
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational and experimental evidence for Sb(V) metal mediated C–H activation and oxidative functionalization of arenes†
Anjaneyulu Koppaka *a,
Shu-Sen Chena,
Dongdong Yanga,
Artem Marchenkoa,
Sanaz Mohammadzadeh Koumleha,
David J. Michaelisa,
Roy A. Periana*b and
Daniel H. Ess*a
*a,
Shu-Sen Chena,
Dongdong Yanga,
Artem Marchenkoa,
Sanaz Mohammadzadeh Koumleha,
David J. Michaelisa,
Roy A. Periana*b and
Daniel H. Ess*a
aDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, Utah 84604, USA. E-mail: Anjaneyulu.Koppaka@byu.edu; dhe@byu.edu
bThe Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology, University of Florida, Jupiter, Florida 33458, USA. E-mail: rperiana@ufl.edu
First published on 21st May 2025
Abstract
Inorganic SbV complexes are one of the most well-known Lewis acids that typically indirectly activate nonpolar hydrocarbon bonds by acting as Brønsted superacids. Here we have used a combination of quantum-chemical calculations and experiments to demonstrate that inorganic SbV complexes can directly activate aromatic sp2 C–H bonds and induce oxidative functionalization to form aryl esters. C–H activation was first demonstrated by reaction of SbV(TFA)5 (TFA = trifluoroacetate) with toluene at moderate temperatures that resulted in SbV–C bond intermediates (TFA)4SbV(para-tolyl) and (TFA)3SbV(para-tolyl)2. Calculations predicted and then experiments confirmed that at a higher temperature reductive functionalization occurs to generate oxidized aryl ester products. Theory and experiment indicate that due to a Curtin–Hammet type equilibrium the initial para C–H activation regioselectivity changes to mainly ortho and meta selectivity for reductive oxy-functionalization.
Introduction
For many decades inorganic SbV complexes have been a quintessential example of a strong Lewis acid that indirectly activates nonpolar hydrocarbon bonds through protonation as a Brønsted superacid (Fig. 1a).1–3 In contrast to this Lewis acid/superacid reactivity and mechanism, we recently disclosed that SbVX5 (X = trifluoroacetate) can react with alkanes to generate alkyl esters without generating additional products that would be expected from a superacid/protonation reaction mechanism, and our calculations supported the idea that the SbV metal center can directly react with nonpolar C–H bonds to generate SbV–C bond intermediates through C–H activation (Fig. 1b).4,5 However, there was no experimental detection of the SbV–C bond intermediates to support the calculated C–H activation/metalation and SbV–C bond reductive oxy-functionalization reaction steps. Therefore, in this work we have used both calculations and experiments to examine the reaction of inorganic SbV complexes with arenes that support the proposal that a SbV metal center can directly activate and oxidize nonpolar C–H bonds (Fig. 1c). | ||
| Fig. 1 (a) Outline of the general mechanism for alkane protonation by inorganic SbV complexes in strong Brønsted acid. The protonated hydrocarbon then generates a carbocation intermediate. (b) Outline of C–H activation and M–R oxy-functionalization reaction steps (R = alkyl or aryl). (c) Brief outline of experiments and calculations reported in this work demonstrating that SbVX5 is capable of chemoselective and regioselective C–H activation and oxy-functionalization reaction steps. X = trifluoroacetate. | ||
In addition to being a vehicle for examining SbV metal mediated C–H functionalization, direct and selective oxidative functionalization of unactivated aromatic sp2 C–H bonds to ester (or alcohol) functional groups is itself an important and surprisingly difficult transformation.6 There are only a few examples where this transformation is promoted by a homogeneous transition metal system. The most prominent examples are Pd catalyzed7–9 and Cu induced arene oxidative functionalization to aryl esters.10–17 Most germane to the work here, Gunnoe reported that Cu acetates can oxidatively functionalize benzene and toluene, and proposed a metal mediated C–H activation and reductive functionalization mechanism.18 As outlined in Fig. 1b, our goal was to determine if p-block main-group SbV complexes can promote arene C–H functionalization through metal mediated C–H activation/metalation and functionalization mechanistic steps. While arene C–H activation and reductive functionalization are established reaction steps for transition metals there is much less evidence for these reaction steps with high-oxidation state p-block main-group metals.
Here we report experimental and theoretical studies that show that at moderate temperatures SbVX5 in trifluoroacetic acid (TFAH) solvent induces innersphere metal mediated chemoselective and regioselective para C–H activation of toluene to give [SbV]-tolyl ([SbV] = SbV with four anion ligands of either TFA or aryl) intermediates (see Fig. 1c). Consistent with theoretical predictions, subsequent experiments at higher temperatures generated aryl esters with selectivity switched to ortho-substituted and meta-substituted esters rather than the expected para-substituted esters. Calculations indicate that para to ortho/meta selectivity change occurs because at a higher temperature there is a Curtin–Hammet type equilibrium between bis-tolyl and mono-tolyl SbV intermediates that occurs before rate limiting oxy-functionalization from the more electron deficient mono-tolyl SbV intermediate that is selective for ortho/meta functionalization. Independent synthesis of (TFA)4SbV(para-tolyl) showed the same selectivity in TFAH. Overall, the combination of calculations and experiments demonstrate that SbVX5 is capable of both C–H activation and SbV–C bond oxy-functionalization, and arenes can be directly functionalized to aryl ester products.
Results and discussion
Experimental evaluation of C–H activation and computational prediction and experimental realization of oxy-functionalization
For sixth-row p-block main-group metals, there are a few previous reports of TlIII complexes reacting with arenes to generate TlIII-aryl intermediates,19–23 but these reactions were often proposed to occur through either electron-transfer type reaction steps or electrophilic aromatic substitution with a carbocation/Wheland type intermediate. However, our calculations showed that benzene and mono-substituted arenes likely undergo metalation with TlIII metal centers through a C–H activation mechanism rather than a carbocation or electron transfer mechanism.24 Added nucleophiles to the TlIII-aryl intermediate were generally necessary to result in functionalization. There are also previous reports of PbIV complexes reacting with arenes. Partch reported that inorganic PbIV complexes can oxidize benzene and toluene.25 For toluene it was reported that the benzylic position was oxidized potentially suggesting a radical mechanism, although no byproducts indicative of a radical reaction were observed. Sternhell characterized PbIV–aryl intermediates from reaction of PbIV complexes with benzene derivatives.26,27While the TlIII and PbIV examples outlined above indicate that oxidative functionalization of unactivated arenes is possible with high oxidation state sixth-row p-block metals there is little precedence for a similar transformation with fifth-row metals, specifically with a SbV metal center. Generally, as outlined in the Introduction, SbV complexes operate as a powerful Lewis acid to generate super Brønsted acids and the innersphere of the metal does not directly participate in bond making and breaking reaction steps with hydrocarbons. However, we have recently demonstrated that inorganic SbV complexes in both TFAH and sulfuric acid solvents can induce oxidative functionalization of light alkanes and based on calculations we proposed that the Sb metal center participates in C–H bond activation and functionalization reaction steps.4,5 But again, there was no experimental evidence to support the calculated C–H activation and SbV–C bond reductive oxy-functionalization reaction steps.
As outlined in the Introduction, our goal was to determine if oxidative functionalization of aromatic C–H bonds can be promoted with SbV complexes using C–H activation and functionalization mechanistic steps. We targeted SbV(TFA)5 because we previously used it to functionalize light alkane C–H bonds and it can be straightforwardly generated from reaction of [SbV(OMe)5]2 with trifluoroacetic anhydride (TFAA) and trifluoroacetic acid (TFAH).4 We examined the reaction of SbV(TFA)5 with toluene because this unactivated aromatic compound provides the ability to evaluate regioselectivity/positional C–H functionalization selectivity as well as sp2 (aromatic) versus sp3 (benzylic) C–H functionalization selectivity. The aromatic versus benzylic positional selectivity provides a probe to compare non-radical versus radical type mechanisms where functionalization at the benzylic position is expected in a radical mechanism.18
Our initial experiments focused on the synthesis of SbV(TFA)5 and identifying reaction conditions where we could monitor toluene C–H activation. Under an argon atmosphere 0.076 mmol of [SbV(OMe)5]2 was added to 0.2 mL of trifluoroacetic anhydride (TFAA). The reaction mixture was stirred at room temperature for 5 minutes after which 0.8 mL of TFAH was added and stirred at room temperature for another 15 min. This resulted in a solution of SbV(TFA)5 indicated from the formation of 5 equivalents of methyl trifluoroacetate (MeTFA) generated from the reaction of [SbV(OMe)5]2 with TFAA. Toluene (∼0.5 equivalents) was then added to the Sb(TFA)5/TFAA/TFAH solution and heated at 60 °C for 1 h. Fig. 2, bottom spectrum, shows the 1H NMR of the crude reaction mixture. To our surprise, this relatively mild reaction condition resulted in conversion (∼50%) of toluene to the para C–H activation/metalation product, (TFA)4SbV(para-tolyl). Trace levels of Sb-tolyl products from C–H activation of the ortho or meta toluene C–H bonds may be present but could not be resolved. We confirmed the signals of the mono-tolyl complex by addition of independently synthesized para-toluenestibonic acid ((OH)2OSbV(para-tolyl)) to TFAH solvent which quantitively generated (TFA)4SbV(para-tolyl).28 The ESI† shows time dependent 1H NMR of the crude reaction mixture from room temperature up to 80 °C and from 10 minutes to 7 hours. At room temperature the same mono-tolyl complex was formed but the reaction is much slower.
 | ||
Fig. 2 Overlay of the 1H NMR spectra for the crude reaction mixtures from reaction of SbV(TFA)5 with toluene in TFAH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TFAA (4:1) taken after 1 hour at 60 °C (bottom spectrum) and after 17 hours at 70 °C (top spectrum). The initial product is the mono-tolyl, (TFA)4SbV(para-tolyl), labeled as * in the bottom spectrum. The bis-tolyl product, (TFA)3SbV(para-tolyl)2, is labeled as $ in the top spectrum. :TFAA (4:1) taken after 1 hour at 60 °C (bottom spectrum) and after 17 hours at 70 °C (top spectrum). The initial product is the mono-tolyl, (TFA)4SbV(para-tolyl), labeled as * in the bottom spectrum. The bis-tolyl product, (TFA)3SbV(para-tolyl)2, is labeled as $ in the top spectrum. | ||
Heating for longer periods or increasing the temperature to 70 °C for 17 hours (upper spectrum) led to loss of the mono-tolyl product and conversion to what we propose as the bis-tolyl intermediate, (TFA)3SbV(para-tolyl)2 (Fig. 2). It was surprising to us that the signals for (TFA)3SbV(para-tolyl)2 would be downfield relative to (TFA)4SbV(para-tolyl). Therefore, we used MN15 (ref. 29)/def2-SVP30 density functional theory (DFT) gauge-independent atomic orbital31 NMR calculations in Gaussian 16 (ref. 32) to calculate the relative chemical shifts of the aryl hydrogens. These calculations confirmed the relative signal assignment of the mono-tolyl versus bis-tolyl complex and indicate that the downfield shift of the bis-tolyl structure is due to the adoption of a trans-like geometry. For both the mono-tolyl and bis-tolyl complexes C–H activation/metalation occurred almost exclusively at the toluene para position. This para selective C–H activation of toluene is consistent with the similar reaction between TlIII(TFA)3 and toluene where McKillop reported a 9:4:87 ratio of ortho:meta:para metalation.20
With the observation of both (TFA)4SbV(para-tolyl) and (TFA)3SbV(para-tolyl)2 and the relative time dependence of these products we considered two general mechanisms for the formation of the mono-tolyl and bis-tolyl complex. The first mechanism involves a metalation/C–H activation reaction step to generate the mono-tolyl intermediate followed by a second metalation/C–H activation reaction step to generate the bis-tolyl product. The most straightforward alternative mechanism involves two of the mono-tolyl intermediates undergoing tolyl for TFA group transfer to generate (TFA)3SbV(tolyl)2 and SbV(TFA)5. To confirm this reaction sequence and potentially distinguish between double C–H activation and disproportion pathways, we independently prepared (TFA)4SbV(para-tolyl), through a route using (OH)2SbV(O)(para-tolyl), and examined its reaction and products in the presence and absence of toluene and/or toluene-d8 in TFAH/TFAA (4:1) solvent (see ESI for results†). This showed that starting with (TFA)4SbV(para-tolyl) generated the same bis-tolyl product as that generated from the reaction of SbV(TFA)5 with toluene at extended times or higher temperatures. This generally confirms that the bis-tolyl complex is generated by conversion from the mono-tolyl complex. Also, this reaction indicates that the TFA for tolyl group transfer pathway is likely viable and that formation of the bis-tolyl complex does not require reaction with free toluene. However, this simple interpretation is complicated because heating (TFA)4SbV(p-tolyl) in TFAH generates low concentrations of free toluene and indicates that protonation is slower but competitive with formation of the bis-tolyl complex and therefore it cannot be ruled out that the free toluene would rapidly react with (TFA)4SbV(para-tolyl) to give the bis-tolyl complex through the double C–H activation pathway (see ESI†). While it is conceivable that the double C–H activation and ligand group transfer pathways could be distinguished based on rate dependence of added toluene unfortunately the poor resolution of the resonances of the mono-tolyl versus the possible bis-tolyl complexes in the 1H NMR spectra limited this type of quantitative rate analysis (see ESI†).
As can be seen from Fig. 2, reaction of SbV(TFA)5 with toluene in TFAH/TFAA at 60 °C and 70 °C generated [SbV]-tolyl products but did not generate oxy-functionalized TFA ester cresol products. In retrospect this was perhaps not surprising since the calculated barriers for toluene C–H activation are relatively low (see later section describing calculations) while the reductive functionalization barriers generating tolyl trifluoroacetate cresol esters are relatively high. This comparison of experiments and calculated barriers prompted us to examine whether SbV(TFA)5 could indeed be coaxed to induce oxy-functionalization of toluene at higher temperatures. Fig. 3 shows the 1H NMR spectrum of the crude reaction mixture of SbV(TFA)5 reaction with toluene in TFAH at 170 °C for 3 hours. Consistent with the computational predictions, the 1H NMR shows the formation of the TFA esters of cresol but with some decomposition to unidentified products. But not predicted, and relatively surprising, by comparison to independently synthesized cresol TFA esters, the major oxy-functionalized products are primarily ortho and meta cresol trifluoroacetates with only traces of the para cresol trifluoroacetate. The formation of ortho and meta cresol trifluoroacetates is surprising because we initially assumed oxy-functionalization would occur from either the para mono-tolyl or bis-tolyl complexes. To determine if the formation of ortho and meta cresol trifluoroacetates resulted from first the formation of the para cresol trifluoroacetate we examined whether this compound could be converted to ortho or meta products under reaction conditions. However, control reactions with independently synthesized para cresol trifluoroacetate showed no such conversion (see ESI†).
 | ||
| Fig. 3 Overlay of 1H NMR spectra of crude reaction mixture of SbV(TFA)5 and toluene in TFAH:TFAA (4:1) taken after 3 hours at 170 °C and neat cresols dissolved in TFAH:TFAA (4:1). # = Unidentified products. ♦ = Cresol esters. | ||
The experimental observation of (TFA)4SbV(para-tolyl) and (TFA)3SbV(para-tolyl)2 from reaction of toluene with SbV(TFA)5 at lower temperatures as well the tolyl trifluoroacetate products at higher temperatures are important for several reasons. First, in our previous examination of SbV(TFA)5 reaction with methane and ethane we did not observe a [SbV]–alkyl intermediate (analogous to the Sb-tolyl intermediate here) presumably because the functionalization reaction step was significantly faster than the C–H activation reaction step. Rather, [SbV]–alkyl intermediates were only predicted based on DFT calculations of the reaction mechanism. These calculations suggested that for reactions with alkanes the C–H activation first step (other mechanisms were computationally ruled out) is rate limiting and there is a subsequent relatively fast reductive functionalization step. However, here observation of the SbV-tolyl bond indicates that C–H activation is a viable mechanistic step for a SbV metal center,33 which contrasts the general view of this metal and oxidation state as being primarily a Lewis acid that generates superacids and is not directly involved with C–H bond making and breaking reaction steps. Also, observation of a SbV–aryl bond rather than a SbV–benzyl bond suggests a non-radical type of mechanism for C–H activation. Second, while transition metal mediated arene C–H activation is well known, the subsequent reductive functionalization to generate C–O bonds is relatively rare, with Pd and Cu being the most prominent examples. Therefore, the observation of tolyl trifluoroacetate products demonstrates a new metal and oxidation state for this type of arene oxy-functionalization reaction, and this can be compared to previous work where TlIII was reported to be capable of arene C–H activation, but functionalized organic products were generally not reported without added nucleophiles. This new reaction can also be compared to work showing that PbIV can also promote oxidative functionalization of arenes,25–27 but has sometimes been proposed to occur through a radical, non-C–H activation mechanism. Third, as mentioned in the previous paragraph, in this reaction the mono-tolyl and bis-tolyl complexes show very high para selectivity, which is not surprising. However, it is surprising that subsequent functionalization generated only ortho and meta cresol trifluoroacetates.
Computational evaluation of C–H functionalization pathways
While the experiments established (TFA)4SbV(para-tolyl) and (TFA)3SbV(para-tolyl)2 as intermediates in route to oxy-functionalization of toluene, it was not clear from experiments the exact mechanism and selectivity of forming these intermediates and their subsequent functionalization. Therefore, we decided to use DFT calculations to determine the energies and structures for reaction mechanisms leading to (TFA)4SbV(tolyl), and the metalation regioselectivity. Also, calculations were used to examine the reaction mechanisms and selectivity for reductive functionalization to determine if functionalization occurs from the mono-tolyl complex or the bis-tolyl complex.Our previous computational evaluation4 of SbV(TFA)5 reaction with ethane established that the likely ground state of this complex involves an equilibrium with solvent coordination to the SbV center to give (TFAH)SbV(TFA)5. Our previous work also examined possible multinuclear ground states and in general while multi-Sb ground states are likely formed they do not change the mechanistic or reactivity model for inorganic SbV TFA complexes.5 Here we used MN15/def2-SVP to optimize all structures in Gaussian 16.32 Frequency calculations and intrinsic reaction coordinate (IRC) calculations were used to establish potential-energy surface connections. All intermediates have all positive vibrational frequencies while transition-state structures have a single imaginary frequency. MN15/def2-TZVPD single point energies were then performed on the MN15/def2-SVP structures. Final energies reported correspond to MN15/def2-TZVPD//MN15/def2-SVP. We also calculated single point energies with the M06 and PWPB95-D3(BJ) functionals. This showed that while there are a range of energies calculated structures for C–H activation and functionalization intermediates and transition states no qualitative conclusions change based on a different functional. Zero-point, thermal, and Gibbs energy corrections were applied from the MN15/def2-SVP calculations. We used a modified version of the SMD34 continuum solvent method for estimation of trifluoroacetic acid (TFAH) solvent stabilization. This was used during both structure optimizations and single point energy evaluations. Additionally, we also included an explicit TFAH solvent in nearly all calculations. For each structure we used CREST/GFN2-xTB35,36 to perform a comprehensive search to identify Sb-ligand conformations and explicit solvent hydrogen bonding. Generally, the 15 lowest energy xTB structures were then optimized with DFT.
Fig. 4a outlines the general mechanisms examined for toluene C–H bond cleavage and formation of (TFA)4SbV(para-tolyl). Pathway A involves the C–H activation pathway where in one step there is proton transfer to a TFA ligand and formation of the SbV–C bond. A two-step electrophilic substitution pathway could be envisioned to occur by Sb-induced dearomatization to generate a Wheland type intermediate (i.e. a carbocation intermediate with a full Sb–C σ bond) followed proton transfer step. However, similar to our previous calculations with TlIII(TFA)3,24 we could not locate a Wheland type intermediate on the potential energy surface because these types of structures are high in energy. Alternative to C–H activation, pathway B describes the possibility of a sequence of electron transfer (ET) and proton transfer (PT) that gives an aryl and SbIV radical pair that then combine to form the SbV–C bond. The one-step process that combines ET and PT is proton-coupled electron transfer (PCET) or formally hydrogen atom transfer. These one-electron, open-shell reaction pathways are reasonable to consider because Kochi previously reported the observation of radical species when TlIII was combined with arenes.22,23 However, our previous calculations suggested that only relatively activated arenes with more than three alkyl groups or more potent electron donor groups (e.g. OMe group) result in open-shell reaction pathways being more favorable than C–H activation.24 Lastly, pathway D involves Sb-induced hydride abstraction to generate a Sb–H anion and an arene cation.
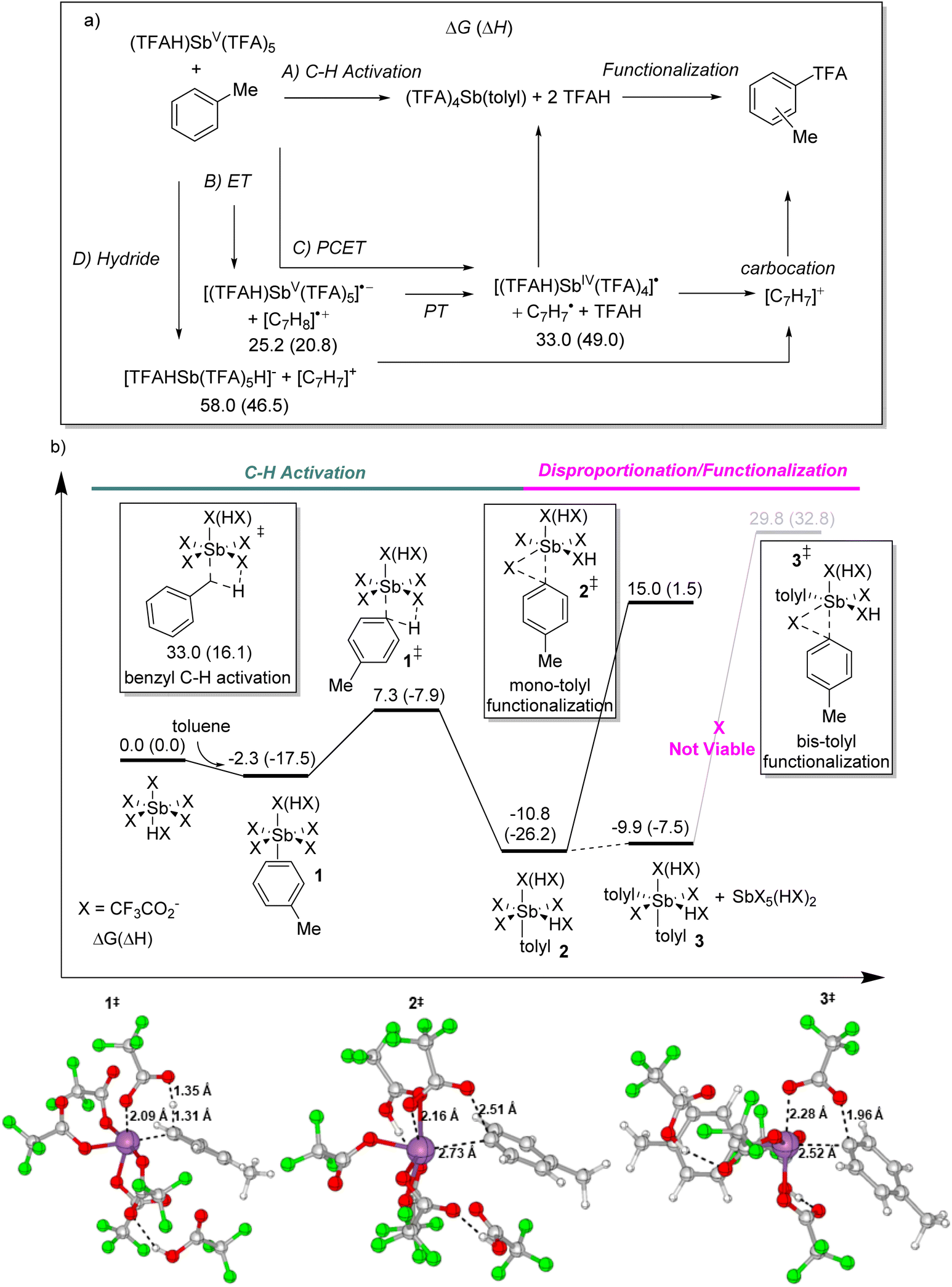 | ||
| Fig. 4 (a) Outline of plausible reaction mechanisms leading to toluene oxy-functionalization promoted by (TFAH)SbV(TFA)5 (energies in kcal mol−1). (b) Relative Gibbs energies and enthalpies (MN15/def2-TZVPD//MN15/def2-SVP; kcal mol−1) for the first and second sp2 aromatic C–H activation reaction steps between (TFAH)SbV(TFA)5 and toluene. Also shown is the benzylic C–H activation transition state and the intramolecular reductive functionalization transition state. | ||
The enthalpy/Gibbs energy change for ET from toluene to (TFAH)SbV(TFA)5 to generate [(TFAH)SbV(TFA)5]˙− and the toluene radical cation is 20.8/25.2 kcal mol−1, which is a relatively low energy process for generating an ion pair and indicates that this SbV is a highly electrophilic species. PCET to generate (TFAH)2SbIV(TFA)4 and the para tolyl radical has a thermodynamic enthalpy/Gibbs energy change of 49.0/33.0 kcal mol−1. While the energies of ET/PT and PCET reaction pathways are not extremely high in energy as they are in the somewhat related reaction with alkanes, they are significantly higher in energy than the C–H activation pathway (see below). As expected, due to the instability of a sp2 carbocation, para hydride abstraction to form [(TFAH)SbV(H)(TFA)5]− and an aryl carbocation is very high in energy at ∼60 kcal mol−1. However, we did locate a one-step reaction pathway where hydride abstraction is coupled with TFA capture of the carbocation and this process requires an activation enthalpy of 46.5 kcal mol−1, but this is still too high to be viable. Compared to these pathways, the C–H activation pathway was calculated to be much lower in energy.
