
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Local coordination geometry within cobalt spinel oxides mediates photoinduced polaron formation†
Erica P.
Craddock
 ,
Jacob L.
Shelton‡
,
Michael T.
Ruggiero
and
Kathryn E.
Knowles
*
,
Jacob L.
Shelton‡
,
Michael T.
Ruggiero
and
Kathryn E.
Knowles
*
Department of Chemistry, University of Rochester, Rochester, NY 14627, USA. E-mail: kknowles@ur.rochester.edu
First published on 12th May 2025
Abstract
Understanding the photophysics of transition metal oxides is crucial for these materials to realize their considerable potential in applications such as photocatalysis and optoelectronics. Recent studies suggest that formation of localized excited states consisting of polarons (quasi-particles comprising a charge carrier strongly coupled to a proximal lattice distortion) plays a crucial role in the photophysics of these materials. Cobalt-containing spinel oxides (Co3O4 and ZnCo2O4) offer a unique opportunity to investigate the influence of local geometry, and cation inversion on photoinduced polaron formation. Here, we use Hubbard-corrected density functional theory (DFT + U) paired with resonance Raman and temperature-dependent optical spectroscopies to demonstrate that low-energy transitions observed in Co3O4 are associated with d–d transitions involving cobalt ions occupying tetrahedral sites within the spinel lattice. These low-energy optical transitions exhibit strong coupling to phonon modes associated with tetrahedral sites. Replacing most tetrahedral cobalt ions with zinc produces the slightly inverted ternary spinel material, ZnCo2O4, in which we observe a phonon-coupled optical transition that occurs at the same energy as observed in Co3O4. We propose that these phonon-coupled optical transitions enable direct access to a polaronic state upon photoexcitation; however, the intensity of this optical transition depends on temperature in Co3O4, whereas no significant temperature dependence is observed in ZnCo2O4. We therefore hypothesize that in Co3O4 the mechanism of polaron formation is coupling of the optical transition to dynamic, thermally-gated lattice distortions, whereas, in ZnCo2O4, the transition couples to static lattice defects that arise from the presence of a small population of tetrahedrally-coordinated cobalt ions.
Introduction
Transition metal oxides have promising characteristics for solar energy conversion technologies because of their visible band gap energies, stability, abundance, cost-effectiveness and low toxicity.1,2 However, these materials contain weakly dispersive bands originating from the metal 3d orbitals that lead to low charge-carrier mobility,3,4 rapid charge recombination,5,6 and formation of localized, “self-trapped” states near the band-edges.7 These states, which comprise one or more localized charge carriers coupled to proximal lattice distortions arising from one or more phonons, are known as polarons.7,8 Small polarons, first described by Holstein,9,10 are described by a short-range carrier–phonon interaction (radiuspolaron ∼ lattice parameter) and carrier mobility that increases with increased temperature.11 Conversely, large polarons have long-range carrier–phonon interactions (radiuspolaron ≫ lattice parameter) with carrier mobility that decreases with increased temperature.12,13 Many transition metal oxides are reported to host small polarons,7,14–19 and in many cases the performance of these materials for electrocatalytic, photoelectrocatalytic, and optoelectronic applications is impacted by small polaron formation.20–25Although conductivity studies have contributed to a thorough understanding of polaron transport mechanisms in transition metal oxides,11,15,17,26,27 there is still a limited understanding of the mechanisms by which polarons form in photoexcited states. In hematite (α-Fe2O3), for example, one proposed mechanism involves indirect population of polaron states via relaxation from an initially excited, higher-energy non-polaronic state.20,28 Our group recently identified an additional mechanism involving the direct population of polaronic excited states at room temperature upon band-edge excitation in α-Fe2O3.29,30Fig. 1 depicts the difference between indirect and direct population of polaronic states via photoexcitation. Importantly, optical population of polaronic states in α-Fe2O3 is proposed to occur even in a pristine, defect-free lattice: the optical transitions couple to intrinsic, thermally-activated phonon distortions within the crystal.29,30 There is also evidence of polaron formation arising from charge carriers coupling to intrinsic lattice distortions in rutile TiO2 and LiNbO4.16,31 These self-trapping mechanisms differ from other descriptions of small polarons forming via charge carriers coupling to dopants or lattice defects.32–35 Distinguishing the mechanism of photoinduced polaron formation (mediated by dynamic thermally activated lattice distortions or static lattice defects) is fundamentally important to the development of strategies for engineering metal oxide materials for photoapplications.
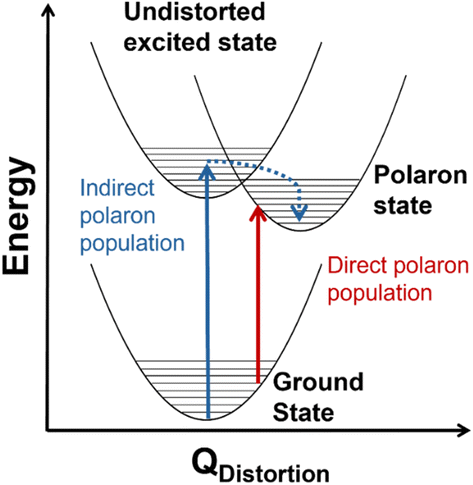 | ||
| Fig. 1 Conceptual configuration coordinate diagram depicting mechanisms of photoexcited polaron formation. The red arrow illustrates direct photoexcitation from a thermally distorted ground state into a polaronic state and the blue arrow shows relaxation into a polaronic state via an undistorted ground state. | ||
Spinel oxides (AB2O4) offer a unique opportunity to understand how the mechanism of photoinduced polaron formation depends on orbital composition of the band edge, coordination geometry of metal centers, and the presence of substitutional defects. These materials are mixed-valent with Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m symmetry and two different site symmetries for metals: tetrahedral (Td) and octahedral (Oh). Ternary spinel oxides, in which A and B are different metals, are described by an inversion parameter x (0.0 < x < 1.0) that quantifies the percentage of the A cations that occupy octahedral sites. When x = 0.0, meaning all of the A cations are in tetrahedral sites, the spinel is considered “normal,” whereas when x = 1.0, meaning all of the A cations are in octahedral sites, the spinel is fully inverted. Intermediate values of x correspond to population of A cations in both tetrahedral and octahedral sites. Spinel oxides containing cobalt are of particular interest because of the difference in crystal field splittings of Co2+ Td and Co3+ Oh (Fig. 2A). Transitions between the e and t2 states within the Td sites are allowed by the Laporte selection rule whereas this rule forbids transitions between the t2g and eg states within the Oh sites. Normal Co3O4, which contains both Co2+ Td and Co3+ Oh, exhibits optical transitions associated with localized d–d transitions at 0.8, 0.9 and 1.6 eV, in addition to an optical transition at 2.5 eV associated with a ligand-to-metal-charge transfer (LMCT) transition.36–40 Normal ZnCo2O4, with only Co3+ Oh, is reported to have only the LMCT-type transition at 2.5 eV.27 The coordination geometry of cobalt in spinel oxides thus impacts their optical spectra; we aim to understand how this coordination geometry impacts photoinduced polaron formation.
m symmetry and two different site symmetries for metals: tetrahedral (Td) and octahedral (Oh). Ternary spinel oxides, in which A and B are different metals, are described by an inversion parameter x (0.0 < x < 1.0) that quantifies the percentage of the A cations that occupy octahedral sites. When x = 0.0, meaning all of the A cations are in tetrahedral sites, the spinel is considered “normal,” whereas when x = 1.0, meaning all of the A cations are in octahedral sites, the spinel is fully inverted. Intermediate values of x correspond to population of A cations in both tetrahedral and octahedral sites. Spinel oxides containing cobalt are of particular interest because of the difference in crystal field splittings of Co2+ Td and Co3+ Oh (Fig. 2A). Transitions between the e and t2 states within the Td sites are allowed by the Laporte selection rule whereas this rule forbids transitions between the t2g and eg states within the Oh sites. Normal Co3O4, which contains both Co2+ Td and Co3+ Oh, exhibits optical transitions associated with localized d–d transitions at 0.8, 0.9 and 1.6 eV, in addition to an optical transition at 2.5 eV associated with a ligand-to-metal-charge transfer (LMCT) transition.36–40 Normal ZnCo2O4, with only Co3+ Oh, is reported to have only the LMCT-type transition at 2.5 eV.27 The coordination geometry of cobalt in spinel oxides thus impacts their optical spectra; we aim to understand how this coordination geometry impacts photoinduced polaron formation.
