
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Heterocyclic carbene-catalyzed enantioselective (dynamic) kinetic resolution for the assembly of inherently chiral macrocycles†
Zhipeng Li‡
,
Jingyang Zhang‡,
Wanmeng Zhu,
Tianyi Wang,
Yefeng Tang and
Jian Wang *
*
School of Pharmaceutical Sciences, Key Laboratory of Bioorganic Phosphorous Chemistry and Chemical Biology, Ministry of Education, Tsinghua University, Beijing, 100084, China. E-mail: wangjian2012@tsinghua.edu.cn
First published on 5th May 2025
Abstract
Heterocalixaromatics play a significant role in supramolecular chemistry and materials science. However, the absence of robust enantioselective synthetic methods has constrained their broader applications. In contrast, the construction of inherently chiral macrocycles via N-heterocyclic carbene (NHC) remains underexplored to date. We herein report an NHC-catalyzed approach for the rapid assembly of inherently chiral macrocycles. This transformation proceeds via a dynamic kinetic resolution (DKR) or kinetic resolution (KR) process, enabling the conversion of racemic substrates into inherently chiral heterocalixaromatics with good to high yields and high to excellent enantioselectivities. DFT calculations were carried out to clarify the chirality control in the related DKR process.
NHCs have advanced significantly to accelerate asymmetric transformations and are capable of assembling a variety of skeletons that are biologically important or medically useful.1 The in-depth promotion of NHC catalysis is attributed to its bench-stable NHC precursors derived from natural sources, multiple activation modes, and mild reaction environments.1c,2 Meanwhile, macrocycles (i.e. rings containing 12 or more atoms) have unique structural properties that distinguish them from their acyclic small molecule counterparts because they offer a compromise between structural pre-organization and sufficient conformational flexibility,3 and to which much of their remarkable biological activities is attributed.4 Compared with the plethora of reports on the creation of central and axial chirality, the asymmetric construction of chiral macrocycles using NHCs as a tool has only been reported in recent years.5 One of the current focuses in this scenario is the asymmetric preparation of [2,2]-paracyclophanes (Fig. 1a, left). Of note, suchcontributions were pioneered by Chi, Jin and Veselý via kinetic resolution,5a parallel kinetic resolution5b and a desymmetrization strategy,6 respectively. Most recently, NHC catalysis has moved its attention to the generation of planar-chiral macrocycles (Fig. 1a, middle). In this context, our group uncovered the first atroposelective synthesis of indole-based planar-chiral macrocycles via NHC-catalyzed macrocyclization.5c Meanwhile, the Chi and Zhao groups disclosed the examples of NHC-catalyzed asymmetric synthesis for the preparation of planar-chiral cyclophanes.5e–g
