
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBismuth drug eradicates multi-drug resistant Burkholderia cepacia complex via aerobic respiration†
Jingru
Li
 ab,
Haibo
Wang
a,
Peng
Gao
c,
Runming
Wang
a,
Chun-Lung
Chan
a,
Richard
Yi-Tsun Kao
d,
Hongyan
Li
*a and
Hongzhe
Sun
*a
ab,
Haibo
Wang
a,
Peng
Gao
c,
Runming
Wang
a,
Chun-Lung
Chan
a,
Richard
Yi-Tsun Kao
d,
Hongyan
Li
*a and
Hongzhe
Sun
*a
aDepartment of Chemistry, The University of Hong Kong, Hong Kong SAR, P. R. China. E-mail: hsun@hku.hk
bChemistry and Chemical Engineering Guangdong Laboratory, Shantou 515031, P. R. China
cApplied Oral Sciences & Community Dental Care Division, Faculty of Dentistry, The University of Hong Kong, Hong Kong SAR, P. R. China
dDepartment of Microbiology, The University of Hong Kong, Hong Kong SAR, P. R. China
First published on 9th May 2025
Abstract
Burkholderia cepacia complex (Bcc) is a group of Gram-negative opportunistic pathogens highly responsible for chronic pulmonary infection in cystic fibrosis (CF). Current therapies involving double or triple antibiotic combinations can rarely eradicate the pathogen in chronically infected patients owing to its intrinsic resistance to a variety of antibiotics. Herein, we show that a bismuth drug (and related compounds) could inhibit the growth of clinically antibiotic-resistant Bcc strains, with MIC (ca. 25 μg mL−1) comparable to that for Helicobacter pylori, and the combination of a bismuth drug and antibiotics also demonstrated excellent activity against biofilm and persisters of Bcc. Importantly, the in vitro antimicrobial activity of a bismuth drug could be well translated into in vivo evidenced by about 50% survival rates in the Galleria mellonella infection model. Transcriptomics analysis shows the dynamic responses of Bcc to bismuth treatment. Using a homemade metalloproteomic approach, we could identify 26 BiIII-binding proteins (15 cytosolic proteins and 11 membrane proteins). Further mechanistic studies reveal that bismuth drugs initially target the TCA cycle through the binding and inactivation of a series of enzymes including malate dehydrogenase (MDH), malate synthase (AceB), and succinyl coenzyme A synthetase (SCS), then interfere oxidative phosphorylation through binding to terminal oxidases, i.e., CyoC and CydA, to disrupt electron transport chain, eventually, disrupt protein translation and ribosome via binding and down-regulation of key proteins. Our studies highlight the great potential of bismuth drugs and/or compounds to treat multidrug-resistant Bcc infections.
Introduction
Antibiotics are the most commonly used drugs to treat bacterial infections in humans. However, the global rise and spread of antibiotic-resistant bacterial strains and the scarcity of new antibiotics currently in the pipeline have forced scientists to develop new therapeutic strategies.1–5 Among these strategies, metal complexes have received increasing attention in recent years either as direct-killing metallo-antimicrobials6–9 or resistance breakers to combat antimicrobial resistance.10–12 This is largely because metal complexes possess unique multi-target modes of action7,13,14 and also can disrupt antibiotic-resistant biofilms.15,16In recent years, metallodrugs have been noted to serve as anti-cancer, antibacterial agents, and contrast agents for magnetic resonance imaging.17,18 Cisplatin has been known to be used for over half of chemotherapy.18 Bismuth compounds are among the few metallo-antimicrobials that are currently in the clinic for the treatment of stomach ulcers caused by Helicobacter pylori infection.19 The bismuth-based quadruple therapy containing a proton-pump inhibitor (PPI), a bismuth drug (bismuth subsalicylate, BSS or colloidal bismuth subcitrate (CBS)) and two antibiotics (e.g. tetracycline and metronidazole) is a well-established first-line therapy, which shows excellent success rates in the eradication of H. pylori even for antibiotic-resistant strains.20,21 Despite being used for over three decades, bismuth-based antimicrobial drugs are surprisingly still effective towards the pathogen, and there is no bismuth-resistant bacterial strain reported.19,22 In addition to anti-H. pylori activity, bismuth compounds have also been demonstrated to possess anti-Leishmania19,23 and antivirus activities.24–27 The use of bismuth as anti-H. pylori agents are highly empirical, rather than rational design.28 Indeed, recent studies also revealed that bismuth drugs could eradicate an oral bacterium, Porphyromonas gingivalis (P. gingivalis),29 methicillin-resistant Staphylococcus aureus (MRSA), as well as revitalize the efficacy of certain antibiotics against drug-resistant bacteria to fight against antimicrobial resistance (AMR).12,30 Such studies open a new horizon for further exploration of the new medicinal application of bismuth.
Burkholderia cepacia complex (Bcc) is a group of catalase-producing, non-spore-forming-bacilli Gram-negative bacteria consisting of at least 18 different subspecies,31,32 which are collectively regarded as human opportunistic pathogens. Certain subspecies including B. cepacia and B. cenocepacia have been identified to be significant causes of pneumonia in immunocompromised individuals and cystic fibrosis (CF) patients.33 Bcc infections, which are responsible for the premature death of about 5–10% of these patients worldwide,34 are difficult to treat clinically owing to their high-level intrinsic resistance to a variety of antibiotics including most cephalosporins, polymyxins, ticarcillin, fosfomycin and aminoglycosides.35,36 Current therapies involving double or triple antibiotics combinations can rarely eradicate the pathogen in chronically infected patients. Therefore, the development of effective treatment against Bcc infections is urgently needed.
In this study, we aimed to further expand the antimicrobial spectrum of bismuth drugs by screening a panel of bismuth compounds against different bacterial strains other than H. pylori. We found that bismuth compounds exhibit excellent activity against the clinical antibiotic-resistant Bcc strains i.e., B. cenocepacia, B. cepacia and B. multivorans, exhibiting an MIC value of 25 μg mL−1 for CBS—comparable to H. pylori strains. Besides, bismuth drug (CBS) could synergistically kill Bcc with multiple antibiotics, while combinations of CBS and antibiotics showed excellent anti-biofilm and anti-persister activity. By using transcriptomics and our in-house metalloproteomics together with bioinformatics and bioassays, we uncovered the molecular mechanism of action of bismuth compounds against Bcc. We show dynamic killing processes of bismuth compounds, i.e., bismuth compounds initially target the TCA cycle, then oxidative phosphorylation, finally disrupting translation and ribosomes through binding or downregulation of key enzymes. Considering the good in vivo efficacy and well-documented safety profile of bismuth drugs, bismuth drugs hold great potential to be repurposed to treat multidrug-resistant Bcc infections in the clinic.
Results
Bismuth compounds are effective in eradicating B. cepacia
To further expand the antimicrobial spectrum of bismuth compounds/drugs, we performed a primary screening on a panel of bismuth compounds, including 7 synthesized compounds (with chemical structures and characterization shown in Fig. S1†) against different Gram-positive and -negative bacterial strains using agar diffusion test. The inhibitory effect was defined as a zone diameter greater than 6 mm, the larger the inhibitory zone, the higher the antimicrobial activity. Silver nanoparticle (AgNP) was used as a positive control. As shown in Table S1,† all the bismuth compounds had no observable inhibitory effects against tested pathogens including E. coli, Pseudomonas aeruginosa (P. aeruginosa), Acinetobacter baumannii (A. baumannii), Klebsiella pneumonia (K. pneumonia) apart from B. cepacia. B. cepacia was thus selected to further evaluate the antimicrobial efficacy of bismuth compounds.The susceptibility of three Bcc subspecies i.e., B. cenocepacia J2315, B. cepacia 6349, and B. multivorans C1576 to bismuth compounds and antibiotics was examined using standard micro-dilution method. As shown in Table S2,† all three strains showed high-level resistance to tetracycline (TCN), amoxicillin, and metronidazole with MIC values of 32 μg mL−1, >256 μg mL−1 and >256 μg mL−1, respectively, and a certain level of resistance to meropenem (MEM) and chloramphenicol. Notably, bismuth thiolates i.e., [Bi(NAC)3] and [Bi(GSH)3] displayed potent anti-Bcc activity with MICs ranging from 8 to 32 μM, which are comparable to those of the most effective antibiotics e.g. levofloxacin and ciprofloxacin with MIC of 6 and 12 μM respectively. Other bismuth compounds including the bismuth drug CBS also possess anti-Bcc activity though with less potency compared with these two fluoroquinolone antibiotics (Table S2†).
