
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the local coordination effect of Cu–N–C single-atom catalysts towards CO adsorption via a gas-phase cluster model approach†
Qing Wangab,
Dingding Lva,
Jian Zhouab,
Detong Konga,
Shanshan Linab,
Lili Zhangab,
Zhen-an Qiao c,
Yuxiao Ding*bd and
Xiaoyan Sun*ab
c,
Yuxiao Ding*bd and
Xiaoyan Sun*ab
aState Key Laboratory of Photoelectric Conversion and Utilization of Solar Energy, Qingdao New Energy Shandong Laboratory, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: sunxy@qibebt.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun, 130012, China
dState Key Laboratory of Low Carbon Catalysis and Carbon Dioxide Utilization, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, 730000, China. E-mail: yuxiaoding@licp.cas.cn
First published on 21st May 2025
Abstract
The current understanding regarding how the coordination environment of single-atom catalysts supported on nitrogen-doped carbon (M–N–C SACs) regulates their reactivity remains controversial, due to the complicated surface chemistry and lack of atomic-level insights. Here we introduce an experimental modeling approach to unambiguously identify the individual contribution of the local environment to the adsorption activity of CO on Cu–N–C systems. The fundamental intrinsic activities of Cu–N–C systems with different N coordination numbers, N coordination geometries (e.g., pyrrolic N and pyridinic N), defect sites (e.g., armchair and zig-zag), as well as S and P dopants, towards CO adsorption can be explicitly obtained and compared at the strictly atomic level, which would be challenging to access via conventional techniques in SACs research. For all kinds of coordination structures, we further identified general rules that control CO adsorption strength and experimental reaction rate. This novel approach is general and can be applied to other SAC metal and reaction systems.
Introduction
The understanding of heterogeneous catalytic systems should go to the atomic level, as the supported active species on different defective sites could have totally different coordination environment and reaction behavior.1–4 The complexity of the support surface chemistry and the supported active species (mostly with a mixture of metal particles, clusters and single species) make this an impossible mission.5,6 Single-atom catalysts (SACs) possess prominent advantages of maximized atomic utilization with a clear definition of the supported single sites, which reduce the complexity of the active sites.7,8 2D metal–nitrogen-doped carbon (M–N–C) materials, as a typical class of SACs, have presented high promise in many reactions.9,10 However, there is still ongoing debate regarding how the different coordination structures of the metal center affect the activity and selectivity. Taking the Cu–N–C catalyzed CO2 reduction reaction (CO2RR) as an example, besides the most identified Cu–N4 configuration,11 Cu–N2 (ref. 12) and Cu–N3 (ref. 13) have also been proposed as active structures for CO2 reduction to CO. Furthermore, theoretical and experimental studies indicated that the configuration of N (e.g., pyrrolic N and pyridinic N),14 defects in the carbon support,15 as well as the presence of other heteroatoms,16 all significantly impact the catalytic process, though no consistent pattern has emerged so far.The root cause resulting in the current disparate understanding can be attributed to the inhomogeneity of most supported M–N–C SACs and the lack of suitable tools for structure–activity correlation studies with atomic resolution. Supports such as N-doped graphene are inherently inhomogeneous with coexistence of many different N configuration structures commonly formed via a pyrolysis process at high temperature.17 The complexity makes it very difficult to precisely identify and quantify each coordination structure in the as-synthesized samples as well as to obtain appropriate structure–performance correlations. This also leads to large differences between the theoretical analysis based on perfect modeling and the real catalytic systems.18 Furthermore, analytical tools for studying M–N–C SACs are generally restricted to spectroscopic methods like X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS), infrared spectroscopy (IR), as well as microscopy-based techniques like high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM), which either provide only averaged information of observable species,19–21 or are difficult to discern changes in the support structure and adsorption of intermediates. This raises an urgent demand to develop new methods for deciphering the atomic understanding of M–N–C SACs.
It has been acknowledged that the chemical reaction is a local event: bond breaking and bond making are confined to the catalytically active sites which involve only a limited number of atoms.22–25 More importantly, SACs differ substantially from metal surfaces and can be considered analogues of coordination compounds.3,26,27 This similarity provided us with new inspiration to study SACs, that is, using definitive coordination models to decouple the individual contribution of local geometry. Therefore, we introduced an innovative mass spectrometry-based approach to investigate the M–N–C SACs system with atomic resolution. We started with the Cu–N–C system and selected CO as the probe molecule, because the adsorption and activation of CO is an essential intermediate step in many important reactions like CO2RR,28 CO reduction29 (CORR), Fischer–Tropsch (F–T) synthesis30 and carbonylation reactions.31 A series of cluster models with defined coordination structures were generated, and their corresponding reactivities towards CO were further examined and compared by virtue of established gas-phase experimental techniques. On this basis, a comprehensive correlation of CO adsorption activities with varied N coordination numbers, N coordination geometries, as well as S and P dopants, can be explicitly obtained. Density functional theory (DFT) calculations revealed the essential factors determining the adsorption kinetics and thermodynamic bonding strength of CO, which provide a guiding principle in designing *CO intermediate-involved selectivity paths. This methodology is not only feasible for Cu–N–C system but is more generally applicable in other SACs.
Results and discussion
Reaction apparatus and generation of Cu(I)–N–C model clusters
In the realistic Cu–N–C SAC systems, the coordination environment of Cu active sites is highly complicated (Fig. 1a), involving different coordination numbers, coordination types, and dopant atoms (such as P, S, etc.). Herein, we utilized a modified linear ion trap mass spectrometer (LTQ-XL MS) apparatus to produce a series of Cu–N–C model clusters resembling the active sites of the catalyst and monitored their reactivity with CO online. As shown in Fig. 1c, electrospray ionization (ESI) was applied as a mild ionization source to facilitate the generation of Cu-based ions with intact ligands.32 A series of ligands (Fig. 1b) with different nitrogen coordination numbers (e.g., pyridine, bipyridine, tripyridine), configurations (e.g., carbazole, acridine), and other heteroatom dopants (e.g., P, S) were dissolved in methanol and co-injected with Cu(NO3)2 into the ESI apparatus. The desired Cu species were thus obtained either through collision-induced dissociation (CID) or direct isolation with precise isotope distributions (Fig. S1–S3†). A high-resolution mass spectrometer (Q Exactive MS) further confirmed the exact elemental composition and quantity of elements of the complex (Fig. S4 and S5†).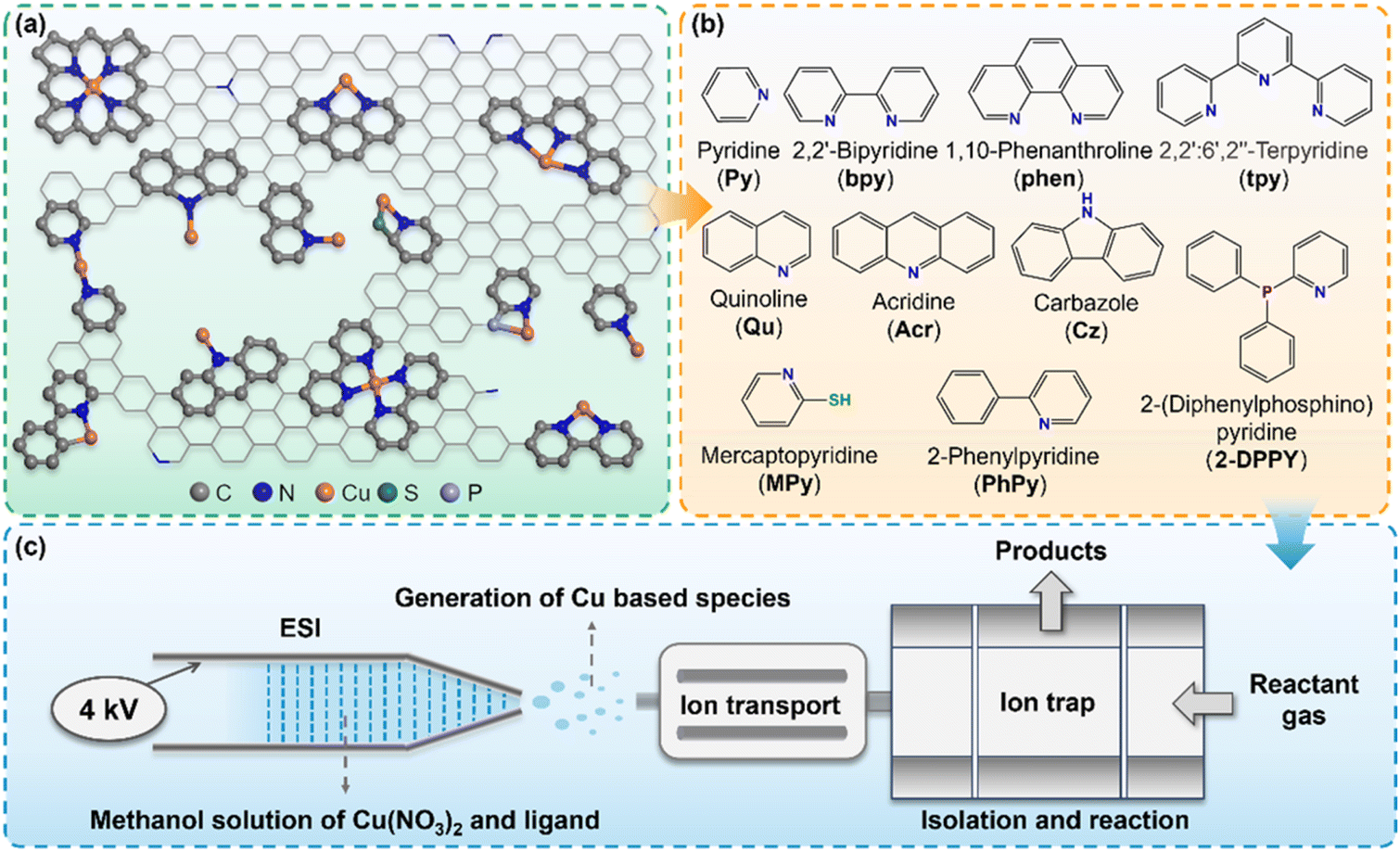 | ||
| Fig. 1 Reaction apparatus and generation of Cu(I)–N–C model clusters. (a) Representative structures of the realistic Cu–N–C catalysts. (b) The ligands used in this study. (c) Schematic diagram of the mass spectrometry setup for the generation and reaction of model clusters. | ||
Given that the ligands coordinated to copper are electrically neutral, except for those with pyrrolic N or P-doped configurations, the resulting complexes can be regarded as nominal Cu(I) ion centers on neutral supports. Herein our goal is to investigate the regulatory role of the Cu(I) center in CO adsorption due to its critical importance.33,34 We believe this strategy can also be applied to investigate other valence states of metal centers by creating models with appropriate ligands and precursor metal salts. Subsequently, CO was introduced into the ion trap reacting with the isolated Cu species, and the products were detected simultaneously without any fragment ions formed during the reaction (Fig. S6†). The pseudo-first-order rate constants (k) for each species can be well-fitted at different reaction times, the value of which are depicted next to the corresponding mass spectra and the detailed calculation methods are provided in the ESI Text, Table S1 and Fig. S7.† Based on this approach, the effect of various N coordination forms and doped atoms towards CO chemisorption can be independently probed and quantitatively compared.
Effects of N coordination number
We first investigated the effect of N-coordination number on the activity of the Cu(I) center. A series of Cu complexes with pyridine (Py), bipyridine (bpy), and terpyridine (tpy) ligands, as well as the naked [Cu]+, were produced to represent local geometry with 0 to 4 pyridinic N atoms (Ns) coordinated to the Cu center. It is notable that Cu active sites with varying N coordination numbers exhibited distinctly different adsorption activities towards CO (Fig. 2a). As compared to the inert bare [Cu]+, [Cu-Py]+ and [Cu-bpy]+ can easily capture CO, suggesting that [Cu]+ is activated through coordination with pyridinic N. Interestingly, although the adsorption rate is higher with bidentate coordination as compared with the monodentate, further increasing the coordination number, i.e., [Cu-tpy]+ and [Cu-bpy2]+, resulted in the inert CO adsorption. In addition, the presence of water in the ion trap competed with CO adsorption, with being most pronounced when coordinated with single pyridinic N. | ||
| Fig. 2 Effects of N coordination number. (a) Mass spectra for the reactions of CO with mass-selected [Cu]+ and [CuL]+ at the reaction time of 30 ms. The rate constants (k in 10−9 (±30%) cm3 molecule−1 s−1) are given in red. Black lines indicate CO-inert species, while colored lines represent species with CO adsorption activity. Peaks marked with asterisks are attributed to the adsorption of water or solvents (CH3OH, CH3CN). (b) Optimized geometry of model clusters and their CO-bound products in the ground state. The bond lengths are provided in Å, with hydrogen atoms omitted for clarity. Unless stated otherwise, the same representations hereinafter. (c) Charge population on Cu atom in the model clusters (blue lines) and the encounter complexes (green lines); charge population on the reactant CO of [OC–CuL]+ (red lines). (d) Variation of charge on Cu in [CuL]+ (blue curve) and ΔH (red curve) with different pyridinic N coordination numbers. (e) Frontier orbitals and corresponding molecular orbital structures of CO, [Cu]+ and [CuL]+. | ||
The geometric structures and the bonding feature between the ligands and Cu center, as well as their interactions with CO were further analyzed, which suggested a covalent bonding nature at both Cu–CO and Cu–N interfaces (Fig. 2b and see the detailed discussion in the ESI Text†). Charge population analysis demonstrated the charge transfer among the ligands, Cu atom and CO (Fig. 2c). An increased number of coordinated pyridinic N resulted in more electrons transferred from the ligand to the Cu center, leading to a lower charge on Cu. This, in turn, reduced the electron transfer from CO to Cu, thereby weakening the Cu–CO bond, aligning with the observed trend in the reaction exothermicity (Table S5† and Fig. 2d). This charge reduction deactivates the Cu center in coordination with 3 or 4 pyridinic Ns, while the [Cu-Py]1+ complex exhibits the strongest bonding affinity with CO (ΔHCO = −138.02 kJ mol−1).
Analysis of the frontier molecular orbitals (FMOs) in [CuL]+ complexes revealed that ligands with different N coordination numbers can regulate CO adsorption rates by modulating the FMOs of the complexes. A comparison of the FMOs of Cu–Nx (x = 0–4) in Fig. 2e shows that effective ligand conjugation narrows the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap of the complex, which favors the energy alignment with the bonding orbital of CO. Thus, a narrower gap in [CuL]+ improves electron transfer efficiency, leading to a higher adsorption rate. However, it's important to note that the FMOs of [CuL]+ are sometimes predominantly localized on the organic ligands (Fig. S10†), whose orbital symmetry mismatched with that of CO, thereby hindering the bond formation. In this case, the low-lying HOMO−n and LUMO+n (n = 0–3) orbitals, that have the more symmetry-matched 5s orbital of Cu (Fig. S11†), should be considered as active orbitals to calculate the “active gap”. Therefore, the narrower active gap of the two pyridinic N-coordinated [Cu-bpy]+ (4.93 eV) compared to the monocoordinated [Cu-Py]+ (5.71 eV) resulted in a substantially higher adsorption rate. In contrast, for [Cu-tpy]+ and [Cu-bpy2]+ with more pyridinic Ns and even small HOMO–LUMO gaps, due to the absence of low-lying active symmetry-matched orbitals, these complexes cannot effectively participate in bonding with CO in the ground state, resulting in an extremely slow or negligible adsorption rate, although [Cu-tpy]+ is not fully coordinatively saturated (16e) with relatively small steric hindrance. Although the FMOs between bare [Cu]+ and CO match well, its CO reactivity remains inert due to the stable 3d10 closed-shell configuration and energy-level mismatch during π-back donation. Additionally, the [Cu–CO]+ complex, with only three vibrational degrees of freedom and the smallest cross-section, struggles to dissipate the heat released during adsorption, preventing stable product formation.
To investigate the effect of different N coordination numbers on the C–O bond properties, we calculated the CO stretching frequencies before and after adsorption. The results show that as the N coordination number increases, the C–O stretching frequency gradually decreases, and the C–O bond length correspondingly increases (Table S6†). Analysis of the donation and back-donation interactions between [CuL]+ and CO indicates that a higher N coordination number and larger conjugated systems donate more electrons to the Cu center, thereby enhancing the Cu → CO π-back donation (Table S7†). This interaction weakens the C–O bond, leading to a red shift in its vibrational frequency. Essentially, the ligands influence the strength of the C–O bond by modulating the charge on the Cu center. In subsequent studies, we also calculated the C–O vibrational frequencies for other systems, and the results are consistent with the above trend (see the detailed discussion in the ESI Text†).
Based on the above analysis, we can conclude that the ligands regulate CO adsorption by modulating the charge state of the Cu center and altering the orbital energy levels. In this context, we propose using the charge on Cu in the model clusters and the “active gap” energy as descriptors to represent the thermodynamic CO adsorption strength and kinetic reaction rate, respectively. In particular, through the following experimental and theoretical data, we will demonstrate that this is a general rule for all coordination structures, not just for the influence of coordination number (Fig. S12†). Thus, using the two descriptors, we can theoretically predict the CO adsorption strength and adsorption rate for various cluster models. In addition, the vibrational degrees of freedom and collision cross-section of the system can also exert a certain degree of influence on the apparent reaction rate (ESI Text†).
Effects of N coordination configuration
The different coordination configurations of nitrogen on M–N–C have also been shown in previous studies to impact reaction activity and selectivity, but remained inconclusively addressed.14 To get a atomic-level insight, we first compared two model clusters with bidentate coordination but different configurations. In contrast to the reactive adsorption for the [Cu-bpy]+ system with ortho-position of two pyridine rings (Fig. 2a), the para-position in [Cu-Py2]+ cannot effectively bond with CO (Fig. 3a). This complex lacked vacant orbitals to form a σ bond with CO due to steric hindrance (Fig. 3b). Additionally, the higher electron-donation ability of para-coordination resulted in a very low charge state of Cu (0.27 |e|), thus making CO adsorption thermodynamically unfavorable (ΔG = 1.78 kJ mol−1) (Table S5†). | ||
| Fig. 3 Effects of N coordination configuration. (a) Mass spectra for the reactions of CO with mass-selected [CuL]+ with rate constants (k in 10−9 (±30%) cm3 molecule−1 s−1) given in red. (b) LUMO + n (n = 0–3) of [Cu-Py2]+. (c) Optimized geometry of model clusters and their CO-bound products in the ground state. | ||
Besides pyridinic N coordination, research on the role of pyrrolic N and graphitic N is also necessary given their important role previously discussed in the related reactions.35 While creating a cluster model with graphitic N is challenging due to the lack of relevant ligands, we turned our attention on the more accessible pyrrolic N to give a comparative study. We began by using [Cu-Acr]+ (Acr for acridine with a pyridinic N) and [Cu-Cz]+ (Cz for carbazole without the hydrogen atom on the pyrrolic N) as examples to compare the oxidation state and charge distribution of Cu. Although the Cu center is in the higher +2 oxidation state when coordinated to pyrrolic N in [Cu-Cz]+, the charge on the Cu center is almost identical in both coordination configurations. Besides, due to the similar size of the conjugated systems, the active gap values of both are also very close. This aligns well with their small difference in CO bonding strength as well as in the experimentally observed adsorption rates (Fig. 3a). This situation holds true upon comparing single-ring pyridine and the simulated single-ring pyrrole (Pyr) coordinated models, wherein [Cu-Py]+ and [Cu-Pyr]+ complexes displayed similar behavior (Fig. 3c and Table S9†). This leads us to conclude that, at the strictly atomic level, no significant difference exists for pyridinic and pyrrolic N coordinated Cu centers, albeit with nominal different oxidation states.
Effects of doping/functional groups on the ortho-carbon
Besides the N coordination number and configuration, modifying coordination environments on the ortho-carbon adjacent to the pyridinic N has been highlighted to indirectly regulate the reactivity of the metal center.36 Therefore, in this part, we specifically investigated the effects of introducing different functional groups solely at this position, including doping atoms (P, S) and the phenyl group. Except for the P-containing ligands, other ligands maintain their integrity when coordinated with Cu, making the ligands electrically neutral, while Cu is assumed to be in the +1 oxidation state. Due to the special adsorption process of the P-containing complex, the charge on Cu is not considered here.In the case of P and pyridinic N co-doped ligand, we observed a remarkable phenomenon: Cu bonded with P instead of N giving an appreciable adsorption rate (Fig. 4a). Orbital analysis revealed that P doping significantly reshaped the FMOs, localizing active orbitals on P atom (Fig. 4c), resulting in the CO adsorption site shifting from Cu to P atom. In addition, the pronounced covalent interaction of the P–C bond contributed to strong adsorption strength with CO (Table S2†). Interestingly, P dopant has been proposed as the active site in the metal-free catalyzed CO2RR.37 This consistent result highlights the potential significance of our molecular model study.
 | ||
| Fig. 4 Effects of doping/functional groups on the ortho carbon. (a) Mass spectra for the reactions of CO with mass-selected [CuL]+ with rate constants (k in 10−9 (±30%) cm3 molecule−1 s−1) given in red. (b) Optimized geometry of model clusters and their CO-bound products in the ground state. (c) FMOs of [Cu–P–Py]+. | ||
Compared with P doping, S has a lower electron-donating ability, allowing it to fine tune the charge on Cu without altering the adsorption site like P, as demonstrated in the case of [Cu-MPy]+ wherein MPy refers to the mercaptopyridine ligand (Fig. 4a and b). This, in turn, affects the CO adsorption strength, resulting in a relatively weaker Cu–CO bond as compared to that of only pyridinic N coordinated [Cu-Py]+. From another perspective, it is possible that, via S doping on the adjacent ortho carbon, the retention of *CO intermediate can be regulated, ultimately affecting the selectivity of active sites.38
For phenyl-pyridine (PhPy) ligands (Fig. 4a), the addition of a benzene ring expanded the conjugated system. As compared with Cu–N1 of [Cu-Py]+, this leads to increased electron transfer to the Cu center (0.53 |e| vs. 0.74 |e|) and a narrowed active gap (4.81 eV vs. 5.71 eV), with the consequence of the decreased CO bonding strength and a much higher adsorption rate (Fig. 4a and Table S5†). In addition, the distinctly altered reaction behavior reminds us that it is also necessary to consider the effect of the size scale of the cluster models, which will be discussed in the next part.
Overall, the intrinsic activity of local active sites affected by the introduction of different functional groups at the ortho-carbon position can be identified at the strictly atomic level, which is unlikely probed via conventional methods of SACs. The real coordination environment is actually more complex.39 Different neighboring micro-environments can be easily mimicked by different ligand structures to get universal regularity, therefore we believe our cluster model approach is encouraging to disclose the underlying mystery on M–N–C SACs with atomic resolution.
Effect of the size scale of the cluster models
To further investigate the impact of the model size on the activity of the Cu center, we focused on the cases of pyridinic Cu–N1 and Cu–N2 structures, as they exhibited relatively higher reactivity (Fig. 2a). For the Cu–N1 coordination, we compared pyridine, quinoline (Qu), and acridine (Acr) as ligands, in which a fused benzene ring adjacent to the pyridine ring is progressively extended (Fig. 2a, 3a and 5a, b). Remarkably, albeit with the same pyridinic N, the larger the conjugated system expanded, the higher the adsorption rate observed experimentally. | ||
| Fig. 5 Effect of the size scale of the cluster models. (a) Mass spectra for the reaction of mass-selected [Cu-Qu]+ and [Cu-phen]+ with CO. The rate constants (k in 10−9 (±30%) cm3 molecule−1 s−1) are given in red. (b) and (d) Optimized geometry of model clusters and their CO-bound products in the ground state. (c) Plots of charge on Cu in [CuL]+ with ΔH, as well as the active gap for [CuL]+ with experimental reaction rate, on different models. (e) Plots of charge on Cu in [CuL]+ as well as the active gaps on different models shown in (d). | ||
FMO analysis provided the reason behind this phenomenon: as the conjugated system enlarges, the active gap narrows, resulting in the enhanced reactivity. Simultaneously, the weakened CO-adsorption strength correlated with the increased electron-donating ability of the ligands, due to the addition of adjacent benzene rings (Fig. 5c). We further computationally investigated the effect of expanding the π-conjugated system in the meta and para directions of pyridine N (Table S10†). The calculations revealed that the charge on Cu and the active gap remained almost identical to those observed with single-ring pyridine N, indicative of similar reactivity patterns, suggesting it is the coordination of the ortho carbon adjacent to the nitrogen that influences the reactivity. Tracing back to the defective structures of carbon support, this may provide new insights into understanding the structure–activity relationship of Cu–N coordination at the zig-zag edges in Cu–N–C systems.
Next, we investigated the cluster size effect on Cu–N2 coordination for CO adsorption. We started with bipyridine (bpy) and compared the adsorption behaviors with phenanthroline (phen) ligand, as well as other theoretical models featuring gradually expanded π-conjugation in different directions (Fig. 2a and 5a, d). Interestingly, in those Cu–N2 coordination structures, which are similar to the armchair edge in graphene, the addition of extra benzene rings in different directions does not have a notable influence on CO adsorption behavior, either in the experimentally observed adsorption rates (i.e., [Cu-bpy]+ and [Cu-phen]+) or in the theoretically calculated charge values and active gap (Fig. 5e and Table S11†). Therefore, we infer that increasing the conjugated system at the armchair edges may have little effect on the adsorption reactivity of Cu–N2 active sites. This is also true for the Cu–N1 coordination on the armchair edge, as demonstrated by the corresponding theoretical models (Table S12†). It is quite different with the aforementioned Cu–N1 coordination at the zigzag edge (Fig. 5b) possibly due to the varied atomic stacking order. This fundamental result is intriguing as it indicates that the charge state of Cu atoms can remain stable on the armchair edge even when the π-conjugated system is extended. This stability is crucial for the design and optimization of the performance of Cu–Nx (x = 1, 2) structures analogous to armchair edges.
Implications for condensed phase catalysis
Note that the ligands we used herein are the common nitrogen-containing precursors for preparing supported SACs in the condensed phase, which, to some extent, can well represent the local geometry of M–N–C SACs.40 Therefore, our approach, from a novel cluster model perspective, may provide an atomic-level mechanistic elaboration for the experimental phenomena reported previously. Taking CO2/CORR as examples, the binding energy of *CO is a crucial descriptor allowing control of the desired pathway. Modifying the *CO adsorption on Cu has been reported to enable the optimization of the reaction kinetics.41 In contrast to other M–N4 configurations, which generally produced various C1 products (CO, CH4, CH3OH),42 the kinetics of the CO2RR on Cu–N4 are relatively slow with a weak adsorption capacity to CO.43 This thus led to the low CO2RR activity with CO as the main product.11 Our study provided a viable explanation for this behavior: firstly, due to the mismatch of orbital symmetry, the bond formation between the Cu–N4 center and CO was kinetically sluggish, which suppressed the accumulation of CO on the catalyst surface for C–C coupling. Secondly, the low charge state of Cu resulted in a weak binding strength of Cu–CO, favoring the desorption of CO rather than further hydrogenation.For the low-coordinated Cu–N2 and Cu–N1 systems, the relatively strong CO-binding affinity of the coordination structures can effectively restrain the desorption of *CO during the reaction.44 With sufficient active hydrogen provided, these systems can further hydrogenate CO to CH4.34 In addition, a high *CO concentration on the catalyst surface facilitates the production of C2+ products.33 In this study, the low coordination endows Cu with a higher charge state and an appropriate active gap, effectively regulating the adsorption rate, allowing the active center to rapidly enrich and stably adsorb *CO. This provides direct evidence at the atomic level for the aforementioned mechanism (Scheme 1).
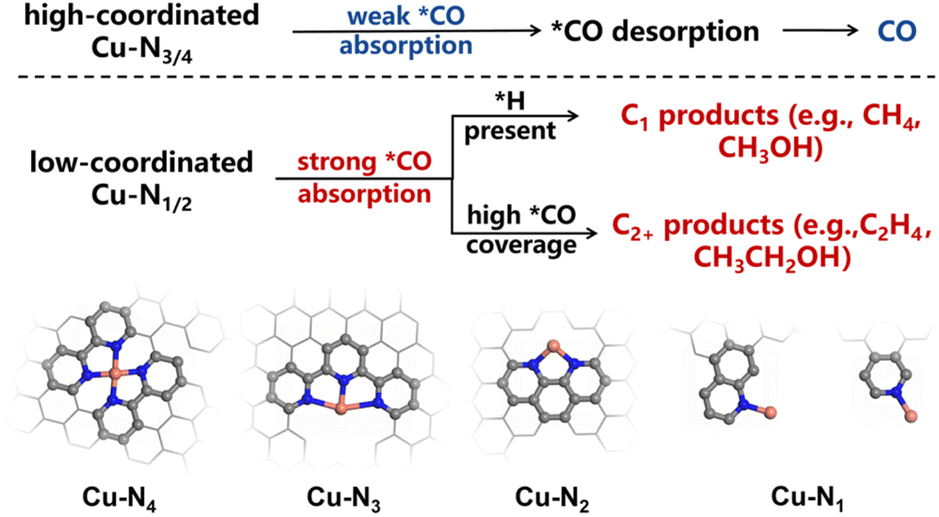 | ||
| Scheme 1 Comparison of the catalytic mechanisms of *CO adsorption at Cu–Nx (x = 1–4) centers. | ||
Beyond giving a comprehensive understanding of the reaction mechanism, our ultimate goal is to leverage this method to offer practical guidance for rationally designing and screening more effective catalysts. Apart from CO2/CORR, in the F–T synthesis, CO dissociation is typically identified as the rate-determining step.45 Thus, stable adsorption is a prerequisite for the further activation of CO. Our joint experimental and theoretical findings suggest a general rule for controlling *CO adsorption via tuning the coordination of the metal center at the atomic level, which could possibly provide design criteria for improved F–T catalysts.
Generality of the cluster model approach
The design concept of cluster models to simulate the active sites of M–N–C systems, along with their interactions with reactants and reaction intermediates, is versatile and generally applicable. In addition to the CO adsorption, the interactions with other reactants, for example, C2H4 and N2, can also be probed, showing the significant differences in the adsorption activity (Fig. S13†), which could possibly provide valuable insights into the related reactions. Moreover, for different metal centers, we prepared a series of Co(I) complexes and optimized their structures, as detailed in Fig. S14,† including [Co-Py]+, [Co-Py2]+, [Co-bpy]+, [Co-tpy]+, and [Co-bpy2]+. Due to Co(I) having a different electron count (8e) compared to Cu(I) complexes (10e), it demonstrates a distinct preference for CO adsorption numbers. Consequently, a remarkably different reactivity pattern in CO adsorption was observed compared to that of the copper-based complexes (Fig. 6). Intriguingly, [Co-Py]+ demonstrated the ability to adsorb one to three CO molecules. Besides, [Co-bpy]+ also showed CO adsorption activity, with a maximum of two CO molecules, while [Co-Py2]+ exhibited a weaker CO adsorption signal, which contrasted with the case of the Cu complexes. This suggests that the cobalt active center possesses an inherent advantage in promoting C–C coupling, which correlated well with its high selectivity toward long-chain hydrocarbons growing in the F–T synthesis.30 The cluster models might only give some partial information of the condensed phase catalysts. The regularity obtained from the model systems could be very inspiring for both heterogeneous and homogeneous catalytic processes. Overall, this general strategy holds great prospects in precisely revealing the underlying mechanism of SACs, which is currently unattainable by other methods. | ||
| Fig. 6 Generality of the cluster model approach. Mass spectra for the reactions of CO with mass-selected [Co-bpy2]+, [Co-tpy]+, [Co-bpy]+, [Co-Py2]+, and [Co-Py]+ at the reaction time of 30 ms, with optimized geometry of model clusters in their triplet ground state. | ||
Conclusions
We proposed an atomic-level cluster model approach to reveal the intrinsic activity of active centers in M–N–C SACs where CO adsorption served as a probe reaction. Diverse nitrogen-coordination environments can be systematically generated and controlled. Based on experimental and theoretical results, we identified a general trend wherein the charge on Cu and the gap between the low-lying symmetry-matched orbitals act as descriptors for CO adsorption strength and experimental adsorption rate, respectively. Thermodynamically, ligands with stronger electron-donating capabilities can transfer more electrons to the Cu center, which in turn weaken the electronic coupling between CO and Cu, with a decreased adsorption strength. From a kinetic perspective, regulating the active gap of the cluster models via coordination, that is, a narrower gap with a closer energy match between the active orbital on Cu and the FMOs of CO, will enhance bonding efficiency and accelerate CO adsorption. Despite certain differences between the gas-phase models and the actual catalysts in terms of structure and external conditions (such as photoexcitation, thermal excitation, electrocatalysis, etc.), which may lead to a decrease in the reaction energy barrier, the general reaction trends should remain inspiring. In this respect, we envision our approach would provide a universal solution for SACs in other related research fields.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: X. S. Methodology: X. S., Q. W., and D. K. Investigation: Q. W., J. Z., S. L., and L. Z. Supervision: X. S. and Y. D. Writing—original draft: Q. W. and D. L. Writing—review & editing: X. S., Y. D., and Z. Q.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to Professor Helmut Schwarz. We appreciate the financial support of QIBEBT (Grant: QIBEBT S202306), Natural Science Foundation of Shandong Province (ZR2022MA040, ZR2023QB049, ZR2023QB266), National Natural Science Foundation of China (22202217, 22302217, 22175071), and Shandong Energy Institute and Enterprise (Grant No. SEI U202308).Notes and references
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem., 2018, 2, 65–81 Search PubMed.
- J. Hulva, M. Meier, R. Bliem, Z. Jakub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Unraveling CO adsorption on model single-atom catalysts, Science, 2021, 371, 375–379 CrossRef CAS PubMed.
- C. Bisio, F. Carniato and M. Guidotti, The Control of the Coordination Chemistry for the Genesis of Heterogeneous Catalytically Active Sites in Oxidation Reactions, Angew. Chem., Int. Ed., 2022, 61, e202209894 Search PubMed.
- B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, The active site of low-temperature methane hydroxylation in iron-containing zeolites, Nature, 2016, 536, 317–321 CrossRef CAS PubMed.
- Y. Xin, N. Zhang, Q. Li, Z. Zhang, X. Cao, L. Zheng, Y. Zeng and J. A. Anderson, Active Site Identification and Modification of Electronic States by Atomic-Scale Doping To Enhance Oxide Catalyst Innovation, ACS Catal., 2018, 8, 1399–1404 CrossRef CAS.
- Q. Qu, S. Ji, Y. Chen, D. Wang and Y. Li, The atomic-level regulation of single-atom site catalysts for the electrochemical CO2 reduction reaction, Chem. Sci., 2021, 12, 4201–4215 RSC.
- X. Shang, X. Yang, G. Liu, T. Zhang and X. Su, A molecular view of single-atom catalysis toward carbon dioxide conversion, Chem. Sci., 2024, 15, 4631–4708 RSC.
- Y. Wang, X. Cui, L. Peng, L. Li, J. Qiao, H. Huang and J. Shi, Metal-Nitrogen-Carbon Catalysts of Specifically Coordinated Configurations toward Typical Electrochemical Redox Reactions, Adv. Mater., 2021, 33, 2100997 CrossRef CAS PubMed.
- M. Wijesingha and Y. Mo, Computational Study of Electrochemical CO2 Reduction on 2D Graphitic Carbon Nitride Supported Single-Atom (Al and P) Catalysts (SACs), ChemPhysChem, 2024, 26, e202400908 CrossRef PubMed.
- S. Wang, L. Wang, D. Wang and Y. Li, Recent advances of single-atom catalysts in CO2 conversion, Energy Environ. Sci., 2023, 16, 2759–2803 RSC.
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Atomically Defined Undercoordinated Active Sites for Highly Efficient CO2 Electroreduction, Adv. Funct. Mater., 2020, 30, 1907658 Search PubMed.
- S. Chen, M. Xia, X. Zhang, L. Pei, Z. Li, X. Ge, M.-J. Lin, W. Zhang and Z. Xie, Guanosine-derived atomically dispersed Cu-N3-C sites for efficient electroreduction of carbon dioxide, J. Colloid Interface Sci., 2023, 646, 863–871 Search PubMed.
- Y. Xiong, H. Li, C. Liu, L. Zheng, C. Liu, J. O. Wang, S. Liu, Y. Han, L. Gu, J. Qian and D. Wang, Single-Atom Fe Catalysts for Fenton-Like Reactions: Roles of Different N Species, Adv. Mater., 2022, 34, 2110653 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Identification of catalytic sites for oxygen reduction in iron- and nitrogen-doped graphene materials, Nat. Mater., 2015, 14, 937–942 Search PubMed.
- Y. Wu, C. Chen, X. Yan, X. Sun, Q. Zhu, P. Li, Y. Li, S. Liu, J. Ma, Y. Huang and B. Han, Boosting CO2 Electroreduction over a Cadmium Single-Atom Catalyst by Tuning of the Axial Coordination Structure, Angew. Chem., Int. Ed., 2021, 60, 20803–20810 CrossRef CAS PubMed.
- P. Christopher, Single-Atom Catalysts: Are All Sites Created Equal?, ACS Energy Lett., 2019, 4, 2249–2250 Search PubMed.
- C. Jin, L. Cheng, G. Feng, R. Ye, Z.-H. Lu, R. Zhang and X. Yu, Adsorption of Transition-Metal Clusters on Graphene and N-Doped Graphene: A DFT Study, Langmuir, 2022, 38, 3694–3710 CrossRef CAS PubMed.
- Y. Lu, J. Wang, L. Yu, L. Kovarik, X. Zhang, A. S. Hoffman, A. Gallo, S. R. Bare, D. Sokaras, T. Kroll, V. Dagle, H. Xin and A. M. Karim, Identification of the active complex for CO oxidation over single-atom Ir-on-MgAl2O4 catalysts, Nat. Catal., 2019, 2, 149–156 Search PubMed.
- M. J. Huelsey, B. Zhang, Z. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. Wu and N. Yan, In situ spectroscopy-guided engineering of rhodium single-atom catalysts for CO oxidation, Nat. Commun., 2019, 10, 1330 Search PubMed.
- L. Cao, Q. Luo, W. Liu, Y. Lin, X. Liu, Y. Cao, W. Zhang, Y. Wu, J. Yang, T. Yao and S. Wei, Identification of single-atom active sites in carbon-based cobalt catalysts during electrocatalytic hydrogen evolution, Nat. Catal., 2019, 2, 134–141 CrossRef CAS.
- E. L. Muetterties, Molecular metal clusters, Science, 1977, 196, 839–848 CrossRef CAS PubMed.
- H. Schwarz, How and Why Do Cluster Size, Charge State, and Ligands Affect the Course of Metal-Mediated Gas-Phase Activation of Methane?, Isr. J. Chem., 2014, 54, 1413–1431 CrossRef CAS.
- S. M. Lang, T. M. Bernhardt, M. Krstić and V. Bonačić-Koutecký, Inside Cover: The Origin of the Selectivity and Activity of Ruthenium-Cluster Catalysts for Fuel-Cell Feed-Gas Purification: A Gas-Phase Approach, Angew. Chem., Int. Ed., 2014, 53, 5218 CrossRef.
- J.-J. Chen, Y.-Z. Liu, Q.-Y. Liu, X.-N. Li and S.-G. He, Single Ti3+ Ion Catalyzes NO Reduction on Stoichiometric Titanium Oxide Cluster Anions (TiO2)n− (n = 1-11), ACS Catal., 2022, 12, 8768–8775 CrossRef CAS.
- M. Borrome and S. Gronert, Gas-Phase Dehydrogenation of Alkanes: C-H Activation by a Graphene-Supported Nickel Single-Atom Catalyst Model, Angew. Chem., Int. Ed., 2019, 58, 14906–14910 CrossRef CAS PubMed.
- G. Di Liberto, L. A. Cipriano and G. Pacchioni, Role of Dihydride and Dihydrogen Complexes in Hydrogen Evolution Reaction on Single-Atom Catalysts, J. Am. Chem. Soc., 2021, 143, 20431–20441 Search PubMed.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design, Adv. Mater., 2019, 31, 1807166 Search PubMed.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Enhancing CO2 Electroreduction to Methane with a Cobalt Phthalocyanine and Zinc-Nitrogen-Carbon Tandem Catalyst, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- Y. Chen, J. Wei, M. S. Duyar, V. V. Ordomsky, A. Y. Khodakov and J. Liu, Carbon-based catalysts for Fischer-Tropsch synthesis, Chem. Soc. Rev., 2021, 50, 2337–2366 Search PubMed.
- J. Mu, G. Long, X. Song, S. Feng, X. Li, Q. Yuan, B. Li, Z. Jiang, L. Yan and Y. Ding, Engineering the Coordination Environment of Single-Rh-Site with N and S Atoms for Efficient Methanol Carbonylation, Adv. Funct. Mater., 2023, 33, 2305823 Search PubMed.
- M. Yamashita and J. B. Fenn, Electrospray Ion-source-another variation on the free-jet theme, J. Phys. Chem., 1984, 88, 4451–4459 CrossRef CAS.
- H. Shi, Y. Liang, J. Hou, H. Wang, Z. Jia, J. Wu, F. Song, H. Yang and X. Guo, Boosting Solar-Driven CO2 Conversion to Ethanol via Single-Atom Catalyst with Defected Low-Coordination Cu-N2 Motif, Angew. Chem., Int. Ed., 2024, 63, e202404884 CrossRef CAS PubMed.
- H. Wu, B. Tian, W. Xu, K. K. Abdalla, Y. Kuang, J. Li and X. Sun, Pressure-Dependent CO2 Electroreduction to Methane over Asymmetric Cu-N2 Single-Atom Sites, J. Am. Chem. Soc., 2024, 146, 22266–22275 Search PubMed.
- Y. Ren, Q.-Y. Liu, Y.-X. Zhao, Q. Yang and S.-G. He, C-C Coupling of Methane Mediated by Atomic Gold Cations under Multiple-Collision Conditions, Acta Phys.-Chim. Sin., 2020, 36, 1904026 Search PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- J. Xie, M. A. Ghausi, J. Wang, X. Wang, W. Wang, R. Yang, M. Wu, Q. Zhang and Y. Wang, Low-Energy CO2 Reduction on a Metal-Free Carbon Material, ChemElectroChem, 2020, 7, 2145–2150 Search PubMed.
- S. Lu, Y. Zhang, M. F. Mady, O. Egwu Eleri, W. Mekonnen Tucho, M. Mazur, A. Li, F. Lou, M. Gu and Z. Yu, Sulfur-Decorated Ni−N−C Catalyst for Electrocatalytic CO2 Reduction with Near 100 % CO Selectivity, ChemSusChem, 2022, 15, e202200870 CrossRef CAS PubMed.
- H. M. Nguyen, H. W. T. Morgan, T. Chantarojsiri, T. A. Kerr, J. Y. Yang, A. N. Alexandrova and N. G. Léonard, Charge and Solvent Effects on the Redox Behavior of Vanadyl Salen–Crown Complexes, J. Phys. Chem. A, 2023, 127, 5324–5334 CrossRef CAS PubMed.
- Y. Wang, X. Cui, L. Peng, L. Li, J. Qiao, H. Huang and J. Shi, Metal-Nitrogen-Carbon Catalysts of Specifically Coordinated Configurations toward Typical Electrochemical Redox Reactions, Adv. Mater., 2021, 33, 2100997 CrossRef CAS PubMed.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Design of a Single-Atom Indiumδ+-N4 Interface for Efficient Electroreduction of CO2 to Formate, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 Search PubMed.
- T. Tang, Z. Wang and J. Guan, Electronic Structure Regulation of Single-Site M-N-C Electrocatalysts for Carbon Dioxide Reduction, Acta Phys.-Chim. Sin., 2022, 39, 2208033 Search PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, Boosting CO2 Electroreduction to CH4 via Tuning Neighboring Single-Copper Sites, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai, X. Li, G. G. Gurzadyan, J. Hou, H. Yang and X. Guo, Atomically Dispersed Indium-Copper Dual-Metal Active Sites Promoting C-C Coupling for CO2 Photoreduction to Ethanol, Angew. Chem., Int. Ed., 2022, 61, e202208904 CrossRef CAS PubMed.
- W. Chen, I. A. W. Filot, R. Pestman and E. J. M. Hensen, Mechanism of Cobalt-Catalyzed CO Hydrogenation: 2. Fischer-Tropsch Synthesis, ACS Catal., 2017, 7, 8061–8071 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01982f |
| This journal is © The Royal Society of Chemistry 2025 |