2H4 (1/1, v/v) | 2.0, 298 | C2H6: 23.6 | C2H4 > 99.95%, 12 L kg−1 | 68 |
| C2H4: 21.4 | |||||
| TYUT-17 | C2H6/C2H4 (1/9, v/v) | 2.0, 298 | C2H6: 27.1 | C2H4 > 99.99%, 77.4 L kg−1 | 66 |
| C2H4: 21.4 | |||||
| ZUL-100 | C2H2/C2H4 (1/99, v/v) | 1.25, 298 | C2H2: 65.3 | C2H4 > 99.9999%, 121.2 mmol g−1 | 119 |
| C2H4: <40 | |||||
| Cu(OH)INA | C2H2/C2H4 (1/99, v/v) | 2.0, 298 | C2H2: 36.1 | C2H4 > 99.99%, 167 mL cm−3 |
120 |
| C2H4: 29.6 | |||||
| MOF-303 | C2H2/C2H4/C2H6 (1/9/90, v/v/v) | 1.25,296 | C2H2: 31.7 | C2H4 > 99.95%, 1.35 mmol g−1 | 123 |
| C2H4: 24.3 | |||||
| C2H6: 25.1 | |||||
| Al-PyDC | C2H2/C2H4 (1/99, v/v) | 1.25, 296 | C2H2: 35.3 | C2H4 > 99.999%, 7.93 mmol g−1 | 83 |
| C2H4: 27.8 | |||||
| C2H6: 30.1 | |||||
| MOF-808-Bzz | C2H2/C2H4/C2H6 (1/1/1, v/v/v) | 1.0, 298 | C2H2: 32.36 | C2H4 ≥ 99.95%, — | 53 |
| C2H4: 26.43 | |||||
| C2H6: 29.87 | |||||
| MAC-4 | C2H6/C3H6/C2H4 (2/10/25, v/v/v) | 7.0, 298 | C2H6: 22.7 | C2H4 ≥ 99.9% 27.4 L kg−1 | 132 |
| C2H4: 17.1 | 1C3H6 ≥ 99.5% 36.2 L kg−1 | ||||
| C3H6: 25.3 | |||||
| MOF-1 | C2H6/C2H2/C2H4 (3/3/10, v/v/v) | 7.0, 298 | C2H6: 31.8 | C2H4 > 99.9%, 4.6 L kg−1 |
133 |
| C2H4: 28.7 | |||||
| C3H6: 23.2 | |||||
| Ni-dcpp-bpy | C2H2/CH4 (1/1, v/v) | 6.0, −298 | C2H2: 33.8 | C2H4 > 99.9%, 6.1 L kg−1 |
134 |
| C2H4: 26.7 | |||||
| SNNU-33 | C2H2/CH4 (5/2, v/v) | 5.0, 298 | C2H2: 40.4 | CH4 > 99.95%, 3.5 mmol g−1 | 135 |
| C2H4: 32.9 | |||||
| NU-57 | C3H8/C3H6 (1/1, v/v) | 3.0, 298 | C3H8: 33.0 | C3H6 > 99.5%, 34.2 L kg−1 | 130 |
| C3H6: 28.5 | |||||
| JNU-3a | C3H6/C3H8 (1/1, v/v) | 1.0, 298 | C3H8: 34.6 | C3H6 > 99.5%, 34.2 L kg−1 | 65 |
| C3H6: 38.9 | |||||
| HIAM-301 | C3H6/C3H8 (95/5, v/v) | 5.0, 298 | C3H6: 27.0 | C3H6 > 99.6%, 38.5 cm3 g−1 | 64 |
| ZJU-75 | C3H6/C3H8 (1/1, v/v) | 2.0, 296 | C3H8: 33.1 | C3H6 > 99.5% 18.7 L kg−1 | 90 |
| C3H6: 65.9 | |||||
| FDC-4 | C3H6/C3H8 (1/1, v/v) | 4.0, 300 | C3H6: 35.0 | C3H6 99.7%, 19.5L kg−1 |
74 |
| GeFSIX-dps-Cu | C3H4/C3H6 (1/9, v/v) and C2H2/C2H4 (1/9, v/v) | 2.0, 298 | C2H2: 56.3 | C2–C3 alkynes > 99.99%, — | 114 |
| Sql-NbOFFIVE-bpe-Cu-AB | C3H6/C3H8 (1/99, v/v) | 1.0, 298 | C3H4:69.0 | C3H6 > 99.9%, 118 mmol g−1 | 136 |
| C3H6 : 53.0 | |||||
| Zn-BPZ-TATB | C2H6/C3H6/C2H4 (2/10/25,54 v/v/v) | 8.0, 298 | C2H6: 23.1 | C3H6 99.5%, 38.2 L kg−1 | 3 |
| C2H4: 18.3 | C2H4 99.9%, 12.7 L kg−1 | ||||
| C3H6: 28.1 | |||||
| Zn-MOF | C3H6/C2H4(1/9, v/v) | 5.0, 298 | C2H4: 25.8 | C2H4 (>99.9%), 98.71 L kg−1 |
137 |
| C3H6: 33.3 | |||||
| Tb-MOF-76(NH2) | C2H6/C2H4/Ar (5/5/90, v/v/v | 7.0, 298 | C2H4: 30.9–29.5 | C2H4, >99.9%, — |
138 |
| C2H6: 32.8–30.7 | |||||
| X-dia-1-Ni0.89Co0.11 | (C2H6/C2H4: 1/9, v/v) | 10, 298 | — | C2H4, >99.9%, — |
56 |
| Y-abtc | C3H8/C3H6: 5/95, v/v) | 4.0, 298 | C3H6: 57.4 | C2H4, 99.5%, — | 6 |
| NKMOF-1-M (M = Cu or Ni) | C3H4/CH2CCH2/C3H6 (0.5/0.5/99, v/v/v) |
2.0, 298 | C3H4: 65.1 (Ni), 67.2 (Cu) | C3H6 99.996%, — | 52 |
| CH2CCH2: 54.0 (Ni), 45.2 (Cu) |
|||||
| C3H6: 38.0 (Ni), 37.2 (Cu) | |||||
| Ca-based MOF | C3H4/CH2CCH2/C3H6 (0.5:0.5:99, v/v/v) |
2.7, 298 | C3H4: 55.5 | C3H6 99.95%, — | 139 |
| CH2CCH2: 51.7 |
|||||
| ZNU-2 | C3H4/C3H6 (1/9 or 1/99, v/v) | 2.0, 298 | C3H4: 43.2 | C3H6 > 99.996%, 37.8 or 52.9 mol kg−1 | 140 |
| C3H6: 35.5 | |||||
| NTU-88 | C3H4/C3H6 (1/1, v/v) | 2.0, 298 | C3H4: 44 | C3H6 > 99.95%, — | 69 |
| NTU-101-NH2 | C3H4/C3H6 (1/99, v/v) | 4.0, 298 | C3H4: 43.5 | C3H6, 99.95%, 15.7 mL g−1 | 62 |
| JNU-9-CH3 | C3H4/CH2CCH2/C3H6/C3H8 (1/1/1/1, v/v/v/v) |
2.0, 298 | C3H4: 34.6 | C3H6 ≥ 99.99%, — | 60 |
| CH2CCH2: 31.9 |
|||||
| C3H6: 27.4 | |||||
| C3H8: 31.1 | |||||
| PCP-IPA | C3H6/C3H8 (1/1, v/v) | 3.7, 298 | C3H8: 50.94 | C3H6 (99.99%), 15.2 L kg−1 | 129 |
| C3H6: 43.36 | |||||
| CuZrF6-TPA | C3H4/CH2CCH2(1/1, v/v) |
0.8, 298 | C3H4: 46.1 | CH2CCH2 > 99.95%, 4.7 mol L−1 |
141 |
| CH2CCH2: 37.1 |
|||||
| NbOFFIVE-1-Ni | C3H6/C3H8 (1/1, v/v) | 4.0, 298 | C3H6: 57.4 | C3H6, ∼0.6 mol kg−1 | 32 |
| NTU-85-WNT | C3H6/C3H8 (1/1, v/v) | 0.5, 298 | C3H6:49.9 | C3H6, > 99.8%, 1.6 mL mL−1 | 37 |
| NTU-68 | C3H6/C3H8 (1:1, v/v) |
1.0, 298 | C3H6: 42.8 | C3H8 > 99.95%, — | 40 |
| DL-mal-MOF | C3H6/C3H8 (1/1 or 95/5 v/v) | 2.0, 298 | C3H6: 64.4 | Highly pure C3H6, 2.1 L kg−1 | 27 |
3.1 C2H4 harvesting from the desorption process
 | ||
| Fig. 12 (a) The crystal structure of guest-free UTSA-280, single-component sorption isotherms of C2H4 and C2H6 at 298 K and breakthrough curves from different scales for an equimolar C2H4/C2H6 mixture at 298 K and 1bar. (b) The internal structures of ZU-901, adsorption isotherms of C2H4 and C2H6 at different temperatures and high pressure, and breakthrough curve of the fresh and regenerated sample via vacuum desorption. (c) The structures of Co(aip)(pyz)0.5, C2H4 and C2H6 adsorption isotherms of Co(aip)(pyz)0.5 at 298 K, and experimental column breakthrough curves for C2H4/C2H6 equimolar binary mixtures at 298 K. Reprinted with permission from ref. 70. Copyright 2018 Springer Nature Limited, ref. 116. Copyright 2023 John Wiley & Sons, ref. 117. Copyright 2023 Elsevier Inc, respectively. | ||
ZU-901 exhibits a unique ‘S’ shaped C2H4 adsorption curve, providing a high C2H4 working capacity of up to 1.36 mmol g−1, at 273 K and 1 bar.116 Interestingly, the material showed a C2H4 uptake of 0.19 mmol g−1 at 0.1 bar and 273 K, followed by a rapid increase to 1.55 mmol g−1 at 1 bar, signifying the C2H4 working capacity of 1.36 mmol g−1. A similar adsorption phenomenon was observed upon increasing the temperature and pressure, to 298 K and 3 bar, respectively. Comparably, the C2H6 capacity of ZU-901 is as low as 0.26 mmol g−1 under both conditions. In breakthrough experiments, the clear separation interval confirmed the ability for C2H4/C2H6 separation. Thanks to the relatively low binding energy, adsorbed C2H4 can be rapidly regenerated and collected with >95% purity. Aspen adsorption simulation revealed that 99.51% purity of C2H4 could be obtained with 75% recovery from a two-bed pressure swing adsorption process. In addition, this material can be prepared via stirred, associated with mother liquor circulation (Fig. 12b). Meanwhile, some other MOFs, such as Co(aip)(pyz)0.5 (Fig. 12c)117 and MOF-808-Bzz,53 have been observed to demonstrate selective capture of C2H6 from C2H4-containing mixtures. Furthermore, we have summarized the top ten C2H4/C2H6 adsorption ratios, as illustrated in Fig. 16a. Notably, MAF-42 achieves an impressive adsorption ratio, up to 301 at 1 bar.118 This summary serves as a valuable reference for efficient light hydrocarbon separation techniques.
3.2 C2H4 harvesting from the adsorption process
 | ||
| Fig. 13 (a) Pore geometry of ZUL-100 with an intralayer, C2H2 and C2H4 adsorption isotherms at 298 K, and experimental column breakthrough curves for the C2H2/C2H4 mixture at 298 K and 1bar. (b) Structure and one-dimensional channel of Cu(OH)INA, adsorption isotherms of C2H2 and C2H4 at various temperatures, and breakthrough experiment with a C2H2/C2H4 mixture at a flow rate of 2 mL min−1 at 298 K. (c) Single-crystal structure of ZJU-300, adsorption isotherms for C2H2 and C2H4 at 296 K, and experimental column breakthrough curves for 1/99 C2H2/C2H4 separation at a flow rate of 5 mL min−1 under ambient conditions. Reprinted with permission from ref. 119. Copyright 2020 Springer Nature Limited, ref. 120. Copyright 2024 John Wiley & Sons, ref. 121. Copyright 2023 The American Association for the Advancement of Science, respectively. | ||
Cu(OH)INA exhibited remarkable C2H2 adsorption capacity performance within the temperature range of 272–313 K, even at lower pressures.120 At 0.01 bar and 298 K, the adsorption amount of C2H2 reached 44.01 cm3 cm−3, while the adsorption amount of C2H4 was found to be considerably lower. The adsorptive selectivity was calculated to be 105–41 and 71–35 at two distinct temperatures (298 K and 313 K), which confirmed the potential of the material for C2H2/C2H4 (1/99, v/v) separation. Breakthrough experiments further validated the material's separation ability by obtaining high purity C2H4 (>99.99%) with a productivity of 167 mL cm−3. Moreover, after the increase in flow rate, the separation efficiency (165 mL cm−3) remained close to that of the low flow rate. Furthermore, the large-scale synthesis, at least, can be scaled up to 8 L under stirring and 1.31 kg of product (91% yield) can be obtained (Fig. 13b).
Despite the fact that many of the MOFs have been the focus of research in regard to their potential for selective C2H2/C2H4 capture, only a limited number of them have been shown to achieve a balance between C2H2 uptake and C2H2/C2H4 selectivity. SIFSIX-2-Cu-i with periodic SiF62− ions demonstrated a high C2H2 adsorption capacity (2.1 mmol g−1, 0.05 bar, 298 K) and selectivity (39.7 to 44.8).34 In contrast, SIFSIX-1-Cu exhibited exceptionally high C2H2 uptake (8.5 mmol g−1) at 298 K and 1.0 bar, yet the superior C2H4 uptake resulted in moderately high C2H2/C2H4 separation selectivity (7.1 to 10.6). These values reflected the emerging separation performance of C2H2/C2H4. In the breakthrough experiments, highly-pure C2H4 broke the sample beds of SIFSIX1-Cu and SIFSIX-2-Cu-i at first, while 0.38 and 0.73 mmol g−1 of C2H2 was captured by from the 1/99 mixture, respectively. After replacing the linear ligand from a 2-c to 4-c linker, ZJU-300 with densely decorated SiF62− ions also exhibited a promising C2H2 uptake of 3.23 mmol g−1 (0.01 bar, 296 K). This result is substantially higher than that of all the SIFSIX materials (Fig. 13c).121
When the pressure was increased to 1 bar, the C2H2 uptake increased to 5.4 mmol g−1, while the C2H4 uptake was only 2.39 mmol g−1. It is noteworthy that the C2H2/C2H4 (1/99) selectivity is as high as 1672. This observation was subsequently confirmed through dynamic breakthrough experiments, which revealed that the C2H4 productivity reached 436.7 mmol g−1. Moreover, the material maintained good stability under humidity and an acidic gas atmosphere.
 | ||
| Fig. 14 (a) Structures of Fe2(O2)(dobdc), adsorption isotherms of C2H6 and C2H4 at 298 K, and experimental column breakthrough curves for a C2H6/C2H4 mixture at 298 K and 1.01 bar. (b) The three-dimensional open framework of Ni-MOF 2, C2H4 and C2H6 single-component adsorption isotherms at 298 K, and dynamic breakthrough curves of equimolar C2H6/C2H4 gas mixtures at 298 K and 1 bar. (c) The structure TYUT-17, single-component gas adsorption isotherms at 298 K, and breakthrough curves of TYUT-17 for C2H6/C2H4 (v/v, 10/90) mixtures at 298 K and 1 bar. (d) C2H6 adsorption induced the structural transformation of X-dia-1-Ni, C2H4 and C2H6 adsorption isotherms for X-dia-1-Ni0.89Co0.11 at 273 K, and cyclic breakthrough separation experiments for C2H4/C2H6 (1/9) mixtures performed at 100 kPa and 263 K. Reprinted with permission from ref. 46. Copyright 2018 The American Association for the Advancement of Science, ref. 63. Copyright 2023 John Wiley & Sons, ref. 66. Copyright 2024 John Wiley & Sons, ref. 56. Copyright 2024 American Chemical Society, respectively. | ||
The two isostructural MOFs Ni-MOF-1 and Ni-MOF-2 have also been investigated for C2H6/C2H4 separation.63 For Ni-MOF-1, nearly the same adsorption isotherms of C2H6 (113 cm3 g−1) and C2H4 (116 cm3 g−1) were observed at 298 K and 1 bar. However, Ni-MOF 2 demonstrated a substantially higher adsorption of C2H6 (133 cm3 g−1) over C2H3 (105 cm3 g−1). Along with an uptake difference of 40 cm3 g−1 at 0.5 bar, Ni-MOF 2 can efficiently separate the equimolar C2H6/C2H4 mixture gas in dynamic penetration experiments, yielding a C2H4 productivity of 12 L kg−1. In addition, cycling experiments demonstrated that Ni-MOF-2 has good reproducibility for this separation (Fig. 14b).
Different from the previously mentioned rigid MOFs, TYUT-17, a flexible-robust framework, exhibited a selective adsorption tendency for C2H6.66 TYUT-17 demonstrated a higher adsorption capacity of 62.6 cm3 g−1 for C2H6, at 298 K and 0.1 bar; however, the C2H4 uptake was found to be 18.7 cm3 g−1, providing an exceptional C2H6/C2H4 selectivity (6.4) and uptake ratio (3.3). It was observed that an increase in temperature resulted in a shift of the gate-opening phenomenon to a high-pressure region, but the PC2H4 always lagged behind the PC2H6, indicating the reality of sample regeneration. Following the introduction of a feed gas comprising C2H6/C2H4 mixtures to the packed TYUT-17, the observation of different retention times indicated unambiguous separation. The productivity of C2H4 of >99.9% purity was recorded at 77.4 L kg−1. Furthermore, the gate-opening of TYUT-17 facilitated the separation of the C2H6/C2H4 mixture at 313 K (Fig. 14c).
To promote the separation temperature, an implication of saving energy in the separation process, we developed two hydrogen-bonded interpenetrated frameworks, NTU-101 and NTU-101-NH2.62 A three H-bond linked interpenetrated porous framework, NTU-101 exhibited a structural response to C2H6 and C2H4 in the temperature range of 288 to 303 K. Comparably, the single H-bond connected framework of NTU-101-NH2 exhibited a structural response to C2H6 and C2H4 in a wider temperature range of 288 to 333 K, particularly the gate-opening pressure under C2H6 is always earlier than that of C2H4. Consequently, NTU-101-NH2 is capable of separating the C2H6/C2H4 (1/9, v/v) mixtures at a relative higher temperature of 328 K. To our knowledge, this is the first example that can selectively capture C2H6 from C2H4 at such a higher temperature and yielded a productivity of polymer-grade C2H4, of up to 15.7 mL g−1. Meanwhile, the robust NTU-101-NH2 can be prepared on a large scale through the stirring method at room temperature.
In addition, the flexible dia coordination network of X-dia-1-Ni0.89Co0.11 exhibited preferred gate-opening for C2H6, but not for C2H4 at 263 K.56 The unique structural change driven the C2H6 adsorption sharply increased at a pressure of 50 kPa, and the maximum adsorption capacity reached 131.4 cm3 g−1. However, the structure did not show evident gate-opening, thus providing a low C2H4 uptake of 44.8 cm3 g−1. The ability for harvesting high-purity (99.9%) C2H4 directly was validated by dynamic breakthrough experiments at 263 K (C2H6/C2H4: 1/9, v/v) (Fig. 14d).
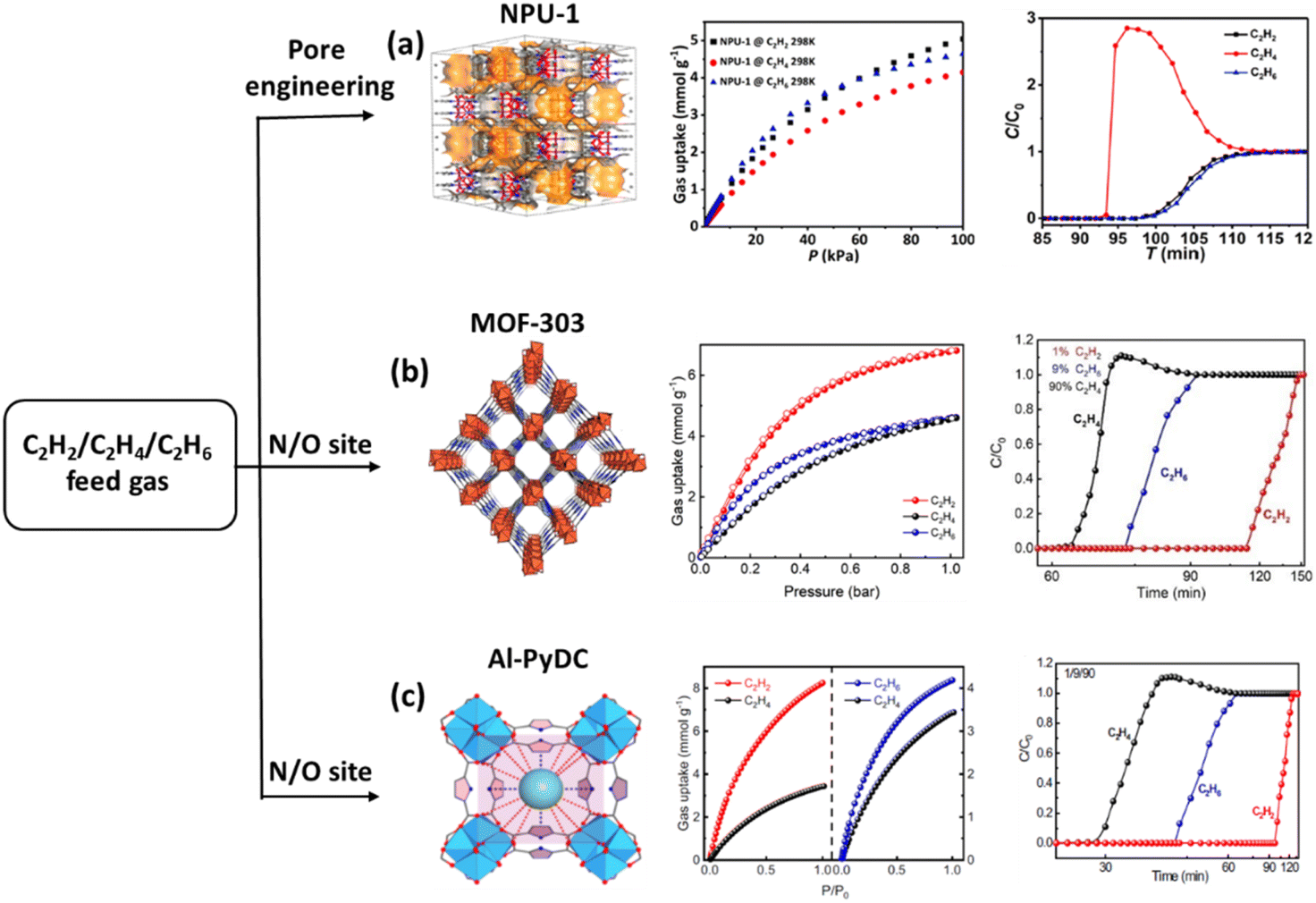 | ||
| Fig. 15 (a) NPU-1 three-dimensional structure; C2 gas sorption isotherms of NPU-1 at 298 K; and experimental breakthrough curves at 298 K for C2 (1:1:1mixture) separation. (b) The framework of MOF-303, C2 adsorption isotherm of MOF-303 at 296–313 K, and experimental column breakthrough curves of MOF-303 for C2 mixtures under ambient conditions. (c) Multiple supramolecular binding sites in the pores of Al-PyDC, C2 adsorption isotherms of Al-PyDC at 296 K, and experimental column breakthrough curves of Al-PyDC for C2 mixtures under ambient conditions. Reprinted with permission from ref. 122. Copyright 2021 American Chemical Society, ref. 123. copyright 2023 John Wiley & Sons, ref. 83. Copyright 2023 Springer Nature Limited, respectively. | ||
The aluminium-based MOF (MOF-303) is a stable, low-cost, and easily scalable separation material.123 It also demonstrated preferred C2H2 (7.94 mmol g−1) and C2H6 (5.01 mmol g−1) adsorption compared with that of C2H4. In the dynamic breakthrough experiment, the binary mixture C2H2/C2H4 (1/99, v/v) can be separated, with an outlet effluent C2H4 yield of 5.07 mmol g−1 and a purity greater than 99.95%. Furthermore, the yield of the C2H4/C2H6 binary mixture is also as high as 0.56 mmol g−1, and the purity is greater than 99.95%. In the ternary mixture (1/9/90, v/v/v), the productivity of C2H4 is as high as 1.35 mmol g−1 (>99.95%), exhibiting excellent separation performance (Fig. 15b).
The low polarity O/N supramolecular binding sites allow Al-PyDC to show both higher C2H2 (8.24 mmol g−1) and C2H6 (4.20 mmol g−1) uptakes in comparison to C2H4 (3.44 mmol g−1) at 296 K and 1 bar,83 along with both a good C2H2/C2H4 (1/99, v/v) selectivity of 4.3 and C2H6/C2H4 (50/50, v/v) selectivity of 1.9. In the dynamic breakthrough experiments, both C2H2/C2H4 and C2H6/C2H4 binary mixtures can be separated. Therefore, Al-PyDC can separate the C2H2/C2H4/C2H6 ternary mixtures for getting polymer-grade C2H4 in one-step with a productivity of 1.61 mmol g−1. Furthermore, the system demonstrates consistent recyclability over 20 cycles, exhibiting no decline in performance under acidic gas conditions (Fig. 15c).
3.3 C3H6 harvesting from the desorption process
In the presence of an orthogonal-array dynamic molecular sieve, both stepwise C3H6 and C3H8 adsorption isotherms were observed in JUN-3a.65 The rapid uptake at low pressure gradually diminished and the step shifted to higher pressures following increased pressure, a phenomenon attributable to the local dynamic of the molecular pocket. Importantly, at an earlier gate opening pressure, the C3H6 uptake reached 58.6 cm3 g−1 at 303 K and 1 bar, which is considerably than that of C3H8 (42.5 cm3 g−1). The density of C3H6 was calculated to be 404 g L−1 based on the pore volume, reflecting its good packing ability of C3H6 compared to the density of gaseous C3H6 (1.707 g L−1). Breakthrough curves revealed that JNU-3a can separate equimolar C3H6/C3H8 at 303 K. C3H8 was eluted from the packed column at about 70 min, while C3H6 penetrated through the column after 35 min. In the desorption stage, JNU-3a was able to obtain high-purity C3H6 with a productivity of 34.2 L kg−1 (Fig. 17b).
In the context of quasi-discrete cavities, DL-mal-MOF demonstrated rapid and different uptakes of C3H6 and C3H8 in the low pressure region, with uptake values of up to 1.68 mmol g−1 and 1.21 mmol g−1 at 298 K and 1 kPa.27 In contrast, both L-mal-MOF and D-mal-MOF exhibited nearly the same adsorption isotherms for C3H6 and C3H8. Remarkably, DL-mal-MOF demonstrated both a high Henry's selectivity of 5.86 and kinetic selectivity of 114.2. The observed disparity in diffusion rates facilitates the dynamic separation of C3H6/C3H8 mixtures (50/50, or 95/5, v/v) at 298 K. During the desorption stage, the polymer-grade C3H6 was attained, with a yield of 2.1 L kg−1 (Fig. 17c).
Dependent on the cooperative global and local dynamics, FDC-4 exhibited gate-opening during C3H6 adsorption, yielding an uptake of 122 cm3 g−1 at 300 K.74 However, C3H8 adsorption in this narrow channel adopts a diffussion controlled manner, thereby resulting in the very low uptake of C3H8 being observed. The substantial differences in both uptake capacity and adsorption kinetics enabled the dynamic separation of an equimolar C3H6/C3H8 mixture, thereby revealing sieving performance and producing C3H6 (19.5 L kg−1) with a purity of 99.7% (Fig. 17d).
NTU-85-WNT, characterized by localized dynamics of a water nanotube within a rigid framework, exhibited rapid C3H6 uptake within the initial pressure range, and reached 20.9 mL mL−1.37 Conversely, C3H8 uptake was observed to be negligible (0.13 mL mL−1). The clear cut-off adsorption yields an extremely high adsorption selectivity of 1570 at 298 K and 1 bar. This sieving phenomenon is very similar to that of the rigid separator of KAUST-1, but the underlying mechanism is totally different. Breakthrough experiments substantiated the exclusive adsorption of C3H6 by NTU-85-WNT, with C3H8 eluting out of the packed column initially. The confined effect of the water nanotube enables the harvesting of adsorbed C3H6 at the desorption stage with a high purity of 98.8%. However, the lower porosity of the water nanotube limits the C3H6 productivity (1.6 mL mL−1) at a certain content (Fig. 17e).
The same group developed a new concept of delicate softness in a temperature-responsive MOF (NTU-68) for efficient separation of C3H6/C3H8 in the next year.40 Quantitatively, NTU-68 exhibited a pronounced uptake of C3H6 (33.3 cm3 g−1 at 298 K) at 2 kPa, which exceeds the total uptake of KAUST-7 at 1 bar. The calculated density of C3H6 is as high as 565.9 mL mL−1, closely approaching the density of liquid C3H6 (606 mL mL−1). In contrast, the MOF exhibited a markedly lower C3H8 uptake (7.5 cm3 g−1) at 298 K and 1 bar. As illustrated by the changing trend (in the temperature range of 273 to 323 K), the temperature-responsive delicate softness around the channel neck results in a thermodynamic preferred C3H6 adsorption (the higher the temperature, the lower the uptake), but a kinetic dominated C3H8 uptake (the higher the temperature, the higher the uptake). Consequently, the breakthrough separation efficiency of this mixture was found to be twofold enhanced upon increasing the temperature from 273 to 298 K. Notably, the separation process remained unaffected by the presence of general impurities.
The ftw-type MOFs consisting of a hexanuclear cluster M6 (M = Zr4+, Hf4+, Ce4+, Y3+) and a four-coordination linker have potential for molecular sieving due to the large cage-like cavities interconnected by narrow windows. For example, the iso-configurational MOFs ftw-MOF-ABTC and HIAM-301 both exhibit excellent molecular sieving in the separation of C3H6/C3H8 mixtures, but the mechanisms of separation are different.64,126 The MOF-ABTC achieves molecular sieving by adjusting the length of the ligand only to change the window size and thus its pore size. In contrast, HIAM-301 has a highly distorted pore due to the inconsistency of the plane created by the mutual rotation of the octahedra and the large aspect ratio of the H4eddi linker; in addition, the use of Y6 clusters instead of Zr6 clusters changes the framework from electrically neutral to an anionic framework and the presence of counterions in the cavities provides additional pore size modulation. The two synergistically regulate the pore size of HIAM-301 to achieve efficient sieving of C3H6/C3H8. In terms of separation performance, HIAM-301 showed better separation performance compared to ftw-MOF-ABTC, with 3.16 mmol g−1 adsorption for C3H6 and negligible adsorption for C3H8 (<0.3 mmol g−1) at 298 K and 1 bar, enabling the complete separation of C3H6 and C3H8. Breakthrough experiments at room temperature also showed that the dynamic adsorption capacity of HIAM-301 for C3H6 is 46.4 cm3 g−1, which enabled the production of polymer-grade C3H6. The single-component adsorption curves and breakthrough experiments of ftw-MOF-ABTC showed that the differences in adsorption amounts of ftw-MOF-ABTC for C3H6 and C3H8 are not significant (2.3 mmol g−1, 2.46 mmol g−1), but the adsorption kinetics of C3H6 is faster than that of C3H8 under the low-pressure condition, so that C3H6/C3H8 is effectively separated in the breakthrough experiment (Fig. 18a and b). Furthermore, we also compared the top ten C3H6/C3H8 adsorption ratios, as shown in Fig. 16b, where NTU-85-WNT reaches an adsorption ratio of 160, which has the potential to completely separate the C3H6/C3H8 mixture.37 This summary provides a reference for efficient light hydrocarbon separation.
 | ||
| Fig. 16 The top ten in terms of uptake ratios for C2H4/C2H6 (a) and C3H6/C3H8 (b). | ||
 | ||
| Fig. 17 (a) The crystal structure of KAUST-7, adsorption isotherms for pure C3H8, pure C3H6, and C3H6/C3H8 equimolar mixtures at 298 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 298 K. (b) The crystal structure of C3H6@JNU-3a, adsorption isotherms for pure C3H8, pure C3H6, and C3H6/C3H8 equimolar mixtures at 303 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 303 K. (c) The ligand racemization strategy is illustrated, adsorption isotherms of C3H6 and C3H8 on DL-mal-MOF and breakthrough experimental curves of C3H6/C3H8 (50/50, v/v) mixtures at 298 K. (d) The crystal structure of FDC-4a, adsorption isotherms for C3H6 and C3H8 at 240 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 300 K. (e) The crystal structure of NTU-85-WNT⊃C3H6, hydrogen bond interactions of C3H6 in one-dimensional nanochannels, adsorption isotherms for C3H6 and C3H8 at 298 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 300 K. (f) The crystal structure of ZU-609, adsorption isotherms for pure C3H8 and C3H6 at 298 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 298 K and 1 bar. Reprinted with permission from ref. 32. Copyright 2016 Science, ref. 65. Copyright 2021 Springer Nature Limited, ref. 27. Copyright 2023 American Chemical Society, ref. 74. Copyright 2024 Springer Nature Limited, ref. 37. Copyright 2023 American Chemical Society, ref. 33. Copyright 2024 Science, respectively. | ||
 | ||
| Fig. 18 (a)The crystal structure of ftw-MOF-ABTC, adsorption isotherms for pure C3H8 and C3H6 at 298 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 298 K. (b) The crystal structure of ftw-HIAM-301, adsorption isotherms for pure C3H8 and C3H6 at different temperatures, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at room temperature. Reprinted with permission from ref. 126. Copyright 2018. The Royal Society of Chemistry, ref. 64. Copyright 2021 American Chemical Society, respectively. | ||
In addition, other MOFs, such as ZJU-75, MAF-23-O, Y-abtc, and Co-MOF-74, have been observed to exhibit preferential adsorption of C3H8 from mixtures containing C3H6.6,22,88,90 However, due to the closed separation mechanism discussed above, we will not discuss them further.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CH)/C3H6/C3H8 feed gas.
The purification of C3H6 from ternary or quaternary C3 hydrocarbons has been considered as the most challenging process in gas separation.127 To recognize the extremely small molecular difference, we developed a group of soft PCPs (NTU-65-FeZr, NTU-65-FeTi, NTU-65-CoZr and NTU-65-CoTi)51 on the basis of a global soft framework of NTU-65 that has a Cu2+ node and electronegative site of a SiF62− anion. The changed strength of the coordination bonds, derived from the changed metal node and the multiple sites, endowed the framework with the capacity to show a system structure response: from solely the C3H4 response at 17.1 kPa in NTU-65-FeZr to the sequential response of C3H4 at 1.6 kPa, C3H6 response at 19.4 kPa and finally C3H8 at 57.2 kPa in NTU-65-CoTi at 273 K. Therefore, such unprecedented gradient structural-response driven NTU-65-CoTi is capable of separating C3H4/C3H6/C3H8 (0.5/49.75/49.75, v/v/v, 2 mL min−1) mixtures. Based on breakthrough results, two beds were connected for the adsorption cycle. From bed B, C3H6 with a purity of 99.5% can be harvested at the desorption stage, while C3H8 with highly purity can be obtained at the adsorption stage (Fig. 19).
CH)/C3H6/C3H8 feed gas.
The purification of C3H6 from ternary or quaternary C3 hydrocarbons has been considered as the most challenging process in gas separation.127 To recognize the extremely small molecular difference, we developed a group of soft PCPs (NTU-65-FeZr, NTU-65-FeTi, NTU-65-CoZr and NTU-65-CoTi)51 on the basis of a global soft framework of NTU-65 that has a Cu2+ node and electronegative site of a SiF62− anion. The changed strength of the coordination bonds, derived from the changed metal node and the multiple sites, endowed the framework with the capacity to show a system structure response: from solely the C3H4 response at 17.1 kPa in NTU-65-FeZr to the sequential response of C3H4 at 1.6 kPa, C3H6 response at 19.4 kPa and finally C3H8 at 57.2 kPa in NTU-65-CoTi at 273 K. Therefore, such unprecedented gradient structural-response driven NTU-65-CoTi is capable of separating C3H4/C3H6/C3H8 (0.5/49.75/49.75, v/v/v, 2 mL min−1) mixtures. Based on breakthrough results, two beds were connected for the adsorption cycle. From bed B, C3H6 with a purity of 99.5% can be harvested at the desorption stage, while C3H8 with highly purity can be obtained at the adsorption stage (Fig. 19).
 | ||
| Fig. 19 The crystal structure of NTU-65-CoTi, adsorption isotherms for C3H4, C3H6 and C3H8 at 273 K, and breakthrough experimental curves of C3H6/C3H8 (0.5/49.75/49.75, v/v/v, 2 mL min−1) mixtures at 273 K. Reprinted with permission from ref. 51. Copyright 2024 The Royal Society of Chemistry. | ||
3.4 C3H6 harvesting from the adsorption process
CH and/or CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CCH2)/C3H6 feed gas.
To remove the trace amount of C3H4 from C3H6, a stronger host–C3H4 interaction is imperative. This is necessary to ensure that C3H6, in its highly pure state, can be effectively harvested from the adsorption process. Typically, ZNU-2, an ultra-stable MOF with cage-like pores (decorated with TiF62− anions) interconnected with each other by contracted necks, has been reported for selective C3H4 capture from C3H4/C3H6 mixtures. Single gas adsorption isotherms revealed that ZNU-2 demonstrated both high C3H4 uptake at 0.01 bar (3.9 mmol g−1) and 1 bar (7.7 mmol g−1) at 298 K, while the uptake of C3H6 reached 0.61 and 5.3 mmol g−1, respectively. Therefore, with the combination of a C3H4/C3H6 selectivity (1/99, v/v) of 12.5, the separation potential, a concept developed by Krishina, of ZNU-2 is as high as 31.0 mmol g−1, the maximum value among the MOFs. After the introduction of the C3H4/C3H6 (1/99, v/v) feed gas into the packed column, pure C3H6 broke out at first, while C3H4 was detected about 150 min later. The clear separation interval indicates a productivity of C3H6 of 42.0 mol kg−1. Similarly, an anionic pillared caged MOF, ZNU-2-M (M = Si, Ti, Nb) was also developed for light hydrocarbon separations (Fig. 20a).128
CCH2)/C3H6 feed gas.
To remove the trace amount of C3H4 from C3H6, a stronger host–C3H4 interaction is imperative. This is necessary to ensure that C3H6, in its highly pure state, can be effectively harvested from the adsorption process. Typically, ZNU-2, an ultra-stable MOF with cage-like pores (decorated with TiF62− anions) interconnected with each other by contracted necks, has been reported for selective C3H4 capture from C3H4/C3H6 mixtures. Single gas adsorption isotherms revealed that ZNU-2 demonstrated both high C3H4 uptake at 0.01 bar (3.9 mmol g−1) and 1 bar (7.7 mmol g−1) at 298 K, while the uptake of C3H6 reached 0.61 and 5.3 mmol g−1, respectively. Therefore, with the combination of a C3H4/C3H6 selectivity (1/99, v/v) of 12.5, the separation potential, a concept developed by Krishina, of ZNU-2 is as high as 31.0 mmol g−1, the maximum value among the MOFs. After the introduction of the C3H4/C3H6 (1/99, v/v) feed gas into the packed column, pure C3H6 broke out at first, while C3H4 was detected about 150 min later. The clear separation interval indicates a productivity of C3H6 of 42.0 mol kg−1. Similarly, an anionic pillared caged MOF, ZNU-2-M (M = Si, Ti, Nb) was also developed for light hydrocarbon separations (Fig. 20a).128
 | ||
| Fig. 20 (a) The crystal structure of C3H4 loaded ZNU-2-Si, C3H4 and C3H6 adsorption isotherms for ZNU-2-Si at 278, 298 and 308 K, and breakthrough experimental curves of equimolar C3H4/C3H6/C3H8 mixtures at 298 K. (b) The crystal structure of NTU-100-NH2, C3H4 and C3H6 adsorption isotherms for ZNU-2-Si at 278, 298 and 308 K, and breakthrough experimental curves of equimolar C3H4/C3H6 mixtures at different inlet rates at 298 K. (c) The crystal structure of NTU-88, adsorption isotherms for C3H4 and C3H6 at different temperatures, and breakthrough curves of NTU-88 for C3H4/C3H6 and C3H4/He at 298 K. (d) The 3D structure of NKMOF-1-M (M = Ni or Cu), adsorption isotherms for C3H4, CH2CCH2 and C3H6 and breakthrough experimental curves of C3H4, CH2CCH2 and C3H6 (0.5/0.5/99, v/v/v) mixtures at 298 K. Reprinted with permission from ref. 128. Copyright 2023 The Royal Society of Chemistry, ref. 84. Copyright 2024 John Wiley & Sons, ref. 69. Copyright 2023 John Wiley & Sons, ref. 52. Copyright 2019 John Wiley & Sons, respectively. | ||
NTU-100-NH2, a highly porous MOF, possesses densely decorated NH2 sites within its framework, resulting in a strong interaction with C3H4. This interaction enables a remarkable uptake capacity of up to 84.5 cm3 g−1 at 1 kPa and 298 K, which is almost two times higher than that of NTU-100-NO2, the same framework with decorated NO2.84 Furthermore, NTU-100-NH2 exhibited significantly improved adsorptive selectivity (1.4 to 11.3) and diffusion selectivity (0.13 to 3.15). Therefore, NTU-100-NH2 has been shown to possess the capacity to remove trace amounts of C3H4 from C3H4/C3H6 (1/99, v/v) mixtures at room temperature, while also being capable of producing polymer-grade C3H6 directly. Further breakthrough experiments revealed that the productivity of pure C3H6 remained almost unchanged at different sweep rates (1, 2, and 4 mL min−1) even with wet feed gas (Fig. 20b).
The phenomenon of co-adsorption has been frequently observed in the context of adsorption separation mechanisms that are reliant upon the interaction between the host and guest molecules. NTU-88, the framework with rotational pyridyl rings that was reported by us, exhibited sieving separation of C3H4/C3H6.69 At 298 K, a two-step C3H4 adsorption process, with a steep increase occurring at 15.6 kPa and a maximum uptake of 86.0 cm3 g−1 was observed. Furthermore, the shutter rotating pressure was observed to shift to a very low value of 2.9 kPa at 273 K. However, the total uptake remained constant within the temperate range of 273 to 328 K. In contrast, negligible C3H6 uptake was observed in NTU-88 within these wide temperature ranges. The observation of nearly the same elution times for C3H4/He and C3H4/C3H6 in the dynamic separation process indicates that the co-adsorption of C3H4 and C3H6 was strongly restricted in NTU-88. Furthermore, breakthrough experiments demonstrated that NTU-88 has excellent ability for selective capture of C3H4 from C3H4/C3H6 mixtures, yielding highly-pure C3H6 with a productivity of 4.10 mmol g−1 at 273 K. Notably, this MOF can be scale-up synthesized via room temperature stirring of the related reagents in methanol in a very short time (Fig. 20c).
NKMOF-1-M (M = Cu or Ni) displayed the preferred adsorption of trace C3H4 from C3H6. Both MOFs demonstrated high adsorption of C3H4, including CH3CCH and CH2CCH2 (over 3.0 mmol g−1 at 298 K, 1 bar), which is considerably higher than that of C3H6 (over 1.8 mmol g−1). Importantly, the steep curves observed at pressures below 1 kPa, across a wide temperature range, indicated strong binding affinity for C3H4. Therefore, their adsorption selectivity is very high, over 100. Breakthrough experiments showed that NKMOF-1-Ni can selectively capture trace amounts of C3H4, yielding highly pure C3H6 (99.996%, 230 mmol L−1) from the feed gas of CH3CCH/CH2CCH2/C3H6 (0.5/0.5/99) mixtures at room temperature. A similar phenomenon has been observed in ultra-microporous MOF-NKMOF-M (Fig. 20d).52
 | ||
| Fig. 21 (a) The 3D structure of PCP-IPA, the adsorption isotherms of different light hydrocarbons at 298 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 298 K and 1 bar. (b) The structure of NU-57, the adsorption isotherms of C3H6 and C3H8 for NU-57 and NU-58, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 298 K. (c) The crystal structure of C3H8 loaded CdIF-13, adsorption isotherms for C3H6 and C3H8 at 298 K, and breakthrough experimental curves of equimolar C3H6/C3H8 mixtures at 298 K and 1 bar. Reprinted with permission from ref. 129. Copyright 2022 Springer Nature Limited, ref. 130. Copyright 2025 John Wiley & Sons, ref. 43. Copyright 2023 American Chemical Society, respectively. | ||
A desymmetrization approach was utilised in the preparation of NU-57, which realized inversion C3H8/C3H6 separation. The maximum C3H8 uptake of NU-57 was found to be superior to that of C3H6, despite the adsorption isotherms of C3H8 and C3H6 demonstrating almost complete overlap in the low-pressure region.130 In addition, the Qst values of NU-57 for C3H8 and C3H6 were 33.0 kJ mol−1 and 28.5 kJ mol−1, respectively, suggesting a stronger interaction between the framework and C3H8 molecules than C3H6 molecules. Harvesting of pure C3H6 from C3H8-containing mixtures was confirmed by breakthrough experiments. A similar approach was adopted in Cu-ASY,131 a new MOF that incorporates infinite Cu-carboxylate rods which can separate C3H8/C3H6 mixtures with a C3H6 productivity of 2.2 L kg−1 (Fig. 21b).
In addition to the aforementioned rigid frameworks, which have been demonstrated to be capable of performing inversion C3H8/C3H6 separation, a soft framework has also been shown to be effective for this task. CdIF-13, prepared from Cd2+ and benzimidazolate, exhibited guest-induced structural changes.43 At 298 K, step-shaped C3H8 adsorption isotherms revealed that gate opening of CdIF-13 occurred at 0.3 bar, which is far earlier than that of C3H6 (0.8 bar). Comparatively, the isostructural ZIF-7 displayed a very small difference in gate-opening pressure of C3H8 (0.008 bar) and C3H6 (0.020 bar) at 298 K. Therefore, highly pure C3H6 was harvested by using a double column packed with CdIF-13 and BPL carbon from equimolar C3H8/C3H6 feed gas (Fig. 21c).
CH and or CH2CCH2)/C3H6/C3H8 feed gas.
The purification of C3H6 from four-component mixtures (C3H4/CH2CCH2/C3H6/C3H8) represents the most challenging process in light hydrocarbon separations. JNU-9-CH3, a hydrolytically stable Cu4I4-triazolate MOF with a spatial distribution of polar and nonpolar sites, was designed.60 The densely packed and readily accessible iodine and nitrogen sites in the 1D channel allowed JNU-9-CH3 to show the single gas uptakes in a sequence of C3H8 > CH3CCH > CH2CCH2 > C3H6. The calculated selectivity of the C3H8/C3H6 (1/1, v/v) mixture is 1.5 at 298 K and 1 bar, while the selectivities of CH3CCH/C3H6 (1/1, v/v) and CH2CCH2/C3H6 (1/1, v/v) are 2.1 and 1.3, respectively. Breakthrough experiments showed that JNU-9-CH3 is able to separate such equimolar quaternary hydrocarbons, yielding highly pure C3H6 of 102.7 mmol L−1 (Fig. 22). In addition, the velocity of the feed gas exerts minimal influence on the separation performance.
 | ||
| Fig. 22 The crystal structure of JNU-9-CH3, adsorption isotherms for C3H4, CH2CCH2, C3H6 and C3H8 at 298 K, and breakthrough experimental curves of C3H4, CH2CCH2, C3H6 and C3H8 (25/25/25/25, v/v/v) mixtures at 298 K. Reprinted with permission from ref. 60. Copyright 2024 American Chemical Society. | ||
4. Large-scale synthesis
Since their first appearance, MOFs have undergone remarkable development and have demonstrated excellent performance in the field of gas separation. If MOFs with gas separation functions can be synthesized on a large scale, it will undoubtedly lead to a significant reduction in production costs and bring significant benefits for the application of MOFs in the market. Despite considerable efforts, only a few of the MOF products with unique structures and potential applications have been synthesized on a large scale. The synthesis of MOFs on a large scale is still challenging due to the high cost of some of the organic ligands and metal salts that are essential components in the synthesis of MOFs. Ensuring consistent quality and reproducibility, a key requirement for large-scale production, remains a formidable task. It is therefore widely recognised in the scientific community that the large-scale synthesis of MOFs should focus on the following key aspects. First, the material cost must be reduced, which can be achieved by exploring alternative raw materials or more efficient synthesis routes. Second, higher yields should be achieved to meet the growing demands of various industries. Thirdly, a relatively fast and simple synthesis process is highly desirable, which can increase production efficiency and reduce production time. Fourthly, the synthesis process should have less impact on the environment, in line with the principles of sustainable development.142,143In addressing the complex issues hindering large-scale synthesis of MOFs, researchers have developed a range of methods, including room-temperature stirring, solvothermal, mechanochemical synthesis, continuous flow production and electrochemical which have been explored with the aim of improving production efficiency.144–147 These large-scale synthesis methods have their own advantages and shortcomings, which are detailed in Table 6. The employment of such methodologies has yielded encouraging outcomes with regard to enhancing the efficiency of the production process. In the context of MOF production, it is imperative to give due consideration to both extrinsic and intrinsic factors. Poor management of these factors can lead to increased costs in the production process, which is a significant concern for both academic research and industrial applications. Despite the difficulties in large-scale production of MOFs, companies such as BASF, Framergy, Nuada or MOF apps are now able to produce a range of MOFs. MIL-100 (Fe), UiO-66 (Zr), Al-fum MOF, ZIF-8, ZIF-67, HKUST-1, PCN-250 (Fe) or MIL-127 (Fe) can be produced on a kilogram scale.148 CALF-20, a MOF material that can efficiently capture CO2, in the presence of moisture after combustion,149 is now being produced by BASF on a multi-ton scale. The success of large-scale synthesis of these examples has shed light on the large-scale synthesis of MOFs (Fig. 23).
| Methods | Pros. | Cons. |
|---|---|---|
| Room temperature stirring | Green, simple, and low energy | Not universally applicable for MOF preparation |
| Mechanochemistry | Environmental friendly, simple, and low-cost | Damaged material and high impurity content |
| Solvothermal | Simple | Long-reaction-cycle |
| High cost of the reactor | ||
| Flow chemistry | High-yield production with excellent batch consistency | Clog-prone piping |
| Electrochemistry | High purity | A relatively high cost |
 | ||
| Fig. 23 (a) Large-scale preparation of NTU-88 via room temperature stirring. (b) Large-scale preparation of Uio-66 using solvothermal methods. (c) Key components of the twin-screw extruder. (d) Demonstration of the continuous flow method for synthesizing MOFs. (e) Synthesis of MOFs using Cu as the electrode material by the electrochemical synthesis method. Reprinted with permission from ref. 69. Copyright 2023 John Wiley & Sons, ref. 162. Copyright 2015 Elsevier, ref. 152. Copyright 2015 The Royal Society of Chemistry, ref. 163. Copyright 2022 The Royal Society of Chemistry, ref. 164. 2006. Copyright The Royal Society of Chemistry, respectively. | ||
4.1 Room-temperature stirring
As an alternative fast and simple method, room-temperature stirring has been a well-established for material synthesis in laboratory and industry, due to its significant advantages of cost-effectiveness, safety, scalability, versatility, simple operation, and almost negligible energy consumption. For MOF synthesis, the main procedure involves the dissolution of suitable organic ligands and metal salts separately in appropriate solvents, followed by the gradual pouring of one of the above solutions into another under stirring conditions, or the simultaneous pouring of both solutions into a third vessel. The gentle nature of this method renders it less likely to result in the rapid precipitation of reactants, thus necessitating extended periods to achieve complete crystallisation at room temperature in comparison to methods conducted at elevated temperatures. The incorporation of reaction promoters, such as formic acid, acetic acid, triethylamine, or ammonia water, has been demonstrated to effectively modulate the reaction rate. It is also crucial to ensure the homogeneity of the mixture throughout the reaction vessel to avoid the formation of undesirable particles due to localised supersaturation.NKMOF-8-Br and NKMOF-8-Me, which possess excellent separation performance for C2H6/C2H4, can be efficiently prepared through room temperature stirring of ligands, CuI, and triethylamine (TEA) in acetonitrile at room temperature within 3 minutes. Both MOFs have high yields (>90%). Notably, the acetonitrile solution can be readily recycled through filtration and then reused in the following reaction cycles without the need for further purification.150 We have recently achieved large-scale synthesis of a series of light hydrocarbon separators, including NTU-88 (60.0 g, stirring in 1 min) and NTU-101-NH2 (50.5 g, 5 min stirring) via room temperature stirring of N containing ligands and Cu2+ or Ni2+.62,69 For them, ammonia or triethylamine was gently added into the stirring system. Meanwhile, a MOF of CeBTB (H3BTB: 1,3,5-Tri(4-carboxyphenyl) benzene) with a carboxylate ligand can also be synthesized by such a method.151 Differently, the promoter 1-methylimidazole was used to regulate the deprotonation ability of H3BTB. CeBTB (uniform size of 150 nm) can be conveniently prepared in a 1 L beaker. As a C3H6/C3H8 separator, ZJU-75 can also be readily scaled up through room temperature stirring of K2[Ni(CN)4]·nH2O pyz-NH2 and Co(NO3)2·6H2O in water/ethanol solution.90 Further kilogram-scale synthesis has also been achieved in this case, giving a high yield of 75% within 8 hours. The calculated space-time yield was found to be 158 kg per m3 per day. It is interesting that MOF-303 can be prepared in water as the sole solvent and can be readily scaled up to the kilogram level with a yield of up to 93%, but a reflux reaction apparatus is required.123
4.2 Mechanochemical synthesis
The concept of mechanochemistry is regarded as one of the important innovations that have had a profound impact on the world.152 The application of mechanical forces can break the intramolecular bonds between reactants, thereby facilitating the formation of new bonds. This process is accompanied by the transformation of mechanical energy into thermal energy through the process of heating.153 The mechanochemical synthesis has been shown to be solvent-free or to require only a minimal amount of solvent, thereby minimising its environmental impact. Furthermore, mechanochemistry has been demonstrated to exhibit advantages over the above-mentioned strategies, including reduced reaction times and enhanced efficiency.154–156 This characteristic is of particular significance for poorly soluble metal precursors or ligands, as it circumvents the issues associated with solubility whilst concomitantly minimizing environmental damage. By reducing the reliance on solvents, there is the potential to substantially reduce production costs, a highly desirable outcome in both academic research and industrial production.A mixture of dried copper acetate and isonicotinic acid was subjected to 10 minutes of milling, resulting in the formation of copper(II) isonicotinic acid and the trapping of acetic acid and water molecules within the pores of the resultant product.157 Notwithstanding the potential of mechanical milling for the synthesis of MOFs, the majority of extant research has focused on gram-scale feeds. In order to find ways to scale-up this technique, a ThermoFisher Process-11 Twin Screw Extruder, a machine with a screw configuration of a sequence of alternating conveying and kneading zones, was used.152 Through the feed port, the MOF precursor can be introduced into a heatable drum containing screws and the exit port can be connected to a mold for collecting the final material, yielding continuous synthesis of various metal complexes, including Ni(salen), Ni(NCS)2(PPh3)2, HKUST-1, ZIF-8, and MAF-4 Al(fumarate)(OH). It is important to note that the space time yields for these methods are as high as 144 × 103 kg m−3 per day, which is significantly greater than the yields for other methods of MOF synthesis. Recent breakthroughs in mechanochemical synthesis have demonstrated the remarkable efficiency of a novel “cage-on-MOF” strategy, enabling the rapid preparation of 28 distinct MOF@PCC composites within just 5 min.158 Particularly noteworthy is the exceptional performance of these materials, with MOF-808@PCC-4 showing dramatic enhancements in CO2/C2H2 separation (exhibiting a 64% increase in C2H2 uptake capacity and 166% improvement in IAST selectivity in breakthrough tests), while MIL-101@PCC-4 achieved outstanding C2H2 adsorption reaching 6.11 mmol g−1 with an IAST selectivity of 2.59. This mechanochemical approach represents a significant advancement compared to conventional solution-based post-synthetic modification methods, which typically require time-consuming heating procedures and multiple purification steps. The “cage-on-MOF” methodology not only achieves gram-scale synthesis (up to 100 g batches) with near-quantitative yields, but also maintains excellent crystallinity and porosity control while completely eliminating organic solvent consumption-offering a synthesis efficiency approximately 300 times greater than that of traditional solution-phase methods while ensuring consistent material quality.
Although mechanochemistry has evident advantages, this strategy is not universally applicable as the generation of excessive mechanical energy may lead to the amorphization of MOFs and the subsequent decomposition and damage of the material.159 In addition, the formation of new bonds is often inconsistent, leading to the production of various impurities that cannot be easily removed. Furthermore, mechanical synthesis is often not suitable for MOFs with high coordination numbers that require a relatively slow and well-controlled crystallization process.
4.3 Solvothermal and hydrothermal synthesis
In 1995, Nalco Chemical Company and the Yaghi group produced the first MOF materials using solvothermal synthesis. A certain proportion of organic ligands and metal salts are dissolved in different solvents, after which the solutions are mixed in a sealed reaction vessel, and then the nucleation and growth of crystals is promoted by heating.160 This method is by far the most widely used method for producing MOF crystals in laboratories.161However, there are significant limitations for the large-scale production of MOFs. In particular, increasing the size of the reaction vessel leads to a significant reduction in the surface-to-volume ratio, which further reduces the efficiency of the reaction, but also requires, for example, the use of hundreds of liters of solvent per batch. In addition, the reaction time is too long, given the high temperature and pressure involved. Finally, the cost of the pressure vessel, especially those for hundreds of liters, is very high.
4.4 Flow chemistry synthesis
Flow chemistry is a process in which the solutions of various components are passed through a reaction apparatus, leading to the occurrence of chemical reactions.163,165,166 This process contrasts with traditional batch-reaction devices, in which different components are placed in separate vessels and react over time. Consequently, flow chemistry can be regulated precisely through parameter change and device design, thereby ensuring reproducibility of high-quality products. In the context of flow chemistry, the most salient benefit of this process is that the rapid separation of MOF crystals and related solvents facilitates the recycling of solvents. This, in turn, has a number of significant benefits, such as environmental friendliness, capital investment and energy input (hot solvent will be transferred into the reactor in time).The utilisation of both a stirred tank reactor and a plug flow reactor has become a widespread practice in the field of flow synthesis. For instance, a microwave-assisted flow chemistry method has been employed to synthesize UiO-66 with a significantly enhanced reaction rate when compared to conventional solvothermal synthesis.162 In addition, MOF-5 with a space time yield of 0.1 ton per m3 per day, has been synthesised via a flow chemistry method.167 After optimization, the flow system, operated at 140 °C and residence time ≥5.5 h, reached a steady state that can continuously produce high surface area MOF-5. In other words, parameter optimization in flow chemistry plays an important role in the manufacture of MOFs with controlled quality.
Following the modification of the heating reactor to incorporate a microwave-assisted heating reactor, a notable enhancement in the rate of MOF growth, accompanied by excellent reproducibility, has been observed in the case of MOF-74(Ni).168 The raw materials of MOF-74(Ni) were introduced into a microwave reactor to facilitate initial nucleation. The solution was subsequently introduced into a reactor for a duration of 8 minutes to complete the growth of the crystals. This process resulted in enhanced crystallinity, accompanied by a space time yield of 2160 kg per m3 per day. Using a similar system, UiO-66, HKUST-1 and MIL-53(Al) were obtained in 7, 1 and 4 minutes of residence time, resulting in high space time yields of 7204 kg per m3 per day, 64800 kg per m3 per day and 3618 kg per m3 per day, respectively.146 In such a system, the separation of the nucleation and growth steps has been shown to accelerate the reaction rate and ensure the quality of the resulting MOFs.
However, it was also noted that the flow chemistry synthesis suffers from a problem of reactor/pipeline fouling and clogging. During the reaction process, precipitates are often generated, and the clogging caused by these precipitates can accumulate in the system. As a consequence, the efficiency and quality of the product are affected. In order to address this issue, it is imperative to implement regular cleaning procedures or to undertake a redesign of the reactor components, such as the introduction of a match screw inside the tube, in order to maintain the functionality of the flow system.
4.5 Electrochemical synthesis
In comparison with the conventional solvothermal synthesis method, the electrochemical synthesis has milder reaction conditions, a significantly reduced reaction time, and the process of MOF synthesis can be regulated by modulating the voltage.148 With regard to electrochemical synthesis, the methods can be categorised into three distinct classifications depending on the direction of electron transfer and the location where the nucleation process occurs: (1) surface electrode nucleation, including anodic dissolution and cathodic deposition, (2) indirect bipolar electrodeposition and (3) electrophoretic deposition.169 The researchers in PASF immersed a copper plate as an anode into an organic solution containing 1,3,5-benzenetricarboxylic acid. When a specific current was applied, Cu ions were transferred from the surface of the anode to the solution and reacted with the organic linker in the solution, ultimately producing an octahedral powder of HKUST-1. Notably, the surface area of the electrochemical synthesis material is 1820 m2 g−1, which is larger than that of HKUST-1 synthesised by the solvothermal method (1550 m2 g−1). Recently, a mechanism for the anodic solvation synthesis of HKUST-1 was proposed, which consists of initial nucleation; growth of HKUST-1 islands; intergrowth; and crystal detachment.170Despite the advances achieved thus far, numerous challenges have been encountered when attempting to realise large-scale production. Initially, a large-scale electrochemical synthesis device is required; however, this task involves complex issues. For example, the electrodes must be uniformly arranged over a large area, and the circulation and stirring of the electrolyte must be well-controlled, which pose significant technical difficulties. Furthermore, when reaction control is considered during large-scale synthesis, ensuring the homogeneity of parameters such as electric field, temperature, and electrolyte concentration throughout the entire reaction system is extremely challenging, as doing so would produce products of unstable quality and poor consistency.
In a word, room-temperature stirring is one of the most practical and effective methods for large-scale synthesis of MOFs, especially holding a unique position in the large-scale synthesis of MOFs for light hydrocarbon separation. Secondly, mechanochemical methods have emerged and shown potential in the large-scale synthesis of light hydrocarbon separation MOFs. Although solvothermal, electrochemical and flow chemistry methods have made some progress in the large-scale synthesis of some MOFs, they still need to be studied more in the large-scale synthesis of light hydrocarbon separation MOFs. It is believed that in the near future, in terms of light hydrocarbon separation, the large-scale production methods are more diversified, the industrialization conditions will be more mature, and the large-scale production of high-efficiency, low-cost and sustainable high-quality light hydrocarbon separation MOFs will be realized. It is encouraging to see that companies like BASF are already striving for excellence and have achieved exciting results in the large-scale production of MOFs. It is encouraging that companies like BASF are already pursuing excellence and achieving exciting results in the large-scale production of MOFs. It is expected that this trend will continue, leading to significant progress in the large-scale production and application of MOFs not only in light hydrocarbon separation but even in other areas.
5. Conclusion and outlook
The development of high-performance MOFs for light hydrocarbon separation has witnessed remarkable progress, driven by innovative design strategies and engineering advancements. Rigid MOFs, functionalized with tailored binding sites, enable selective guest interactions through precise control of pore environments, while flexible MOFs leverage dynamic structural responses under external stimuli to achieve molecular recognition. These advancements, coupled with emerging scale-up synthesis techniques, underscore the potential of MOFs to revolutionize industrial separation processes. However, critical challenges remain in achieving cost-effective, one-step separation processes, precise material design for specific applications, and scalable production. Addressing these challenges will require interdisciplinary collaboration and sustained innovation in both fundamental research and applied technologies.To propel MOFs toward practical implementation in light hydrocarbon separation, future research should prioritize the following directions.
5.1 Pore aperture engineering
Precise control over pore size and geometry is essential for molecular sieving. While rigid MOFs allow tunability via ligand and metal node selection, flexible MOFs offer dynamic pore adjustment in response to guest molecules. Integrating computational tools—such as artificial intelligence (AI) and molecular simulations—will accelerate the prediction of optimal MOF architectures for target separations. These approaches can minimize trial-and-error experimentation and guide the design of frameworks with pore environments tailored to specific gas pairs.5.2 Enhanced host–guest interactions
Synergistic integration of multiple interaction mechanisms (e.g., van der Waals forces, open metal sites, hydrogen bonding, and π-complexation) within a single MOF can amplify selectivity. Future work should focus on multifunctional frameworks that combine complementary binding sites, such as Lewis acidic metal clusters and functionalized organic linkers, to enable multi-mechanism adsorption. Additionally, exploring stimuli-responsive systems (e.g., light- or temperature-triggered structural changes) could further refine dynamic separation processes.5.3 Elucidating mechanisms
A deeper understanding of the interplay between thermodynamic equilibria and kinetic diffusion is critical. Advanced characterization techniques (e.g., in situ spectroscopy and operando diffraction) combined with molecular dynamics (MD) simulations can unravel molecular transport pathways and adsorption site competition. Investigating the nano-confinement effects within MOF pores may also reveal strategies to manipulate guest molecule behavior, particularly under industrially relevant high-temperature conditions.5.4 Scalability and stability
Bridging the gap between laboratory synthesis and industrial deployment demands scalable, low-cost production methods. Room temperature stirring synthesis and solvent-free routes should be optimized to reduce energy consumption and material waste. Concurrently, improving MOF stability under harsh operational conditions (e.g., moisture, high pressure, and thermal cycling) is imperative for long-term viability.5.5 Interdisciplinary collaboration
Cross-disciplinary efforts integrating chemistry, materials science, chemical engineering, and data science will be vital to overcome existing barriers. Partnerships with industry can accelerate technology transfer, while open-access databases for MOF properties and performance metrics will streamline material discovery.In conclusion, MOFs represent a transformative platform for light hydrocarbon separation, offering unprecedented control over molecular recognition and process efficiency. While challenges in design precision, stability, and scalability persist, the convergence of computational modeling, advanced synthesis, and mechanistic insights holds immense promise. By addressing these hurdles through targeted research and collaborative innovation, MOFs may soon transition from academic breakthroughs to industrial mainstays, ushering in a new era of sustainable and energy-efficient chemical separations.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
J. D. conceived the idea of this project. H. Z., J. T. and C. Y. collected literatures, wrote and revised the paper. All authors offered constructive comments on the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are thankful for the financial support from the National Natural Science Foundation of China (22171135 and 22471235), the Natural Science Foundation of Jiangsu Province (BK20231269), the Outstanding Graduate Student Innovation Project of Xinjiang University (51252500201) and the State Key Laboratory of Materials-Oriented Chemical Engineering (SKL-MCE-23A18).References
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- W. D. Fan, X. Wang, X. R. Zhang, X. P. Liu, Y. T. Wang, Z. X. Kang, F. N. Dai, B. Xu, R. M. Wang and D. F. Sun, ACS Cent. Sci., 2019, 5, 1261–1268 CrossRef CAS.
- G.-D. Wang, Y.-Z. Li, W.-J. Shi, L. Hou, Y.-Y. Wang and Z. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202311654 CrossRef CAS.
- S. A. Chernyak, M. Corda, J. P. Dath, V. V. Ordomsky and A. Y. Khodakov, Chem. Soc. Rev., 2022, 51, 7994–8044 RSC.
- P. Hu, J. L. Hu, H. Liu, H. Wang, J. Zhou, R. Krishna and H. B. Ji, ACS Cent. Sci., 2022, 8, 1159–1168 CrossRef CAS.
- H. Wang, X. Dong, V. Colombo, Q. Wang, Y. Liu, W. Liu, X.-L. Wang, X.-Y. Huang, D. M. Proserpio, A. Sironi, Y. Han and J. Li, Adv. Mater., 2018, 30, 1805088 CrossRef.
- J. Q. Liu, Y. J. Liu, D. K. Talay, E. Calverley, M. Brayden and M. Martinez, Carbon, 2015, 85, 201–211 CrossRef CAS.
- Q. Z. Xue, X. F. Li, X. Chang, C. C. Ling, L. Zhu and W. Xing, Appl. Surf. Sci., 2018, 444, 772–779 CrossRef CAS.
- Y. F. Yuan, Y. S. Wang, X. L. Zhang, W. C. Li, G. P. Hao, L. Han and A. H. Lu, Angew. Chem., Int. Ed., 2021, 60, 19063–19067 CrossRef CAS PubMed.
- Y. Chai, X. Han, W. Li, S. Liu, S. Yao, C. Wang, W. Shi, I. da-Silva, P. Manuel, Y. Cheng, L. D. Daemen, A. J. Ramirez-Cuesta, C. C. Tang, L. Jiang, S. Yang, N. Guan and L. Li, Science, 2020, 368, 1002–1006 CrossRef CAS.
- J. G. Min, K. C. Kemp and S. B. Hong, Sep. Purif. Technol., 2020, 250, 117146 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- X. Wang, W. Lu, Z. Y. Gu, Z. Wei and H. C. Zhou, Chem. Commun., 2016, 52, 1926–1929 RSC.
- K. S. Walton, Nat. Chem., 2014, 6, 277–278 CrossRef CAS.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Tobbens, S. Zander, R. S. Pillai, G. Maurin, F. X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS.
- S. H. Yang, X. Lin, W. Lewis, M. Suyetin, E. Bichoutskaia, J. E. Parker, C. C. Tang, D. R. Allan, P. J. Rizkallah, P. Hubberstey, N. R. Champness, K. M. Thomas, A. J. Blake and M. Schröder, Nat. Mater., 2012, 11, 710–716 CrossRef CAS.
- L. Wen, P. Cheng and W. B. Lin, Chem. Sci., 2012, 3, 2288–2292 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167–170 CrossRef CAS PubMed.
- J. Duan, M. Higuchi, R. Krishna, T. Kiyonaga, Y. Tsutsumi, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, Chem. Sci., 2014, 5, 660–666 RSC.
- Z.-J. Liang, F.-D. Dong, L. Ye, K. Zheng, D.-Y. Hu, X. Feng, W.-Y. Su, Z.-S. Wang, M.-Y. Zhou, Z.-L. Fang, D.-D. Zhou, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2025, 16, 3307–3312 RSC.
- Y. S. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Snurr, Angew. Chem., Int. Ed., 2012, 51, 1857–1860 CrossRef CAS PubMed.
- Y.-L. Peng, T. Wang, C. Jin, C.-H. Deng, Y. Zhao, W. Liu, K. A. Forrest, R. Krishna, Y. Chen, T. Pham, B. Space, P. Cheng, M. J. Zaworotko and Z. Zhang, Nat. Commun., 2021, 12, 5768 CrossRef CAS PubMed.
- X. W. Gu, J. X. Wang, E. Y. Wu, H. Wu, W. Zhou, G. D. Qian, B. L. Chen and B. Li, J. Am. Chem. Soc., 2022, 144, 2614–2623 CrossRef CAS PubMed.
- Z. Z. Lu, H. G. W. Godfrey, I. da Silva, Y. Q. Cheng, M. Savage, P. Manuel, S. Rudic, A. J. Ramirez-Cuesta, S. H. Yang and M. Schroder, Chem. Sci., 2018, 9, 3401–3408 RSC.
- S. Horike, Y. Inubushi, T. Hori, T. Fukushima and S. Kitagawa, Chem. Sci., 2012, 3, 116–120 RSC.
- X. Huang, F. Chen, H. Sun, L. Yang, Q. Yang, Z. Zhang, Y. Yang, Q. Ren and Z. Bao, J. Am. Chem. Soc., 2023, 146, 617–626 CrossRef PubMed.
- C.-X. Chen, Z.-W. Wei, T. Pham, P. C. Lan, L. Zhang, K. A. Forrest, S. Chen, A. M. Al-Enizi, A. Nafady, C.-Y. Su and S. Ma, Angew. Chem., Int. Ed., 2021, 60, 9680–9685 CrossRef CAS PubMed.
- H. Angewandte ChemieChen, B. Wang, B. Zhang, J. Chen, J. Gui, X. Shi, W. Yan, J. Li and L. Li, Chem. Sci., 2023, 14, 7068–7075 RSC.
- Q. Min Wang, D. Shen, M. Bülow, M. Ling Lau, S. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217–230 CrossRef.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137–140 CrossRef CAS PubMed.
- J. Cui, Z. Zhang, L. Yang, J. Hu, A. Jin, Z. Yang, Y. Zhao, B. Meng, Y. Zhou, J. Wang, Y. Su, J. Wang, X. Cui and H. Xing, Science, 2024, 383, 179–183 CrossRef CAS.
- X. Cui, K. Chen, H. Xing, Q. Yang, R. Krishna, Z. Bao, H. Wu, W. Zhou, X. Dong, Y. Han, B. Li, Q. Ren, M. J. Zaworotko and B. Chen, Science, 2016, 353, 141–144 CrossRef CAS PubMed.
- Q. Ding, Z. Zhang, C. Yu, P. Zhang, J. Wang, X. Cui, C.-H. He, S. Deng and H. Xing, Sci. Adv., 2020, 6, eaaz4322 CrossRef CAS.
- Q. Dong, Y. Huang, K. Hyeon-Deuk, I. Y. Chang, J. Wan, C. Chen, J. Duan, W. Jin and S. Kitagawa, Adv. Funct. Mater., 2022, 32, 2203745 CrossRef CAS.
- Q. Dong, Y. Huang, J. Wan, Z. Lu, Z. Wang, C. Gu, J. Duan and J. Bai, J. Am. Chem. Soc., 2023, 145, 8043–8051 CrossRef CAS PubMed.
- C. Gücüyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef PubMed.
- A. N. Hong, H. Yang, T. Li, Y. Wang, Y. Wang, X. Jia, A. Zhou, E. Kusumoputro, J. Li, X. Bu and P. Feng, ACS Appl. Mater. Interfaces, 2021, 13, 52160–52166 CrossRef CAS.
- Y. Huang, J. Wan, T. Pan, K. Ge, Y. Guo, J. Duan, J. Bai, W. Jin and S. Kitagawa, J. Am. Chem. Soc., 2023, 145, 24425–24432 CrossRef CAS PubMed.
- Y. Jiang, J. Hu, L. Wang, W. Sun, N. Xu, R. Krishna, S. Duttwyler, X. Cui, H. Xing and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200947 CrossRef CAS PubMed.
- J. Luo, G. Yang, G. Zhang, Z. Huang, J. Peng, Y. Luo, X. Wang, C. Yang, J. Jiang, D. Cao, B. Chen, H. Tang and J. Xiao, Chem, 2024, 10, 3148–3158 CAS.
- R. A. Klein, L. W. Bingel, A. Halder, M. Carter, B. A. Trump, E. D. Bloch, W. Zhou, K. S. Walton, C. M. Brown and C. M. McGuirk, J. Am. Chem. Soc., 2023, 145, 21955–21965 CrossRef CAS.
- B. Li, X. Cui, D. O'Nolan, H.-M. Wen, M. Jiang, R. Krishna, H. Wu, R.-B. Lin, Y.-S. Chen, D. Yuan, H. Xing, W. Zhou, Q. Ren, G. Qian, M. J. Zaworotko and B. Chen, Adv. Mater., 2017, 29, 1704210 CrossRef PubMed.
- K. Li, D. H. Olson, J. Seidel, T. J. Emge, H. Gong, H. Zeng and J. Li, J. Am. Chem. Soc., 2009, 131, 10368–10369 CrossRef CAS.
- L. Li, R.-B. Lin, R. Krishna, H. Li, S. Xiang, H. Wu, J. Li, W. Zhou and B. Chen, Science, 2018, 362, 443–446 CrossRef CAS.
- L. Li, R.-B. Lin, R. Krishna, X. Wang, B. Li, H. Wu, J. Li, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 7733–7736 CrossRef CAS.
- Y. Li, Y. Wu, J. Zhao, J. Duan and W. Jin, Chem. Sci., 2024, 15, 9318–9324 RSC.
- P.-Q. Liao, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 8697 CrossRef PubMed.
- C. X. Zhao, X. Liu, J. N. Liu, J. Wang, X. Wan, C. Wang, X. Y. Li, J. Shui, L. Song, H. J. Peng, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202313028 CrossRef.
- S. Liu, Y. Huang, J. Wan, J.-J. Zheng, R. Krishna, Y. Li, K. Ge, J. Tang and J. Duan, Chem. Sci., 2024, 15, 6583–6588 RSC.
- Y. L. Peng, C. He, T. Pham, T. Wang, P. Li, R. Krishna, K. A. Forrest, A. Hogan, S. Suepaul, B. Space, M. Fang, Y. Chen, M. J. Zaworotko, J. Li, L. Li, Z. Zhang, P. Cheng and B. Chen, Angew. Chem., Int. Ed., 2019, 58, 10209–10214 CrossRef CAS PubMed.
- C. Song, F. Zheng, Y. Liu, Q. Yang, Z. Zhang, Q. Ren and Z. Bao, Angew. Chem., Int. Ed., 2023, 62, e202313855 CrossRef CAS.
- J. Cui, Z. Zhang, L. Yang, J. Hu, A. Jin, Z. Yang, Y. Zhao, B. Meng, Y. Zhou, J. Wang, Y. Su, J. Wang, X. Cui and H. Xing, Science, 2024, 383, 179–183 CrossRef CAS.
- Y.-J. Tian, C. Deng, L. Zhao, J.-S. Zou, X.-C. Wu, Y. Jia, Z.-Y. Zhang, J. Zhang, Y.-L. Peng, G. Chen and M. J. Zaworotko, Nat. Chem., 2025, 17, 141–147 CrossRef CAS.
- S.-M. Wang, M. Shivanna, S.-T. Zheng, T. Pham, K. A. Forrest, Q.-Y. Yang, Q. Guan, B. Space, S. Kitagawa and M. J. Zaworotko, J. Am. Chem. Soc., 2024, 146, 4153–4161 CrossRef CAS PubMed.
- Y. Jiang, J. Hu, L. Wang, W. Sun, N. Xu, R. Krishna, S. Duttwyler, X. Cui, H. Xing and Y. Zhang, Angew. Chem., Int. Ed., 2022, 134, e202200947 CrossRef.
- Y. Hu, Y. Jiang, J. Li, L. Wang, M. Steiner, R. F. Neumann, B. Luan and Y. Zhang, Adv. Funct. Mater., 2023, 33, 2213915 CrossRef CAS.
- S.-C. Xiang, Z. Zhang, C.-G. Zhao, K. Hong, X. Zhao, D.-R. Ding, M.-H. Xie, C.-D. Wu, M. C. Das, R. Gill, K. M. Thomas and B. Chen, Nat. Commun., 2011, 2, 204 CrossRef PubMed.
- X.-J. Xie, Z.-H. Zhang, Q.-Y. Cao, Y.-L. Huang, D. Luo, H. Zeng, W. Lu and D. Li, J. Am. Chem. Soc., 2024, 146, 30155–30163 CrossRef CAS PubMed.
- H. Yang, F. Peng, D. E. Schier, S. A. Markotic, X. Zhao, A. N. Hong, Y. Wang, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2021, 60, 11148–11152 CrossRef CAS PubMed.
- W. Yang, J. Wang, K. Tan, H. L. Zhou, M. Zhang, R. Krishna, J. Duan and L. Huang, Angew. Chem., Int. Ed., 2025, 64, e202425638 CrossRef CAS PubMed.
- Y. Ye, Y. Xie, Y. Shi, L. Gong, J. Phipps, A. M. Al-Enizi, A. Nafady, B. Chen and S. Ma, Angew Chem. Int. Ed. Engl., 2023, 62, e202302564 CrossRef CAS PubMed.
- L. Yu, X. Han, H. Wang, S. Ullah, Q. Xia, W. Li, J. Li, I. da Silva, P. Manuel, S. Rudić, Y. Cheng, S. Yang, T. Thonhauser and J. Li, J. Am. Chem. Soc., 2021, 143, 19300–19305 CrossRef CAS PubMed.
- H. Zeng, M. Xie, T. Wang, R.-J. Wei, X.-J. Xie, Y. Zhao, W. Lu and D. Li, Nature, 2021, 595, 542–548 CrossRef CAS PubMed.
- L. Zhang, B. Yu, M. Wang, Y. Chen, Y. Wang, L.-B. Sun, Y.-B. Zhang, Z. Zhang, J. Li and L. Li, Angew. Chem., Int. Ed., 2025, 64, e202418853 CrossRef CAS PubMed.
- P. Zhang, Y. Zhong, Y. Zhang, Z. Zhu, Y. Liu, Y. Su, J. Chen, S. Chen, Z. Zeng, H. Xing, S. Deng and J. Wang, Sci. Adv., 2022, 8, eabn9231 CrossRef CAS PubMed.
- Y.-B. Wang, T.-F. Zhang, Y.-X. Lin, J.-X. Wang, H.-M. Wen, X. Zhang, G. Qian and B. Li, J. Mater. Chem. A, 2024, 12, 25812–25819 RSC.
- J. Wan, H. L. Zhou, K. Hyeon-Deuk, I. Y. Chang, Y. Huang, R. Krishna and J. Duan, Angew. Chem., Int. Ed., 2023, 62, e202316792 CrossRef CAS PubMed.
- R.-B. Lin, L. Li, H.-L. Zhou, H. Wu, C. He, S. Li, R. Krishna, J. Li, W. Zhou and B. Chen, Nat. Mater., 2018, 17, 1128–1133 CrossRef CAS PubMed.
- N. Nijem, H. Wu, P. Canepa, A. Marti, K. J. Balkus Jr., T. Thonhauser, J. Li and Y. J. Chabal, J. Am. Chem. Soc., 2012, 134, 15201–15204 CrossRef CAS PubMed.
- C. Gu, N. Hosono, J. J. Zheng, Y. Sato, S. Kusaka, S. Sakaki and S. Kitagawa, Science, 2019, 363, 387–391 CrossRef CAS PubMed.
- Y. Wang, T. Li, L. Li, R.-B. Lin, X. Jia, Z. Chang, H.-M. Wen, X.-M. Chen and J. Li, Adv. Mater., 2023, 35, 2207955 CrossRef CAS.
- Y. Su, K.-I. Otake, J.-J. Zheng, P. Wang, Q. Lin, S. Kitagawa and C. Gu, Nat. Commun., 2024, 15, 2898 CrossRef CAS.
- S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739–1753 CrossRef CAS.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- U. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- L. Alaerts, M. Maes, M. A. van der Veen, P. A. Jacobs and D. E. De Vos, Phys. Chem. Chem. Phys., 2009, 11, 2903–2911 RSC.
- S. H. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schroder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS.
- Y. Duan, Y. Huang, C. Wang, Q. Wang, K. Ge, Z. Lu, H. Wang, J. Duan, J. Bai and W. Jin, Chem. Sci., 2023, 14, 4605–4611 RSC.
- H. G. Hao, Y. F. Zhao, D. M. Chen, J. M. Yu, K. Tan, S. Q. Ma, Y. Chabal, Z. M. Zhang, J. M. Dou, Z. H. Xiao, G. Day, H. C. Zhou and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16067–16071 CrossRef CAS PubMed.
- E. Y. Wu, X. W. Gu, D. Liu, X. Zhang, H. Wu, W. Zhou, G. D. Qian and B. Li, Nat. Commun., 2023, 14, 6146 CrossRef CAS PubMed.
- Y. H. Huang, Y. F. Feng, Y. Li, K. Tan, J. Tang, J. F. Bai and J. G. Duan, Angew. Chem., Int. Ed., 2024, 63, e202403421 CrossRef CAS.
- Z. Q. Zhang, S. B. Peh, Y. X. Wang, C. J. Kang, W. D. Fan and D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 18927–18932 CrossRef CAS PubMed.
- S. Noro, S. Kitagawa, M. Kondo and K. Seki, Angew. Chem., Int. Ed., 2000, 39, 2082–2084 CrossRef CAS.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Q. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS.
- Y. Wang, N.-Y. Huang, X.-W. Zhang, H. He, R.-K. Huang, Z.-M. Ye, Y. Li, D.-D. Zhou, P.-Q. Liao, X.-M. Chen and J.-P. Zhang, Angew. Chem., Int. Ed., 2019, 58, 7692–7696 CrossRef CAS PubMed.
- G. D. Wang, Y. Z. Li, W. F. Zhang, L. Hou, Y. Y. Wang and Z. H. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 58862–58870 CrossRef CAS.
- D. Liu, J. Pei, X. Zhang, X.-W. Gu, H.-M. Wen, B. Chen, G. Qian and B. Li, Angew. Chem., Int. Ed., 2023, 62, e202218590 CrossRef CAS PubMed.
- R. B. Lin, L. B. Li, H. Wu, H. Arman, B. Li, R. G. Lin, W. Zhou and B. L. Chen, J. Am. Chem. Soc., 2017, 139, 8022–8028 CrossRef CAS PubMed.
- Q. Dong, X. Zhang, S. Liu, R. B. Lin, Y. Guo, Y. Ma, A. Yonezu, R. Krishna, G. Liu, J. Duan, R. Matsuda, W. Jin and B. Chen, Angew. Chem., Int. Ed., 2020, 59, 22756–22762 CrossRef CAS PubMed.
- S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2005, 127, 13519–13521 CrossRef CAS PubMed.
- P. L. Llewellyn, P. Horcajada, G. Maurin, T. Devic, N. Rosenbach, S. Bourrelly, C. Serre, D. Vincent, S. Loera-Serna, Y. Filinchuk and G. Ferey, J. Am. Chem. Soc., 2009, 131, 13002–13008 CrossRef CAS PubMed.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- M. K. Taylor, T. Runcevski, J. Oktawiec, M. I. Gonzalez, R. L. Siegelman, J. A. Mason, J. X. Ye, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2016, 138, 15019–15026 CrossRef CAS PubMed.
- S. Gao, C. G. Morris, Z. Z. Lu, Y. Yan, H. G. W. Godfrey, C. Murray, C. C. Tang, K. M. Thomas, S. H. Yang and M. Schröder, Chem. Mater., 2016, 28, 2331–2340 CrossRef CAS.
- L. B. Upi, R. Krishna, Y. Wang, X. Q. Wang, J. F. Yang and J. P. Li, Eur. J. Inorg. Chem., 2016, 2016, 4457–4462 CrossRef.
- R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, K. Kindo, Y. Mita, A. Matsuo, M. Kobayashi, H. C. Chang, T. C. Ozawa, M. Suzuki, M. Sakata and M. Takata, Science, 2002, 298, 2358–2361 CrossRef CAS PubMed.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS PubMed.
- K. Kishida, Y. Watanabe, S. Horike, Y. Watanabe, Y. Okumura, Y. Hijikata, S. Sakaki and S. Kitagawa, Eur. J. Inorg. Chem., 2014, 2014, 2747–2752 CrossRef CAS.
- Y. W. Chen, Z. W. Qiao, D. F. Lv, C. X. Duan, X. J. Sun, H. X. Wu, R. F. Shi, Q. B. Xia and Z. Li, Chem. Eng. J., 2017, 328, 360–367 CrossRef CAS.
- H. Zeng, M. Xie, Y.-L. Huang, Y. Zhao, X.-J. Xie, J.-P. Bai, M.-Y. Wan, R. Krishna, W. Lu and D. Li, Angew. Chem., Int. Ed., 2019, 58, 8515–8519 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- Y. Su, K. Otake, J. J. Zheng, S. Horike, S. Kitagawa and C. Gu, Nature, 2022, 611, 289–294 CrossRef CAS PubMed.
- H. Zeng, X.-J. Xie, T. Wang, M. Xie, Y. Wang, R.-J. Wei, W. Lu and D. Li, Nat. Chem. Eng., 2024, 1, 108–115 CrossRef.
- Q. J. Wang, J. B. Hu, L. F. Yang, Z. Q. Zhang, T. Ke, X. L. Cui and H. B. Xing, Nat. Commun., 2022, 13, 2955 CrossRef CAS PubMed.
- L. B. Li, H. M. Wen, C. H. He, R. B. Lin, R. Krishna, H. Wu, W. Zhou, J. P. Li, B. Li and B. L. Chen, Angew. Chem., Int. Ed., 2018, 57, 15183–15188 CrossRef CAS PubMed.
- W.-G. Cui, T.-L. Hu and X.-H. Bu, Adv. Mater., 2020, 32, 1806445 CrossRef CAS PubMed.
- C. Y. Chuah, H. Lee and T.-H. Bae, Chem. Eng. J., 2022, 430, 132654 CrossRef CAS.
- S. Q. Yang and T. L. Hu, Coord. Chem. Rev., 2022, 468, 214628 CrossRef CAS.
- L. Wang, S. Wu, J. Hu, Y. Jiang, J. Li, Y. Hu, Y. Han, T. Ben, B. Chen and Y. Zhang, Chem. Sci., 2024, 15, 5653–5659 RSC.
- Y. B. He, R. Krishna and B. L. Chen, Energy Environ. Sci., 2012, 5, 9107–9120 RSC.
- T. Ke, Q. Wang, J. Shen, J. Zhou, Z. Bao, Q. Yang and Q. Ren, Angew. Chem., Int. Ed., 2020, 59, 12725–12730 CrossRef CAS PubMed.
- L. Yang, X. Cui, Y. Zhang, Q. Yang and H. Xing, J. Mater. Chem. A, 2018, 6, 24452–24458 RSC.
- C. Yu, Z. Guo, L. Yang, J. Cui, S. Chen, Y. Bo, X. Suo, Q. Gong, S. Zhang, X. Cui, S. He and H. Xing, Angew. Chem., Int. Ed., 2023, 62, e202218027 CrossRef CAS PubMed.
- S. Tu, D. X. Lin, J. J. Huang, L. Yu, Z. W. Liu, Z. Li and Q. B. Xia, Microporous Mesoporous Mater., 2023, 354, 112532 CrossRef CAS.
- X.-W. Zhang, H. He, Y.-W. Gan, Y. Wang, N.-Y. Huang, P.-Q. Liao, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2024, 63, e202317648 CrossRef CAS PubMed.
- J. Shen, X. He, T. Ke, R. Krishna and Q. Ren, Nat. Commun., 2020, 11, 6259 CrossRef CAS PubMed.
- X. Zhang, Q. C. Chen, X. F. Bai, Y. L. Zhao and J. R. Li, Angew. Chem., Int. Ed., 2024, 63, e202411744 CrossRef CAS PubMed.
- X.-W. Gu, E. Wu, J.-X. Wang, H.-M. Wen, B. Chen, B. Li and G. Qian, Sci. Adv., 2023, 9, eadh0135 CrossRef CAS PubMed.
- B. Zhu, J.-W. Cao, S. Mukherjee, T. Pham, T. Zhang, T. Wang, X. Jiang, K. A. Forrest, M. J. Zaworotko and K.-J. Chen, J. Am. Chem. Soc., 2021, 143, 1485–1492 CrossRef CAS PubMed.
- H. M. Wen, C. Y. Yu, M. Y. Liu, C. Y. Lin, B. Y. Zhao, H. Wu, W. Zhou, B. L. Chen and J. Hu, Angew. Chem., Int. Ed., 2023, 62, e202309108 CrossRef CAS PubMed.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed.
- Y. Y. Xue, J. Lei, H. J. Lv, P. Liang, L. Q. Li and Q. G. Zhai, Small, 2024, 20, 2311555 CrossRef CAS PubMed.
- D.-X. Xue, A. Cadiau, Ł. J. Weseliński, H. Jiang, P. M. Bhatt, A. Shkurenko, L. Wojtas, C. Zhijie, Y. Belmabkhout, K. Adil and M. Eddaoudi, Chem. Commun., 2018, 54, 6404–6407 RSC.
- L. F. Yang, X. L. Cui, Q. W. Yang, S. H. Qian, H. Wu, Z. B. Bao, Z. G. Zhang, Q. L. Ren, W. Zhou, B. L. Chen and H. B. Xing, Adv. Mater., 2018, 30, 1705374–1705381 CrossRef PubMed.
- Y. J. Jiang, L. Y. Wang, T. A. Yan, J. B. Hu, W. Q. Sun, R. Krishna, D. M. Wang, Z. L. Gu, D. H. Liu, X. L. Cui, H. B. Xing and Y. B. Zhang, Chem. Sci., 2023, 14, 298–309 RSC.
- P. Zhang, L. Yang, X. Liu, J. Wang, X. Suo, L. Chen, X. Cui and H. Xing, Nat. Commun., 2022, 13, 4928 CrossRef CAS PubMed.
- C. Zhang, F. Formalik, D. Lv, F. Sha, K. O. Kirlikovali, X. Wang, X. Tang, S. Su, H. Xie, Y. Chen, Z. Li, R. Q. Snurr and O. K. Farha, Angew. Chem., Int. Ed., 2025, 64, e202424260 CrossRef CAS PubMed.
- W. Gong, Y. Xie, A. Yamano, S. Ito, E. W. Reinheimer, J. Dong, C. D. Malliakas, D. M. Proserpio, Y. Cui and O. K. Farha, Angew. Chem., Int. Ed., 2024, 63, e202318475 CrossRef CAS PubMed.
- G. D. Wang, Y. Z. Li, R. Krishna, W. Y. Zhang, L. Hou, Y. Y. Wang and Z. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202319978 CrossRef CAS PubMed.
- G.-D. Wang, Y.-Z. Li, W.-J. Shi, L. Hou, Y.-Y. Wang and Z. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202205427 CrossRef CAS PubMed.
- Z.-H. Guo, X.-Q. Wu, Y.-P. Wu, D.-S. Li, G.-P. Yang and Y.-Y. Wang, Angew. Chem., Int. Ed., 2025, 64, e202421992 CrossRef CAS PubMed.
- Y.-Y. Xue, J. Lei, H.-J. Lv, P. Liang, L. Li and Q.-G. Zhai, Small, 2024, 20, 2311555 CrossRef CAS PubMed.
- M.-Y. Gao, A. A. Bezrukov, B.-Q. Song, M. He, S. J. Nikkhah, S.-Q. Wang, N. Kumar, S. Darwish, D. Sensharma, C. Deng, J. Li, L. Liu, R. Krishna, M. Vandichel, S. Yang and M. J. Zaworotko, J. Am. Chem. Soc., 2023, 145, 11837–11845 CrossRef CAS PubMed.
- Y.-Z. Li, G.-D. Wang, R. Krishna, Q. Yin, D. Zhao, J. Qi, Y. Sui and L. Hou, Chem. Eng. J., 2023, 466, 143056 CrossRef CAS.
- G. D. Wang, R. Krishna, Y. Z. Li, W. J. Shi, L. Hou, Y. Y. Wang and Z. Zhu, Angew. Chem., 2022, 61, e202213015 CrossRef CAS PubMed.
- L. Li, L. Guo, F. Zheng, Z. Zhang, Q. Yang, Y. Yang, Q. Ren and Z. Bao, ACS Appl. Mater. Interfaces, 2020, 12, 17147–17154 CrossRef CAS PubMed.
- Y. Jiang, L. Wang, T. Yan, J. Hu, W. Sun, R. Krishna, D. Wang, Z. Gu, D. Liu, X. Cui, H. Xing and Y. Zhang, Chem. Sci., 2023, 14, 298–309 RSC.
- Y. Jiang, L. Wang, J. Hu, R. Krishna, B. Chen and Y. Zhang, Adv. Mater., 2024, 36, 2311140 CrossRef CAS PubMed.
- J. W. Ren, X. Dyosiba, N. M. Musyoka, H. W. Langmi, M. Mathe and S. J. Liao, Coord. Chem. Rev., 2017, 352, 187–219 CrossRef CAS.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- G. R. Cai, X. Ma, M. Kassymova, K. Sun, M. L. Ding and H. L. Jiang, ACS Cent. Sci., 2021, 7, 1434–1440 CrossRef CAS PubMed.
- D. Chakraborty, A. Yurdusen, G. Mouchaham, F. Nouar and C. Serre, Adv. Funct. Mater., 2024, 34, 2309089 CrossRef CAS.
- M. Taddei, D. A. Steitz, J. A. van Bokhoven and M. Ranocchiari, Chem.–Eur. J., 2016, 22, 3245–3249 CrossRef CAS PubMed.
- T. Gao, H. J. Tang, S. Y. Zhang, J. W. Cao, Y. N. Wu, J. Chen, Y. Wang and K. J. Chen, J. Solid State Chem., 2021, 303, 122547 CrossRef CAS.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastre, ChemInform, 2006, 37, 626–636 Search PubMed.
- J.-B. Lin, T. T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Durekova, F. Akhtar, R. K. Mah, O. Ghaffari-Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. K. H. Shimizu, Science, 2021, 374, 1464–1469 CrossRef CAS PubMed.
- S. Geng, E. Lin, X. Li, W. Liu, T. Wang, Z. Wang, D. Sensharma, S. Darwish, Y. H. Andaloussi, T. Pham, P. Cheng, M. J. Zaworotko, Y. Chen and Z. Zhang, J. Am. Chem. Soc., 2021, 143, 8654–8660 CrossRef CAS PubMed.
- B. Zhou, Q. Li, Q. Zhang, J. Duan and W. Jin, J. Membr. Sci., 2020, 597, 117772 CrossRef CAS.
- D. Crawford, J. Casaban, R. Haydon, N. Giri, T. Mcnally and S. L. James, Chem. Sci., 2015, 6, 1645–1649 RSC.
- C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Chem. Commun., 2015, 51, 6698–6713 RSC.
- M. Vadivelu, S. Sugirdha, P. Dheenkumar, Y. Arun and K. Karthikeyan, Green Chem., 2017, 19, 3601–3610 RSC.
- Y. Wang, H. Wang, Y. Jiang, C. Zhang, J. Shao and D. Xu, Green Chem., 2017, 19, 1674–1677 RSC.
- Q. Cao, R. T. Fallis, I. A. Browne and L. Duncan, ChemSusChem, 2019, 12, 2554–2557 CrossRef CAS PubMed.
- A. Pichon, A. Lazuen-Garay and S. L. James, CrystEngComm, 2006, 8, 211–214 RSC.
- Y. Liang, G. Xie, K.-K. Liu, M. Jin, Y. Chen, X. Yang, Z.-J. Guan, H. Xing and Y. Fang, Angew. Chem., Int. Ed., 2025, 64, e202416884 CrossRef CAS PubMed.
- T. D. Bennett, S. Cao, J. C. Tan, D. A. Keen, E. G. Bithell, P. J. Beldon, T. Friscic and A. K. Cheetham, J. Am. Chem. Soc., 2011, 133, 14546–14549 CrossRef CAS PubMed.
- O. M. Yaghi, R. Jernigan, H. Li, C. E. Davis and T. L. Groy, J. Chem. Soc., Dalton Trans., 1997, 2383–2384 RSC.
- S.-N. Kim, Y.-R. Lee, S.-H. Hong, M.-S. Jang and W.-S. Ahn, Catal. Today, 2015, 245, 54–60 CrossRef CAS.
- T. K. Vo, V. N. Le, K. S. Yoo, M. Song, D. Kim and J. Kim, Cryst. Growth Des., 2019, 19, 4949–4956 CrossRef CAS.
- K. Pobłocki, J. Drzeżdżon, B. Gawdzik and D. Jacewicz, Green Chem., 2022, 24, 9402–9427 RSC.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626–636 RSC.
- J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250 RSC.
- M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910 RSC.
- C. McKinstry, R. J. Cathcart, E. J. Cussen, A. J. Fletcher, S. V. Patwardhan and J. Sefcik, Chem. Eng. J., 2016, 285, 718–725 CrossRef CAS.
- G. H. Albuquerque, R. C. Fitzmorris, M. Ahmadi, N. Wannenmacher, P. K. Thallapally, B. P. McGrail and G. S. Herman, CrystEngComm, 2015, 17, 5502–5510 RSC.
- H. Ren and T. X. Wei, Chemelectrochem, 2022, 9, e202200196 CrossRef CAS.
- N. Campagnol, T. R. C. Van Assche, M. Y. Li, L. Stappers, M. Dinca, J. F. M. Denayer, K. Binnemans, D. E. De Vos and J. Fransaer, J. Mater. Chem. A, 2016, 4, 3914–3925 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |