
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic chaotropic effect and defect engineering promoting ultrahigh ionic conductivity in MOFs†
Dongbo Liua,
Xiao-Min Li *a,
Junchao Jiaa,
Xingyu Longa,
Junpeng Yana,
Mengyang Xiaoa,
Aziz Bakhtiyarovich Ibragimovb and
Junkuo Gao*a
*a,
Junchao Jiaa,
Xingyu Longa,
Junpeng Yana,
Mengyang Xiaoa,
Aziz Bakhtiyarovich Ibragimovb and
Junkuo Gao*a
aChina-Uzbekistan Joint Laboratory on Advanced Porous Materials, School of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China. E-mail: lixm@zstu.edu.cn; jkgao@zstu.edu.cn
bInstitute of General and Inorganic Chemistry, Uzbekistan Academy of Sciences, Tashkent 100170, Uzbekistan
First published on 26th May 2025
Abstract
The development of aqueous electrolytes exhibiting high ionic conductivities through solvent-free methods is of great significance for the progress of aqueous battery technology. This study presents a high-performance ionic conductor engineered through a synergistic chaotropic effect and defective structure using a solvent-free approach. The combination of localized acidification at vacancy sites and doping chaotropic LiI enhances ionic diffusion in D-UiO-66-LiI. The generated cationic substructure promotes ion confinement effects within the D-UiO-66-LiI pores, establishing efficient conduction pathways. Furthermore, temperature-dependent analysis reveals that thermal energy increases ion movement and weakens the hydration of Li+ ions, not only increasing ion mobility but also maintaining the water networks through dynamic hydrogen bonding reconstruction. These coordinated effects enable D-UiO-66-LiI to achieve ultrahigh ionic conductivities across wide temperature and humidity ranges.
Introduction
The continuous consumption of non-renewable resources and the resulting environmental pollution have prompted researchers to actively explore alternative sustainable energy technologies, such as the widely used rechargeable lithium ion (Li-ion) batteries.1–3 However, Li-ion batteries pose significant risks to the environment and human safety due to the use of organic electrolytes with characteristics of volatility, flammability, and toxicity, which has spurred efforts to find safer electrolyte materials that can replace organic-media systems.4–7 Recently, aqueous electrolytes have emerged as a promising option due to their low cost, non-toxicity, non-flammability, and safety benefits.8–11 On the other hand, solid electrolytes, as emerging electrolyte materials, exhibit enhanced safety and energy density.12,13 In the research of water-based electrolytes, the chaotropic effect is generally employed to improve ionic conductivity. The chaotropic effect of chaotropic salts can increase their affinity for water molecules, thereby improving overall ion conductivity.14 While research on aqueous solid electrolytes is still in its early stages, more experimental examples are needed to develop excellent electrolyte materials with high ionic conductivities and good performance durability. Therefore, it is desirable to not only broaden the synthetic strategies available of aqueous solid electrolytes but also comprehend their structure–activity relationships.Metal–organic frameworks (MOFs) have emerged as potential candidates for electrolyte materials due to several key advantages. Firstly, the porous structures of MOFs offer clear pathways for ion transport.15,16 Secondly, the tunable pores of MOFs, regarded as ion sieves, facilitate the selective promotion of cation transfer.17,18 Thirdly, the well-defined structures of MOFs enable a detailed exploration of the structure–activity relationship.19,20 Fourthly, the functionalized frameworks of MOFs allow for diverse synthetic strategies.21,22 Previous studies mainly focused on immersing lithium salts into the pores of MOFs to form lithium transport channels composed of high concentrations of lithium ions.23 Typically, MOFs characterized by cationic structures are employed to immobilize negatively charged ions present in lithium salts within the framework via electrostatic interactions. This arrangement facilitates the mobility of positively charged lithium ions within the pores, thereby establishing ion transport pathways. However, these MOF-based solid electrolytes have insufficient ionic conductivities comparing with liquid organic electrolytes.24 A recent study has demonstrated an enhancement in ion conductivity attributed to the cooperative effect of protons and lithium ions within the MOF pores,8 but this improvement requires high humidity conditions. On the other hand, current research efforts primarily focus on enhancing ion transport by utilizing metal open sites in MOFs as anchors for cation ions or by loading cations into MOF pores.25–29 While defect engineering is a relatively novel research avenue in MOFs, its application in ion transport, particularly in Li-ion conduction, remains limited. Notably, the aforementioned materials derived from MOFs are typically synthesized via the solvothermal technique, which often involves the use of organic solvents that can have adverse effects on both the environment and human health.30,31 Therefore, there is a need to investigate ion conduction within defective MOFs using solvent-free approaches to enhance the environmental sustainability of green energy technologies.
Considering the above, we have developed a strategy on the shoulders of giants to improve ion conductivity, especially under low humidity conditions. Specifically, we chose defective UiO-66 as the research platform. Compared to UiO-66 without defects, defective UiO-66 (referred to as D-UiO-66) has higher acidity (Scheme 1a), which is beneficial for ion transport.32 Moreover, UiO-66 is a classic and easily prepared MOF. The technique for the solvent-free synthesis of defective UiO-66 has reached a level of maturity and can be readily implemented. Lithium iodide (LiI) was chosen as the encapsulated molecule for ion transport due to the chaotropic effect of lithium halides that can increase their affinity for water molecules,33 thereby improving ion conductivity. Notably, LiI exhibits the weakest ionic bond strength, resulting in Li+ being the most readily dissociated.34 According to the published work, we synthesized D-UiO-66 using a solvent-free method.35 Subsequently, LiI was introduced into the D-UiO-66 channels, also employing a solvent-free approach (Scheme 1b). Physical mixing has the potential to enhance the interfacial area between reactants, consequently mitigating diffusion constraints.36,37 The obtained product (referred to as D-UiO-66-LiI) is expected to have higher ion conductivity due to its higher acidity and increased hydrophilicity. After performance evaluation, D-UiO-66-LiI shows ultrahigh ionic conductivities under wide range conditions. D-UiO-66-LiI exhibits a high ionic conductivity of 4.09 × 10−3 S cm−1 even at 30 °C and 40% relative humidity (RH). Under 70 °C and 98% RH, D-UiO-66-LiI shows an ultrahigh ionic conductivity of 5.03 × 10−1 S cm−1. This solvent-free green preparation strategy achieves efficient ion transport under wide range conditions. This study lays a foundation for the development of eco-friendly solid electrolytes.
 | ||
| Scheme 1 (a) Illustration of UiO-66 without defects. (b) Schematic illustration of the preparation of D-UiO-66-LiI. | ||
Results and discussion
The initial material of D-UiO-66 and the target product of D-UiO-66-LiI were synthesized. The powder X-ray diffraction (PXRD) patterns demonstrate that D-UiO-66 and D-UiO-66-LiI have the same structural framework as UiO-66 (Fig. 1a). As shown in Fig. S1 and S2, ESI,† it can be seen that D-UiO-66-LiI has additional elements Li and I after D-UiO-66 is modified by LiI. To demonstrate the defect in D-UiO-66, we also prepared UiO-66 without defects. The PXRD pattern of UiO-66 is consistent with that of simulated UiO-66, showing its successful synthesis (Fig. S3, ESI†). It can be seen from N2 adsorption–desorption data that D-UiO-66 has the largest N2 adsorption capacity and D-UiO-66-LiI has the lowest N2 adsorption capacity (Fig. S4, ESI†). The former is due to the larger pores generated by the missing ligands in D-UiO-66, while the latter is because of the occupation of pores after encapsulating LiI. The wavenumber interval in the Fourier Transform Infrared (FT-IR) spectrum can determine the coordination mode between ligands and metals in MOFs. As displayed in Fig. 1b, there are a strong splitting of 185 cm−1 and a weak splitting of 80 cm−1 in the FT-IR spectra of D-UiO-66 and D-UiO-66-LiI. The former is considered to be the bridging effect of the ligand, while the latter is due to the bidentate ligand.35,38 That is to say, the coordination mode of H2BDC in D-UiO-66 and D-UiO-66-LiI is mainly bridging, and includes a small part of bidentate coordination. The zeta potential results suggest that the introduction of defects and the incorporation of LiI result in the positive charges for D-UiO-66 and D-UiO-66-LiI, respectively (Fig. S5, ESI†). The positively charged MOFs enable the migration of H+ and Li+ away from the frameworks, leading to their accumulation within the pores. This phenomenon promotes the orderly ionic transport throughout the channels. | ||
| Fig. 1 (a) PXRD patterns of simulated UiO-66, D-UiO-66, and D-UiO-66-LiI. (b) FT-IR spectra of D-UiO-66 and D-UiO-66-LiI. (c) TGA curves of UiO-66 and D-UiO-66. The end weight of ZrO2 is normalized to 100%. (d) Acid–base titration curve and first derivative curve for D-UiO-66. | ||