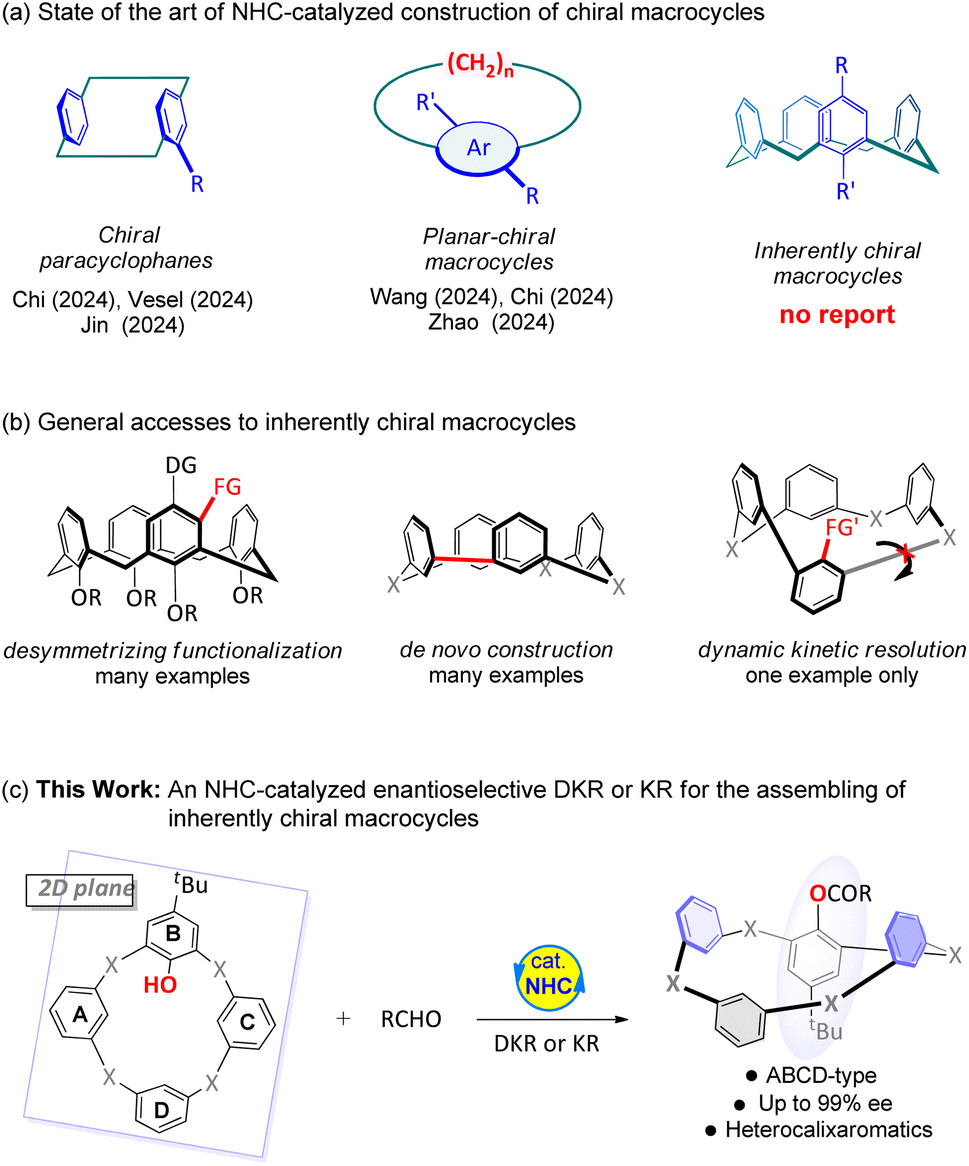 | ||
| Fig. 1 Background and our design. (a) Overview of the NHC-catalyzed enantioselective synthesis of chiral macrocycles. (b) Background of the synthesis of inherently chiral macrocycles. (c) Our design. | ||
In fact, NHC catalysis has gained mature experience for the synthesis of atropisomeric compounds, including planar-chiral macrocyclic atropisomeres.1j,7 Despite these significant advancements, NHC-mediated reactions have not yet been utilized in the generation of inherently chiral macrocycles. Unlike conventional molecules that possess central, axial, planar, or helical chirality elements, inherently chiral molecules lack explicit chiral elements. In contrast, this class of molecules is given chirality by the rigid conformation of whole structures.8 Therefore, it would be very interesting for us to investigate whether NHCs can exhibit similar potential in building central, axial, and planar chirality when establishing inherently chiral macrocycles, and to gain insight into the mechanisms by which this chiral control occurs.
In recent years, the advancement in asymmetric catalytic methodologies has led to the emergence of diverse enantioselective approaches for building inherently chiral macrocycles.9 The distinctive properties of inherently chiral compounds make it feasible to construct themselves through a desymmetric functionalization strategy mediated by directional groups within these molecules (Fig. 1b, left). Recently, chiral phosphoric acid (CPA)-catalyzed Povarov reaction,9a,b metal-catalyzed annulation,9c–g and sulfide-catalyzed sulfenylation9h have been reported. De novo synthesis represents a robust and versatile strategy for constructing inherently chiral macrocycles, specifically ABCD-type macrocycles (Fig. 1b, middle). The incorporation of metal-catalyzed intramolecular coupling reactions9i,k and CPA-catalyzed SNAr reactions9j further enhances the utilization of this strategy. DKR appears to be a versatile strategy for constructing various types of chirality. However, only one case has been reported to synthesize the inherently chiral macrocycles using this approach successfully so far.9l There is no doubt that the racemization of inherently chiral macrocycles just by ring inversion poses a significant challenge to distinguish between the two enantiomers.
Herein, we disclose an NHC-catalyzed DKR or KR approach to afford the inherently chiral macrocycles in good to high yields and with high to excellent enantioselectivities (Fig. 1c). Despite the advancements in the carbene-catalyzed DKR process, the “conformational capture” of nucleophiles exhibiting distinct configurational changes just through benzene ring flipping to acyl azolium intermediates remains a topic of significant interest.