Although CBS did not exhibit the best anti-B. cepacia activity, the susceptibility of Burkholderia strains to CBS (MIC: 25 μg mL−1) is comparable to H. pylori strains according to our previous data (MIC50 value of 20 μg mL−1), and importantly, no neurotoxicity and cardiotoxicity were reported in clinic.37–39 We therefore selected CBS as the drug candidate in this study for further evaluation (Table 1).
| Bismuth complex | MIC (μM) | ||
|---|---|---|---|
| B. cenocepacia J2315 | B. cepacia 6349 | B. multivorans C1576 | |
| a CBS represents colloidal bismuth subcitrate (CBS); Bi(Hino)3 and Bi(GSH)3 represent the complex of one molar equivalent of CBS with three molar equivalents of hinokitiol and glutathione respectively. Other bismuth compounds were synthesized, their chemical structures and solubilities were described in Fig. S1. The cytotoxicity of the compounds above is reported in Table S3. | |||
| CBSa | 62.5 (25 μg mL−1) | 62.5 (25 μg mL−1) | 62.5 (25 μg mL−1) |
| Bi(Hino)3a | 16 | 16 | 16 |
| Bi(NAC)3 | 8 | 8 | 8 |
| Bi(GSH)3a | 16 | 32 | 16 |
| Bi(Tro-NH2)3 | 32 | 64 | 32 |
| Bi(DTT)OTf | 64 | 32 | 16 |
| Bi(p-PySH)3(NO3)3 | 32 | 32 | 32 |
| Bi(TS)3 | 64 | 32 | 64 |
Bismuth compounds potentiate bactericidal antibiotics both in vitro and in cell-infection models and suppress Bcc resistance evolution
Despite metallo-antimicrobials possessing bactericidal or bacteriostatic activity via a multi-targeted mode of action,7,13,14 it is difficult to predict their combined activity with antibiotics. For example, silver and bismuth can potentiate bactericidal antibiotics;7,10 while gallium shows synergistic interactions only with certain antibiotics e.g. colistin and piperacillin/tazobactam, but antagonistic to tobramycin's activity.9 We asked whether bismuth could potentiate bactericidal antibiotics and synergistically kill Bcc. We assessed the combined activity of bismuth (as CBS) with a range of antibiotics from different families including kanamycin (KAN), ampicillin (AMP), tobramycin (TOB), gentamycin (GEN), and tetracycline (TCN) by calculating the log reduction of bacterial contents (CFU) in single-drug treated groups and combination-treated groups. As shown in Fig. 1A, bismuth significantly potentiated antibacterial activities of all antibiotics, especially in combination with tobramycin (TOB), leading to around 5 log reductions in bacterial load compared to single drug-treated groups (treated with 12.5 μg mL−1 CBS or 64 μM TOB), indicating the potential synergism between bismuth and TOB. We further evaluated synergistic effects between CBS and antibiotics by the Bliss model, in which the degree of synergy (S) is applied for quantification.10 As shown in Fig. S2,† the degree of synergy of five antibiotics ranged from 0.524 to 0.835, in which the combination of TOB and CBS exhibited the highest score, indicating these antibiotics all show synergistic effects with bismuth and the synergism between tobramycin and CBS is the greatest, consistent with reduced log CFU data. We therefore selected the combination of TOB and CBS as a showcase and examined its synergy by time-kill assay. We treated B. cepacia 6349 cells with either CBS or TOB alone, or their combination at 0.5× MIC for 24 h and observed significantly enhanced bactericidal effects of the drug combinations in comparison to single components evidenced by more than 1000-fold decrease in CFU (Fig. 1B). | ||
| Fig. 1 Bismuth potentiates antibiotic activity and suppresses the development of high-level resistant B. cepacia. (A) The change in colony-forming units (CFU) mL−1 of B. cepacia in the presence of 0.5× MIC of CBS (12.5 μg mL−1), AMP (100 μg mL−1), GEN (64 μM), TOB (64 μM), TCN (16 μg mL−1), KAN (100 μg mL−1) and combinations of CBS with the respective antibiotics. (B) Time-dependent killing of B. cepacia by the combination of CBS and TOB. (C) A549 cell-associated bacterial load in the presence of the combination of CBS and TOB. (D) Biofilm inhibitory assay of the combination of CBS and TOB by sonication. (E) Time-kill curves for CBS and TOB and their combination against B. cepacia persisters. (F) Resistance acquisition curves during serial passage in the presence of sub-MIC concentrations of meropenem or CBS individually or in combination with CBS and meropenem. | ||
We next examined the antibacterial effect of the combinatorial TOB and CBS in a cell infection model using mammalian cell A549 as a showcase. Before this, we evaluated the potential cytotoxicity of the combination in A549 cells. After 24 h incubation of a high dose of TOB (128 μM) and CBS (100 μg mL−1) individually and bactericidal concentration of the co-therapy (64 μM TOB and 12.5 μg mL−1 CBS), nearly 90% of cells remained viable, which is comparable to the cell viability at the high dose of CBS (Fig. S3†), suggesting no apparent toxicity of the combination therapy in mammalian cells. A549 cells were infected with B. cepacia at a multiplicity (MOI) of 500 for 4 h and then exposed to TOB, CBS, or their combination. We observed a nearly 1000-fold decrease in the cell-associated bacterial loads, i.e., from 107 CFU to 104 CFU, for the combination therapy (Fig. 1C)—indicating that bismuth drugs combined with antibiotics are capable of invading mammalian cells and killing cell-associated bacteria simultaneously.
Bismuth compounds exhibit anti-biofilm activity and also kill persisters
We further evaluated whether a bismuth drug in combination with antibiotics using TOB as a showcase could prevent biofilm formation in B. cepacia 6349. The bacterium was first inoculated into the high-affinity binding plate for 8 h for attachment, followed by the addition of CBS, TOB, or their combination for another 48 h incubation. We observed a significant reduction in the CFU (from 109 to 104) of the bacterium treated with the combination of TOB and CBS at 0.5× MIC (64 μM TOB and 12.5 μg mL−1 CBS), while the CFU dropped from 109 to 106 for CBS or TOB treatment individually, indicating that CBS could work in tandem with tobramycin to inhibit biofilm formation (Fig. 1D). Moreover, we found that the combination therapy showed relatively good biofilm-killing activity by reducing bacterial loads from 1010 CFU to 105 CFU, in comparison, TOB alone failed to kill biofilm-associated bacteria and CBS only exhibited weak anti-biofilm ability (Fig. S4†), indicating that CBS could potentiate the anti-biofilm activity of tobramycin. We next examined the feasibility of combinatorial CBS with antibiotics in killing B. cepacia persister cells. We first isolated persister cells by exposing stationary-phase B. cepacia cells to 5 mg mL−1 ciprofloxacin (5× MIC) for 4 h, then further treated the persister by the bactericidal concentration of TOB and CBS individually or their combination and monitored the growth of persister cells. The time-kill curves showed that the combination therapy consisting of 12.5 μg mL−1 CBS and 64 μM TOB could inhibit the growth of persister cells rapidly in the first 4 h and then gradually eradicated persisters, at 24 h all the persisters were completely eradicated, in contrast, the single components of CBS or TOB inhibited the growth of persister cells to a less extent (Fig. 1E).We also examined resistance development in the absence or presence of CBS by in vitro serial passage assay using meropenem (MER) as a model antibiotic, for which Bcc strains showed less resistance (Table S2†). As shown in Fig. 1F, the MIC of MER was increased by 32-fold for the 20th passaged B. cepacia, compared to only an 8-fold increase in the MIC value of CBS. However, we observed only a 2-fold increase in the MIC of MER combined with 12.5 μg mL−1 CBS against B. cepacia 6349, indicating that bismuth could slow down antibiotic resistance development in Bcc.
Transcriptomics analysis reveals dynamic killing of CBS to B. cepacia
To explore the antimicrobial mechanism of bismuth compounds (as CBS) against Burkholderia, we first analyzed the transcriptome profiles of B. cepecia 6349 upon the treatment of CBS (50 μg mL−1) for 10 min, 30 min, 1 h, 2 h, and 4 h by RNA sequencing (RNA-seq). The growth inhibition curve of B. cepacia (1 × 108 CFU mL−1) treated with 50 μg mL−1 CBS showed a moderate inhibitory effect of CBS compared to the control group (Fig. S5†), demonstrating that good quality RNAs were extracted, and bacteria were not in a severely damaged state. The number of differentially expressed genes (DEGs) was increased significantly with the increase of treatment time and 107 genes were continuously affected by CBS in the whole process of eradication (Fig. 2A and S6†). | ||
| Fig. 2 Transcriptomics analysis of B. cepacia response to CBS treatment. (A) Genes are significantly up- or down-regulated upon treatment with 50 μg mL−1 CBS for 30, 60, 120, and 240 min. (B) Bubble plot of KEGG pathway enrichment analysis of DEGs treated with 50 μg mL−1 CBS for 10, 30, 60, 120, 240 min. “DEG number” indicates the number of DEG induced by CBS in each pathway. “Fold of change” indicates the median fold of change of DEGs in each pathway. (C) Heatmap showing the expression level of key DEGs in the TCA cycle, oxidative phosphorylation, and ribosome and translation. | ||
To further analyze these DEGs, KEGG was applied to reveal the most enriched pathways. As shown in Fig. 2B, five pathways, i.e., ribosome and translation, TCA cycle, oxidative phosphorylation, glyoxylate metabolism and flagellar assembly were most significantly altered upon exposure to CBS (50 μg mL−1). Among these five pathways, TCA cycle, oxidative phosphorylation, ribosome, and translation showed increasing trends in DEGs numbers with time. Besides, the TCA cycle was the only up-regulated pathway in the presence of CBS. At the first 10 min treatment, the number of DEGs in each pathway ranged from 9 to 28 and no DEG was enriched in ribosome and translation, indicating that treatment of bacterium by CBS for 10 min only induced weak response of B. cepacia. With the incubation time increasing, the DEG number and median fold change in glyoxylate metabolism and flagellar assembly did not exhibit drastic change and remained relatively stable, ranging from −1.13 to −1.77 with a DEG number less than 30. On the contrary, the other three pathways exhibited obvious and dynamic changes with time. A significant increase in DEG number was observed in the TCA cycle, reaching the highest fold of change (1.76) at 30 min and disappearing at 2 h treatment, implying that CBS might target the TCA cycle in the initial phase. The drastic change in oxidative phosphorylation occurred in the middle phase (from 1 to 2 h), with DEGs number increasing from 65 to 89 and fold of change from −2.75 to −3.24 respectively, indicating that treatment of bacteria with CBS for 1 to 2 h resulted in oxidative phosphorylation being attacked. In the last phase (from 2 h to 4 h), DEG number and fold of change in ribosome and translation pathway were all significantly greater than those in other pathways and the trend remained increasing, suggesting that CBS perturbed ribosome and translation in the final stage. These results collaboratively suggest that CBS may target the TCA cycle at the initial phase, then oxidative phosphorylation and eventually concentrate on translation and ribosome.
Several key genes of these three pathways were also analyzed. As shown in Fig. 2C, malate synthase A (aceB), dihydrolipoyllysine-residue acetyltransferase (aceF), and 2-oxoglutarate dehydrogenase E1 component (sucA) were significantly up-regulated in the first 2 h CBS treatment, implying that key enzymes encoded by these three genes may be inhibited by CBS. Meanwhile, key DEGs in oxidative phosphorylation were mainly responsible for encoding two enzymes, NADH-quinone oxidoreductase subunit (encoded by nuoJ, nuoH, and nuoE) and ATP synthase (encoded by atpABGDEF). The increasing fold of change in ATP synthase implies that CBS may interfere with energy production in B. cepacia. DEGs in ribosome and translation were related to the assembly of large ribosome subunit 50S and elongation, and the significant down-regulation of these genes was indicative of CBS-mediated disruption of translation and ribosomal structure.
Proteome-wide tracking BiIII-binding proteins in B. cepacia by GE-ICP-MS
To further unveil the molecular mechanism of antimicrobial activity of CBS against Bcc at a proteome level, we next applied our home-made gel electrophoresis. We inductively coupled plasma mass spectrometry (GE-ICP-MS)—allowing metal ions and their associated proteins to be identified simultaneously40—to identify BiIII-binding proteins in Bcc at a proteome-wide scale. By carefully optimizing the conditions of the column gel system, we identified 15 cytosolic and 11 membrane-associated BiIII-binding proteins (Fig. 3A, B, Tables S4 and S5†) respectively. The integrations of the BiIII-binding protein peaks showed that the alkyl hydroperoxide reductase (AhpC) had the highest bismuth content, followed by universal stress protein A (UspA) and single-stranded DNA-binding protein (SSB), indicating that these proteins have relatively higher expression levels or higher binding affinity to bismuth owing to their high ratios of Cys and His, for which bismuth exhibits a higher binding preference.41. | ||
| Fig. 3 Metalloproteomics reveals key biological pathways disrupted by bismuth in B. cepacia. Profiles of BiIII-binding proteins in B. cepacia extracted from cytosol (A) and membrane (B). (C) Protein–protein interaction of identified Bi-binding proteins. Three functional categories are viewed by Cytoscape. (D) Comparison of the functional pathways enriched by bismuth-regulated differently expressed genes and BiIII-binding proteins, as revealed by transcriptomics and metalloproteomics. | ||
The identified BiIII-binding proteins were subjected to the Gene Ontology (GO) analysis with the Database for Annotation, Visualization, and Integrated Discovery (DAVID).42 These binding proteins are mainly involved in oxidative phosphorylation, TCA cycle, ribosome, pyruvate metabolism, and bacterial membrane (Fig. S7†). To further understand the relationships among the identified BiIII-binding proteins, three functional categories are viewed in protein–protein interaction networks (Fig. 3C). The largest numbers of BiIII-binding proteins were found in the network of translation and transcription, followed by TCA cycle and ETC, indicative of multiple pathways being disrupted by bismuth at a protein level.
We next integrated our transcriptomics and metalloproteomics data to evaluate the effects of CBS on B. cepacia. We identified the TCA cycle, oxidative phosphorylation, translation, and ribosome to be the three common enriched pathways, which may contribute to the toxicity of CBS in B. cepacia (Fig. 3D).