In order to verify defect engineering in D-UiO-66, thermogravimetric analysis (TGA) and acid–base titration experiment were conducted. The final residue of UiO-66 and D-UiO-66 is ZrO2, so the weight of residual ZrO2 is normalized to 100% for the convenience of TG comparison. As shown in Fig. 1c, there are two weight loss stages in the TGA curves of UiO-66 and D-UiO-66, which are attributed to the loss of guest molecules and the collapse of the frameworks, respectively. It can be noted that no solvent was added during the synthesis of D-UiO-66, so the weight loss in the first stage is attributed to the water molecules adsorbed at the defect sites,35 which also indicates the formation of defect sites. The collapse of the framework is related to the amount of the ligand. The weight loss observed during the second stage in UiO-66 exceeds that of D-UiO-66, indicating the decreased ligands in D-UiO-66, which provides evidence for the formation of defects in D-UiO-66. Another method to determine defect sites is acid–base titration experiments. The acid–base reaction involves three distinct types of protons of μ3-OH, –OH2, and –OH in D-UiO-66. The acid–base titration curve of D-UiO-66 shown in Fig. 1d exhibits three inflection points, which are indicative of the above three proton types. To ascertain the equivalence points within the titration curve, the first derivative of the curve is employed, in which the initial point is attributed to the μ3-OH proton, and the remaining protons are associated with defect sites within the node structure (Zr–OH2 and Zr–OH protons). After calculation, the number of defects for D-UiO-66 is 1.24. These findings suggest the successful preparation of defect engineering in D-UiO-66. Additionally, Fig. S6, ESI,† shows the TGA curves of D-UiO-66 and D-UiO-66-LiI, proving their sufficient thermal stabilities for further ionic conductivity measurements.
The incorporation of defects within MOF leads to an enhancement in ionic conductivity by modifying various properties, including porosity, water adsorption capacity, and acidity. Defect vacancies can accommodate additional ion carriers, while an increased pore volume facilitates ion migration. The ionic conductivities of D-UiO-66 and D-UiO-66-LiI were evaluated through AC impedance. The humidity-dependent ionic conductivities were measured at 30 °C and different humidities of 40–98% RH (Fig. S7, ESI† and 2a). The result shows the positive relationship between humidity and ionic conductivity. The data presented in Fig. 2b indicate that, in comparison to D-UiO-66, D-UiO-66-LiI exhibits a marked superiority in terms of ionic conductivity. Additionally, the detection of water vapor adsorption of D-UiO-66 and D-UiO-66-LiI highlights that the introduction of LiI significantly enhances the ability of D-UiO-66-LiI to adsorb water molecules (Fig. 2c). Due to the increased acidity caused by defect engineering39 and the strong affinity with water molecules resulting from LiI with a chaotropic effect, D-UiO-66-LiI has an ultrahigh ionic conductivity of 7.02 × 10−2 S cm−1 at 30 °C and 98% RH. As shown in Fig. 2d, the frameworks of D-UiO-66-LiI have positively charged layers due to its defects. The repulsive forces between charges result in the concentration and arrangement of Li+ ions and H+ ions within the pores of D-UiO-66-LiI, thereby facilitating efficient ion transport. It is worth noting that D-UiO-66-LiI demonstrates a high ionic conductivity of 4.09 × 10−3 S cm−1 at 30 °C and 40% RH. This finding suggests that D-UiO-66-LiI shows superior ion conduction behavior under a wide range of conditions, including environmental conditions.
 | ||
| Fig. 2 (a) Nyquist plots of D-UiO-66-LiI at 30 °C and different humidities varying from 40% to 98% RH. (b) Ionic conductivities of D-UiO-66 and D-UiO-66-LiI at 30 °C and different humidities varying from 40% to 98% RH. (c) H2O vapor adsorption–desorption isotherms of D-UiO-66 and D-UiO-66-LiI at 298 K. (d) Schematic illustration of ionic transport in D-UiO-66-LiI under humidity conditions. | ||
To investigate the influence of temperature on ion conduction, the temperature-dependent ionic conductivities of D-UiO-66 and D-UiO-66-LiI at 98% RH were assessed (Fig. S8, ESI† and 3a). The result displayed in Fig. 3b shows that the ionic conductivities of D-UiO-66 and D-UiO-66-LiI increase with rising temperature. Until 70 °C, the ionic conductivities can reach 1.78 × 10−2 S cm−1 and 5.03 × 10−1 S cm−1 for D-UiO-66 and D-UiO-66-LiI, respectively. It is desirable to achieve such high ionic conductivity of D-UiO-66-LiI, which validates the usefulness of the proposed methodology. The performance comparison of D-UiO-66-LiI with some other representative MOF-based ion conducting materials indicates that the target material of this work exhibits a significant advantage in ion conductivity, which also proves the usefulness of this strategy (Table S1, ESI†). Notably, the structural integrity of D-UiO-66 and D-UiO-66-LiI was maintained after performance tests, which is crucial for potential applications (Fig. S9–S11, ESI†).
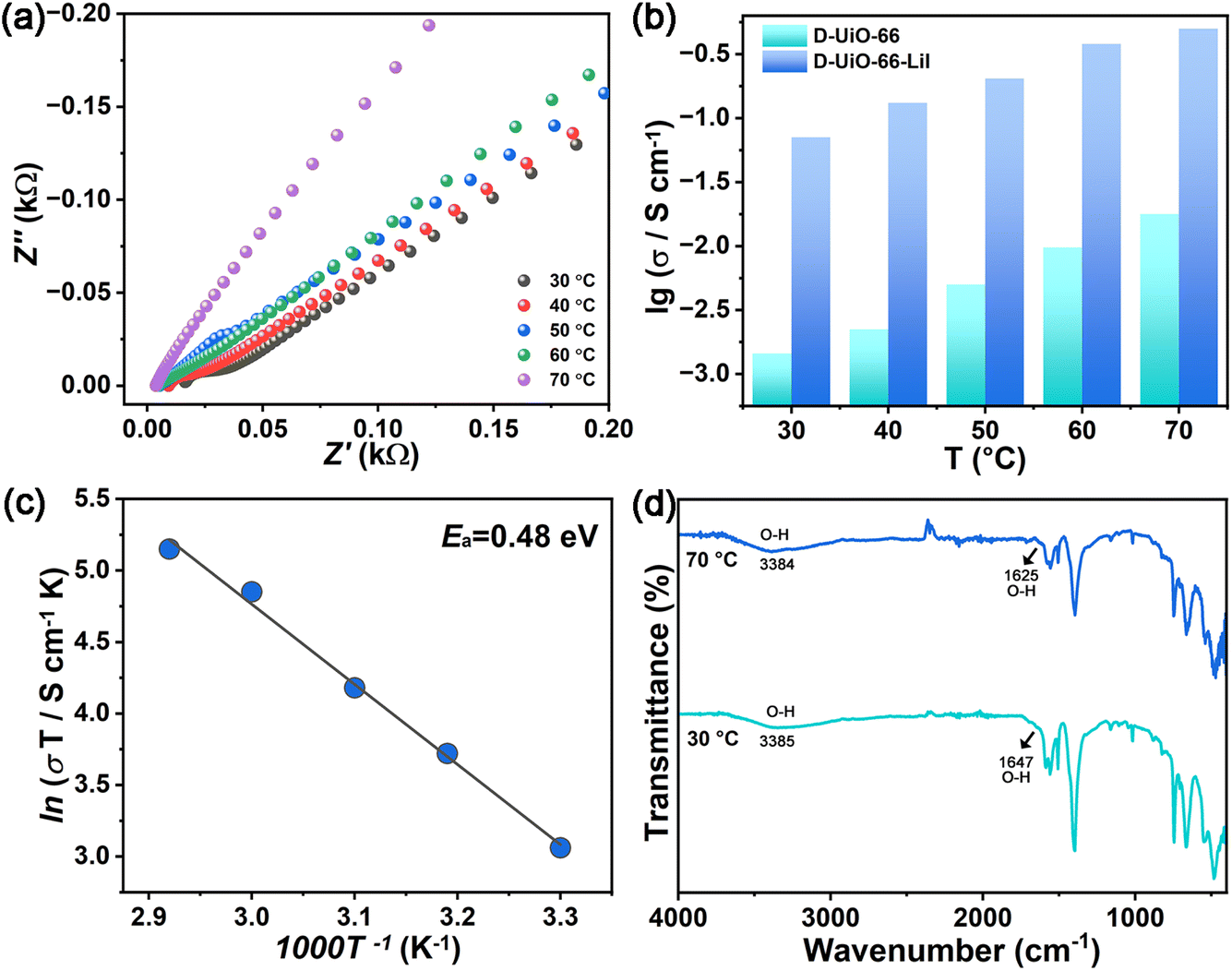 | ||
| Fig. 3 (a) Nyquist plots of D-UiO-66-LiI at 98% RH and different temperatures varying from 30 to 70 °C. (b) Ionic conductivities of D-UiO-66 and D-UiO-66-LiI at 98% RH and different temperatures varying from 30 to 70 °C. (c) Arrhenius plot of D-UiO-66-LiI at 98% RH and in the temperature range of 30–70 °C. (d) Temperature-dependent FT-IR spectra of D-UiO-66-LiI. | ||
Furthermore, the ionic transport modes of D-UiO-66 and D-UiO-66-LiI were determined by fitting the Arrhenius plots under 98% RH and in the temperature range of 30–70 °C. As shown in Fig. S12, ESI† and 3c, the activation energies (Ea) of D-UiO-66 and D-UiO-66-LiI are 0.61 eV and 0.48 eV, respectively, which belong to the vehicle mechanism.40 This result is consistent with the concept shown in Fig. 2d. Specifically, the integrated entity of H3O+ and Li+ is arranged within the channels of D-UiO-66-LiI for transport through free diffusion. In order to better understand the ion transport behavior, temperature-dependent FT-IR spectra of D-UiO-66-LiI were measured. As the temperature rises, the thermal motion of H+ ions and Li+ ions intensifies, facilitating the ionic migration within the channels of D-UiO-66-LiI. Meanwhile, it can be observed from Fig. 3d that the stretching and bending vibrations of O–H from free water molecules41 undergo redshifts with the increasing temperature. This phenomenon may be attributed to the enhanced hydrogen bonds of O–H⋯I− induced by I− with increasing temperature, while the bound water molecules released by Li+ participate in the formation of O–H⋯O hydrogen bonds. Additionally, the 7Li VT-SSNMR spectra of D-UiO-66-LiI have been supplemented. The increase in temperature results in a shift of 7Li signal towards lower frequency, indicating that rapid Li+ movement has taken place within the channels of the MOF42 (Fig. S13, ESI†).