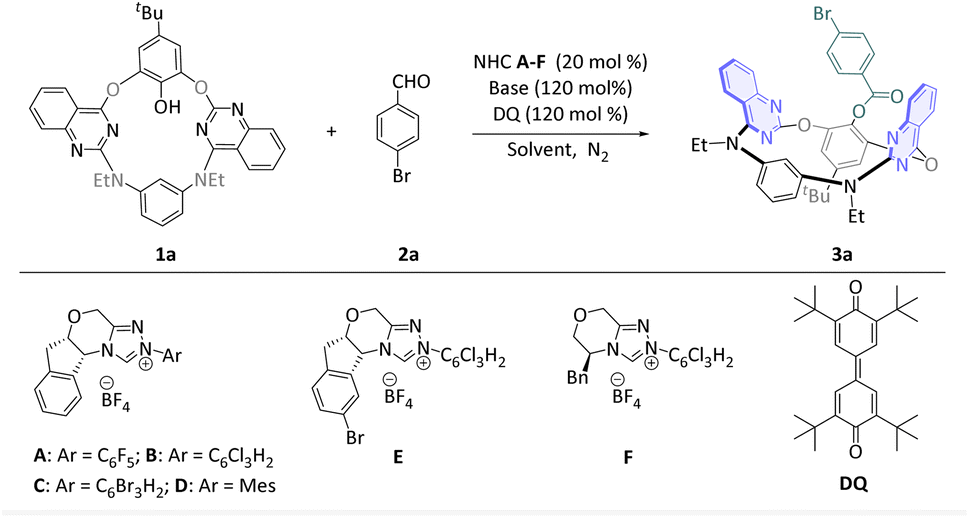
In recent years, the types of inherently chiral macrocycles achieved through asymmetric catalysis have primarily focused on calix[4]arenes. Owing to differences in bridging atoms, nitrogen- and oxygen-bridged calixaromatics exhibit distinct spatial conformations compared to calix[4]arenes, as well as offering varied application potentials in supramolecular chemistry.10 So our study was focused on heteroatom-bridged calix[4](het)arenes. Diazadioxacalix[2]-arene[2]quinazoline (1a) and 4-bromobenzaldehyde (2a) were selected as model substrates to verify whether the DKR process can occur. Preliminary studies (Table 1) have shown that N-pentafluorophenyl substituted NHC pre-cat. A provided the corresponding product 3a in moderate yield but only 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 e.r. under conditions of K2CO3 as base, tetra-tert-butyldiphenylquinone (DQ) as oxidant, tetrahydrofuran (THF) as solvent, and room temperature (entry 1). Gratifyingly, N-1,3,5-trichlorophenyl substituted NHC pre-cat. B, N-1,3,5-tribromophenyl substituted NHC pre-cat. C and N-mesityl substituted NHC pre-cat. D all contributed to different degrees of e.r. value, but not a significant increase in yield (entries 2–4). A small increase in the e.r. value was achieved by introducing an additional bromide functional group on the NHC pre-cat. B (entry 5, NHC pre-cat. E). The conversion of the NHC scaffold to a chiral morpholine skeleton did not result in a substantial enhancement in e.r. or yield (entry 6, NHC pre-cat. F). In addition, a solvent survey (entries 7–9) indicates that the yield was slightly improved in dichloromethane (DCM) (entry 7, 61%). Inorganic and organic bases other than K2CO3 that we evaluated failed to improve the reaction outcomes (entries 10–12). By adjusting the amount of substrate 2a to 3.0 equivalents, the yield was enhanced to 86%, albeit with a concomitant decrease in the e.r. value (entries 13 and 14). We subsequently investigated the impact of temperature and discovered that elevating the temperature resulted in an increased e.r. value of 3a (entries 15 and 16). This phenomenon may be attributed to the enhanced racemization rate of 1a. When the reaction proceeds at 50 °C, 88% yield and 93:7 e.r. were achieved (entry 16). Finally, we introduced an equal volume of n-hexane to DCM into the reaction system to dilute the reactants, thereby reducing the reaction concentration. Ultimately, this approach enabled us to complete the screening with an optimal result of 91% yield and 96:4 e.r. (entry 17) Furthermore, 20 mol% of NHC pre-cat. E was essential. (entry 18)
:18 e.r. under conditions of K2CO3 as base, tetra-tert-butyldiphenylquinone (DQ) as oxidant, tetrahydrofuran (THF) as solvent, and room temperature (entry 1). Gratifyingly, N-1,3,5-trichlorophenyl substituted NHC pre-cat. B, N-1,3,5-tribromophenyl substituted NHC pre-cat. C and N-mesityl substituted NHC pre-cat. D all contributed to different degrees of e.r. value, but not a significant increase in yield (entries 2–4). A small increase in the e.r. value was achieved by introducing an additional bromide functional group on the NHC pre-cat. B (entry 5, NHC pre-cat. E). The conversion of the NHC scaffold to a chiral morpholine skeleton did not result in a substantial enhancement in e.r. or yield (entry 6, NHC pre-cat. F). In addition, a solvent survey (entries 7–9) indicates that the yield was slightly improved in dichloromethane (DCM) (entry 7, 61%). Inorganic and organic bases other than K2CO3 that we evaluated failed to improve the reaction outcomes (entries 10–12). By adjusting the amount of substrate 2a to 3.0 equivalents, the yield was enhanced to 86%, albeit with a concomitant decrease in the e.r. value (entries 13 and 14). We subsequently investigated the impact of temperature and discovered that elevating the temperature resulted in an increased e.r. value of 3a (entries 15 and 16). This phenomenon may be attributed to the enhanced racemization rate of 1a. When the reaction proceeds at 50 °C, 88% yield and 93:7 e.r. were achieved (entry 16). Finally, we introduced an equal volume of n-hexane to DCM into the reaction system to dilute the reactants, thereby reducing the reaction concentration. Ultimately, this approach enabled us to complete the screening with an optimal result of 91% yield and 96:4 e.r. (entry 17) Furthermore, 20 mol% of NHC pre-cat. E was essential. (entry 18)