Fig. 4b shows the C–H activation pathway (pathway A) energy landscape for reaction of (TFAH)SbV(TFA)5 with toluene to give the para mono SbV-tolyl intermediate 2. The sequence of reaction steps involves π-coordination of toluene to give 1, which is only slightly exergonic. There is a weak sp2 C–H σ-coordination structure prior to the C–H activation transition state 1‡. In this transition state there is concerted formation of the SbV–aryl bond and proton transfer to the non-coordinating oxygen atom of a TFA ligand (see the bottom of Fig. 4). This C–H activation barrier is surprisingly low with a ΔG‡ value of only 9.6 kcal mol−1 relative to 1. However, this low barrier is consistent with some conversion observed at room temperature. Nearly all density functional methods surveyed gave a low barrier height (e.g. M06). This suggests that the barrier is likely underestimated because we used a mononuclear Sb ground state model. Most important, this C–H activation barrier is much lower in energy than ET/PT or PCET pathways. The [SbV]-tolyl intermediate 2 is exergonic by 10.8 kcal mol−1. We also calculated the analogous C–H activation barrier at the toluene benzyl position. Consistent with the lack of experimental observation of a [SbV]–benzyl intermediate, this sp3 C–H bond position has a much larger barrier with a ΔG‡ value of 33.0 kcal mol−1. Further support for the C–H activation mechanism is the calculated metalation regioselectivity. Transition state 1‡ provided correct modelling of the experimental regioselectivity with para selectivity. For 1‡ the ortho and meta versions are 3.8 and 1.0 kcal mol−1 higher in Gibbs energy than the para transition state.
As previously discussed, after 2 is formed there is the possibility of either a second C–H activation reaction step or ligand exchange to form (TFAH)(TFA)3SbV(tolyl)2 (3). The calculated Gibbs energy for 2 (TFAH)(TFA)4SbV(tolyl) (2) → (TFAH)(TFA)3SbV(tolyl)2 (3) + (TFAH)SbV(TFA)5 is −3.0 kcal mol−1, and this small energy change is consistent with a thermodynamic equilibrium giving both mono-tolyl and bis-tolyl complexes and favoring the latter. Despite significant searching efforts exploring the potential energy surface we could not locate a definitive transition state for this ligand exchange process and therefore cannot easily estimate the kinetic barrier for disproportionation. In contrast, we did locate the second C–H activation transition state between 2 and toluene to give 3. The barrier for this reaction step is 12.1 kcal mol−1. This is not a large barrier and could be accessible at room temperature and therefore unfortunately the calculations cannot definitively determine whether the mono-tolyl to bis-tolyl conversion occurs through the double C–H activation pathway or the disproportionation pathway. Regardless of the kinetic pathway for ligand exchange, the calculated thermodynamics accurately model the mono-tolyl versus bis-tolyl thermodynamic equilibrium.
Fig. 4 shows that the functionalization transition state 3‡ from the bis-tolyl involves a formal reductive elimination generating SbIII and the aryl ester through an intramolecular substitution type mechanism where the TFA coordinated to the Sb metal center nucleophilically displaces the Sb metal center. The barrier for 3‡ is >45 kcal mol−1, which while potentially feasible seemed too high to see significant conversion even at 170 °C. As a note, we also calculated the aryl–aryl coupling transition state from 2. This transition state is >10 kcal mol−1 higher in energy than 3‡ and explains why no C–C bond forming product is found. These large barriers from the bis-tolyl prompted us to examine the reductive functionalization transition state 2‡ from the mono-tolyl complex. This reductive functionalization transition state requires a Gibbs barrier of only 28.0 kcal mol−1 (relative to 2) that is more consistent with the experimental observation of oxy-functionalization at 170 °C. This lower barrier for functionalization of the mono-tolyl intermediate versus the bis-tolyl intermediate is the result of a much larger reduction potential for the SbV to SbIII conversion. Therefore, the lowest energy pathway for functionalization of the bis-tolyl complex is first reversion back to the mono-tolyl complex followed by transition state 2‡.
The potential energy surface shown in Fig. 4b with a relatively large barrier for 2‡ provides an explanation for the ortho/meta functionalization selectivity that is opposite to that of the para C–H activation selectivity and that there was no para-tolyl trifluoroacetate observed. At the lower temperatures (room temperature to 70 °C) the C–H activation transition state 1‡ controls the selectivity, and as described above the calculated barrier for para C–H activation is lower than ortho and meta C–H activation. In contrast, at high temperatures the conversion of 2 back to 1 and toluene through 1‡ will occur at a much faster rate than 2 proceeding through 2‡. This suggests a Curtin–Hammett type selectivity model where at higher temperatures there is rapid equilibration of the mono-tolyl and bis-tolyl complexes (and potentially even a tris-tolyl complex) with SbV(TFA)5 and toluene followed by slow functionalization. Consistent with the selectivity model, calculation of the ortho 2‡, meta 2‡, and para 2‡ shows ortho and para transition states of nearly equal energy and para 2‡ is about 2 kcal mol−1 higher in energy, which is just high enough in energy not to be competitive.