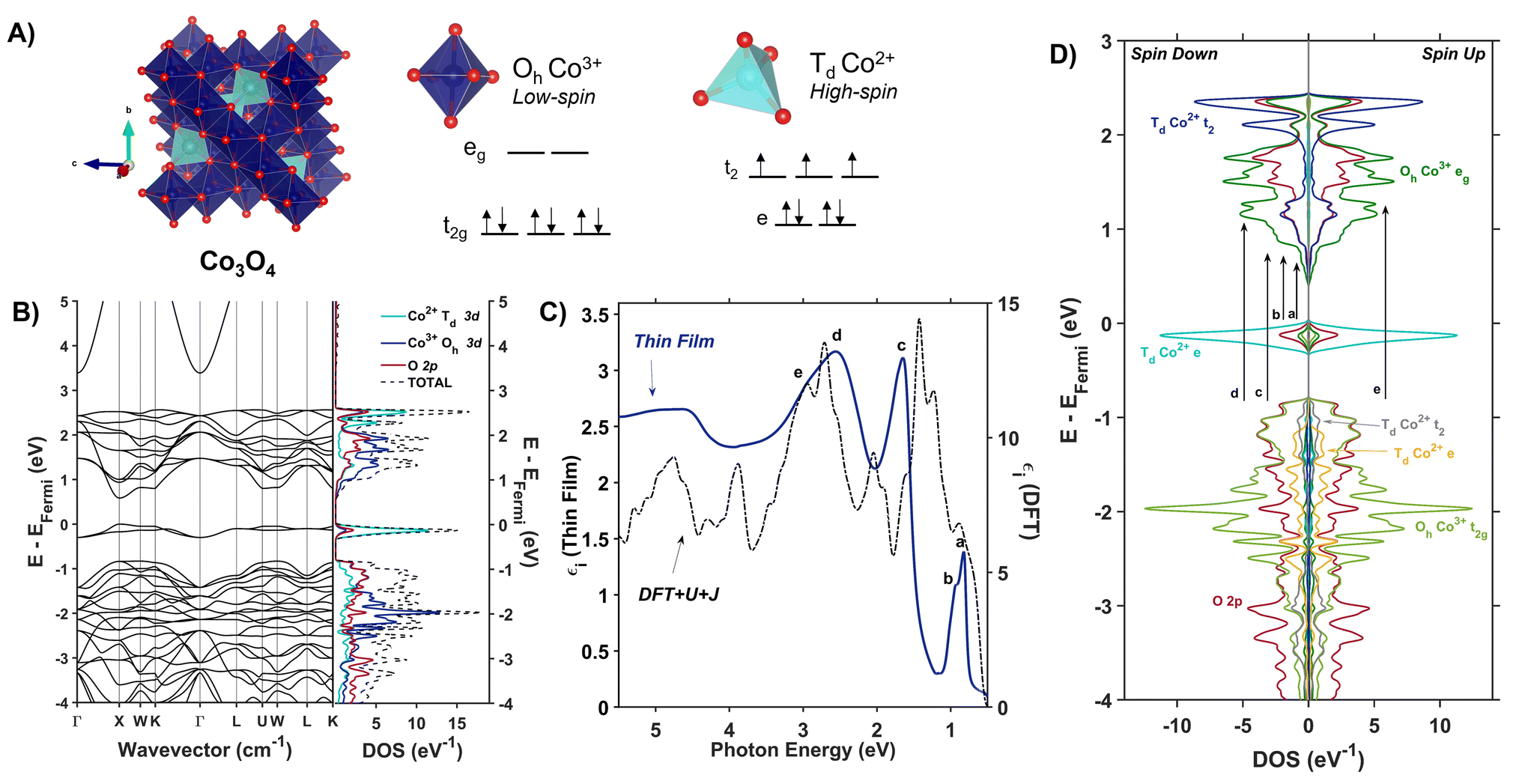 | ||
| Fig. 2 (A) The Co3O4 unit cell (Fdm) with associated d-splitting diagrams for Co3+ (Oh) and Co2+ (Td). (B) Electronic band structure and projected density of states of Co3O4 calculated with Hubbard-corrected DFT. (C) Plot of experimentally (solid blue line) and computationally (dashed black line) determined imaginary dielectric spectra of Co3O4. Labels a–e correlate with arrows in (D), the spin-symmetrized projected density of states arising from the primitive cell of Co3O4. | ||
The formation of small polarons in Co spinel oxides (Co3O4 and ZnCo2O4) has been inferred from the observation of thermally activated charge transport in these materials;26,27 however, as with other transition metal oxides, descriptions of photoinduced polarons in these materials are scarce. Transient absorption (TA) studies of Co3O4 have reported that the strongest TA signal is induced by thermal effects,41,42 which mirrors the behavior of hematite, a material known to undergo photoinduced polaron formation.29,30,43,44 In other work using extreme ultraviolet (XUV) spectroscopy, Zhang, et al. describe indirect formation of small polarons from self-trapped photocarriers in Co3O4.45 Using a combined approach of resonance Raman spectroscopy, temperature-dependent optical spectroscopy and Hubbard-corrected density functional theory, we investigate the influence of cobalt coordination geometry on photoinduced polaron formation in Co3O4 and ZnCo2O4. We report evidence that Td Co in Co3O4 mediates resonance Raman enhancement of specific phonon modes, indicating the presence of phonon-coupled optical transitions that lead to polaron formation. Parallel studies of partially inverted ZnCo2O4, in which the majority of the Td Co ions are replaced with Zn, confirm the involvement of Td Co in phonon-coupled transitions; however, in ZnCo2O4 these transitions do not depend on temperature. This observation suggests that photoinduced polarons in ZnCo2O4 form at static defects rather than dynamic lattice distortions. The fundamental understanding of photophysical properties as a function of coordination geometry presented here is crucial to designing transition metal oxides for optical applications.
Results and discussion
Assignment of the optical spectra of Co3O4
Co3O4 adopts a spinel crystal structure (space group Fdm) with two different metal sites: Co3+ ions occupy octahedral sites and Co2+ ions occupy tetrahedral sites (Fig. 2A). The two coordination sites give rise to two distinct crystal field splittings of the 3d orbitals as shown in Fig. 2A. Transitions between the e and t2 states within the Td sites are allowed by the Laporte selection rule whereas this rule forbids transitions between the t2g and eg states within the Oh sites. The unpaired electrons of adjacent Td Co2+ atoms in Co3O4 are antiferromagnetically coupled.46 Density Functional Theory with Hubbard and Hund corrections (DFT + U + J) was used to calculate the ground-state electronic structure of Co3O4 within the Born–Oppenheimer approximation, in which nuclear motion is neglected. Hubbard (electron correlation correction)47 and Hund (local magnetization correction)48 corrections are used in highly correlated materials such as Co3O4 to mitigate self-interaction errors.30 Using a linear response method,30,47U and J parameters were calculated for Co3O4 from first principles via perturbation of the local environments of open-shell ions (Td Co2+ and Oh Co3+ in the case of Co3O4, see ESI† for more computational details). The DFT + U + J-computed band structure and projected density of states (pDOS) shown in Fig. 2B indicate that 3d orbitals associated with Co2+ Td atoms are the primary contributors to an energetically isolated band at the valence band maximum (VBM), whereas both Co2+ Td and Co3+ Oh 3d orbitals contribute to the conduction band minimum (CBM). These computed results suggest isolated valence bands with Co2+ Td character participate in the low-energy transitions observed in the experimental imaginary dielectric spectrum at 0.82 and 0.92 eV (labeled a, b in Fig. 2C).
Fig. 2C plots the DFT + U + J-computed dielectric spectrum overlaid with the experimental dielectric spectrum extracted from transmission and reflection spectra measured from a 53.7-nm thick Co3O4 film (see ESI† for details of dielectric spectrum determination and powder X-ray diffraction pattern). We applied a rigid shift of +0.3 eV to all conduction band eigenvalues to bring the computed dielectric function into alignment with the measured spectrum. Herein, this shift is applied to all electronic band diagrams and electronic density of states plots of Co3O4. In order to fulfill the f-sum rule governing total oscillator strength, all computed optical spectra are subsequently renormalized by a factor of (1 − (0.3 eV/ħω)).49,50 The computed single-particle dielectric spectrum shows good agreement with the measured spectrum, confirming the features at 0.82, 0.92 and 1.64 eV involve Co2+ Td bands (a–c). From the spin-resolved density of states separated into band contributions from t2g and eg (Oh), e and t2 (Td) and O 2p (Fig. 2D), optical transitions are assigned while also considering spatial wavefunction overlap. The onsets of the conduction bands derived from Oh Co3+ eg, Td Co2+ t2, and O 2p orbitals occur at the same energy; however, the largest projected density of states comes from Oh Co3+ eg. Thus, when considering transitions from Td Co2+ e to the conduction band, these DFT + U + J computations exhibit no energetic difference among transitions to Oh Co3+ eg, Td Co2+ t2, and O 2p. The oscillator strength of the experimental dielectric peak at 0.82 eV is higher than the 0.92 shoulder, indicating the higher likelihood of the 0.82-eV transition; however, the shoulder is not resolved in the imaginary dielectric computed by DFT + U + J. Therefore, to assign this shoulder, we qualitatively assessed the spatial overlap of orbital wavefunctions by mapping the contributions of specific atoms to specific projected densities of states (Fig. S10†). This approach reveals that the most probable transition contributing to the 0.82 eV feature is an intra-atomic transition of Co2+ Td that satisfies the spin transition selection rule (Fig. S10A and B†). The spatial overlap of the orbitals involved in an intra-atomic transition is greater than the overlap of orbitals between a Td Co and an Oh Co (inter-sublattice charge transfer), further supporting the assignment of the 0.82-eV optical transition to an intra-atomic d-to-d transition in Td Co2+ (e → t2). From the spin-resolved density of states combined with our spatial overlap analysis, the feature at 0.92 eV is best described as a charge transfer transition from the tetrahedral sublattice to the octahedral sublattice (Td Co2+ e → Oh Co3+ eg). Similarly, the transition at 1.62 eV can be described as an inter-sublattice charge transfer from Oh Co3+ t2g → Td Co2+ t2. The covalency between Td Co2+ and O 2p, as well as Oh Co3+ and O 2p, allows these metal-to-metal (inter-sublattice charge transfer) transitions to occur. The feature at 2.55 eV and its shoulder at 2.90 eV arise from ligand-to-metal charge transfer (LMCT) type transitions. The assignment of these two LMCT transfers is resolved from comparing the experimental spectrum of Co3O4 with ZnCo2O4, which has majority Oh Co3+ (Fig. S12†). It becomes apparent that the O 2p → Oh Co3+ eg transition is slightly higher in energy than the O 2p → Td Co2+ t2 transition, leading to the following assignments: O 2p → Td Co2+ t2 (2.55 eV) and O 2p → Oh Co3+ eg (2.90 eV). All transitions are spin-conserved (Fig. 2D and Table 1).
| Peak center (eV) | Label | Transition | Description |
|---|---|---|---|
| 0.82 | a | Td Co2+ e → t2 | Intra-atomic transition |
| 0.92 | b | Td Co2+ e → Oh Co3+ eg | Inter-sublattice charge transition |
| 1.64 | c | Oh Co3+ t2g → Td Co2+ t2 | Inter-sublattice charge transition |
| 2.55 | d | O 2p → Td Co2+ t2 | Ligand-to-metal charge transition |
| 2.90 | e | O 2p → Oh Co3+ eg | Ligand-to-metal charge transition |
The electronic density of states and band structure of Co3O4 have been previously calculated using many approaches including, but not limited to GGA + U, hybrid functional PBE0, range-separated exchange–correlation functional HSE06, and many-body Green's function GW approximation.27,51,52 Commonly, the density of states shows some degree of Co 3d and O 2p wavefunction overlap at the band edges; however, depending on the computational approach, the band gap varies from 0.78 to 1.6 eV.27,51,52 Singh, et al. explored many different DFT approaches to calculating the electronic structure of Co3O4, including PBE, PBE + Ueff, HSE06, and many-body Green's function using the GW approximation (Sc-GW0).52 Of these, the Sc-GW0 method produces the most accurate representation of the electronic structure of Co3O4 based on computed electronic bands, density of states, and absorption spectra.52 Although the absorption spectrum computed with Sc-GW0 exhibits a high oscillator strength between ∼0.5 and 2.0 eV,52 unlike our DFT + U + J approach, it cannot resolve the two distinct transitions at 0.8 and 1.6 eV reported in experimental optical spectra.27,36,37,41,45
The experimental presence of the 0.8-eV optical transition and discrepancies in the band gap energies computed with various approaches has led to debate over defining the optical band gap of Co3O4: some report it as 0.8 eV,27,39,53 while others define it to be 1.6 eV.41,54 The incongruity in reported band gap energies is scrutinized by Smart, et al. in their work modelling Co3O4 optical transitions with DFT + U and a hybrid functional that includes a fraction of Hartree–Fock exchange.55 They propose the 0.8-eV optical transition arises from photoinduced formation of a small hole polaron, and that this transition becomes most apparent upon application of uniaxial lattice strain.55 Our calculation of the ground-state electronic structure of Co3O4 using a Hubbard- and Hund- corrected plane-wave pseudopotential approach demonstrates that the low-energy transition at 0.8 eV (and its 0.9-eV shoulder) originate from localized Co2+ Td valence bands (e). Additionally, the corresponding empty Co2+ Td conduction bands (t2) participate in the observed transition at 1.64 eV, indicating the importance of Td Co2+ electronic character to this transition as well. Although the computational approach used here is a single-particle method that neglects many-body perturbations and nuclear motion, its ability to capture all the features observed in the experimental dielectric spectrum supports its accuracy in describing the nature of the bands that contribute to optical transitions in Co3O4. Because the dielectric spectrum reported here is calculated within the Born–Oppenheimer approximation, transitions originating from ground states containing nuclear displacements are not captured, contrasting the description of the 0.8 eV transition as involving a lattice strain-induced small hole polaron by Smart, et al.55
Optical phonon enhancement in Co3O4
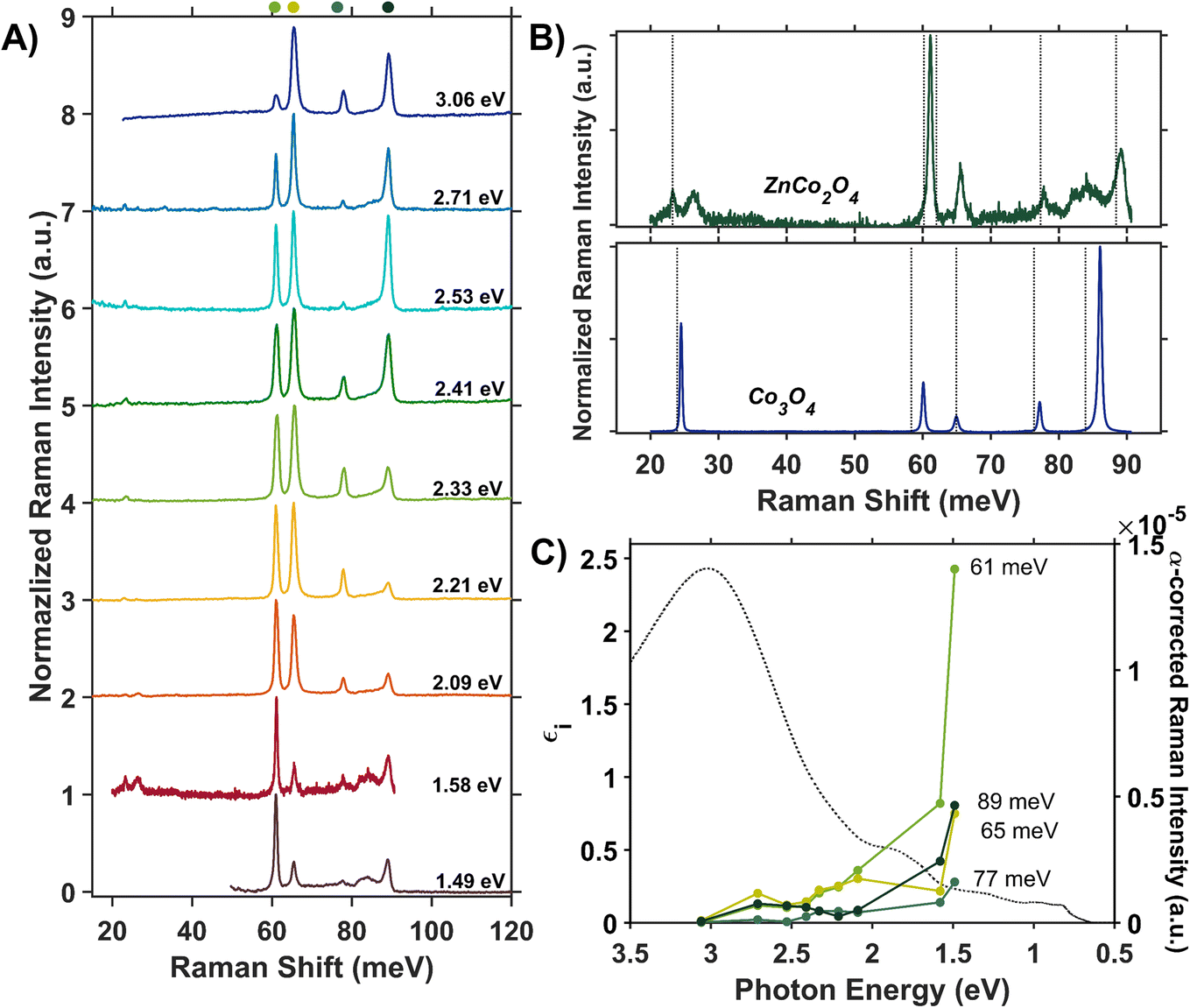
We assess phonon coupling to various optical transitions in Co3O4 using Stokes resonance Raman spectroscopy. Fig. 3A plots a series of resonance Raman spectra collected for a 428-nm thick Co3O4 film deposited on a sapphire substrate using a variety of excitation lasers with photon energies ranging from 1.49 to 3.06 eV, which spans the Co3O4 absorption spectrum (Fig. 3B). These Raman spectra of Co3O4 each contain five phonon modes, consistent with previous reports.56,57 The phonon mode at 86 meV (oxygen breathing about Td Co2+, Fig. 4D) is the most intense at all excitation energies, except hνexc = 1.88 eV, which corresponds to the inter-sublattice charge transfer transition (Oh Co3+ t2g → Td Co2+ t2). When Co3O4 is excited with a photon energy of 1.58 eV, which correspond to the lower-energy edge of this inter-sublattice charge transfer peak, the 86 meV phonon mode becomes most intense again. Fig. 3C plots the excitation spectrum for each phonon mode corrected for scattering cross section and sample absorption, which enables the comparison of phonon mode intensities across different excitation energies. This quantitative analysis of relative intensities reveals that there is amplified resonance enhancement of all modes upon excitation at 1.49 eV, indicating strong phonon coupling to this optical transition. This analysis was repeated on a thin film of Co3O4 deposited on quartz, and the same trends are apparent (Fig. S7†). Given that the 86-meV phonon mode is most intense at an excitation energy of 1.49 eV, we propose it is this phonon mode that most strongly couples to the optical transition at 1.49 eV. | ||
| Fig. 3 (A) Internally normalized Stokes Raman spectra of Co3O4 collected with various excitation energies that span the absorption spectrum plotted in (B). Raman spectra are vertically offset for clarity. Because the spectra are internally normalized, only relative changes in phonon mode intensity can be assessed as a function of excitation energy. Note that the spectrum excited with 1.49 eV begins at a Raman shift of 30 meV due to instrumental constraints, primarily the bandwidth of the filter used to remove Rayleigh scattering. (C) Plot of the intensities of the various Raman modes shown in part A corrected for scattering cross section and sample absorption superimposed on the imaginary dielectric spectrum. Note the corresponding color point of each phonon mode indicated above the relevant Raman peak in A. For the full correction, see ESI Fig. S5.† | ||
 | ||
| Fig. 4 (A) Raman spectrum of Co3O4 collected with a excitation photon energy of 1.58 eV overlaid with the energies of DFT + U + J-computed Raman-active phonon modes. The atomic displacements corresponding to the modes at 24 meV (B), 60 meV (C) and 86 meV (D) are shown. The circled atoms demonstrate the vector displacements of the phonon modes in a 2 × 2 × 1 super cell, where Td sites are most shifted at 24 meV, Oh sites at 60 meV and oxygens surrounding Td sites at 86 meV. Note that the extra oxygen atoms in the 24 meV and 86 meV vector-displacement images appear from the displacement of the neighboring super-cell. | ||
DFT + U + J was used to calculate the displacement vectors of all 39 optical phonon modes in Co3O4 at k-point Γ (Table S2†). White and DeAngelis determined through evaluating the Raman selection rules that there are five Raman active modes in normal spinel oxides.58 The symmetry of these modes was compared with the displacement vectors computed with DFT + U + J to identify the Raman active modes, the energies of which are overlaid with the Raman spectrum in Fig. 4A. From the resonance Raman profile (Fig. 3A and C), the phonon modes with energies of 24, 60, and 86 meV exhibit the most significant resonance enhancement upon excitation at 1.49 eV, corresponding to the onset of the absorption feature centered at 1.64 eV. These modes correspond to motion of tetrahedral Co2+ (24 meV), octahedral Co3+ (60 meV), and oxygen stretching symmetrically about Co2+ Td atoms (86 meV, Fig. 4B–D).
The observed enhancement of the 24-meV (Td Co2+motion) and 60-meV (Oh Co3+ motion) phonons at a Raman excitation energy of 1.49 eV is related to the contribution of both Td Co2+ and Oh Co3+ electronic character to this optical transition, which corresponds to population of empty Td Co2+ t2 conduction band states from Oh Co3+ t2g valence band states. Although both Td Co2+ and Oh Co3+ phonon modes exhibit resonance enhancement because of associated electronic character in the optical transition centered at 1.64 eV, it is the oxygen breathing mode about Co2+ Td ions at 86 meV that exhibits the most enhancement. The degree of energetic overlap between the O 2p and Co2+ Td projected density of states in the conduction band at the energy of the Oh Co3+ t2g →Td Co2+ t2 transition is significant (Fig. 2B and D) and supports the observed enhancement of the 86-meV phonon, which corresponds to motion of oxygen atoms along their bond axes to Td Co2+.
Temperature dependence of optical transitions
To further understand the role of phonon coupling in the optical transitions of Co3O4, we measured the dependence of its dielectric spectrum on temperature between 82 and 470 K. Fig. 5A and B plot the resulting thermal difference spectra (TDS) calculated according to eqn (1).| Δεi(T) = εi,T − εi,294K | (1) |
 | ||
| Fig. 5 Thermal difference imaginary dielectric spectra of Co3O4 collected at temperatures above (A) and below (B) room temperature (294 K). (C) Absolute value of the thermal difference spectra integrated from 0.68 to 1.86 eV and normalized to the integrated intensity of the spectrum collected at 82 K plotted versus temperature (black circles). The solid blue line plots the temperature-dependent change in Bose–Einstein population of a phonon with an energy of 24 meV. Bose–Einstein distributions corresponding to the energies of the other Raman active phonon modes are plotted as dotted lines. | ||
We assessed the impact of temperature on the dielectric spectrum by integrating the intensity of the thermal difference spectra. Fig. 5C (blue) plots the absolute value of the thermal difference spectra integrated from 0.68 to 1.86 eV and normalized to the integrated intensity obtained from the spectrum collected at 82 K. To account for sign changes, the integrands of spectra collected below room temperature are shown as negative, and those collected above room temperature are positive. For phonon-coupled optical transitions, we expect the change in intensity with changing temperature to be proportional to the change in population of the coupled phonons. The thermal population of phonon modes is dictated by the Bose–Einstein distribution shown in eqn (2), where ħΩ is the phonon energy. Normalization of the differential spectra to an arbitrary temperature, in this case 82 K, leads to eqn (3), where we compare the change in intensity of the dielectric spectrum (Δε defined in eqn (1)) to the change in population of the Raman active phonon modes at various temperatures.
 | (2) |
 | (3) |
The temperature dependence of the TDS intensity overlays well with the change in population of the 24-meV Raman-active phonon with temperature predicted by the Bose–Einstein distribution (eqn (3)). This agreement suggests that the low energy optical transitions (1.64 eV, 0.96 eV and 0.82 eV) are coupled strongly to the thermal population of this phonon. Interestingly, the 24-meV phonon mode is not the most enhanced in resonance Raman spectra collected with an excitation photon energy of 1.49 eV; however, both the 24-meV and 86-meV phonon modes are described by displacement vectors primarily of or around Td Co2+ions (Fig. 4B and D). Evidence of thermally activated optical transitions combined with strong phonon coupling to optical transitions observed in resonance Raman spectra (vide supra) suggests an optically accessed polaronic state related to Td Co2+. Compared to resonance Raman measurements, thermal difference spectra are not as precise in determining the energies of the specific phonon modes coupling to the optical transition, as there may be multiple phonon modes contributing to thermal activation. However, the Bose–Einstein distribution corresponding to a phonon energy of 24 meV overlays the temperature-dependent TDS intensities much better than the distributions corresponding to other Raman-active phonon modes (Fig. 5C). We therefore interpret the threshold phonon energy of 24 meV to be where thermal activation occurs. Similar temperature-dependent behavior is observed in α-Fe2O3: at the excitation energy where maximal Raman enhancement is observed (∼2.2 eV), the strongest temperature-dependence in the optical spectrum is also present.29 Thermal activation of optical transitions in α-Fe2O3 is the proposed mechanism by which direct excitation into intrinsic polaronic states occurs.29,30 The similarities in the trends of Raman enhancement and the temperature-dependence of the dielectric spectrum observed for Co3O4 and α-Fe2O3 support the presence of an optically accessible polaronic state in Co3O4 arising from coupling to intrinsic, dynamic lattice distortions (phonons) (Fig. 1, red arrow).
Co3O4 compared to ZnCo2O4: assessing the role of Td Co in spinel oxides
The resonance Raman and thermal difference spectra of Co3O4 suggest that Co2+ ions occupying Td sites are involved in the phonon-coupled optical transitions. To further understand the role of Td Co2+ in the optical spectra of Co3O4, we assessed the optical properties of ZnCo2O4, where Zn2+ replaces Td Co2+ (Fig. 6A). We computed the imaginary dielectric spectrum for normal ZnCo2O4 with DFT + U (see ESI for details†). Within normal ZnCo2O4, the Td Zn2+ 3d orbitals are completely filled, and the Oh Co3+ 3d orbitals have a low-spin configuration, termed “quasi close-shelled”.59,60 With no unpaired electrons present, the Hund parameter, J, is not implemented in these calculations. The DFT + U calculations of normal ZnCo2O4 reveal two absorption bands at and above ∼2.5 eV, while the experimental spectrum only has one transition in this region. Literature reports assign the experimentally observed feature at 3.0 eV to an LMCT-type transition,27,38 which matches the lower energy peak (2.5 eV) observed in the DFT + U dielectric spectrum. The 3.5 eV peak in the DFT + U dielectric spectrum computed here is associated with an intra-sublattice charge transfer (Oh Co3+ t2g → Oh Co3+ eg). Because this is a charge transfer between two different lattice sites, the Laporte selection rule does not apply. Due to the hybridized nature of the Co 3d and O 2p orbitals in both the valence and conduction bands, we assign the experimental dielectric peak at 3.0 eV to a combination of LMCT and intra-sublattice charge transfer transitions (see Fig. S11 in the ESI†). Interestingly, when comparing the experimental peak shape of the dielectric transition at 3.0 eV in ZnCo2O4 (Fig. 6B) to the analogous transition in Co3O4 (Fig. 2C and S12†), it becomes apparent that the shoulder in Co3O4 (labeled e in Fig. 2C) is at the same energy as the peak center of the experimental ZnCo2O4 transition. This energetic alignment in the experimental dielectric spectra, used to assign the 2.90-eV shoulder in Co3O4 as excitation into Oh Co3+ eg bands, has been observed previously in a Zn dopant study of Co3O4.38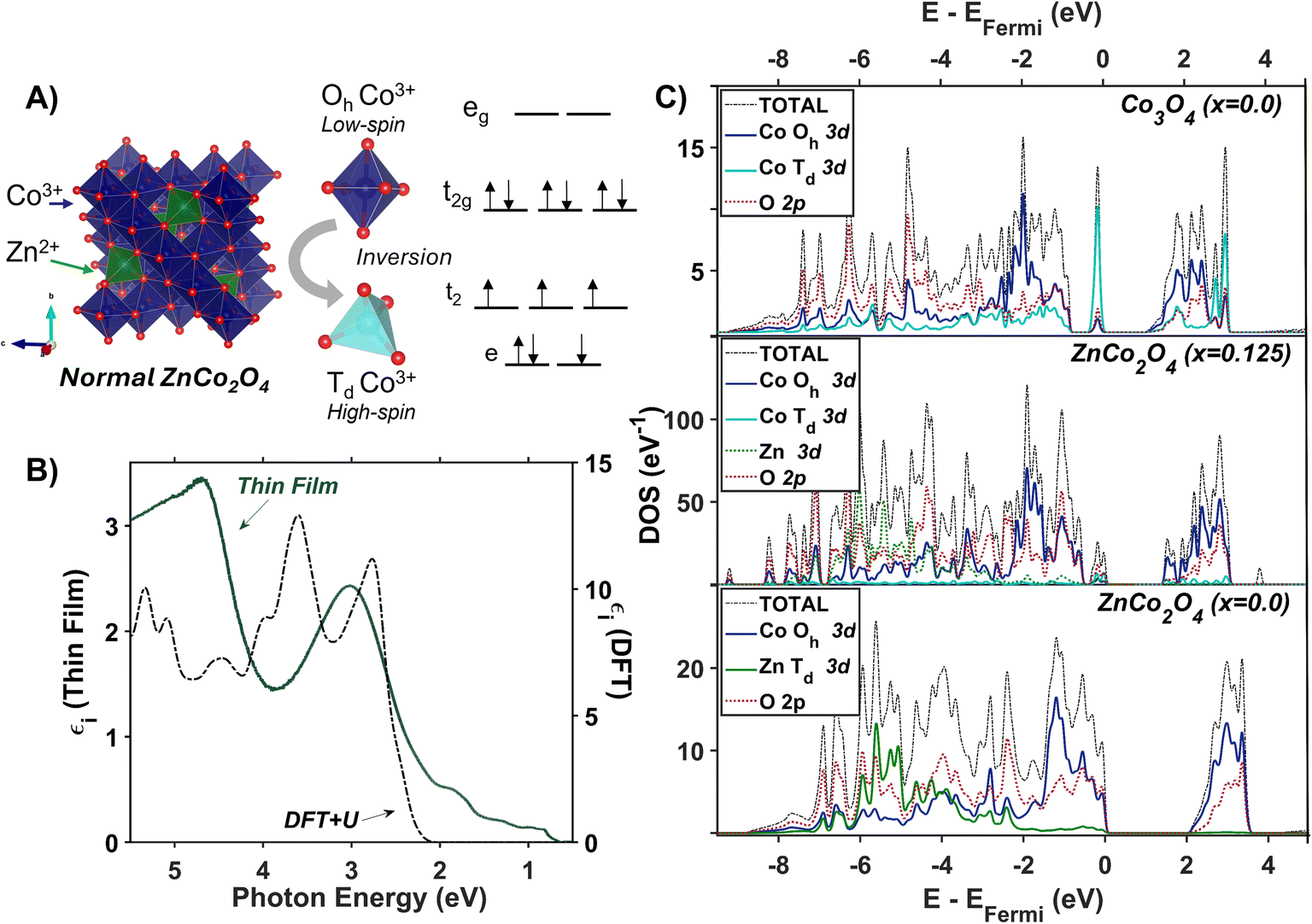 | ||
| Fig. 6 (A) Unit cell of normal ZnCo2O4 with associated crystal field splitting diagrams of Co3+ in octahedral and tetrahedral coordination. (B) Plot of experimentally (solid green line) and computationally (dashed black line) determined imaginary dielectric spectra of ZnCo2O4. The computed dielectric spectrum was calculated for normal ZnCo2O4. (C) The electronic density of states calculated with Hubbard-corrected DFT for normal Co3O4 (top), ZnCo2O4 with an inversion factor of 0.125 (middle), and normal ZnCo2O4 (bottom). | ||
The lower-energy region of the experimental dielectric spectrum of ZnCo2O4 contains broad and weak features at 1.79, 0.98 and 0.82 eV, which coincide with where Co3O4 has transitions associated with Co2+ Td. X-ray fluorescence analysis indicates the stoichiometry of the ZnCo2O4 film used to produce the dielectric spectrum is 1.94 ± 0.04 Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn, and the powder X-ray diffraction pattern indicates the film is phase-pure spinel (Fig. S3†). We therefore suspected that our ZnCo2O4 film may be slightly inverted and contain a small population of cobalt in tetrahedral sites, consistent with previous reports.61 In an effort to control cation distribution in ZnCo2O4, we changed the temperature at which ZnCo2O4 films were annealed following spin-coating. We observe that annealing above 600 °C induces phase separation as features associated with ZnO become apparent in the X-ray diffraction pattern and Raman spectra (Fig. S13 and S14†). Additionally, as annealing temperature increases, the low energy peaks observed in the dielectric spectrum increase in intensity (Fig. S15†). We therefore suspect that the films containing ZnO also contain Co3O4, which accounts for the presence of the intense low-energy peaks in the dielectric spectrum (Fig. S15†). With no ZnO peaks present in X-ray diffraction or ZnO phonon modes observed in Raman for the films annealed at 600 °C, we hypothesize that the weak, low-intensity features observed in the dielectric spectra of these films arise from cation inversion, whereby a fraction of the Co ions occupy Td sites and a fraction of the Zn ions occupy Oh sites. We describe the Co ions in Td sites as substitutional lattice defects.
:Zn, and the powder X-ray diffraction pattern indicates the film is phase-pure spinel (Fig. S3†). We therefore suspected that our ZnCo2O4 film may be slightly inverted and contain a small population of cobalt in tetrahedral sites, consistent with previous reports.61 In an effort to control cation distribution in ZnCo2O4, we changed the temperature at which ZnCo2O4 films were annealed following spin-coating. We observe that annealing above 600 °C induces phase separation as features associated with ZnO become apparent in the X-ray diffraction pattern and Raman spectra (Fig. S13 and S14†). Additionally, as annealing temperature increases, the low energy peaks observed in the dielectric spectrum increase in intensity (Fig. S15†). We therefore suspect that the films containing ZnO also contain Co3O4, which accounts for the presence of the intense low-energy peaks in the dielectric spectrum (Fig. S15†). With no ZnO peaks present in X-ray diffraction or ZnO phonon modes observed in Raman for the films annealed at 600 °C, we hypothesize that the weak, low-intensity features observed in the dielectric spectra of these films arise from cation inversion, whereby a fraction of the Co ions occupy Td sites and a fraction of the Zn ions occupy Oh sites. We describe the Co ions in Td sites as substitutional lattice defects.
To investigate the impact of cation inversion on the optical spectra of ZnCo2O4, we computed the electronic structure of inverted ZnCo2O4 using DFT + U + J (see ESI for details†). Inverted ZnCo2O4 (i-ZnCo2O4) was modeled by switching two Oh Co3+ ions with two Td Zn2+ ions within a 2×2×2 supercell of normal ZnCo2O4 to produce an inversion factor of x = 0.125 while maintaining charge balance and stoichiometry. The resulting two Td Co3+ ions were modeled to be antiferromagnetically coupled to maintain net-zero magnetization. Compared to normal ZnCo2O4 (n-ZnCo2O4), i-ZnCo2O4 contains an isolated state at the valence band-edge with primarily Td Co and O 2p character (Fig. 6C, middle and bottom). The conduction band edge in i-ZnCo2O4 appears at a lower energy than that in n-ZnCo2O4 (similar to Co3O4), and i-ZnCo2O4 contains an isolated region at the conduction band-edge of primarily Oh Co and O 2p character (Fig. 6C, middle and top). The band-edge character of i-ZnCo2O4, despite the 3+ oxidation state of the cobalt ion in Td sites, has more similarities to that of Co3O4 than n-ZnCo2O4 (Fig. 6C). Thus, the presence of Co (Td) in spinel oxides results in low-energy d-to-d transitions, regardless of its oxidation state (+2 or +3). Because there is no evidence of phase separation (i.e. presence of Co3O4) to describe the low-energy features in the dielectric, we conclude the synthesized ZnCo2O4 contains at least a small degree of inversion.
The Raman spectrum of ZnCo2O4 has five distinct modes (Fig. 7A and B) consistent with previous reports.62,63 The phonon mode frequencies are similar to those observed in Co3O4, which is expected, as both materials adopt the spinel crystal structure. Additionally, the 23-meV, 61-meV, and 89-meV phonon modes in ZnCo2O4 are described by the same phonon motions as in Co3O4: motion of Td metal center motion of Oh metal center, and oxygen stretching about Td sites respectively (Fig. 4B–D). Fig. 7B (top) plots the experimental Raman spectrum of ZnCo2O4 overlaid with the computed phonon modes of normal ZnCo2O4. While the five prominent phonon modes are accounted for by DFT + U phonon calculations, there are two broad and weak features at 26 meV and 85 meV that do not appear in calculations. Both phonons have similar energies to corresponding modes observed in Co3O4, which are both related predominantly to Td sites (Fig. 7B, bottom). The discrepancy between the energies of experimentally observed Raman-active optical phonon modes and the computed modes of normal ZnCo2O4 can be explained by the sample crystallizing with a degree of inversion. With a small percentage of tetrahedral sites occupied by Co3+, and the rest by Zn2+, the phonon modes dominated by tetrahedral motion (∼24 meV and ∼89 meV) split into two distinct distributions. This phenomenon has been observed in other inverted spinel oxides64 and is further evidence of the ZnCo2O4 films crystallizing with a small percent occupation of cobalt in tetrahedral sites (Fig. 7B).
 | ||
| Fig. 7 (A) Internally normalized Raman spectra of a ZnCo2O4 thin film deposited on a sapphire substrate collected with excitation photon energies that span its absorption spectrum. Because the spectra are internally normalized, only relative changes in phonon mode intensity can be assessed as a function of excitation energy (see ESI† for full work-up of Raman data). (B) Raman spectra of ZnCo2O4 (top) and Co3O4 (bottom) excited at a photon energy of 1.58 eV are overlaid with the Raman-active phonon modes computed for normal ZnCo2O4 and Co3O4, respectively. (C) Plot of the intensities of the various Raman modes shown in part A corrected for scattering cross section and sample absorption superimposed on the imaginary dielectric spectrum of ZnCo2O4. Note the corresponding color point of each phonon mode indicated above the relevant Raman peak in A. | ||
Unlike Co3O4, the resonance Raman profile of ZnCo2O4 does not exhibit increased resonance enhancement of the phonons associated with Td motion (23 and 89 meV) with decreasing excitation energy. In contrast, the 61-meV phonon, related to Co Oh motion (described by Fig. 4B), exhibits enhanced intensity in ZnCo2O4 relative to all the other modes as excitation photon energy decreases (Fig. 7A). Interestingly, when the Raman profile is corrected for scattering cross section and sample absorption, the 61-meV mode is most enhanced at an excitation photon energy of 1.49 eV – the same excitation photon energy at which the most significant resonance enhancement is observed in Co3O4 (Fig. 3C and Fig. 7C). This analysis was repeated on a thin film of ZnCo2O4 deposited on quartz, and the same trends are apparent (Fig. S8†). The similarity in excitation photon energy where resonance enhancement is observed in ZnCo2O4 and Co3O4 indicates that the onset of the optical transition centered at ∼1.6 eV is significant for both materials.
Based on the calculated density of states of inverted ZnCo2O4 (Fig. 6C), the experimental optical transition observed at 1.6 eV is assigned to metal-to-metal charge transfer (MMCT) from bands with Td Co3+ character (arising from the Td Co substitutional defects) to bands with Oh Co3+ character. The most enhanced Raman spectrum (hνexc = 1.49 eV) occurs upon excitation at the onset of the 1.6-eV transition, implying that the presence of the Co3+ Td substitutional defect is crucial for the observed enhancement of the Oh phonon in ZnCo2O4. The conduction band character associated with the 1.6-eV transition arises from 3d orbitals associated with Co3+ Oh, the same site associated with the main vibrational motion of the 61-meV phonon (Fig. 4C). Although the strong resonance enhancement observed for the 61-meV phonon in ZnCo2O4 is evidence of strong coupling between this mode and the optical transition at 1.6 eV, we do not observe significant temperature dependence of this transition in thermal difference spectra when compared to Co3O4 (see Fig. S16 in ESI†). In fact, unlike Co3O4, the thermal difference spectra collected for ZnCo2O4 at elevated temperatures do not exhibit any well-defined features corresponding to spectral features observed in the dielectric spectrum at room temperature.
The spectral and computational results reported here for ZnCo2O4 and Co3O4 indicate that Co 3d character at the band edge mediates phonon-coupled optical transitions (Fig. 3C, 4C, D and 7C). However, the contrast in temperature-dependence of the optical transition where both materials exhibit the strongest phonon coupling (1.6 eV) suggests fundamentally different processes dictating the observed enhancement. We interpret our findings in Co3O4 as direct population of an intrinsic polaronic state via photoexcitation, which is strongly influenced by the contributions of Co2+ Td ions to band-edge states. In ZnCo2O4, we interpret the experimental ZnCo2O4 sample to have a small percent occupation of tetrahedral sites by Co based on the presence of low-energy optical transitions associated with Td Co. Although the occupation of Co3+ in tetrahedral sites enables the optical transition and phonon enhancement at 1.6 eV, the lack of significant temperature dependence of this optical transition indicates that the phonon-coupled optical transitions leading to Raman enhancement in ZnCo2O4 are fundamentally different from those observed in Co3O4.
We suspect that the differences observed in the thermal difference spectra of Co3O4 and ZnCo2O4 are related to the disruption in the translational symmetry of the lattice induced by cation inversion in ZnCo2O4. From assignment of optical transitions in Co3O4, the low-energy optical transitions are either highly localized transitions between neighboring Co atoms or intra-atomic transitions in Td Co (Table 1). With fewer Co atoms in Td sites, these optical transitions become suppressed, as observed in ZnCo2O4 (Fig. 6B); however, the localized transitions still exhibit phonon coupling, leading to the observed resonance Raman enhancements (Fig. 7C). The lack of temperature dependence of these transitions in ZnCo2O4 suggests that, although these transitions access localized phonon-coupled states, the localization is related to the static lattice defect of tetrahedral cobalt rather than thermally induced dynamic lattice displacements within a pristine, translationally symmetric lattice. The lack of thermal dependence in ZnCo2O4, but presence of resonance Raman enhancement of Oh phonon modes upon excitation of a transition from Td Co to Oh Co, is strong evidence of an optically accessed polaronic state. We therefore conclude that in both Co3O4 and ZnCo2O4 there is evidence of an optically accessible polaronic state. However, in Co3O4 formation of the polaronic state is due to intrinsic, dynamic lattice deformations caused by thermally activated phonons (exactly analogous to the mechanism we observe in hematite), whereas in ZnCo2O4, photoinduced polaron formation is mediated by static lattice defects arising from cation inversion.
Conclusions
The optical characterization of Co3O4 and ZnCo2O4 reported here shows that low energy transitions at 0.8, 0.9 eV and 1.6 eV arise from tetrahedrally coordinated cobalt ions, regardless of oxidation state (Co2+, Co3+). The d–d transition observed at 1.6 eV in both Co3O4 and ZnCo2O4 is a phonon-coupled optical transition. In Co3O4, the combination of temperature-dependent intensity and resonance Raman enhancement at the onset of the Oh Co3+ → Td Co2+ optical transition (1.64 eV) is evidence that this transition directly populates an intrinsic polaron state coupled to thermally activated phonons. Although the resonance Raman profile of ZnCo2O4 exhibits a similar Raman enhancement upon excitation of the 1.6-eV transition, the observed inversion and lack of temperature dependence suggests that this photoexcited polaron forms due to the presence of tetrahedral cobalt substitutional defects within the lattice. We conclude that Td-coordinated cobalt is a significant factor in phonon-coupled transitions for cobalt-containing spinel oxides. The contrast in the mechanism of polaron formation (intrinsic or self-trapped polaron vs. defect-mediated polaron) observed for these similar materials is an important insight that we anticipate will help uncover mechanisms of photoinduced polaron formation in other oxide materials. Although both mechanisms of polaron formation observed here have been previously reported, understanding the spectral signatures that distinguish them is crucial for further development of oxide materials for photo-applications.Data availability
Data supporting this article have been included as part of the ESI.† Primary data are available from the authors upon reasonable request.Author contributions
E. P. C., J. L. S., and K. E. K. contributed to conceptualization of the project and interpretation of the data; K. E. K. supervised the project. E. P. C. performed most experiments and computations. J. L. S. contributed to some computations for Co3O4. M. T. R. collected low-frequency Raman spectra. E. P. C. wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the University of Rochester via a University Research Award. M. T. R. thanks the USA National Science Foundation for financial support (DMR-2348765). The authors acknowledge the University of Rochester Raman Facility and thank David McCamant for discussion about Raman collection. We are grateful to the computational resources and technical assistance from the University of Rochester Center for Integrated Research Computing. For X-ray diffraction and energy dispersive X-ray emission measurements, we acknowledge the Chemical Analysis Laboratory Material Science Facilities at Rochester Institute of Technology and thank Tom Allston for guidance on measurements. We thank Todd Krauss and URNano for access to atomic force microscopes (AFM) and thank Alex Searle for collection and processing of AFM data, as well as Sean O'Neill and Lisbeth Compton for discussions regarding AFM collection and work-up. We acknowledge URNano for profilometry measurements and thank Gregory Madjeski for discussions regarding data collection.References
- D. Franchi and Z. Amara, Applications of Sensitized Semiconductors as Heterogeneous Visible-Light Photocatalysts in Organic Synthesis, ACS Sustainable Chem. Eng., 2020, 8(41), 15405–15429 CrossRef CAS.
- J. Luo, S. Zhang, M. Sun, L. Yang, S. Luo and J. C. Crittenden, A Critical Review on Energy Conversion and Environmental Remediation of Photocatalysts with Remodeling Crystal Lattice, Surface, and Interface, ACS Nano, 2019, 13(9), 9811–9840 CrossRef CAS PubMed.
- F. J. Morin, Electrical Properties of α-Fe2O3, Phys. Rev., 1954, 93(6), 1195–1199 CrossRef CAS.
- F. J. Morin, Electrical Properties of NiO, Phys. Rev., 1954, 93(6), 1199–1204 CrossRef CAS.
- J. Husek, A. Cirri, S. Biswas and L. R. Baker, Surface Electron Dynamics in Hematite (α-Fe2O3): Correlation between Ultrafast Surface Electron Trapping and Small Polaron Formation, Chem. Sci., 2017, 8(12), 8170–8178 RSC.
- S. Biswas, J. Husek, S. Londo and L. R. Baker, Ultrafast Electron Trapping and Defect-Mediated Recombination in NiO Probed by Femtosecond Extreme Ultraviolet Reflection–Absorption Spectroscopy, J. Phys. Chem. Lett., 2018, 9(17), 5047–5054 CrossRef CAS.
- C. Franchini, M. Reticcioli, M. Setvin and U. Diebold, Polarons in Materials, Nat. Rev. Mater., 2021, 6(7), 560–586 CrossRef CAS.
- L. Landau, Über die Bewegung der Elektronen im Kristallgitter, Phys. Z. Sowjetunion, 1933, 3, 644–645 Search PubMed.
- T. Holstein, Studies of Polaron Motion: Part I. The Molecular-Crystal Model, Ann. Phys., 1959, 8(3), 325–342 CAS.
- T. Holstein, Studies of Polaron Motion: Part II. The “Small” Polaron, Ann. Phys., 1959, 8(3), 343–389 CAS.
- P. Nagels, M. Denayer and J. Devreese, Electrical Properties of Single Crystals of Uranium Oxide, Solid State Commun., 1963, 1(2), 35–40 CrossRef CAS.
- H. Fröhlich, Electrons in Lattice Fields, Adv. Phys., 1954, 3(11), 325–361 CrossRef.
- X. Mettan, J. Jaćimović, O. S. Barišić, A. Pisoni, I. Batistić, E. Horváth, S. Brown, L. Rossi, P. Szirmai, B. Farkas, H. Berger and L. Forró, Tailoring Thermal Conduction in Anatase TiO2, Commun. Phys., 2019, 2(1), 123 CrossRef.
- M. Reticcioli, U. Diebold, G. Kresse, and C. Franchini, Small Polarons in Transition Metal Oxides, in Handbook of Materials Modeling, W. Andreoni, and S. Yip, Springer International Publishing, Cham, 2020, pp 1035–1073 Search PubMed.
- I. G. Austin, A. J. Springthorpe, B. A. Smith and C. E. Turner, Electronic Transport Phenomena in Single-Crystal NiO and CoO, Proc. Phys. Soc., 1967, 90(1), 157–174 CrossRef CAS.
- S. Yang, A. T. Brant, N. C. Giles and L. E. Halliburton, Intrinsic Small Polarons in Rutile TiO2, Phys. Rev. B, 2013, 87(12), 125201 CrossRef.
- H. L. Tuller and A. S. Nowick, Small Polaron Electron Transport in Reduced CeO2 Single Crystals, J. Phys. Chem. Solids, 1977, 38(8), 859–867 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Electron Localization Determines Defect Formation on Ceria Substrates, Science, 2005, 309(5735), 752–755 CrossRef CAS PubMed.
- R. Karsthof, M. Grundmann, A. M. Anton and F. Kremer, Polaronic Interacceptor Hopping Transport in Intrinsically Doped Nickel Oxide, Phys. Rev. B, 2019, 99(23), 235201 CrossRef CAS.
- S. Biswas, S. Wallentine, S. Bandaranayake and L. R. Baker, Controlling Polaron Formation at Hematite Surfaces by Molecular Functionalization Probed by XUV Reflection-Absorption Spectroscopy, J. Chem. Phys., 2019, 151(10), 104701 CrossRef PubMed.
- N. Mansourian-Hadavi, S. Wansom, N. H. Perry, A. R. Nagaraja, T. O. Mason, L. Ye and A. J. Freeman, Transport and Band Structure Studies of Crystalline ZnRh2O4, Phys. Rev. B, 2010, 81(7), 075112 CrossRef.
- T. Feng, L. Li, Q. Shi, S. Dong, B. Li, K. Li and G. Li, Evidence for the Influence of Polaron Delocalization on the Electrical Transport in LiNi0.4+x Mn0.4−x Co0.2 O2, Phys. Chem. Chem. Phys., 2020, 22(4), 2054–2060 RSC.
- N. C. Verissimo, F. A. Pires, I. Rodríguez-Gutiérrez, J. Bettini, T. E. R. Fiuza, C. A. Biffe, F. E. Montoro, G. R. Schleder, R. H. R. Castro, E. R. Leite and F. L. Souza, Dual Modification on Hematite to Minimize Small Polaron Effects and Charge Recombination for Sustainable Solar Water Splitting, J. Mater. Chem. A, 2024, 12(11), 6280–6293 RSC.
- S. Bandaranayake, A. Patnaik, E. Hruska, Q. Zhu, S. Das and L. R. Baker, Effect of Surface Electron Trapping and Small Polaron Formation on the Photocatalytic Efficiency of Copper(I) and Copper(II) Oxides, ACS Appl. Mater. Interfaces, 2024, 16(31), 41616–41625 CrossRef CAS.
- H. Gajapathy, S. Bandaranayake, E. Hruska, A. Vadakkayil, B. P. Bloom, S. Londo, J. McClellan, J. Guo, D. Russell, F. M. F. de Groot, F. Yang, D. H. Waldeck, M. Schultze and L. R. Baker, Spin Polarized Electron Dynamics Enhance Water Splitting Efficiency by Yttrium Iron Garnet Photoanodes: A New Platform for Spin Selective Photocatalysis, Chem. Sci., 2024, 15(9), 3300–3310 RSC.
- C.-S. Cheng, M. Serizawa, H. Sakata and T. Hirayama, Electrical Conductivity of Co3O4 Films Prepared by Chemical Vapour Deposition, Mater. Chem. Phys., 1998, 53(3), 225–230 CrossRef CAS.
- X. C. Huang, J. Y. Zhang, M. Wu, S. Zhang, H. Y. Xiao, W. Q. Han, T.-L. Lee, A. Tadich, D.-C. Qi, L. Qiao, L. Chen and K. H. L. Zhang, Electronic Structure and p-Type Conduction Mechanism of Spinel Cobaltite Oxide Thin Films, Phys. Rev. B, 2019, 100(11), 115301 CrossRef CAS.
- L. M. Carneiro, S. K. Cushing, C. Liu, Y. Su, P. Yang, A. P. Alivisatos and S. R. Leone, Excitation-Wavelength-Dependent Small Polaron Trapping of Photoexcited Carriers in α-Fe2O3, Nat. Mater., 2017, 16(8), 819–825 CrossRef CAS.
- J. L. Shelton and K. E. Knowles, Thermally Activated Optical Absorption into Polaronic States in Hematite, J. Phys. Chem. Lett., 2021, 12(13), 3343–3351 CrossRef CAS PubMed.
- J. L. Shelton and K. E. Knowles, Polaronic Optical Transitions in Hematite (α-Fe2O3) Revealed by First-Principles Electron–Phonon Coupling, J. Chem. Phys., 2022, 157(17), 174703 CrossRef CAS.
- F. Freytag, G. Corradi and M. Imlau, Atomic Insight to Lattice Distortions Caused by Carrier Self-Trapping in Oxide Materials, Sci. Rep., 2016, 6(1), 36929 CrossRef CAS.
- Y. Yamada, O. Hino, S. Nohdo, R. Kanao, T. Inami and S. Katano, Polaron Ordering in Low-Doping La1−xSrxMnO3, Phys. Rev. Lett., 1996, 77(5), 904–907 CrossRef CAS PubMed.
- G. Allodi, M. Cestelli Guidi, R. De Renzi, A. Caneiro and L. Pinsard, Ultraslow Polaron Dynamics in Low-Doped Manganites from 139La NMR-NQR and Muon Spin Rotation, Phys. Rev. Lett., 2001, 87(12), 127206 CrossRef CAS PubMed.
- S. X. Zhang, D. C. Kundaliya, W. Yu, S. Dhar, S. Y. Young, L. G. Salamanca-Riba, S. B. Ogale, R. D. Vispute and T. Venkatesan, Niobium Doped TiO2: Intrinsic Transparent Metallic Anatase versus Highly Resistive Rutile Phase, J. Appl. Phys., 2007, 102(1), 013701 CrossRef.
- E. Possenriede, H. Kröse, T. Varnhorst, R. Scharfschwerdt and O. F. Schirmer, Shallow Acceptor and Electron Conduction States in BaTiO3, Ferroelectrics, 1994, 151(1), 199–204 CrossRef CAS.
- J. G. Cook and M. P. van der Meer, The Optical Properties of Sputtered Co3O4 Films, Thin Solid Films, 1986, 144(2), 165–176 CrossRef CAS.
- K. M. E. Miedzinska, B. R. Hollebone and J. G. Cook, An Assignment of the Optical Absorption Spectrum of Mixed Valence Co3O4 Spinel Films, J. Phys. Chem. Solids, 1987, 48(7), 649–656 CrossRef CAS.
- K. J. Kim and Y. R. Park, Optical Investigation of Charge-Transfer Transitions in Spinel Co3O4, Solid State Commun., 2003, 127(1), 25–28 CrossRef CAS.
- L. Qiao, H. Y. Xiao, H. M. Meyer, J. N. Sun, C. M. Rouleau, A. A. Puretzky, D. B. Geohegan, I. N. Ivanov, M. Yoon, W. J. Weber and M. D. Biegalski, Nature of the Band Gap and Origin of the Electro-/Photo-Activity of Co3O4, J. Mater. Chem. C, 2013, 1(31), 4628–4633 RSC.
- C.-M. Jiang, L. R. Baker, J. M. Lucas, J. Vura-Weis, A. P. Alivisatos and S. R. Leone, Characterization of Photo-Induced Charge Transfer and Hot Carrier Relaxation Pathways in Spinel Cobalt Oxide (Co3O4), J. Phys. Chem. C, 2014, 118(39), 22774–22784 CrossRef CAS.
- M. M. Waegele, H. Q. Doan and T. Cuk, Long-Lived Photoexcited Carrier Dynamics of d–d Excitations in Spinel Ordered Co3O4, J. Phys. Chem. C, 2014, 118(7), 3426–3432 CrossRef CAS.
- P. Aroonratsameruang, P. Chakthranont and P. Pattanasattayavong, The Cause of Limited Photoelectrochemical Water Reduction Performance of Co3O4 Photocathodes, Mater. Chem. Phys., 2021, 270, 124834 CrossRef CAS.
- D. Hayes, R. G. Hadt, J. D. Emery, A. A. Cordones, A. B. F. Martinson, M. L. Shelby, K. A. Fransted, P. D. Dahlberg, J. Hong, X. Zhang, Q. Kong, R. W. Schoenlein and L. X. Chen, Electronic and Nuclear Contributions to Time-Resolved Optical and X-Ray Absorption Spectra of Hematite and Insights into Photoelectrochemical Performance, Energy Environ. Sci., 2016, 9(12), 3754–3769 RSC.
- A. S. M. Ismail, Y. Uemura, S. H. Park, S. Kwon, M. Kim, H. Elnaggar, F. Frati, Y. Niwa, H. Wadati, Y. Hirata, Y. Zhang, K. Yamagami, S. Yamamoto, I. Matsuda, U. Halisdemir, G. Koster, B. M. Weckhuysen and F. M. F. de Groot, Direct Observation of the Electronic States of Photoexcited Hematite with Ultrafast 2p3d X-Ray Absorption Spectroscopy and Resonant Inelastic X-Ray Scattering, Phys. Chem. Chem. Phys., 2020, 22(5), 2685–2692 RSC.
- Y. Zhang, C. Zhang, X. Huang, Z. Yang, K. H. L. Zhang and Y. Yang, Barrierless Self-Trapping of Photocarriers in Co3O4, J. Phys. Chem. Lett., 2021, 12(50), 12033–12039 CrossRef CAS.
- W. L. Roth, The Magnetic Structure of Co3O4, J. Phys. Chem. Solids, 1964, 25(1), 1–10 CrossRef CAS.
- M. Cococcioni and S. de Gironcoli, Linear Response Approach to the Calculation of the Effective Interaction Parameters in the LDA + U Method, Phys. Rev. B, 2005, 71(3), 035105 CrossRef.
- B. Himmetoglu, R. M. Wentzcovitch and M. Cococcioni, First-Principles Study of Electronic and Structural Properties of CuO, Phys. Rev. B, 2011, 84(11), 115108 CrossRef.
- M. Zacharias, C. E. Patrick and F. Giustino, Stochastic Approach to Phonon-Assisted Optical Absorption, Phys. Rev. Lett., 2015, 115(17), 177401 CrossRef.
- Y. Kang, H. Peelaers, K. Krishnaswamy and C. G. Van De Walle, First-Principles Study of Direct and Indirect Optical Absorption in BaSnO3, Appl. Phys. Lett., 2018, 112(6), 062106 CrossRef.
- J. Chen, X. Wu and A. Selloni, Electronic Structure and Bonding Properties of Cobalt Oxide in the Spinel Structure, Phys. Rev. B, 2011, 83(24), 245204 CrossRef.
- V. Singh, M. Kosa, K. Majhi and D. T. Major, Putting DFT to the Test: A First-Principles Study of Electronic, Magnetic, and Optical Properties of Co3O4, J. Chem. Theory Comput., 2015, 11(1), 64–72 CrossRef CAS.
- C. Lohaus, J. Morasch, J. Brötz, A. Klein and W. Jaegermann, Investigations on RF-Magnetron Sputtered Co3O4 Thin Films Regarding the Solar Energy Conversion Properties, J. Phys. D: Appl. Phys., 2016, 49(15), 155306 CrossRef.
- A. F. Lima, Interpretation of the Optical Absorption Spectrum of Co3O4 with Normal Spinel Structure from First Principles Calculations, J. Phys. Chem. Solids, 2014, 75(1), 148–152 CrossRef CAS.
- T. J. Smart, T. A. Pham, Y. Ping and T. Ogitsu, Optical Absorption Induced by Small Polaron Formation in Transition Metal Oxides: The Case of Co3O4, Phys. Rev. Mater., 2019, 3(10), 102401 CrossRef CAS.
- V. G. Hadjiev, M. N. Iliev and I. V. Vergilov, The Raman Spectra of Co3O4, J. Phys. C: Solid State Phys., 1988, 21(7), L199–L201 CrossRef.
- B. Rivas-Murias and V. Salgueiriño, Thermodynamic CoO–Co3O4 Crossover Using Raman Spectroscopy in Magnetic Octahedron-shaped Nanocrystals, J. Raman Spectrosc., 2017, 48(6), 837–841 CrossRef CAS.
- W. B. White and B. A. DeAngelis, Interpretation of the Vibrational Spectra of Spinels, Spectrochim. Acta, Part A, 1967, 23(4), 985–995 CrossRef CAS.
- Z. Wang, P. K. Nayak, J. A. Caraveo-Frescas and H. N. Alshareef, Recent Developments in p-Type Oxide Semiconductor Materials and Devices, Adv. Mater., 2016, 28(20), 3831–3892 CrossRef CAS.
- S. Lany, Semiconducting Transition Metal Oxides, J. Phys.: Condens. Matter, 2015, 27(28), 283203 CrossRef PubMed.
- M. Dekkers, G. Rijnders and D. H. A. Blank, ZnIr2O4, a p-Type Transparent Oxide Semiconductor in the Class of Spinel Zinc-d6-Transition Metal Oxide, Appl. Phys. Lett., 2007, 90(2), 021903 CrossRef.
- V. Venkatachalam, A. Alsalme, A. Alswieleh and R. Jayavel, Double Hydroxide Mediated Synthesis of Nanostructured ZnCo2O4 as High Performance Electrode Material for Supercapacitor Applications, Chem. Eng. J., 2017, 321, 474–483 CrossRef CAS.
- W. Wang, Facile Hydrothermal Synthesis of ZnCo2O4 Nanostructures: Controlled Morphology and Magnetic Properties, J. Mater. Sci.: Mater. Electron., 2021, 32(12), 16662–16668 CrossRef CAS.
- Z. Ž. Lazarević, Č. Jovalekić, A. Milutinović, D. Sekulić, V. N. Ivanovski, A. Rečnik, B. Cekić and N. Ž. Romčević, Nanodimensional Spinel NiFe2O4 and ZnFe2O4 Ferrites Prepared by Soft Mechanochemical Synthesis, J. Appl. Phys., 2013, 113(18), 187221 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental procedures for film fabrication and structural characterization, computational methods, additional resonance Raman spectra, thermal difference spectra of ZnCo2O4, and tables of computed phonon energies. See DOI: https://doi.org/10.1039/d5sc01909e |
| ‡ Current address: National Renewable Energy Laboratory, Chemistry and Nanoscience Center, Golden, CO, 80401, USA. |
| This journal is © The Royal Society of Chemistry 2025 |