|
|||
|---|---|---|---|
| Entrya | Conditions | Yield [%]b | e.r.c |
| a Reaction conditions: 1a (0.033 mmol), 2a (0.04 mmol), DQ (0.04 mmol), NHC pre-cat. A–E (20 mol%), base (1.2 equiv), solvent (0.6 mL) at room temperature for 24 h under a N2 atmosphere.b Isolated yields.c The e.r. values were determined via chiral phase HPLC analysis.d 2a (0.1 mmol), DQ (0.1 mmol).e 2a (0.16 mmol), DQ (0.16 mmol).f DCM (0.6 mL) + Hex. (0.6 mL).g 5 mol% of NHC pre-cat. E, 48 h. | |||
| 1 | NHC A, K2CO3, THF, rt | 43 | 82:18 |
| 2 | NHC B, K2CO3, THF, rt | 53 | 93:7 |
| 3 | NHC C, K2CO3, THF, rt | 45 | 92:8 |
| 4 | NHC D, K2CO3, THF, rt | 51 | 86:14 |
| 5 | NHC E, K2CO3, THF, rt | 55 | 94:8 |
| 6 | NHC F, K2CO3, THF, rt | 45 | 13:87 |
| 7 | NHC E, K2CO3, DCM, rt | 61 | 94.5:5.5 |
| 8 | NHC E, K2CO3, 1,4-dioxane, rt | 38 | 94:6 |
| 9 | NHC E, K2CO3, toluene, rt | Trace | ND |
| 10 | NHC E, Cs2CO3, DCM, rt | 68 | 89.5:10.5 |
| 11 | NHC E, NaHCO3, DCM, rt | 37 | 85:15 |
| 12 | NHC E, DIPEA, DCM, rt | 73 | 84.5:15.5 |
| 13d | NHC E, K2CO3, DCM, rt | 86 | 85.5:14.5 |
| 14e | NHC E, K2CO3, DCM, rt | 85 | 75:25 |
| 15d | NHC E, K2CO3, DCM, 0 °C | 70 | 77:23 |
| 16d | NHC E, K2CO3, DCM, 50 °C | 88 | 93:7 |
| 17d,f | NHC E, K2CO3, DCM/Hex. (1:1), 50 °C |
91 | 96:4 |
| 18 d,f,g | NHC E, K2CO3, DCM/Hex. (1:1), 50 °C |
72 | 96:4 |
The scope of this DKR process was systematically investigated. Aromatic (hetero)aldehydes, specifically including quinoline-4-carbaldehyde, 2-naphthaldehyde, 1-methyl-1H-pyrrole-2-carbaldehyde, and benzofuran-2-carbaldehyde, were evaluated as esterification reagents. These substrates afforded their corresponding desired products in excellent yields and enantioselectivities (3a–3f). Anthracene-9-carbaldehyde, despite its significant steric hindrance, was also found to be compatible (Fig. 2, 3g). It is noteworthy that N-mesityl substituted NHC pre-cat. D exhibits superior reactivity with alkyl aldehyde substrates. Asymmetric esterification reactions of various alkyl aldehydes, including cyclohexanecarbaldehyde, 3-phenylpropanal, (Z)-dec-4-enal, isobutyraldehyde, 3,3-dimethylbutanal, and cyclopentanecarbaldehyde, consistently yielded the corresponding products (3h–3m) with high e.r. values across all examined examples. The absolute configuration of compound 3l was determined by X-ray crystallography (Fig. 2), and the configurations of other products were deduced by analogy.11 Subsequently, the DKR of heterocalix[2]arene[2]quinazolines bearing ethyl and allyl substituents on the bridging nitrogen atom afforded the products in excellent yields and with high e.r. values (3n–3p). In general, alkyl aldehydes exhibit superior compatibility in this reaction. The involvement of aromatic aldehydes with varying degrees of steric hindrance results in minor variations in the efficacy of chirality control.
 | ||
| Fig. 2 Scope of aldehydes and heterocalix[2]arene[2]quinazolines. aReaction conditions: 1a (0.033 mmol), 2a (0.1 mmol), DQ (0.1 mmol), NHC pre-cat. E (20 mol%), K2CO3 (1.2 equiv.), DCM (0.6 mL) + Hex. (0.6 mL) at 50 °C for 36 h under a N2 atmosphere; isolated yields; the e.r. values were determined via chiral-phase HPLC analysis. bNHC D instead of E. | ||
To build more structurally diversified calix[4](het)arene structures, we prepared a series of multifunctional substituted calix[4](het)arenes (Fig. 3, 4a–4d) as starting materials. These molecules do not undergo racemization at room temperature and can be effectively separated by high performance liquid chromatography (HPLC). It is expected that they will only be able to participate in asymmetric transformations through kinetic resolution. When phenylpropanal was used as the reaction partner, rac-4a was converted to enantioenriched corresponding product 5a (97:3 e.r., 42%) and recovered the ent-4a (96:4 e.r., 38%), respectively. When long-chained aldehydes or rigid aldehydes were employed, the reaction still proceeded smoothly and afforded moderate to good yields and high e.r. values (5b–5e). Interestingly, the utilization of tert-butyl 4-formylpiperidine-1-carboxylate led to a decrease in e.r. (5f), suggesting that the KR process exhibits a greater sensitivity to the steric substituents compared to the DKR process. Finally, the KR of calix[4](het)arenes bearing ethyl, allyl and benzyl substituents on the oxygen atom afforded the products in good yields and with high e.r. values (5g, 5h, and 5i).