Conclusions
The reaction between SbV(TFA)5 and toluene showed that [SbV] is capable of C–H activation/metalation and leads to NMR detected (TFA)4SbV(para-tolyl) and (TFA)3SbV(para-tolyl)2 intermediates. This importantly confirms our previous hypothesis of a C–H activation mechanism between SbV(TFA)5 and light alkanes where we could not observe this type of intermediate. This toluene reaction was kinetically selective for para sp2 C–H activation over ortho and meta sp2 C–H activation as well as benzylic sp3 C–H activation. Experiments and theory indicate that (TFA)4SbV(tolyl) is formed through a metal mediated C–H activation transition state where the SbV–C bond is formed simultaneous to proton transfer to a TFA ligand. Subsequently (TFA)4SbV(para-tolyl) converts to (TFA)3SbV(para-tolyl)2 through a ligand exchange reaction step, although we cannot rule out sequential double C–H activation reaction steps. However, because the barriers for reductive functionalization are relatively high there is a Curtin–Hammett type scenario where there is rapid equilibrium between the mono-tolyl and bis-tolyl complexes with SbV(TFA)5 and toluene. Because of this equilibrium and the functionalization transition states the regioselectivity changes to ortho/meta selectivity for oxy-functionalization. Overall, the combination of calculations and experiments more firmly establish a new mode of reactivity for [SbV] beyond the normal Lewis acid induced super Brønsted acidity. Specifically, [SbV] is capable of both C–H activation and [SbV]–C bond oxy-functionalization reaction steps. Knowledge of these reaction steps will hopefully propel new reaction discovery and catalysis based on complexes with a SbV metal center and dovetails with efforts by Gabbai and others demonstrating that complexes with a SbV metal center can operate in non-innocent reactivity modes.37,38Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. Ess designed the experimental and computational studies, analyzed data, and wrote the manuscript. A. Koppaka designed experimental studies, executed experimental studies, interpreted data and wrote the manuscript. S. Chen, D. Yang, and S. Koumleh carried out calculations. A. Marchenko assisted in experimental studies. D. Michaelis and R. Periana analyzed and interpreted data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Brigham Young University and the Office of Research computing, especially the Fulton Supercomputing Lab. This work was fully supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Catalysis Science Program, under award # DE-SC0018329.Notes and references
- G. A. Olah, Electrophilic methane conversion, Acc. Chem. Res., 1987, 20, 422–428 CrossRef CAS.
- G. A. Olah and R. H. Schlosberg, Chemistry in super acids. I. Hydrogen exchange and polycondensation of methane and alkanes in FSO3H-SbF5 ("magic acid") solution. Protonation of alkanes and the intermediacy of CH5+ and related hydrocarbon ions. The high chemical reactivity of "paraffins" in ionic solution reactions, J. Am. Chem. Soc., 1968, 90, 2726–2727 CrossRef CAS.
- G. A. Olah, G. K. S. Prakash, Á. Molnár and J. Sommer, Superacid Chemistry, John Wiley & Sons, Inc., Hoboken, New Jersey, 2nd edn, 2009, pp. 511–517 Search PubMed.
- A. Koppaka, S. H. Park, B. G. Hashiguchi, N. J. Gunsalus, C. R. King, M. M. Konnick, D. H. Ess and R. A. Periana, Selective C-H Functionalization of Methane and Ethane by a Molecular Sb(V) Complex, Angew. Chem., Int. Ed., 2019, 58, 2241–2245 CrossRef CAS PubMed.
- S.-S. Chen, A. Koppaka, R. A. Periana and D. H. Ess, Theory and Experiment Demonstrate that Sb(V)-Promoted Methane C-H Activation and Functionalization Outcompetes Superacid Protonolysis in Sulfuric Acid, J. Am. Chem. Soc., 2021, 143, 18242–18250 CrossRef CAS PubMed.
- R. J. Schmidt, Industrial catalytic processes—phenol production, Appl. Catal., A, 2005, 280, 89–103 Search PubMed.
- S. R. Neufeldt and M. S. Sanford, Controlling Site Selectivity in Palladium-Catalyzed C–H Bond Functionalization, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- T. Yoneyama and R. H. Crabtree, Pd(II) catalyzed acetoxylation of arenes with iodosyl acetate, J. Mol. Catal. A: Chem., 1996, 108, 35–40 CrossRef CAS.
- Z. Li, Z. Wang, N. Chekshin, S. Qian, J. X. Qiao, P. T. Cheng, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, A Tautomeric Ligand Enables Directed C–H Hydroxylation with Molecular Oxygen, Science, 2021, 372, 1452–1457 Search PubMed.
- W. Wang, F. Luo, S. Zhang and J. Cheng, Copper(II)-Catalyzed Ortho-Acyloxylation of the 2-Arylpyridines sp2 C−H Bonds with Anhydrides, Using O2 as Terminal Oxidant, J. Org. Chem., 2010, 75, 2415–2418 CrossRef CAS PubMed.
- J. Gallardo-Donaire and R. Martin, Cu-Catalyzed Mild C(sp2)–H Functionalization Assisted by Carboxylic Acids en Route to Hydroxylated Arenes, J. Am. Chem. Soc., 2013, 135, 9350–9353 CrossRef CAS PubMed.
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, Divergence between Organometallic and Single-Electron-Transfer Mechanisms in Copper(II)-Mediated Aerobic C–H Oxidation, J. Am. Chem. Soc., 2013, 135, 9797–9804 CrossRef CAS PubMed.
- S.-Z. Sun, M. Shang, H.-L. Wang, H.-X. Lin, H.-X. Dai and J.-Q. Yu, Cu(II)-Mediated C(sp2)–H Hydroxylation, J. Org. Chem., 2015, 80, 8843–8848 CrossRef CAS PubMed.
- B. K. Singh and R. Jana, Ligand-Enabled, Copper-Promoted Regio- and Chemoselective Hydroxylation of Arenes, Aryl Halides, and Aryl Methyl Ethers, J. Org. Chem., 2016, 81, 831–841 Search PubMed.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, Cu(II)-Catalyzed Functionalizations of Aryl C−H Bonds Using O2 as an Oxidant, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed.