Performance stability is also important for practical applications. Temperature cycling and durability assessments were conducted to evaluate the performance stability. As shown in Fig. S14 and S15, ESI,† the ionic conductivities of D-UiO-66-LiI at the same temperature during the heating and cooling stages are basically the same, indicating that it has good temperature cycling stability. The consistent Ea values during the heating and cooling stages indicate the stable ion transport mode in D-UiO-66-LiI (Fig. S16, ESI†). As shown in Fig. S17, ESI,† the ionic conductivity of D-UiO-66-LiI at 70 °C and 98% RH can be maintained for at least 24 h, indicating its good durability in performance. Importantly, the PXRD pattern and FT-IR spectrum of D-UiO-66-LiI after performance tests are basically consistent with those before tests, demonstrating its structural integrity (Fig. S18 and S19, ESI†). Additionally, the synthesized D-UiO-66-LiI has excellent long-term stability. As shown in Fig. S20, ESI,† the PXRD pattern of D-UiO-66-LiI stored for 1 year is basically identical with that of simulated UiO-66, indicating its excellent long-term stability. Moreover, the ionic conductivities of the sample stored for 1 year were measured, and the results show its structural stability and the consistent performance with that of the fresh sample (70 °C and 98% RH: 5.02 × 10−1 S cm−1) (Fig. S21, ESI†).