Bismuth drug primarily targets enzymes in the TCA cycle
TCA cycle, as the second step of aerobic respiration, is capable of completely oxidizing nutrients, which produces electron donors, e.g. nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) to further feed electron transport chain (ETC), thereby preparing for energy production. Three key enzymes in TCA cycle, i.e., malate dehydrogenase (MDH), malate synthase (AceB) and succinyl-CoA synthase (SCS), were identified to be BiIII-binding protein and the genes related to TCA cycle (e.g. aceB, sucA and aceF) were significantly regulated by CBS from 10 min to 2 h treatment, implying the important role of TCA cycle in the bacterial eradication by CBS at the initial phase (Fig. 3A and 2C). To further evaluate the biological consequence of the binding of bismuth to these enzymes, we measured the enzymatic activities of two enzymes, i.e., MDH, and SCS in the absence or presence of CBS in cells by malate dehydrogenase assay kit and succinyl-CoA synthase assay kit. As shown in Fig. 4A, even a low dose (2 μg mL−1) of CBS could decrease enzymatic activities of MDH and SCS, while sub-inhibitory concentration (20 μg mL−1) of CBS could reduce MDH and SCS activity to around 55% and 75% respectively. Considering AceB catalyzes the conversion of glyoxylate to malate, followed by the further dehydrogenation catalyzed by MDH with the reduction of NAD+ to NADH, the activities of AceB and MDH in bacterial cells were also assessed by NADH level in bacterial cells. As shown in Fig. 4B, even 10 μg mL−1 of CBS led to a decrease in NADH level from 52.5 to 36.9 pmoles per mg, demonstrating the strong inhibitory effect of bismuth on enzymatic activities of AceB. We further studied the in vitro binding of bismuth to the identified BiIII-binding proteins using B. cepacia malate dehydrogenase (denoted as BcMDH) as a showcase. The recombinant BcMDH was overexpressed and purified as described in the ESI (Fig. S8†). The binding of BiIII (as Bi(NTA)) to BcMDH was monitored by UV-vis spectroscopy and the appearance and increase in the absorption band centered at 340 nm were assignable to BiIII–S ligand-to-metal charge transfer transition (LMCT) (Fig. 4C) with the addition of Bi(NTA) indicated the binding of BiIII to the enzyme. The absorption band at 340 nm levelled off at a molar ratio of [Bi(NTA)]/[BcMDH] of 0.5, suggesting that two BcMDH bound to one BiIII (Fig. 4C). Subsequently, the change in enzyme kinetics of BcMDH induced by different BiIII (as CBS) was also examined and the Lineweaver Burk plot is shown in Fig. 4D. With the increase of BiIII concentration from 0 to 40 μg mL−1, the apparent Vmax decreased from 0.370 to 0.196 μM s−1; while Km values were maintained at ∼27.3 μM, indicating that BiIII (as CBS) inhibits BcMDH via a non-competitive mode. We further examined the effect of CBS at different concentrations on the enzymatic activity of MDH and observed significant declines in BcMDH activity upon treatment of 10 μM BcMDH with 1, 2, and 5 molar equivalents of CBS for 2 h. Even at one molar equivalent of CBS, nearly 40% inhibition of enzymatic activity was induced (Fig. 4E), thereby indicating that bismuth can effectively inhibit BcMDH. Given that SCS is responsible for converting succinyl-CoA to succinate, succinate is also an important byproduct generated by the TCA cycle for electron transport.43 We also examined the level of succinate to assess whether bismuth affected the enzymatic activity of SCS upon the binding. We observed a dose-dependent decrease in succinate level and even a low dose of CBS (2 μg mL−1) could induce a nearly 30% succinate decrease, from 35.24 ng mg−1 to 22.75 ng mg−1 (Fig. 4F), indicative of strong inhibition of CBS on SCS. Besides, glucose as the key fuel feeding the TCA cycle, can be considered as an indicator to evaluate the TCA cycle function by calculating its consumption rate.44 As shown in Fig. S9,† the sub-inhibitory concentration of CBS (20 μg mL−1) decreased glucose consumption to a level lower than 1000 (μg mL−1) from 2000 (μg mL−1) after 2 h incubation, underscoring the significant inhibition of bismuth on TCA cycle. | ||
| Fig. 4 Bismuth targets enzymes in the TCA cycle. (A) The decrease in enzymatic activities of MDH and SCS in the presence of CBS in bacterial cells. (B) NADH level of B. cepacia in the presence of different concentrations of CBS. (C) The difference UV-vis spectra of BcMDH upon the addition of different molar equivalents of bismuth (as Bi(NTA)), and the insert shows the increases in absorption at 350 nm upon the addition of different molar equivalents of CBS. The binding affinity (Kd) was calculated by fitting the data. (D) Double reciprocal plot substrate-dependent enzyme kinetics on inhibition of CBS on BcMDH. (E) In vitro enzymatic activity of MDH with different molar equivalents of BiIII (as CBS). (F) The succinate level of B. cepacia in the presence of different concentrations of CBS. | ||
Bismuth targets two key enzymes in ETC to impede energy production
Oxidative phosphorylation is the last step of aerobic respiration and collaborates with the TCA cycle to generate adequate energy to support various reactions and functions in bacterial cells. In this biological process, the electron transport chain (ETC) plays a crucial role, responsible for electron transport through enzyme complexes, i.e., complex I, II, III, and IV.45 Two terminal oxidases in complex IV, cytochrome bo(3) ubiquinol oxidase (CyoC) and cytochrome bd(1) ubiquinol oxidase subunit I (CydA), which are responsible for catalyzing the oxidation of ubiquinol to ubiquinone and producing proton motive force (PMF) respectively, were found to be bound by BiIII (Fig. 3B). We first examined whether the binding of bismuth to these enzymes resulted in the dissipation of PMF by using intracellular pH probe BCECF-AM, which detects the fluorescent signals of B. cepacia cells treated with or without different concentration of CBS. CCCP was used as a positive control since it is capable of transporting protons across biological membranes to dissipate proton gradient partially.46 As shown in Fig. 5A, we observed a dose-dependent decrease in fluorescence signals, while at 40 μg mL−1 CBS, the fluorescence signal decreased to the level of the group treated with 40 μg mL−1 CCCP, indicative of the drastic dissipation of PMF in B. cepacia by CBS. Meanwhile, we also measured membrane potential, which was regarded as the indicator of CyoC for the oxidation of ubiquinol, by potential-sensitive membrane dye DiSC3(5) using 1% SDS as a positive control. We observed a significant increment of fluorescence in the groups treated with CBS at concentrations ranging from 2 μg mL−1 to 20 μg mL−1 for 2 h, confirming that bismuth dissipated membrane potential via depolarizing the membrane (Fig. 5B). Besides, oxygen, as the terminal electron acceptor, can also be considered as an indicator of normal functions of the electron transport chain.47 We next measured the oxygen level of B. cepacia treated with or without CBS by the extracellular oxygen consumption assay (ab197243; Abcam, Cambridge, MA, USA). The higher the fluorescence signal, the less oxygen is in the sample. As shown in Fig. S10,† a low dose of CBS (10 μg mL−1) did not cause a drastic decrease in total oxygen consumption within 24 h, but significantly attenuated the oxygen consumption rate, leading to oxygen depletion saturated at around 18 h, compared to 7.5 h in the control. However, a more drastic decrease in oxygen consumption was seen at 20 μg mL−1 (sub-inhibitory concentration) CBS treatment, further confirming oxygen consumption in B. cepacia was greatly and continuously affected by bismuth drugs, and ETC is impaired. | ||