- X. Li, Y.-H. Liu, W.-J. Gu, B. Li, F.-J. Chen and B.-F. Shi, Copper-Mediated Hydroxylation of Arenes and Heteroarenes Directed by a Removable Bidentate Auxiliary, Org. Lett., 2014, 16, 3904–3907 CrossRef CAS PubMed.
- M. Shang, Q. Shao, S.-Z. Sun, Y.-Q. Chen, H. Xu, H.-X. Dai and J.-Q. Yu, Identification of monodentate oxazoline as a ligand for copper-promoted ortho-C–H hydroxylation and amination, Chem. Sci., 2017, 8, 1469–1473 RSC.
- F. Kong, S. Chen, J. Chen, C. Liu, W. Zhu, D. A. Dickie, D. H. Ess, S. Zhang and T. B. Gunnoe, Cu(II) Carboxylate Arene C–H Functionalization: Tuning for Non-Radical Pathways, Sci. Adv., 2022, 8, eadd1594 CrossRef CAS PubMed.
- A. McKillop, J. S. Fowler, M. J. Zelesko, J. D. Hunt, E. C. Taylor and G. McGillivray, Thallium in organic synthesis. IX. Facile thallation of aromatic compounds with thallium(III) trifluoroacetate, Tetrahedron Lett., 1969, 10, 2423–2426 CrossRef.
- A. McKillop, J. D. Hunt, M. J. Zelesko, J. S. Fowler, E. C. Taylor, G. McGillivray and F. Kienzle, Thallium in organic synthesis. XXII. Electrophilic aromatic thallation using thallium(III) trifluoroacetate. Simple synthesis of aromatic iodides, J. Am. Chem. Soc., 1971, 93, 4841–4844 Search PubMed.
- E. C. Taylor, F. Kienzle, R. L. Robey, A. McKillop and J. D. Hunt, Thallium in organic synthesis. XXIII. Electrophilic aromatic thallation. Kinetics and applications to orientation control in the synthesis of aromatic iodides, J. Am. Chem. Soc., 1971, 93, 4845–4850 CrossRef CAS.
- I. H. Elson and J. K. Kochi, Thallium(III) in one-electron oxidation of arenes by electron spin resonance, J. Am. Chem. Soc., 1973, 95, 5060–5062 CrossRef CAS.
- W. Lau and J. K. Kochi, Kinetics and mechanism of aromatic thallation. Identification and proof of competing electrophilic and electron-transfer pathways, J. Am. Chem. Soc., 1984, 106, 7100–7112 CrossRef CAS.
- C. R. King, S. J. Gustafson, B. R. Black, S. K. Butler, M. K. Konnick, R. A. Periana, B. M. Hashiguchi and D. H. Ess, Arene C-H Functionalization by p-Block Metal Tl(III) Occurs at the Borderline of C-H Activation and Electron Transfer, Organometallics, 2017, 36, 109–113 CrossRef CAS.
- R. E. J. Partch, Comparative Lead(IV) Chemistry. Reactions of Lead Tetra(trifluoroacetate). I, J. Am. Chem. Soc., 1967, 89, 3662–3663 CrossRef CAS.
- J. R. Kalman, J. T. Pinhey and S. Sternhell, Reactions of Lead Tetrakistrifluoroacetate with Aromatic and Metalloaromatic Compounds. A New Route to Phenols, Tetrahedron Lett., 1972, 18, 5369–5372 CrossRef.
- J. R. Campbell, J. R. Kalman, J. T. Pinhey and S. Sternhell, Trifluoroacetoxylation of Benzene Derivatives with Lead Tetrakistrifluoroacetate (LiTFA) in Trifluoroacetic Acid (TFA), Tetrahedron Lett., 1972, 18, 1763–1766 CrossRef.
- G. O. Doak and H. G. Steinman, The Preparation of Stibonic Acids by the Scheller Reaction, J. Am. Chem. Soc., 1946, 68, 1987–1989 CrossRef CAS PubMed.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, MN15: A Kohn-Sham Global-Hybrid Exchange-Correlation Density Functional with Broad Accuracy for Multi-Reference and Single-Reference Systems and Noncovalent Interactions, Chem. Sci., 2016, 7, 5032–5051 Search PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Ditchfield, Self-consistent perturbation theory of diamagnetism. 1. Gauge-invariant LCAO method for N.M.R. chemical shifts, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko; , R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. R. King, A. Holdaway, G. Durrant, J. Wheeler, L. Suaava, M. M. Konnick, R. A. Periana and D. H. Ess, Supermetal: SbF5-Mediated Methane Oxidation Occurs by C-H Activation and Isobutane Oxidation Occurs by Hydride Transfer, Dalton Trans., 2019, 48, 17029–17036 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- S. Spicher and S. Grimme, Robust Atomistic Modeling of Materials, Organometallic, and Biochemical Systems, Angew. Chem., Int. Ed., 2020, 59, 15665–15673 CrossRef CAS PubMed.
- J. S. Jones and F. P. Gabbai, Coordination- and Redox-Noninnocent Behavior of Ambiphilic Ligands Containing Antimony, Acc. Chem. Res., 2016, 49, 857–867 CrossRef CAS PubMed.
- J. M. Lipschultz, G. Li and A. T. Radosevich, Main Group Redox Catalysis of Organopnictogens: Vertical Periodic Trends and Emerging Opportunities in Group 15, J. Am. Chem. Soc., 2021, 143, 1699–1721 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02048d |
| This journal is © The Royal Society of Chemistry 2025 |