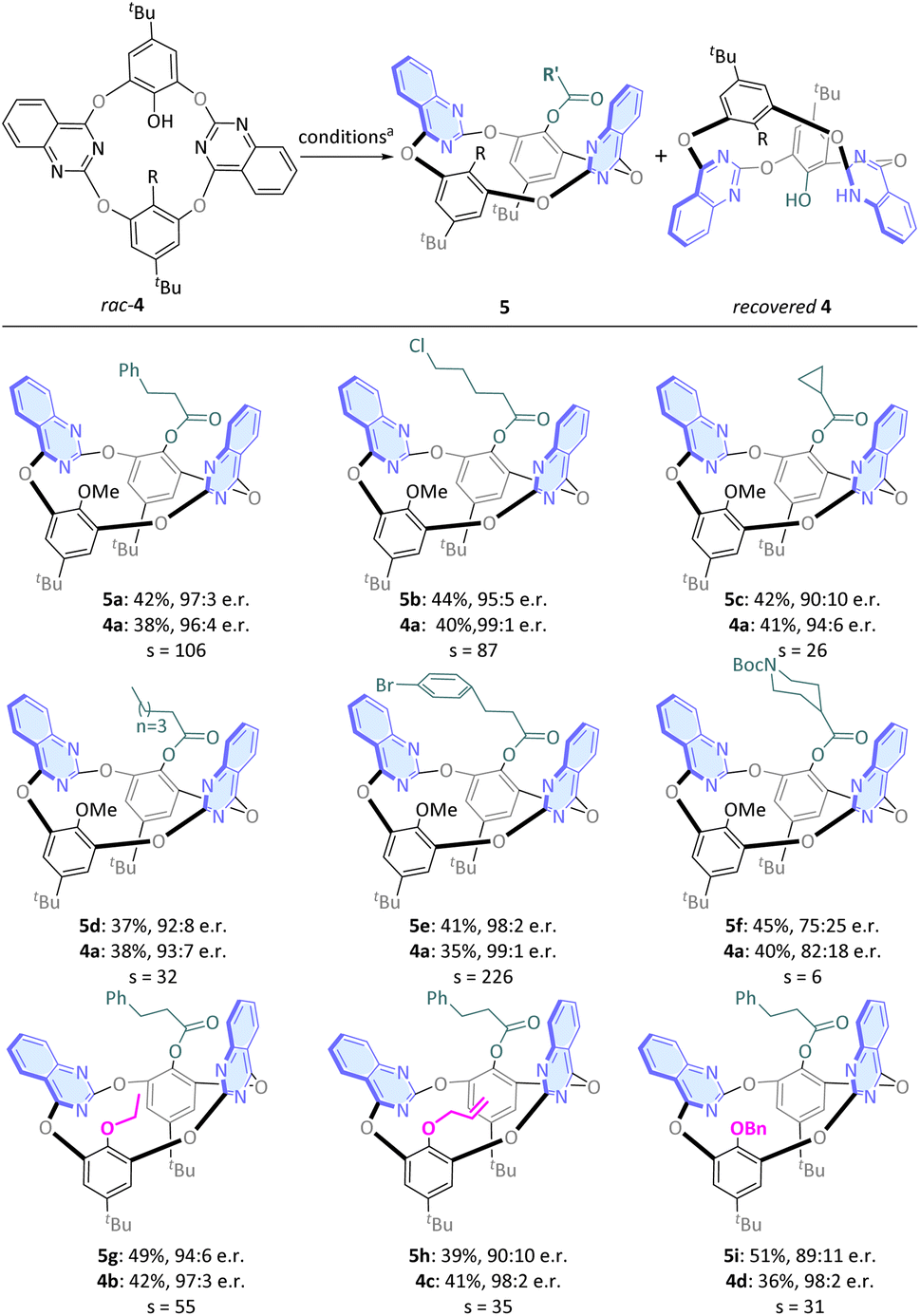 | ||
| Fig. 3 Kinetic resolution of racemic calix[4](het)arene 4. aReaction conditions: 4 (0.1 mmol), 2 (0.08 mmol), DQ (0.055 mmol), NHC pre-cat. E (20 mol%), K2CO3 (0.7 equiv.), DCM (2.0 mL) for 12–24 h under a N2 atmosphere; isolated yields; the e.r. values were determined via chiral-phase HPLC analysis; selectivity factor (s) = ln[(1 − C)(1 − ee1)]/ln[(1 − C)(1 + ee1)], C = ee1/(ee1 + ee2). | ||
To further demonstrate the synthetic utility of this protocol, it was successfully applied to the 1 mmol-scale synthesis of product 3o, yielding 3o in an isolated yield of 86% and with excellent enantioselectivity (99:1 e.r.). To enhance the potential utility of this process, we further modified the inherently chiral macrocycles (Fig. 4). The recovered compound 4a yielded the inherently chiral O-alkylated product 6a (85% yield and 97:3 e.r.). Moreover, the enantiomerically enriched acetylene 6b was efficiently obtained from 3a via Sonogashira cross-coupling directly, and retained an excellent e.r. value (96:4). Furthermore, to validate the broad-spectrum applicability of this asymmetric esterification strategy, we successfully synthesized tetraoxacalix[2]arene[2]pyridines 1d. Preliminary studies have shown that N-1,3,5-tricyclohexyl substituted NHC pre-cat. G provided the corresponding product 6c in moderate yield and 91:9 e.r. This result demonstrates that the NHC-catalytic system remains applicable to the DKR process of other types of heterocalixaromatics.
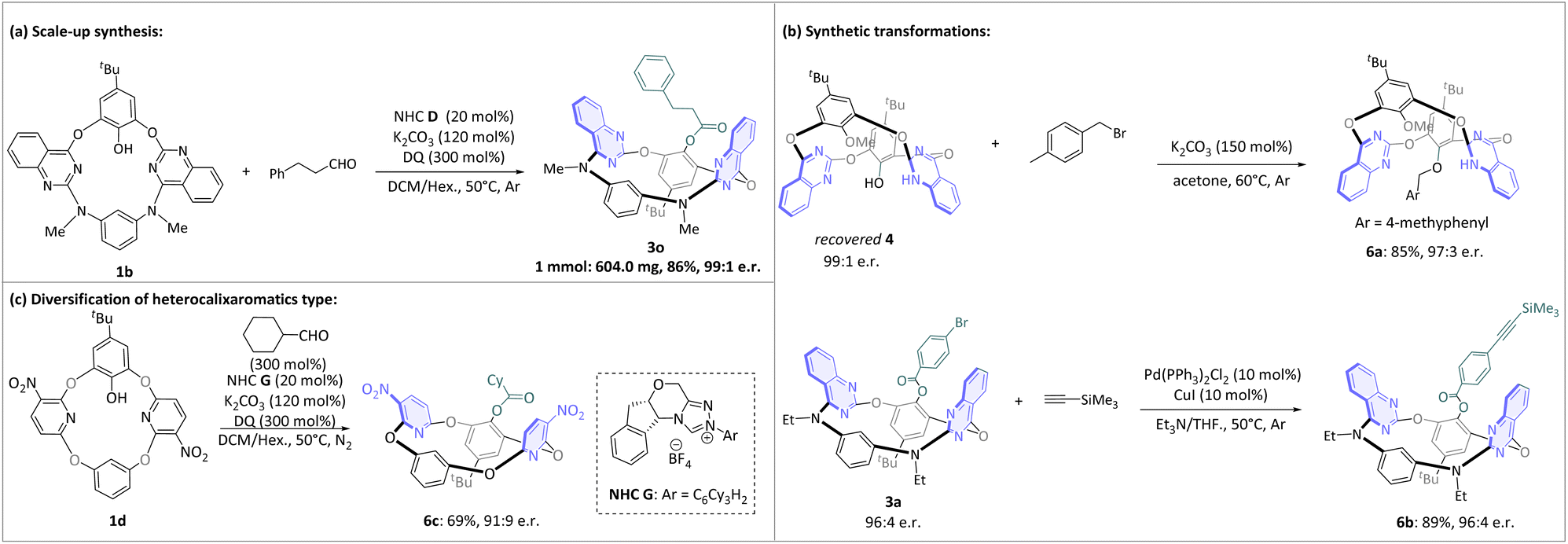 | ||
| Fig. 4 Follow-up chemistry. | ||
For an in-depth understanding of the chirality control mode in the DKR process, we conducted a DFT analysis of TS-(Sp)-1a and TS-(Rp)-1a at the level of SMD(DCM)-M06-2X/def2-TZVP. The three-dimensional structures and key atomic distances of these two transition states are specifically investigated. As illustrated in Fig. 5, the energy barrier of the addition of a hydroxyl group to acyl azolium in TS-(Sp)-1a is 9.2 kcal mol−1 lower than that in TS-(Rp)-1a. Thus, the formation of S-configuration products is kinetically favoured, which aligns with our experimental observations. Furthermore, the analysis of atomic distances and angles indicates that TS-(Sp)-1a occurs at an earlier stage and exhibits lesser deformation. To further vividly demonstrate the difference in non-bonding interactions in transition states, we conducted a brief reduced density gradient analysis (Fig. 5), which showed that it had more weak interactions in TS-(Sp)-1a, especially in the region near trichlorobenzene of the catalyst. In summary, we have successfully achieved the highly enantioselective synthesis of inherently chiral macrocycles through NHC-catalyzed asymmetric esterification of heterocalixaromatics. The steric hindrance imposed by the aromatic ring substituents enables the reaction to proceed via kinetic resolution or dynamic kinetic resolution (28 examples, up to 99:1 e.r.). Applications of these inherently chiral scaffolds in library inclusion and asymmetric catalysis are currently under investigation in our laboratory.