| Fig. 5 Bismuth impairs energy production through binding to CyoC and CydA. (A) The dissipation of PMF in WT strain. CCCP was used as a positive control. (B) DiSC3(5) membrane potential measurement for B. cepacia WT strain in the presence of different concentrations of CBS. 1% SDS was used as a positive control. (C) Bacterial growth curves of the wild-type and two variant strains (6349-pcyoC and 6349-pcydA represent cyoC-overexpression strain and cydA-overexpression strain respectively) in the presence of 25 μg mL−1 CBS. (D) The dissipation of PMF in the two variant strains by CBS. (E) DiSC3(5) membrane potential measurement in the two variant strains. (F) ATP measurement in B. cepacia WT strain. | ||
Given that CyoC and CydA, two important enzymes in ETC for energy production, were identified as BiIII-binding proteins, we examined whether these enzymes served as targets of CBS. We first generated two B. cepacia 6349 strains, i.e., 6349-pcyoC and 6349-pcydA constructed with plasmid pMLBAD48 with the genes encoding CyoC and CydA overexpressed respectively, and examined the effect of CBS on the growth of these variant B. cepacia strains. As shown in Fig. 5C, two variant strains showed similar growth curves and reached stationary phase 4 h earlier than wild type (WT) strain, indicating that the overexpression of two terminal oxidases enhances bacterial aerobic respiration, thereby accelerating growth rate. After reaching the stationary phase, OD600 values of the three strains were the same, suggesting that the variants only accelerated growth rate but did not change final bacterial content. However, treatment of both 6349-pcyoC and 6349-pcydA strains with an inhibitory concentration of CBS (25 μg mL−1) could only inhibit their growth to a certain extent. In contrast, the growth of the WT strain was completely inhibited, indicating that the binding of bismuth to CyoC and CydA functionally disrupts ETC, accountable for bismuth antimicrobial activity against B. cepacia. In addition, we also examined the membrane potential and PMF, the two important indicators of ETC function, of the variant strains. As shown in Fig. 5D, declines in fluorescence due to dissipated PMF could not be observed in the cyoC-overexpressed strain treated with a sub-inhibitory concentration of CBS (20 μg mL−1) either, while a high dose of CBS (40 μg mL−1) could induce slight fluorescence increase in cydA-overexpressed strain, compared with drastic fluorescence increment in WT strain. Similarly, both sub-inhibitory concentration (20 μg mL−1) and high dose of CBS (50 μg mL−1) failed to induce fluorescence increment, an indicator of altered membrane potential in two variant strains (Fig. 5E). These results collaboratively suggest that the bindings of bismuth to CyoC and CydA result in altered membrane potential and dissipated PMF. As a key component of oxidative phosphorylation, ETC is responsible for transporting electrons to ATP synthase (ATPase) and accumulating PMF.49 In ATPase, PMF is utilized to drive specific conformational change, thereby completing oxidative phosphorylation to generate plenty of ATP.50,51 According to transcriptomics analysis (Fig. S11†), a group of DEGs encoding ATPase, i.e., atpA, atpD, atpG, atpB, atpE, atpF, were significantly down-regulated. The in vitro expression level of these genes was further examined by RT-PCR. As shown in Fig. S12,† the relative units of mRNA of these genes were all lower than 0.5, especially atpA with a relative unit of 0.036, implying the ATP synthesis might be severely inhibited by CBS treatment. We therefore examined ATP production and observed a rapid dose-dependent decline in ATP content upon exposure of B. cepacia cells to CBS, confirming that the ability of bacteria to generate energy is drastically damaged by bismuth drugs (Fig. 5F). In contrast, even the high dose of CBS (40 μg mL−1) only induced minor decreases in ATP levels in the 6349-pcyoC and 6349-pcydA strains (Fig. S13†), by 10.7% and 16.0%, respectively. This result underscores CyoC and CydA as targets of bismuth drugs to disrupt energy production.
Bismuth drug inhibits translation at the later stage of B. cepacia eradication
Ribosome is the organelle responsible for protein synthesis and a main target of antimicrobial agents.52 Using the metalloproteomics approach, we have identified three BiIII-binding proteins, RpoA, RplA, and RpmA (Fig. 3B), which are related to ribosome, indicating that bismuth may also interfere with ribosome function in B. cepacia. Meanwhile, our transcriptomics data revealed that DEGs related to ribosomes occurred from 30 min, and that the number and fold change of DEGs were all altered more greatly with the incubation time. This finding indicates that the regulation of CBS on ribosomes requires time to execute. Moreover, all these genes were significantly down-regulated by CBS, implying that the pathogen may not be able to defend this sort of stress induced by bismuth drugs. About 56 down-regulated ribosome-related genes were seen, which are thought to be responsible for various functions related to ribosome and translation, e.g. tRNA binding, rRNA binding, mRNA binding, structural assemble of ribosome etc.53 We first examined the in vitro expression levels of five genes with largest fold of changes by RT-PCR, i.e., ribosomal protein L27 (rpmA), 50S ribosomal protein L4, L1 (rplD, rplA), DNA-directed RNA polymerase subunit alpha (rpoA) and 30S ribosomal protein S17 (rpsQ) upon exposure of CBS (Fig. S11†). As shown in Fig. 6A, a dose-dependent decrease in the expression levels of all five genes was noted, in particular, the expression level of rpsQ was even decreased to 0.06, strongly indicative of bismuth-induced down-regulation of ribosome genes.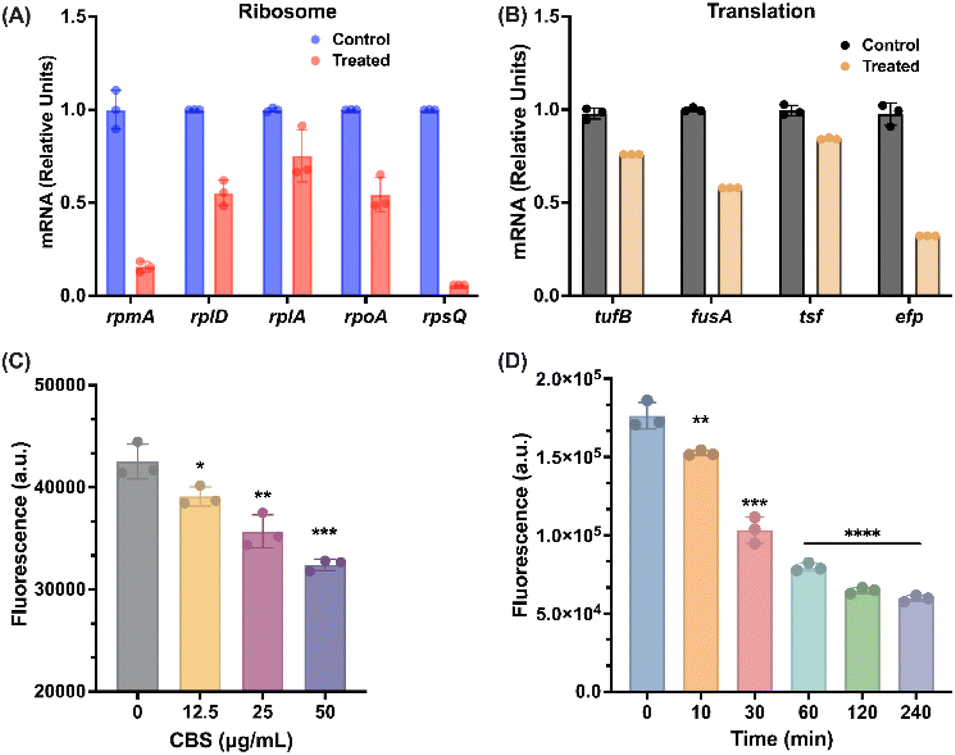 | ||
| Fig. 6 Bismuth down-regulates the ribosome-related genes and inhibits bacterial translation. (A) Nascent mRNA level of genes in ribosome in B. cepacia with or without CBS. (B) Nascent mRNA level of genes in translation in B. cepacia with or without CBS. (C) GFP expression in B. cepacia in the presence of different concentrations of CBS. (D) GFP expression in B. cepacia incubated with CBS for 0, 10, 30, 60, 120, 240 min. | ||
Apart from structural ribosomal proteins, translation is another important pathway that involves DEGs with the large fold of change induced by bismuth. As shown in Fig. S11,† translation elongation factor Tu 2 (tufB), elongation factor G (fusA), and protein chain elongation factor EF-Ts and EF-P (tsf, efp), were significantly down-regulated after 4 h CBS treatment. The down-regulation of these genes by bismuth was also confirmed by in vitro the expression level using RT-PCR (Fig. 6B). We next evaluated the CBS inhibitory effect on B. cepacia promoter of translation genes by using green fluorescent protein (GFP) as a reporter. The plasmid was constructed with the vector pMLS7 as reported previously48 and the promoter evaluated in this study was a constitutive promoter of S7 ribosomal protein, which was also demonstrated to be down-regulated by CBS.48 With the increase in CBS concentration, we observed a dose-dependent decline in fluorescence signals compared to the control group, indicating that bismuth inhibits bacterial translation in a dose-dependent manner (Fig. 6C). Besides when the bacterium was treated with a high dose of CBS (50 μg mL−1), we observed rapid decrease in fluorescence from 0 min to 120 min but only slight changes in the fourth hour of the treatment, further confirming that bismuth-induced translational down-regulation takes time to act (Fig. 6D).
Bismuth shows excellent activity against B. cepacia in vivo
We finally investigated the therapeutic potential of bismuth drugs against Bcc in the Galleria mellonella-infected model. The infection was first induced by injecting 1 × 105 CFU B. cenocepacia J2315 into the larvae of G. mellonella. G. mellonella was then treated with vehicle (PBS), a low dose of CBS (10 mg kg−1), or a high dose of CBS (20 mg kg−1), 1 h post-infection. We observed for 5 days to count the survival rate, and larvae used for bacterial-load counting were collected after 24 h of treatment (Fig. 7A). As shown in Fig. 7B, 50% larvae were rescued by 20 mg kg−1 CBS within 5 days, in comparison, all larvae in the control group died within 24 h. Meanwhile, CBS either at a high dose or a low dose was able to reduce bacterial load within larvae, by ca. 3-fold and 2-fold respectively compared with the control group (Fig. 7C). This indicated the great potential of bismuth drugs in the treatment of Bcc infection in clinic. | ||
| Fig. 7 CBS shows a great therapeutic potential in vivo. (A) Scheme of the experimental protocol of the G. mellonella infection model. (B) Survival rates of G. mellonella larva. Infected larvae (n = 8) were injected with 1 × 105 CFU B. cenocepacia J2315 at the right proleg under the treatment of 10 mg kg−1 or 20 mg kg−1 CBS at the left proleg. (C) Bacterial load in G. mellonella infection model. (D) Survival rates of G. mellonella larvae (n = 8) treated with 10 mg kg−1 CBS individually, 30 mg kg−1 (64 mM kg−1) TOB individually, or the combination of 10 mg kg−1 CBS and 30 mg kg−1 (64 mM kg−1) TOB at left proleg. (E) Bacterial load in G. mellonella infection model. The dosage of CBS and TOB in combination therapy were 10 mg kg−1 and 30 mg kg−1, respectively. The P value of the survival model was a two-sided, log[rank] (Mantel–Cox) test, while the P value was determined by a two-sided, Mann–Whitney U-test. | ||
The in vivo efficacy of the combination therapy was also evaluated by G. mellonella infection. As shown in Fig. 7D, monotherapy of CBS (10 mg kg−1) or low dose of TOB (30 mg kg−1) showed 25% and 40% survival of larvae within 5 days, respectively. While the group treated with combined treatment of 10 mg kg−1 CBS and 30 mg kg−1 TOB simultaneously showed a high survival rate (62.5%). At the same time, only ∼2-fold of reduction in bacterial load within larvae could be observed in mono-drug treated groups (CBS or TOB individually). In contrast, the combination therapy exhibited significantly enhanced antibacterial activity with a 4-fold reduction in bacterial load within larvae (Fig. 7E). This is indicative of the clinical potential of BiIII-containing combination therapy.
Discussion
The emergence and rapid development of multidrug-resistant (MDR) bacteria and the lack of antibiotics with new molecular targets pose a significant challenge to public healthcare systems globally. Alternative strategies are urgently needed to tackle the AMR crisis. Recently, metals and metallodrug-based approaches have received increasing attention either as metallo-antibiotics9,10,17,54 or as adjuvants of antibiotics12,30,55 to combat AMR due to their multiple targeted mode of action, which is less likely to develop resistance. Amongst all the antimicrobial metals, bismuth is one of the few metallodrugs used in clinics to treat H. pylori infection when combined with antibiotics. Very recently, we demonstrated that a combination of bismuth drugs with antibiotics could revitalize the antimicrobial activity of antibiotics against multidrug-resistant P. aeruginosa because bismuth disrupts iron homeostasis to facilitate antibiotics accumulation in P. aeruginosa.30 This study strongly suggests the great potential of bismuth in tackling AMR and its medicinal application could be further expanded.Herein we found that bismuth drugs and compounds exhibit outstanding antimicrobial activity against drug-resistant B. cepacia with an MIC value of 25 μg mL−1 for CBS, comparable to that for H. pylori. CBS also demonstrated synergistic effects with antibiotics from beta-lactam, aminoglycoside, and tetracycline antibiotic family. The combination of CBS and TOB could inhibit biofilm formation, eradicate biofilm-associated bacteria also kill persister cells. Moreover, CBS in combination with antibiotics could slow down the development of high-level resistance, which is possibly attributed to pathogens that are usually less likely to generate mutations to multiple targets simultaneously. Importantly, the in vitro activity could be well translated into in vivo evidenced by about 50% survival rates of G. mellonella larva in the infected model (Fig. 7B).
The molecular mechanism of action of bismuth (using CBS as a showcase) was studied by transcriptomics and GE-ICP-MS-based metalloproteomics. Transcriptomics analysis revealed a dynamic killing process of CBS against B. cepacia. KEGG analysis showed that CBS-induced DEGs were mainly enriched in glyoxylate metabolism, flagellar assembly, TCA cycle, oxidative phosphorylation, and ribosome and translation (Fig. 2B). Among these five pathways, DEGs in glyoxylate metabolism and flagellar assembly remained relatively stable, while those in other three pathways exhibited obvious changes with time. TCA cycle was the only up-regulated pathway in the presence of CBS and only differentially expressed in the first 2 h. Oxidative phosphorylation and ribosome and translation all demonstrated an increasing trend in DEG number, while the former showed the greatest down-regulation from 1 h to 2 h and the dominant role of the latter occurred in the last 2 h (Fig. 2B). By using GE-ICP-MS,40 15 and 11 BiIII-binding proteins were identified from cytosol and membrane respectively (Fig. 3A and B), providing fundamental basis for further studying the molecular mechanism of bismuth against Bcc. By integrating targets from two approaches, the TCA cycle, oxidative phosphorylation, and ribosome and translation were considered to be the most important three pathways in the eradication of B. cepacia by CBS. These results collaboratively suggest that CBS may initially target the TCA cycle, then oxidative phosphorylation and eventually concentrate on ribosome and translation.
As the initial target of CBS, several enzymes, i.e. MDH, SCS, and AceB, were identified to be BiIII-binding proteins, while DEGs e.g. sucA, aceB, and aceF, were up-regulated by CBS for the first 2 h (Fig. 3A and 2C). The inhibition of these three enzymes by bismuth drugs resulted in a decrease in the level of two-electron donor of ETC, i.e. NADH and succinate, while the non-competitive binding of BiIII to BcMDH monitored by UV-vis spectrometry showed that two BcMDH bound to one bismuth with a Kd of 1.80 × 10−22 M (Fig. 4C and E), implying that the relatively strong interaction between bismuth and enzymes, leading to dysfunction of TCA cycle, which produced reducing equivalents, e.g. NADH, to feed ETC.56 In addition, membrane proteins including outer membrane protein A (OmpA) and outer membrane protein assembly factor D (BamD), were identified to be bound by bismuth by metalloproteomics and outer membrane protein W (ompW) was significantly down-regulated by 4 h treatment of CBS, implying that CBS may also disrupt membrane integrity (Fig. 3B and S11†), leading to increased membrane permeability, which may also contribute to membrane potential alteration (Fig. S14†).
With the increase in incubation time to 1 h, CBS began to target oxidative phosphorylation. Two terminal oxidases, i.e., CyoC and CydA, were identified as BiIII-binding proteins, such bindings resulted in altered membrane potential and dissipated PMF (Fig. 5A and B), indicative of disruption of electron transport by CBS. Meanwhile, transcriptomics exhibited significant BiIII-induced down-regulations on ATPase, implying BiIII-induced interferences on energy production (Fig. S11†). The altered membrane potential coupled with dissipated PMF contributed to the decreased ATP level (Fig. 5A, B, and F), while the impeded energy production was rescued in cyoC- and cydA-overexpressed strains (Fig. S13†), indicating that bindings of BiIII to these two enzymes disrupted energy production in B. cepacia.
In the last phase (2–4 h) of exposure of B. cepacia to CBS, B. cepacia with deprived energy supply exhibited more down-regulated genes in ribosome and translation with median fold of change of −3.73 (Fig. 2C). BiIII-induced dose-dependent and time-dependent declines in fluorescence of GFP and significant down-regulated of mRNA level of key DEGs in this pathway indicate that bismuth inhibits the transcription of ribosomal protein and bacterial translation (Fig. 6A–D), which is attributed to accumulated energy loss in previous two phases.
Taken together, bismuth primarily targeted the TCA cycle, the second step of aerobic respiration, via binding to key enzymes, thereby resulting in inhibition of the production of electron carrier (NADH) and key metabolite (succinate). Subsequently, bismuth bound to two terminal oxidases, i.e., CyoC and CydA, leading to altered membrane potential and dissipated PMF, thereby hindering oxidative phosphorylation of Bcc, which is the last step of aerobic respiration.
The inhibition of bismuth on the second and third steps of aerobic respiration greatly interfered with oxygen consumption and abolished energy production in B. cepacia. Finally, bismuth bound to and down-regulated several ribosome-related proteins, resulting in energy deprivation, and bacterial translation is greatly inhibited (Scheme 1).
 | ||
| Scheme 1 Proposed major biological pathways in B. cepacia disrupted by bismuth drugs. Bismuth binds to MDH and SCS, resulting in TCA cycle dysfunction, including inhibiting the generation of electron carriers (NADH) and decreasing succinate levels. In ETC, less electron carrier and down-regulated NADH-quinone oxidoreductase affected electron transport in complex I, while decreased succinate level induced inactivation of complex II. When electrons were transported to complex IV, two terminal oxidases, i.e., Cyo and Cyd were inactivated by bismuth, leading to altered membrane potential and dissipated PMF. The whole process of electron transport completes at complex IV and chemiosmosis prepared for oxidative phosphorylation is impaired, while the expression of complex V (ATPase) was also down-regulated by bismuth, thus, energy production in B. cepacia is greatly suppressed by bismuth drugs, thereby resulting in bacterial fundamental function to be abolished. | ||
Conclusions
We show that bismuth drugs and compounds possessed excellent antimicrobial activity against B. cepacia including inhibition of biofilm formation and eradication of planktonic bacterial cells and persisters. We also demonstrated excellent in vivo antimicrobial activity against Bcc. Our multi-omics and bioassays reveal that bismuth-induced impaired metabolic pathways, i.e., TCA cycle, oxidative phosphorylation, and translation through binding to key enzymes and regulating key genes in these pathways. Specifically, bismuth significantly impaired functions of ETC, thereby depriving energy supply for bacterial survival. Further studies on the identified targets may bring more perspectives and understandings on the antimicrobial mechanism of action of bismuth drugs, thereby better promoting the clinical applications of bismuth drugs in the fight against AMR. Metallo-agents may represent a new arsenal for our fight against antimicrobial resistance, the silence pandemic.17,54Data availability
All experimental data and procedures are provided in the ESI.† Other data are available from the corresponding author upon reasonable request.Author contributions
J. L., P. G., H. L., and H. S. designed research; J. L., H. W., R. W., C. -L. C., and R. Y. K. performed research, and J. L., H. L., and H. S. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Research Grants Council of Hong Kong (17304323, SRFS2122-7S04, 17306323, and R7080-18), Hong Kong SAR, and the University of Hong Kong (URC and Norman & Cecilia Yip Foundation).References
- K. Lewis, Nature, 2012, 485, 439–440 CrossRef CAS PubMed.
- M. Lakemeyer, W. Zhao, F. A. Mandl, P. Hammann and S. A. Sieber, Angew Chem. Int. Ed. Engl., 2018, 57, 14440–14475 CrossRef CAS PubMed.
- M. Claudel, J. V. Schwarte and K. M. Fromm, Chemistry, 2020, 2, 51 CrossRef.
- A. Frei, Antibiotics, 2020, 9, 90 CrossRef CAS PubMed.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS PubMed.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627–2639 RSC.
- H. B. Wang, M. J. Wang, X. H. Xu, P. Gao, Z. L. Xu, Q. Zhang, H. Y. Li, A. X. Yan, R. Y. T. Kao and H. Z. Sun, Nat. Commun., 2021, 12, 3331 CrossRef CAS PubMed.
- Y. C. Wang, B. J. Han, Y. X. Xie, H. B. Wang, R. M. Wang, W. Xia, H. Y. Li and H. Z. Sun, Chem. Sci., 2019, 10, 6099–6106 RSC.
- C. H. Goss, Y. Kaneko, L. Khuu, G. D. Anderson, S. Ravishankar, M. L. Aitken, N. Lechtzin, G. Zhou, D. M. Czyz, K. McLean, O. Olakanmi, H. A. Shuman, M. Teresi, E. Wilhelm, E. Caldwell, S. J. Salipante, D. B. Hornick, R. J. Siehnel, L. Becker, B. E. Britigan and P. K. Singh, Sci. Transl. Med., 2018, 10, 6099–6101 Search PubMed.
- J. R. Morones-Ramirez, J. A. Winkler, C. S. Spina and J. J. Collins, Sci. Transl. Med., 2013, 5, 190ra181 Search PubMed.
- H. Z. Sun, Q. Zhang, R. M. Wang, H. B. Wang, Y. T. Wong, M. J. Wang, Q. Hao, A. X. Yan, R. Y. T. Kao, P. L. Ho and H. Y. Li, Nat. Commun., 2020, 11, 5263 CrossRef CAS PubMed.
- R. Wang, T. P. Lai, P. Gao, H. Zhang, P. L. Ho, P. C. Woo, G. Ma, R. Y. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439 CrossRef PubMed.
- H. Y. Li, R. M. Wang and H. Z. Sun, Acc. Chem. Res., 2019, 52, 216–227 CrossRef CAS PubMed.
- H. Wang, A. Yan, Z. Liu, X. Yang, Z. Xu, Y. Wang, R. Wang, M. Koohi-Moghadam, L. Hu, W. Xia, H. Tang, Y. Wang, H. Li and H. Sun, PLoS Biol., 2019, 17, e3000292 CrossRef PubMed.
- C. F. Garcia, M. Kretschmer, C. N. Lozano-Andrade, M. Schonleitner, A. Dragos, A. T. Kovacs and O. Lieleg, npj Biofilms Microbiomes, 2020, 6, 1 CrossRef PubMed.
- E. Banin, A. Lozinski, K. M. Brady, E. Berenshtein, P. W. Butterfield, M. Moshe, M. Chevion, E. P. Greenberg and E. Banin, Proc. Natl. Acad. Sci. USA, 2008, 105, 16761–16766 CrossRef CAS PubMed.
- C. Wang, X. Wei, L. Zhong, C. L. Chan, H. Li and H. Sun, J. Am. Chem. Soc., 2025, 147, 12361–12380 CrossRef CAS PubMed.
- K. J. Franz and N. Metzler-Nolte, Chem. Rev., 2019, 119, 727–729 CrossRef CAS PubMed.
- D. M. Griffith, H. Li, M. V. Werrett, P. C. Andrews and H. Sun, Chem. Soc. Rev., 2021, 50, 12037–12069 RSC.
- F. Megraud, Thera. Adv. Gastroenter., 2012, 5, 103–109 CrossRef CAS PubMed.
- D. Y. Graham and L. Fischbach, Gut, 2010, 59, 1143–1153 CrossRef CAS PubMed.
- M. M. Gerrits, A. H. van Vliet, E. J. Kuipers and J. G. Kusters, Lancet Infect. Dis., 2006, 6, 699–709 CrossRef CAS PubMed.
- P. C. Andrews, R. Frank, P. C. Junk, L. Kedzierski, I. Kumar and J. G. MacLellan, J. Inorg. Biochem., 2011, 105, 454–461 CrossRef CAS PubMed.
- S. F. Yuan, R. M. Wang, J. F. W. Chan, A. J. X. Zhang, T. F. Cheng, K. K. H. Chik, Z. W. Ye, S. Y. Wang, A. C. Y. Lee, L. J. Jin, H. Y. Li, D. Y. Jin, K. Y. Yuen and H. Z. Sun, Nat. Microbiol., 2020, 5, 1439–1448 CrossRef CAS PubMed.
- N. Yang, J. A. Tanner, B. J. Zheng, R. M. Watt, M. L. He, L. Y. Lu, J. Q. Jiang, K. T. Shum, Y. P. Lin, K. L. Wong, M. C. M. Lin, H. F. Kung, H. Z. Sun and J. D. Huang, Angew. Chem., Int. Ed., 2007, 46, 6464–6468 CrossRef CAS PubMed.
- N. Yang, J. A. Tanner, Z. Wang, J. D. Huang, B. J. Zheng, N. Y. Zhu and H. Z. Sun, Chem. Commun., 2007, 4413–4415 RSC.
- J. Chen, Y. Zhou, X. Wei, X. Xu, Z. Qin, C. P. Ong, Z. W. Ye, D. Y. Jin, B. Boitrel, S. Yuan, J. F. Chan, H. Li and H. Sun, ACS Infect. Dis., 2024, 10, 858–869 CrossRef CAS PubMed.
- B. J. Marshall, C. S. Goodwin, J. R. Warren, R. Murray, E. D. Blincow, S. J. Blackbourn, M. Phillips, T. E. Waters and C. R. Sanderson, Lancet, 1988, 2, 1437–1442 CrossRef CAS PubMed.
- T. Cheng, Y. T. Lai, C. Wang, Y. Wang, N. Jiang, H. Li, H. Sun and L. Jin, Metallomics, 2019, 11, 1207–1218 CrossRef CAS PubMed.
- Y. Xia, X. Wei, P. Gao, C. Wang, A. de Jong, J. H. K. Chen, M. J. Rodriguez-Sanchez, A. Rodriguez-Nogales, P. Diez-Echave, J. Galvez, F. Garcia, W. Wu, R. Y. Kao, H. Li, R. Cebrian, O. P. Kuipers and H. Sun, Nat. Microbiol., 2024, 9, 2600–2613 CrossRef CAS PubMed.
- E. Mahenthiralingam, T. A. Urban and J. B. Goldberg, Nat. Rev. Microbiol., 2005, 3, 144–156 CrossRef CAS PubMed.
- A. Shukla, S. Rodriguez and T. Brennan-Krohn, J. Antimicrob. Chemother., 2024, 79, 2867–2876 CrossRef CAS PubMed.
- V. G, P. Shunmuga Sundaram and T. Rajendran, J. Infect. Chemother., 2024, 31, 102545 CrossRef PubMed.
- T. M. Egan, F. C. Detterbeck, M. R. Mill, M. S. Bleiweis, R. Aris, L. Paradowski, G. Retsch-Bogart and B. S. Mueller, Eur J Cardiothorac Surg., 2002, 22, 602–609 CrossRef CAS PubMed.
- S. Bonacorsi, F. Fitoussi, S. Lhopital and E. Bingen, Antimicrob. Agents Chemother., 1999, 43, 213–217 CrossRef CAS PubMed.
- T. Coenye, P. Vandamme, J. J. LiPuma, J. R. W. Govan and E. Mahenthiralingam, J. Clin. Microbiol., 2003, 41, 2797–2798 CrossRef PubMed.
- R. Ge, X. Sun, Q. Gu, R. M. Watt, J. A. Tanner, B. C. Wong, H. H. Xia, J. D. Huang, Q. Y. He and H. Sun, J. Biol. Inorg. Chem., 2007, 12, 831–842 CrossRef CAS PubMed.
- R. Wang, J. F. Chan, S. Wang, H. Li, J. Zhao, T. K. Ip, Z. Zuo, K. Y. Yuen, S. Yuan and H. Sun, Chem. Sci., 2022, 13, 2238–2248 RSC.
- S. P. Lee, Scand. J. Gastroenterol. Suppl., 1991, 185, 1–6 CAS.
- L. Hu, T. Cheng, B. He, L. Li, Y. Wang, Y. T. Lai, G. Jiang and H. Sun, Angew Chem. Int. Ed. Engl., 2013, 52, 4916–4920 CrossRef CAS PubMed.
- Y. Wang, C. N. Tsang, F. Xu, P. W. Kong, L. Hu, J. Wang, I. K. Chu, H. Li and H. Sun, Chem. Commun., 2015, 51, 16479–16482 RSC.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2008, 4, 44–57 CrossRef PubMed.
- M. Ahmad, A. Wolberg and C. I. Kahwaji, in StatPearls, 2024 Search PubMed.
- S. Hui, J. M. Ghergurovich, R. J. Morscher, C. Jang, X. Teng, W. Lu, L. A. Esparza, T. Reya, Z. Le, J. Yanxiang Guo, E. White and J. D. Rabinowitz, Nature, 2017, 551, 115–118 CrossRef PubMed.
- V. R. I. Kaila and M. Wikstrom, Nat. Rev. Microbiol., 2021, 19, 319–330 CrossRef CAS PubMed.
- J. M. Griffith, P. J. Basting, K. M. Bischof, E. P. Wrona, K. S. Kunka, A. C. Tancredi, J. P. Moore, M. R. L. Hyman and J. L. Slonczewski, Appl. Environ. Microbiol., 2019, 85, 027922 CrossRef PubMed.
- G. F. White, M. J. Edwards, L. Gomez-Perez, D. J. Richardson, J. N. Butt and T. A. Clarke, Adv. Microb. Physiol., 2016, 68, 87–138 CrossRef CAS PubMed.
- M. D. Lefebre and M. A. Valvano, Appl. Environ. Microbiol., 2002, 68, 5956–5964 CrossRef CAS PubMed.
- G. Reguera, Proc. Natl. Acad. Sci. USA, 2018, 115, 5632–5634 CrossRef CAS PubMed.
- J. I. Kishikawa, Y. Nishida, A. Nakano, T. Kato, K. Mitsuoka, K. I. Okazaki and K. Yokoyama, Nat. Commun., 2024, 15, 9883 CrossRef CAS PubMed.
- M. Yoshida, E. Muneyuki and T. Hisabori, Nat. Rev. Mol. Cell Biol., 2001, 2, 669–677 CrossRef CAS PubMed.
- D. N. Wilson, Nat. Rev. Microbiol., 2014, 12, 35–48 CrossRef CAS PubMed.
- M. W. Webster, A. Chauvier, H. Rahil, A. Graziadei, K. Charles, N. Miropolskaya, M. Takacs, C. Saint-Andre, J. Rappsilber, N. G. Walter and A. Weixlbaumer, Science, 2024, 386, eado8476 CrossRef CAS PubMed.
- A. Frei, A. D. Verderosa, A. G. Elliott, J. Zuegg and M. A. T. Blaskovich, Nat. Rev. Chem., 2023, 7, 202–224 CrossRef CAS PubMed.
- Q. Zhang, R. Wang, M. Wang, C. Liu, M. Koohi-Moghadam, H. Wang, P. L. Ho, H. Li and H. Sun, Proc. Natl. Acad. Sci. USA, 2022, 119, e2119417119 CrossRef CAS PubMed.
- W. K. Kwong, H. Zheng and N. A. Moran, Nat. Microbiol., 2017, 2, 17067 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02049b |
| This journal is © The Royal Society of Chemistry 2025 |