Conclusions
In summary, we have developed an excellent ion conductor utilizing chaotropic effect and defect engineering. The increased acidity caused by defect sites and the introduction of LiI with chaotropic effect enhance the ion transport in D-UiO-66-LiI. On the other hand, the cation framework resulting from defects facilitates the aggregation of ions within the pores, thereby improving ion transport. Moreover, as the temperature rises, the binding energy between Li+ ions and water molecules decreases, and some bound water molecules detach from the hydration layers and reintegrate into the hydrogen bonding networks. This phenomenon, coupled with the accelerated ion mobility induced by the elevated temperature, contributes to an enhancement in the ionic conductivity of D-UiO-66-LiI. These factors enable D-UiO-66-LiI to exhibit ultrahigh ionic conductivities under wide range conditions. This solvent-free strategy has broadened the methodologies available for ion conduction and enhanced the comprehension of ion transport mechanisms. However, MOFs are difficult to be directly prepared into ion conductive membranes due to their poor processability. Currently, most MOF membranes use polymers as substrates, which may reduce their ion conductivities. Therefore, utilizing the structural designability of MOFs and strategically selecting polymers for chemical grafting onto frameworks to produce films may represent a promising avenue for further research.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. Liu and X.-M. Li designed the study and wrote the manuscript. D. Liu and J. Jia synthesized the materials and performed the experimental measurements. D. Liu analyzed the data. X. Long, J. Yan, and M. Xiao conducted some characterization tests. X.-M. Li, A. B. Ibragimov, and J. Gao supervised the research. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project of the National Natural Science Foundation of China (No. 22301277), the Baima Lake Laboratory Joint Fund of the Zhejiang Provincial Natural Science Foundation of China under Grant No. LBMHY25A040001, Zhejiang Provincial Natural Science Foundation of China (No. LY20E020001), Research Initiation Fund Project from Zhejiang Sci-Tech University (No. 22212154-Y) and the Fundamental Research Funds of Zhejiang Sci-Tech University (No. 22212290-Y).Notes and references
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- K. Xu and C. Wang, Nat. Energy, 2016, 1, 16161 CrossRef CAS.
- J. Xie and Y.-C. Lu, Adv. Mater., 2025, 37, 2312451 CrossRef CAS PubMed.
- Y. Huang and J. Li, Adv. Energy Mater., 2022, 12, 2202197 CrossRef CAS.
- J. Xie and Y.-C. Lu, Nat. Commun., 2020, 11, 2499 CrossRef CAS PubMed.
- K. Jeong, S. Park and S.-Y. Lee, J. Mater. Chem. A, 2019, 7, 1917–1935 RSC.
- M. K. Sarango-Ramírez, M. Donoshita, Y. Yoshida, D.-W. Lim and H. Kitagawa, Angew. Chem., Int. Ed., 2023, 62, e202301284 CrossRef PubMed.
- S. Kang, A. Singh, K. G. Reeves, J.-C. Badot, S. Durand-Vidal, C. Legein, M. Body, O. Dubrunfaut, O. J. Borkiewicz, B. Tremblay, C. Laberty-Robert and D. Dambournet, Chem. Mater., 2020, 32, 9458–9469 CrossRef CAS.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- X. Wu, J. J. Hong, W. Shin, L. Ma, T. Liu, X. Bi, Y. Yuan, Y. Qi, T. W. Surta, W. Huang, J. Neuefeind, T. Wu, P. A. Greaney, J. Lu and X. Ji, Nat. Energy, 2019, 4, 123–130 CrossRef CAS.
- Z. Tu, K. Chen, S. Liu and X. Wu, Small Methods, 2025, 2401912, DOI:10.1002/smtd.202401912.
- C. Luo, X. Ji, J. Chen, K. J. Gaskell, X. He, Y. Liang, J. Jiang and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 8567–8571 CrossRef CAS PubMed.
- J. Brus, J. Czernek, M. Urbanova, J. Rohlíček and T. Plecháček, ACS Appl. Mater. Interfaces, 2020, 12, 47447–47456 CrossRef CAS PubMed.
- C. Wang, F.-F. Liu, Z. Tan, Y.-M. Chen, W.-C. Hu and X.-H. Xia, Adv. Funct. Mater., 2020, 30, 1908804 CrossRef CAS.
- Y. Xiang, N. Yu, J. Li, H. Xu, S. Chen, Y. Xia, Z. Luo, X. Li, Z. Liu, M. Xu, Y. Jiang and X. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202424288 CrossRef CAS PubMed.
- S. Zheng, Z. Li, L. Chen, Y. Huang, J. Shi, S. Wang, Y. Liu, Y. Liu, Y.-P. Cai and Q. Zheng, ACS Mater. Lett., 2023, 5, 1136–1144 CrossRef CAS.
- L. Shen, H. B. Wu, F. Liu, J. L. Brosmer, G. Shen, X. Wang, J. I. Zink, Q. Xiao, M. Cai, G. Wang, Y. Lu and B. Dunn, Adv. Mater., 2018, 30, 1707476 CrossRef PubMed.
- X.-M. Li, J. Jia, D. Yang, J. Jin and J. Gao, Chin. Chem. Lett., 2024, 35, 108474 CrossRef CAS.