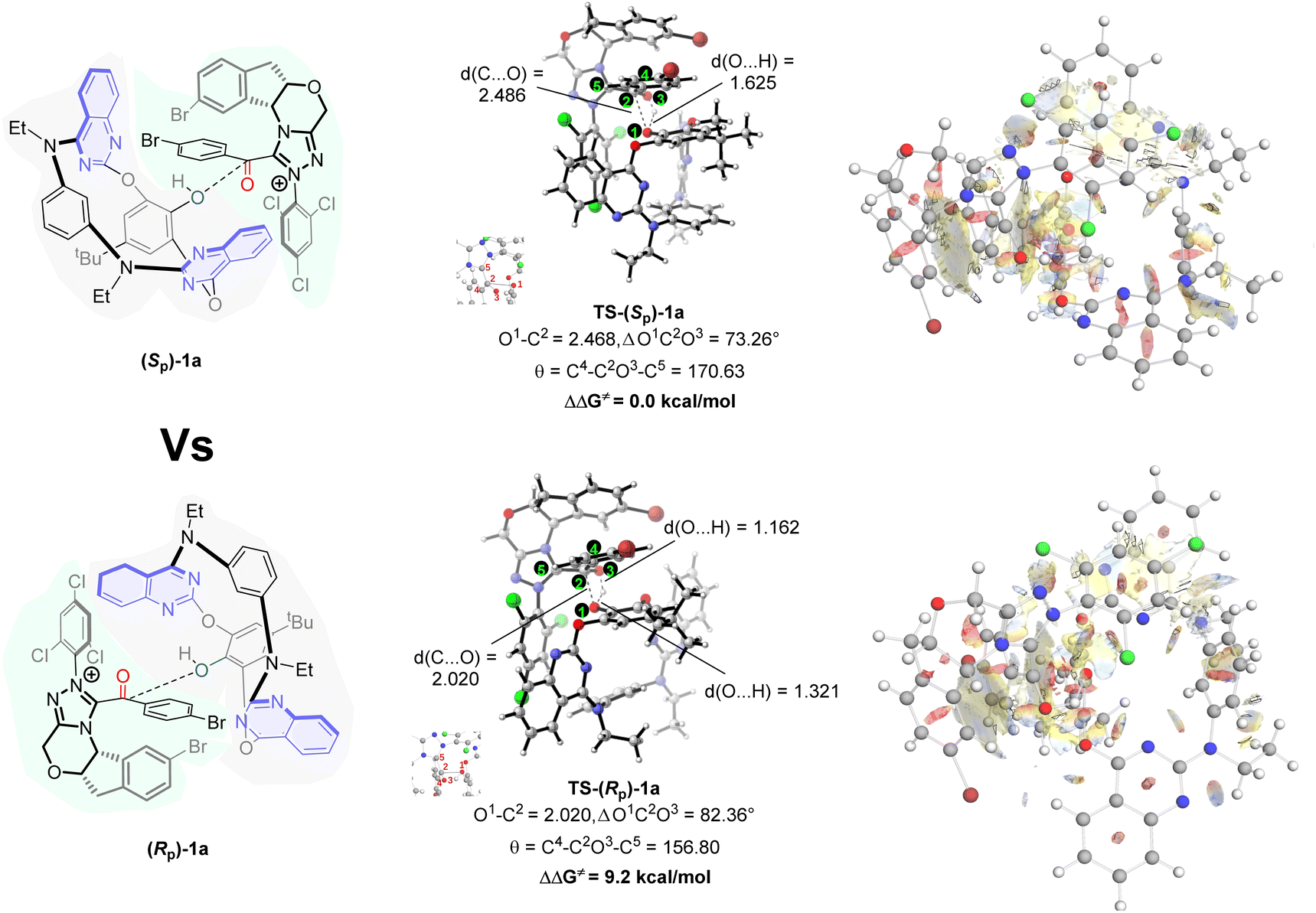 | ||
| Fig. 5 Calculated energy profile of TS-(Sp)-1a and TS-(Rp)-1a and key distances between atoms (in angstrom). | ||
Data availability
All data supporting the findings of this study are available within the article and its ESI.†Author contributions
Z. P. L. conducted the main experiments. J. Y. Z. conducted the DFT calculations. W. M. Z. and T. Y. W. prepared several starting materials, including substrates. J. W. conceptualized and directed the project and drafted the manuscript with assistance from the co-authors. All authors contributed to discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support for this work was provided by the National Natural Science Foundation of China (21871160, 21672121, and 22071130), the Bayer Investigator fellowship, and the fellowship of Tsinghua-Peking Centre for Life Sciences (CLS).Notes and references
- For a review: (a) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed; (b) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS PubMed; (c) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC; (d) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS; (e) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS; (f) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS; (g) R. S. Menon, A. T. Biju and V. Nair, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (h) X.-Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862–3873 CrossRef CAS; (i) H. Ohmiya, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS; (j) S. Chakraborty, S. Barik and A. T. Biju, Chem. Soc. Rev., 2025, 54, 1102–1124 RSC.