- L. Meng, T. Lan, J. Xu, P. Zhao and J. Lei, J. Membr. Sci., 2024, 694, 122409 CrossRef CAS.
- X.-M. Li, J. Jia, M. Zhao, D. Liu, J. Gao and Y.-Q. Lan, Chem. Commun., 2024, 60, 6777–6780 RSC.
- G. Cai, A. A. Chen, S. Lin, D. J. Lee, K. Yu, J. Holoubek, Y. Yin, A. U. Mu, Y. S. Meng, P. Liu, S. M. Cohen, T. A. Pascal and Z. Chen, Nano Lett., 2023, 23, 7062–7069 CrossRef CAS PubMed.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- B. M. Wiers, M.-L. Foo, N. P. Balsara and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14522–14525 CrossRef CAS PubMed.
- S. Wang, Z. Li, F. Shen, Z. Ruan, Y. Huang, Y. Liu, Y. Liu, L. Chen, Y.-Q. Lan and Q. Zheng, J. Mater. Chem. A, 2023, 11, 14025–14033 RSC.
- J. Zhang, Z. Zhang, T. Wu and X. Luo, J. Electroanal. Chem., 2023, 946, 117708 CrossRef CAS.
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 86 CrossRef.
- S. S. Park, Y. Tulchinsky and M. Dincă, J. Am. Chem. Soc., 2017, 139, 13260–13263 CrossRef CAS PubMed.
- M. Vazquez, M. Liu, Z. Zhang, A. Chandresh, A. B. Kanj, W. Wenzel and L. Heinke, ACS Appl. Mater. Interfaces, 2021, 13, 21166–21174 CrossRef CAS PubMed.
- K. Užarević, T. C. Wang, S.-Y. Moon, A. M. Fidelli, J. T. Hupp, O. K. Farha and T. Friščić, Chem. Commun., 2016, 52, 2133–2136 RSC.
- Q. Wu, X. Wang, G. Qi, Q. Guo, S. Pan, X. Meng, J. Xu, F. Deng, F. Fan, Z. Feng, C. Li, S. Maurer, U. Müller and F.-S. Xiao, J. Am. Chem. Soc., 2014, 136, 4019–4025 CrossRef CAS PubMed.
- O. Basu, S. Mukhopadhyay, S. Laha and S. K. Das, Chem. Mater., 2022, 34, 6734–6743 CrossRef CAS.
- Q. Dong, H. Ao, Z. Qin, Z. Xu, J. Ye, Y. Qian and Z. Hou, Small, 2022, 18, 2203347 CrossRef CAS PubMed.
- C. R. Gopikrishnan, D. Jose and A. Datta, AIP Adv., 2012, 2, 012131 CrossRef.
- G. Ye, D. Zhang, X. Li, K. Leng, W. Zhang, J. Ma, Y. Sun, W. Xu and S. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 34937–34943 CrossRef CAS PubMed.
- S. Horike, Y. Kamitsubo, M. Inukai, T. Fukushima, D. Umeyama, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2013, 135, 4612–4615 CrossRef CAS PubMed.
- J. Cattermull, S. Wheeler, K. Hurlbutt, M. Pasta and A. L. Goodwin, Chem. Commun., 2020, 56, 7873–7876 RSC.
- K. B. Lausund and O. Nilsen, Nat. Commun., 2016, 7, 13578 CrossRef CAS PubMed.
- K. Zhang, G.-H. Wen, X.-J. Yang, D.-W. Lim, S.-S. Bao, M. Donoshita, L.-Q. Wu, H. Kitagawa and L.-M. Zheng, ACS Mater. Lett., 2021, 3, 744–751 CrossRef CAS.
- G. A. Ludueña, T. D. Kühne and D. Sebastiani, Chem. Mater., 2011, 23, 1424–1429 CrossRef.
- Z.-H. Li, H. Zeng, G. Zeng, C. Ru, G. Li, W. Yan, Z. Shi and S. Feng, Angew. Chem., Int. Ed., 2021, 60, 26577–26581 CrossRef CAS PubMed.
- W.-L. Xue, W.-H. Deng, H. Chen, R.-H. Liu, J. M. Taylor, Y.-k. Li, L. Wang, Y.-H. Deng, W.-H. Li, Y.-Y. Wen, G.-E. Wang, C.-Q. Wan and G. Xu, Angew. Chem., Int. Ed., 2021, 60, 1290–1297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01923k |
| This journal is © The Royal Society of Chemistry 2025 |