- (a) X. Chen, H. Wang, Z. Jin and Y. R. Chi, Chin. J. Chem., 2020, 38, 1167–1202 CrossRef CAS; (b) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS PubMed.
- (a) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS; (b) J. J. Ciardiello, H. L. Stewart, H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Bioorg. Med. Chem., 2017, 25, 2825–2843 CrossRef CAS.
- (a) L. A. Wessjohann, E. Ruijter, D. Garcia-Rivera and W. Brandt, Mol. Divers., 2005, 9, 171–186 CrossRef CAS PubMed; (b) K. C. Nicolaou, S. Ninkovic, F. Sarabia, D. Vourloumis, Y. He, H. Vallberg, M. R. V. Finlay and Z. Yang, J. Am. Chem. Soc., 1997, 119, 7974–7991 CrossRef CAS.
- For selected examples: (a) Y. Lv, C. Mou, Q. Liu, L. Shu, Y. Cai, X. Lv, Z. Jin and Y. R. Chi, Org. Lett., 2024, 26, 1584–1588 CrossRef CAS PubMed; (b) Q. Liu, K. Teng, Y. Zhang, Y. Lv, Y. R. Chi and Z. Jin, Angew. Chem., Int. Ed., 2024, 63, e202406386 CrossRef CAS; (c) G. Yang, Y. He, T. Wang, Z. Li and J. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316739 CrossRef CAS; (d) G. Yang, S. Liu, S. Ji, X. Wu and J. Wang, Chem. Sci., 2024, 15, 19599–19603 RSC; (e) J. Li, Z. Dong, Y. Chen, Z. Yang, X. Yan, M. Wang, C. Li and C. Zhao, Nat. Commun., 2024, 15, 2338 CrossRef CAS; (f) J. Wang, M. Wang, Y. Wen, P. Teng, C. Li and C. Zhao, Org. Lett., 2024, 26, 1040–1045 CrossRef CAS; (g) X. Lv, F. Su, H. Long, F. Lu, Y. Zeng, M. Liao, F. Che, X. Wu and Y. R. Chi, Nat. Commun., 2024, 15, 958 CrossRef CAS PubMed.
- V. Dočekal, F. Koucký, I. Císařová and J. Veselý, Nat. Commun., 2024, 15, 3090 CrossRef.
- (a) M. Zhang, X. Yang, X. Peng, X. Li and Z. Jin, Sci. China:Chem., 2024, 68, 815–825 CrossRef; (b) J. Wang, C. Zhao and J. Wang, ACS Catal., 2021, 11, 12520–12531 CrossRef CAS.
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef.
- For selected examples: (a) Y.-K. Jiang, Y.-L. Tian, J. Feng, H. Zhang, L. Wang, W.-A. Yang, X.-D. Xu and R.-R. Liu, Angew. Chem., Int. Ed., 2024, 63, e202407752 CrossRef CAS; (b) S. Yu, M. Yuan, W. Xie, Z. Ye, T. Qin, N. Yu and X. Yang, Angew. Chem., Int. Ed., 2024, 63, e202410628 CrossRef CAS PubMed; (c) Y.-Z. Zhang, M.-M. Xu, X.-G. Si, J.-L. Hou and Q. Cai, J. Am. Chem. Soc., 2022, 144, 22858–22864 CrossRef CAS; (d) X. Zhang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2023, 14, 827–832 RSC; (e) T. Li, Y. Zhang, C. Du, D. Yang, M.-P. Song and J.-L. Niu, Nat. Commun., 2024, 15, 7673 CrossRef CAS PubMed; (f) P.-F. Qian, G. Zhou, J.-H. Hu, B.-J. Wang, A.-L. Jiang, T. Zhou, W.-K. Yuan, Q.-J. Yao, J.-H. Chen, K.-X. Kong and B.-F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202412459 CrossRef CAS; (g) G. Giovanardi, G. Scarica, V. Pirovano, A. Secchi and G. Cera, Org. Biomol. Chem., 2023, 21, 4072–4083 RSC; (h) X.-Y. Zhang, D. Zhu, R.-F. Cao, Y.-X. Huo, T.-M. Ding and Z.-M. Chen, Nat. Commun., 2024, 15, 9929 CrossRef CAS PubMed; (i) S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed; (j) X.-C. Li, Y. Cheng, X.-D. Wang, S. Tong and M.-X. Wang, Chem. Sci., 2024, 15, 3610–3615 RSC; (k) Y.-F. Jiang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2024, 15, 12517–12522 RSC; (l) Q.-L. Lu, X.-D. Wang, S. Tong, J. Zhu and M.-X. Wang, ACS Catal., 2024, 14, 5140–5146 CrossRef CAS.
- M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS.
- CCDC 2421502 (3l) contains the supplementary crystallographic data for this paper..
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2421502. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01773d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |