
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of perfluoro(methyl vinyl ether) at Rh(I) complexes: metal-centered versus phosphine-mediated decarbonylation†
Soodeh
Mollasalehi
 ,
Mike
Ahrens
and
Thomas
Braun
*
,
Mike
Ahrens
and
Thomas
Braun
*
Department of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor Str. 2, 12489 Berlin, Germany. E-mail: thomas.braun@cms.hu-berlin.de
First published on 12th May 2025
Abstract
This study investigates the reactivity of perfluoro(methyl vinyl ether) [PMVE, CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF(OCF3)] towards rhodium(I) phosphine complexes. The reaction pathways are characterized by C–O and C–F bond cleavage reactions and decarbonylation steps. On using the complex [Rh(H)(PEt3)3] (1), unprecedented reactivity pathways were observed that are distinct from those found for previously studied fluoroolefins. Reactivity of an excess PMVE at Rh is initiated by coordination to the Rh center in 1, followed by its insertion into the Rh–H bond and a β-OCF3 elimination. This process ultimately results in OCF3 ligand transformation to give trans-[Rh(F)(CO)(PEt3)3] (4) and Et3PF2. Reactions of stoichiometric amounts of PMVE with [Rh(H)(PEt3)3] (1) or an excess amount of it with [Rh(F)(PEt3)3] (6) led to olefin complex formation to yield trans-[Rh(F)(η2-CF2CFH)(PEt3)2] (7) and trans-[Rh(F)(CF(OCF3)CF2)(PEt3)2] (8), respectively. In contrast, a remarkable insertion into the Rh–F bond at [Rh(F)(CO)(PEt3)2] (4) was observed leading to the formation of trans-[Rh(CO)(CF(OCF3)CF3)(PEt3)2] (5). Decarbonylation of PMVE proceeds not only at Rh, but also via a metal-free, phosphine-mediated process. The latter is characterized by oxidative addition of PMVE at PEt3 to form the fluorophosphoranes E/Z-(F3CO)CFCF(PFEt3), which subsequently convert into Et3PF2, CO and presumably tetrafluoroethene.
CF(OCF3)] towards rhodium(I) phosphine complexes. The reaction pathways are characterized by C–O and C–F bond cleavage reactions and decarbonylation steps. On using the complex [Rh(H)(PEt3)3] (1), unprecedented reactivity pathways were observed that are distinct from those found for previously studied fluoroolefins. Reactivity of an excess PMVE at Rh is initiated by coordination to the Rh center in 1, followed by its insertion into the Rh–H bond and a β-OCF3 elimination. This process ultimately results in OCF3 ligand transformation to give trans-[Rh(F)(CO)(PEt3)3] (4) and Et3PF2. Reactions of stoichiometric amounts of PMVE with [Rh(H)(PEt3)3] (1) or an excess amount of it with [Rh(F)(PEt3)3] (6) led to olefin complex formation to yield trans-[Rh(F)(η2-CF2CFH)(PEt3)2] (7) and trans-[Rh(F)(CF(OCF3)CF2)(PEt3)2] (8), respectively. In contrast, a remarkable insertion into the Rh–F bond at [Rh(F)(CO)(PEt3)2] (4) was observed leading to the formation of trans-[Rh(CO)(CF(OCF3)CF3)(PEt3)2] (5). Decarbonylation of PMVE proceeds not only at Rh, but also via a metal-free, phosphine-mediated process. The latter is characterized by oxidative addition of PMVE at PEt3 to form the fluorophosphoranes E/Z-(F3CO)CFCF(PFEt3), which subsequently convert into Et3PF2, CO and presumably tetrafluoroethene.
Introduction
The increasing interest in hydrofluoroolefins (HFOs) stems from their significantly lower potential for ozone depletion and global warming compared to chlorofluorocarbons and hydrofluorocarbons.1–3 This has led to their adoption as refrigerants in automotive air conditioning systems.4–6 Generally, fluorinated olefins show promise as precursors for synthesizing new fluorinated building blocks.7–10 An interesting strategy involves the development of methods for functionalization that are mediated by main group elements or transition metals, including C–F and C–H bond activation reactions.11–14The fluoroolefin perfluoro (methyl vinyl ether) [PMVE, CF2CF(OCF3)] can be classified as part of the PFAS family (per- and polyfluoroalkyl substances) and, therefore, understanding its reactivity will provide insights concerning its depletion.15–17 It is, however, valuable as a monomer for producing high-value fluorinated polymers.18–22 It has also been suggested as an alternative replacement for SF6.23,24 PMVE is formally an analogue of hexafluoropropene with a formal substitution of the CF3 group in hexafluoropropene with the OCF3 group, which leads to a different reactivity. Note that the trifluoromethoxy group OCF3 imparts significant biological properties to molecules, making OCF3 compounds highly valuable targets in the pharmaceutical and agrochemical industries.25,26 This is primarily due to the strong electron-withdrawing effect and high lipophilicity caused by this group.27–32 To ensure these compounds are used safely in industrial applications, thorough research into their properties and reactivity is essential.17,33
Investigations into the reactivity of PMVE towards transition metal complexes are scarce, despite extensive research on fluoroolefins bearing a CF3 group. Noticeably, Baker et al. previously reported a copper catalysed hydrodefluorination of PMVE leading to the formation of difluoroethylene isomers via β-OCF3 elimination.34 In another recent work, they described a copper-mediated fluoroalkyl –CF(OCF3)(CF2H) transfer to organic electrophiles via insertion of the PMVE into Cu–H bonds of Stryker's reagent.35 Note that the hydroamination of PMVE with secondary amines, as well as the addition reaction of azoles to PMVE have been reported.36,37
The notable reactivity of rhodium(I) phosphine complexes [Rh(E)(PEt3)3] (E = H, F, boryl, germyl, silyl) towards fluorinated olefins, such as hexafluoropropene, has been well-documented.8,38,39 The identity of the anionic ligand E holds a significant importance in the activation process for several reasons, such as the formation of strong element-fluorine bonds like H–F, B–F, Ge–F, or Si–F bonds, as well as a kinetic control of regio- and chemoselectivities.8,38–43
On the other hand, examples of C(sp2)–F bond functionalization reactions by simple tertiary phosphines, without the need for activation by metals, metalloids, or a Lewis acid are still rare. Burton and colleagues demonstrated that tertiary phosphines such as PPh3 and PnBu3 could attack perfluorinated cyclic alkenes and perfluorinated linear terminal alkenes leading to the production of stable phosphonium ylides and the generation of a fluorophosphorane nBu3P(F)CFCF(CF3), respectively.44,45 Additionally, García and colleagues reported the formation of the difluorophosphorane Et3PF2 in the hydrodefluorination of a range of polyfluoro(hetero)aromatics, using PEt3 as the sole defluorinating agent.46 Furthermore, the generation of fluorophosphoranes Et3P(F)CFC(X)CF3 (X = H, F) or Et3PF2 through the reaction of various fluorinated olefins with the [Rh(E)(PEt3)3] has been reported.38,41,47 Dissociation of phosphine during the C–F bond activation step at the Rh complex might be essential for these transformations.
Herein we present studies on the reactivity of PMVE towards the rhodium(I) phosphine complexes [Rh(H)(PEt3)3] (1) and [Rh(F)(PEt3)3] (6). PMVE serves as CO source and fluorinating agent. The observed reactivity patterns are very distinctive and involve coordination of the olefin, insertion into Rh–H and Rh–F bonds, β-OCF3 elimination steps as well as oxidative addition at liberated PEt3.
Results and discussion
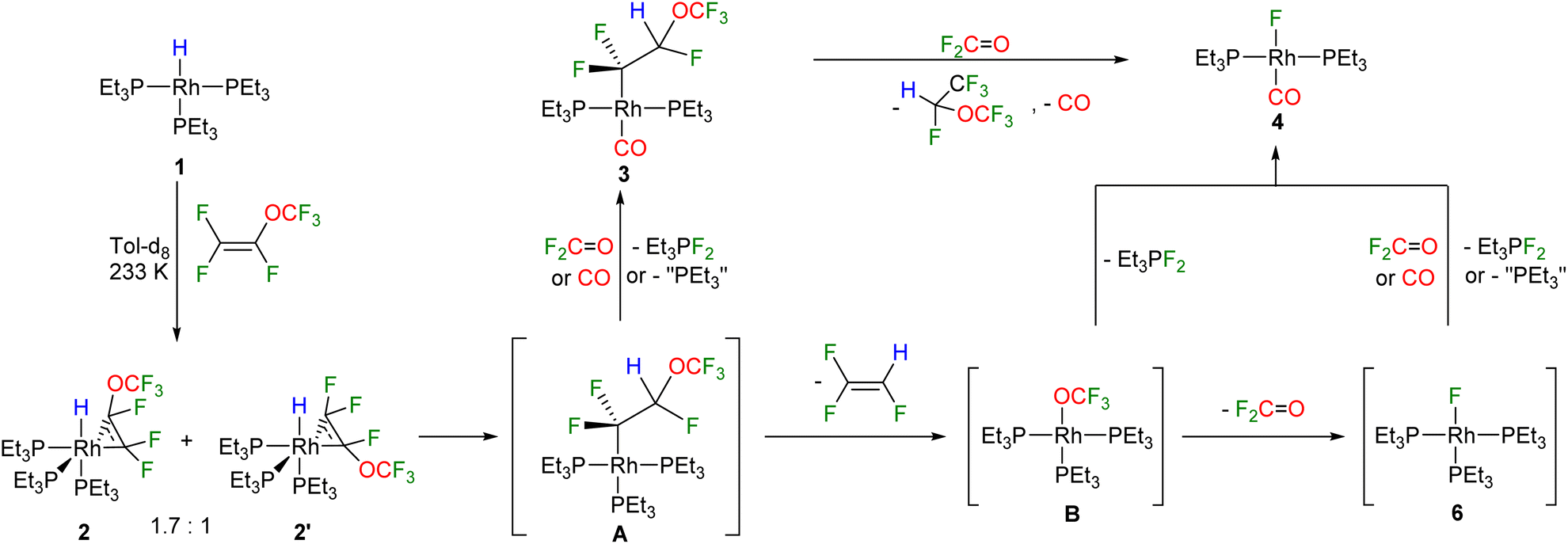
Treatment of the rhodium(I) hydrido complex [Rh(H)(PEt3)3] (1) with an excess of perfluoro (methyl vinyl ether) [PMVE, CF2CF(OCF3)] yielded after 30 minutes the fluoroalkyl complex trans-[Rh(CF2CFH(OCF3))(CO)(PEt3)2] (3) and the Vaska type fluorido complex trans-[Rh(F)(CO)(PEt3)2] (4) in a ratio of 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as well as 1,1,2-trifluoroethylene (Scheme 1). The source of the CO ligand in 3 and 4 is the OCF3 moiety of PMVE, and its rhodium as well as phosphine mediated-transformation is described below.
:1, as well as 1,1,2-trifluoroethylene (Scheme 1). The source of the CO ligand in 3 and 4 is the OCF3 moiety of PMVE, and its rhodium as well as phosphine mediated-transformation is described below.
 | ||
| Scheme 1 Reactivity of complex 1 towards PMVE. | ||
The compounds 3 and 4 were not stable at room temperature and a slow conversion into the rhodium(l) perfluoroalkyl complex trans-[Rh(CO)(CF(OCF3)CF3)(PEt3)2] (5) was found (Scheme 1). The formation of complex 5 suggests an insertion of PMVE, which is present in an excess, into the rhodium–fluorine bond in complex 4. An independent reaction of 4 with PMVE confirmed the formation of 5via the insertion of the olefin into the Rh–F bond. Note that examples for an insertion of fluorinated olefins into metal–fluorine bonds are rare, and have been reported previously at group 10 and 11 transition metal-complexes, but not at rhodium.35,48,49
Compound 4 has been previously synthesised and fully characterised.50 Complexes 3 and 5 were characterised by 1H, 31P{1H} and 19F NMR spectroscopy. For complex 3, in the 31P{1H} spectrum a doublet of triplets is observable due to the coupling to the rhodium centre (138.3 Hz) and the CF2 moiety (24.4 Hz). In the 19F NMR spectrum, the resonance of the OCF3 moiety appears at −58.4 as a broad signal, while for the CF2 group two resonances were found with a geminal F,F coupling constant of 295 Hz, which is in accordance with the C(sp3) character of the CF2 group.34,51 For the CFH group a doublet with 2JF,H of 57 Hz at −134.9 ppm in 19F NMR spectrum and at 5.8 ppm in 1H NMR spectrum are visible. The IR spectrum of 3 reveals an intense absorption band at 1945 cm−1, which can be assigned to the CO stretching vibration and the data are comparable with those of known rhodium carbonyl complexes.50 For complex 5 on the other hand, a doublet appears in 31P{1H} NMR spectrum at 24.3 ppm with a P,Rh coupling constant of 120.9 Hz. In the 19F NMR spectrum, a doublet of quartets at −53.7 ppm corresponds to the CF3 moiety, which couples to the CF group with 10 Hz and to the OCF3 group with 1 Hz, while another doublet of quartets at −81.5 ppm with a coupling constant of 3 Hz to the CF group can be assigned to the OCF3 moiety. Both signals correlate with the quartet of quartets at −129.5 ppm for the CF moiety. Furthermore, a CO stretching vibration band at 1938 cm−1 in the IR spectrum is observable for compound 5.
Moreover, the formation of the fluorophosphorane Et3PF2 and 1,2,2,2-tetrafluoroethyl trifluoromethyl ether, CHF(OCF3)(CF3) was observed (see also below, Scheme 2).47 The presence of CHF(OCF3)(CF3) was revealed by the 19F and 1H NMR spectra data, which were in accordance with the literature.52
 | ||
| Scheme 2 Mechanistic proposal for the reactivity of complex 1 towards PMVE. | ||
The reaction of 1 with PMVE was then monitored by NMR spectroscopy at 233 K and the isomeric rotational intermediates fac-[Rh(H)(η2-CF2C(OCF3)F)(PEt3)3] (2/2′) in a ratio of 1.7:1 were identified (Scheme 2). These olefin complexes were only stable below 273 K, therefore, identification was possible by 1H, 31P{1H} and 19F NMR spectroscopy. Another identified intermediate at this temperature was a fluorophosphorane, Z-(F3CO)CFCF(PFEt3), which indicates the reaction of PMVE with PEt3 (for further explanation see below). Above 273 K the transformation of this compound together with 2 and 2′ into 3, 4, trifluoroethylene, CHF(OCF3)(CF3), and Et3PF2 was observed. Previously, coordination of fluorinated olefins to [Rh(H)(PEt3)3] (1) at low temperatures has been described.38,43,53 Characteristic features in the 1H NMR spectrum are the signals for the hydrido ligands at −12.1 ppm with a large H,Ptrans coupling constant of 152.3 Hz for 2 and at −11.9 ppm for 2′ with 152.8 Hz. Additionally, in the 1H{31P} NMR spectrum, a doublet of doublets appears for 2 due to the coupling to Rh and the one fluorine atom in syn position, while for 2′ a doublet of triplets due to the coupling to Rh and two fluorine atoms in syn position is visible. Due to the high structural similarities of the two isomers, the three sets of signals for the inequivalent phosphine ligands at 14.0–12.8 ppm, 11.2–9.4 ppm and 4.8–2.6 ppm, which integrate in a 1:1:1 ratio, overlap in the 31P{1H} NMR spectrum (242 MHz) at 273 K. However, the Rh,P coupling constants are in accordance with the oxidation state +III at rhodium and a metallacyclopropane structure.38,43 In order to further support the metallacyclopropyl configuration of the complexes 2 and 2′, the structures of the corresponding PMe3 derivatives 2* and 2′* were optimized by DFT calculations and a C–C bond distance of 1.472 Å for 2* and 2′* was obtained (Fig. 1).54–56 Calculations on the PEt3 complexes 2 and 2′ were not conclusive as the energy differences of the converged structures were highly dependent on slight structural differences in the PEt3 ligand sphere, thus making it hard to identify the global minima. However, based on the DFT calculations on the simplified PMe3 derivatives, complex 2* is more stable than complex 2′* by 10.7 kJ mol−1.54–56
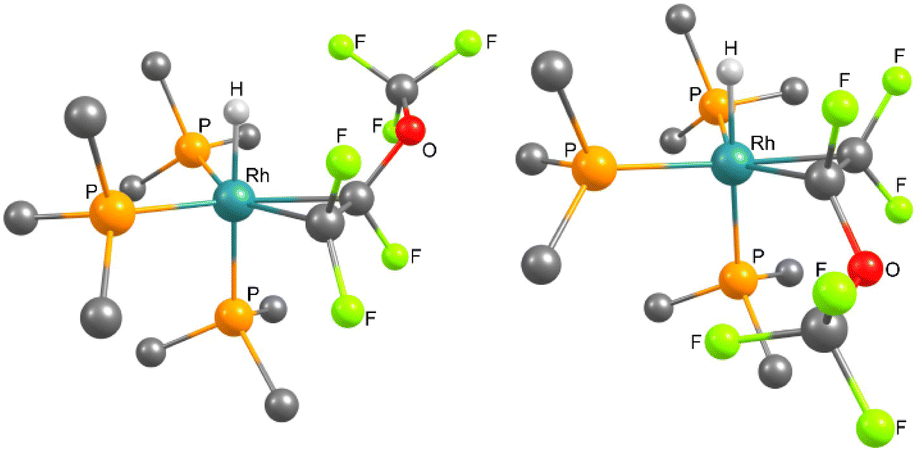 | ||
| Fig. 1 DFT optimised structures ((B3LYP/cc-pvtz and cc-pvdz), see ESI†) of left: fac-[Rh(H){η2-CF2CF(OCF3)}(PMe3)3] (2*), and right: (2′*). Hydrogen atoms of the phosphine ligands are omitted for clarity. | ||
Mechanistically, the transformation of intermediates 2 and 2′ into complexes 3 and 4 requires an insertion of the olefin into the Rh–H bond to yield the intermediate A (Scheme 2). A then undergoes a β-OCF3 elimination accompanied by the release of the trifluoroethylene. It is likely that the complex [Rh(OCF3)(PEt3)3] (B) is then formed as an unstable intermediate, which converts into 4 and F2PEt3. Alternatively, the fluorido complex [Rh(F)(PEt3)3] (6) and fluorophosgene OCF2 are generated. The rapid decomposition of a trifluoromethoxide anion OCF3− into fluorophosgene OCF2 and fluoride has been reported in literature.31 Fluorophosgene can react with A or 6 to yield Et3PF2 as well as the carbonyl complexes 3 and 4, respectively. In addition, it is also feasible that complex 3 reacts with fluorophosgene to convert into 4 upon the release of the observed CHF(OCF3)(CF3) and CO (Scheme 2). Note that olefin insertion into the metal–hydrogen bond and β-OCF3 elimination steps have been proposed for a copper complex that mediates the catalytic hydrodefluorination of PMVE to yield a mixture of cis- and trans-1,2-difluoroethylene in a 4:1 ratio.34
Furthermore, it is important to consider that for the conversion of A into 3 and for the formation of the fluorido complex 4 from [Rh(F)(PEt3)3] (6) at least two equivalents of PMVE as an excess source of CO are required. Hence, initially the role of olefin was investigated further by the reaction of [Rh(H)(PEt3)3] (1) with stoichiometric amounts of PMVE at room temperature, which led to the formation of trans-[Rh(F)(η2-CF2CFH)(PEt3)2] (7) as well as the fluorophosphorane Et3PF2 (Scheme 3). Formation of 7 implies the generation of the reaction products as described above, whereas subsequently a phosphine ligand is substituted by trifluoroethylene at [Rh(F)(PEt3)3] (6) (Scheme 2). The latter step was confirmed independently by treatment of the rhodium fluorido complex [Rh(F)(PEt3)3] (6) with trifluoroethylene to yield complex 7 (Scheme 3). Thus, the result demonstrates that a replacement of the phosphine ligand in [Rh(F)(PEt3)3] (6) by an olefin can compete with the substitution of the phosphine by CO, when less amounts of PMVE as a CO source are provided. Note that the dissociation of a phosphine ligand upon the coordination of a fluoroolefin to the fluorido complex 6 has been previously reported for 1,1,3,3,3-pentafluoropropene in a similar manner.38
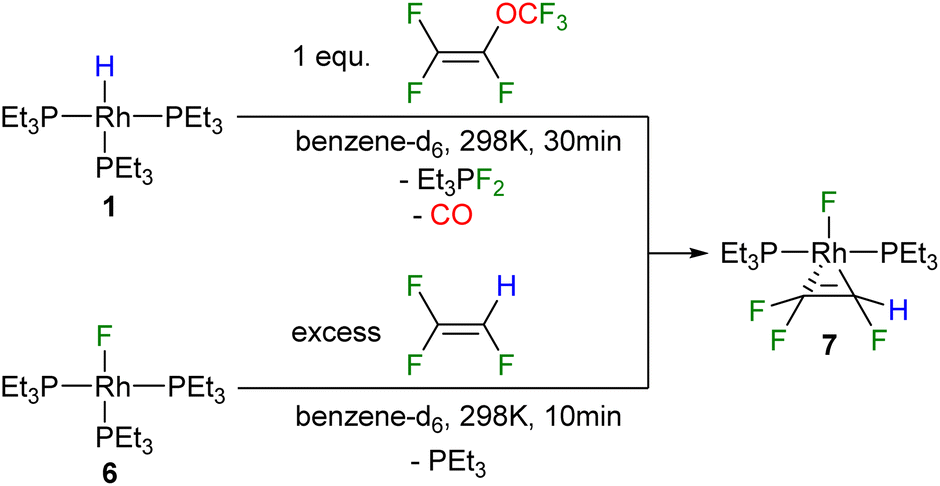 | ||
| Scheme 3 Reactivity of complex 1 towards one equivalent of PMVE (top), and reactivity of complex 6 towards trifluoroethylene (bottom). | ||
The resonances of the two inequivalent phosphine ligands of complex 7 appear at 32.3 and 25.9 ppm in the 31P{1H} NMR spectrum, and are coupled to each other by trans2Jp,p = 368.2 Hz. The phosphorus–rhodium coupling constants of 138.9 Hz and 133.3 Hz, respectively, demonstrate the presence of a Rh(I) complex. The signal at 25.9 ppm represents a doublet of doublet of doublet of doublets due to its coupling to phosphorus, rhodium, one fluorine atom of the CF2 moiety and the fluorido ligand, while the second signal displays a doublet of doublet of triplets due to couplings to phosphorus and rhodium, as well as coupling to one fluorine atom of the CF2 moiety and the fluorine atom of the CFH group. In the 19F NMR spectrum, a geminally coupled pair is observed at −89.4 and −90.8 ppm (2JF,F = 109 Hz) associated with the CF2 fluorine resonances along with an upfield CFH peak at −194.9 ppm and the signal of the fluorido ligand at −218.3 ppm. A distinctive doublet of doublet of doublets appears at 5.5 ppm in the 1H NMR spectrum, corresponding to the CFH hydrogen atom (2JFH = 73.3 Hz).
The reactivity described above of the fluorido complex [Rh(F)(CO)(PEt3)2] (4) towards PMVE (Scheme 1) prompted us to also study the behaviour of the fluorido compound [Rh(F)(PEt3)3] (6) towards PMVE, in order to assess a possible coordination versus insertion in a Rh–F bond, as the latter step was found for [Rh(F)(CO)(PEt3)2] (4). However, treatment of 6 with PMVE gave after 3 hours a mixture of complexes [Rh(F)(η2-CF(OCF3)CF2)(PEt3)2] (8) and 4 in a 2.4:1 ratio, and the Et3PF2 (Scheme 4). At the beginning of this reaction the phosphorane Z-(F3CO)CFCF(PFEt3) was present, but there was no indication for the formation of complex 4. The latter was formed upon depletion of the phosphonium salt indicating a crucial role of Z-(F3CO)CFCF(PFEt3) as an alternative CO source (see below). It can be presumed that initially Z-(F3CO)CFCF(PFEt3) was generated by reaction of liberated PEt3 with PMVE, which indicates that a dissociation of phosphine takes place and that any rebinding process is slower than the reactivity of the free phosphine towards the excess of olefin. Note that when 6 was treated with PMVE in the presence of oxygen only trans-[Rh(F)(η2-CF(OCF3)CF2)(PEt3)2] (8) was generated as the liberated phosphine can be trapped as Et3PO.
 | ||
| Scheme 4 Reactivity of complex 6 towards PMVE. | ||
The 31P{1H} NMR spectrum (202 MHz) of complex 8 displayed two resonances for inequivalent phosphorus atoms at 34.1 and 30.5 ppm. Both signals show a trans P–P coupling of 366.7 Hz and P–Rh coupling constants typical of Rh(I) complexes (133.4 and 134.1 Hz). The signal at 30.5 ppm appears as a doublet of doublet of doublet of doublets due to the couplings to phosphorus and rhodium as well as coupling to one of the fluorine atoms at the CF2 moiety and the fluorido ligand. In contrast, the second resonance shows a doublet of doublet of doublet of triplet of doublets due to the additional coupling to the CF moiety. In the 19F NMR spectrum, five inequivalent resonances for the OCF3 moiety at −57.1 ppm, the two fluorine atoms of the CF2 group at −96.8 and −98.2 ppm, the CF group at −116.1 ppm and the rhodium bound fluorido ligand at −206.3 ppm are displayed. A correlation between the olefinic fluorine signals and fluorido ligand was confirmed by 19F,19F-COSY NMR spectroscopy. The C–C bond distance of the olefin obtained by DFT calculations (1.44 Å) suggests a metallacyclopropane configuration (Fig. 2).54–56 Note that the NMR as well as the DFT calculation results are in agreement with those that have been previously reported for 1,1,3,3,3-pentafluoropropene.38
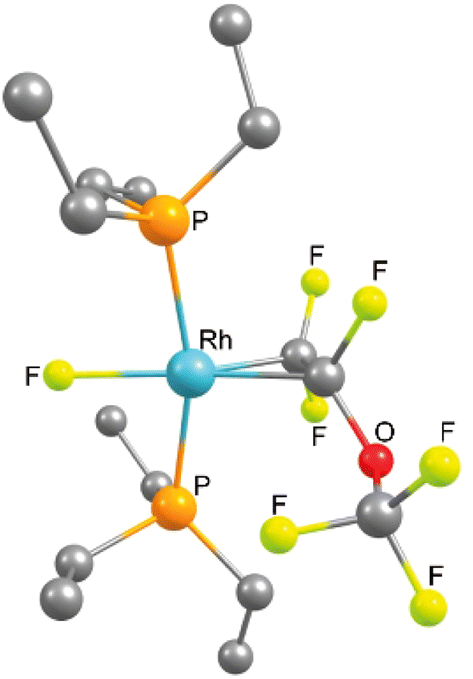 | ||
| Fig. 2 DFT optimised structure (B3LYP/cc-pvdz) of complex 8. Hydrogen atoms of the phosphine ligands are omitted for clarity. | ||
Independent reactivity studies on the reactivity of PEt3 towards PMVE demonstrated that Z/E-(F3CO)CFCF(PFEt3) in a 10:1 ratio can be formed by an oxidative addition at the phosphorus center (Scheme 5). A similar reactivity pattern of phosphines with other fluoroolefins, for instance by Burton and Röschenthaler, or fluoroaromatics was previously reported.38,41,45,47,57,58 Monitoring the reaction solution after the generation of Z/E-(F3CO)CFCF(PFEt3) at room temperature revealed a conversion into F2PEt3, CO, and presumably C2F4 along with other decomposition products. The latter could not be identified, possibly because of oligomerization. The formation of the CO gas was confirmed by 13C{1H} NMR spectroscopy as well as gas chromatography. In the 31P{1H} NMR spectrum of Z/E-(F3CO)CFCF(PFEt3) two signals at −63.8 ppm with a 1JP,F coupling constant of 598.8 Hz and 592.2 Hz are visible. The corresponding signals in the 19F NMR appeared at −19.6 and −22.3 ppm for the Z and E isomers, respectively. Additionally, resonances for the OCF3 moiety and the two CF groups with a 3JF,Ftrans of 116 Hz (for the Z isomer) and a 3JF,Fcis of 25 Hz (for the E isomer) are displayed.
 | ||
| Scheme 5 Reactivity of triethylphosphine towards: (i) perfluoro (methyl vinyl ether), and (ii) fluorophosgene. | ||
Mechanistically it can be assumed that (F3CO)CFCF(PFEt3) converts initially into the phosphonium salt (F3CO)CFCF(PEt3)+F− (Scheme 6). A nucleophilic attack of the fluoride at the vinyl carbon yields the ylide CF2(OCF3)CF(PEt3). Then CF2CF(PFEt3) and OCF2 might be generated via CF2CF(PEt3)+OCF3−. The former CF2CF(PFEt3) might convert into F2PEt3, CO, and C2F4. Note that an independent reaction shows that F2PEt3 and CO can also obtained by the reaction of the phosphine with fluorophosgene (Scheme 5).59
 | ||
| Scheme 6 Mechanistic proposal for the decomposition of the fluorophosphorane Z-(F3CO)CFCF(PFEt3). | ||
Conclusions
The paper demonstrates that the rhodium(I) hydrido complex [Rh(H)(PEt3)3] (1) exhibits distinct reactivity pathways towards PMVE when compared to the reactivity of other fluorinated olefins. These pathways lead to metal- and phosphine-mediated decarbonylation reactions. Both processes involve the generation of a OCF3− moiety that can convert into CO, fluoride and fluorophosgene. While at Rh trifluoroethylene is generated, the phosphine-mediated process presumably leads to the formation of tetrafluoroethene. The studies provide valuable insights into the reactivity of PMVE which is considered a PFAS.15 Decarbonylation processes allow for its decomposition and open up new avenues for transforming fluorinated fragments. Remarkably, It is shown that the formation of the rhodium carbonyl fluorido complex 4 allows the insertion of PMVE into the Rh–F bond forming the –CF(OCF3)(CF3) ligand. Additionally, this paper highlights that phosphines might be suitable tools for a transformation of fluorinated compounds.14Data availability
Details of the experimental procedures, characterization of the complexes and DFT calculations can be found in the ESI.†Author contributions
Conceptualization, S. M. and T. B.; investigation, S. M. and M. A.; writing – original draft preparation, S. M.; writing – review and editing, S. M. and T. B.; funding acquisition, T. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the DFG (Deutsche Forschungsgemeinschaft) for financial support (BR 2065/13-1). S. M. thanks Dr M. Talavera and R. Jaeger for fruitful discussions, N. Limberg for his support with the COF2 experiment, and Dr A. Dallmann for the high-frequency NMR measurements.Notes and references
- E. A. Heath, Int. Leg. Mater., 2017, 56, 193–205 CrossRef.
- T. J. Wallington, M. P. Sulbaek Andersen and O. J. Nielsen, Chemosphere, 2015, 129, 135–141 CrossRef CAS PubMed.
- A. J. Sicard and R. T. Baker, Chem. Rev., 2020, 120, 9164–9303 CrossRef CAS PubMed.
- S. E. Manahan, Fundamentals of environmental and toxicological chemistry: sustainable science, CRC press, 2013 Search PubMed.
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity and Applications, Wiley-VCH, Weinheim, 2nd edn, 2013 Search PubMed.
- P. Javidmand and K. A. Hoffmann, Int. J. Refrig., 2016, 69, 114–135 CrossRef CAS.
- G. Coates, H. Y. Tan, C. Kalff, A. J. P. White and M. R. Crimmin, Angew Chem. Int. Ed. Engl., 2019, 58, 12514–12518 CrossRef CAS.
- M. Talavera and T. Braun, Synlett, 2020, 31, 1760–1774 CrossRef CAS.
- B. J. Murray, L. T. Boulton and G. Sandford, J. Fluorine Chem., 2021, 245, 109774 CrossRef CAS.
- B. Ameduri and H. Sawada, Fluorinated polymers: volume 1: synthesis, properties, Processing and Simulation, Royal Society of Chemistry, 2016 Search PubMed.
- M. Ohashi and S. Ogoshi, Top. Organomet. Chem., 2015, 52, 197–215 CrossRef CAS.
- M. Talavera, S. Mollasalehi and T. Braun, Chem. Sci., 2024, 15, 8472–8477 RSC.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS.
- J. M. Bayne and D. W. Stephan, Chem.–Eur. J., 2019, 25, 9350–9357 CrossRef CAS.
- K. M. Wollin, M. Batke, G. Damm, A. Freyberger, U. Gundert-Remy, A. Mangerich, J. G. Hengstler, F. Partosch, T. Schupp, A. Sonnenburg and H. Foth, Arch. Toxicol., 2023, 97, 3305–3312 CrossRef CAS.
- M. A. Adi and M. Altarawneh, Atmos. Environ., 2023, 307, 119843 CrossRef CAS.
- L. Vereecken, J. N. Crowley and D. Amedro, Phys. Chem. Chem. Phys., 2015, 17, 28697–28704 RSC.
- F. Reis da Cunha, I. Davidovich, Y. Talmon and B. Ameduri, Polym. Chem., 2020, 11, 2430–2440 RSC.
- Q. Quan, Y. Zhao, K. Chen, H. Zhou, C. Zhou and M. Chen, ACS Catal., 2022, 12, 7269–7277 CrossRef CAS.
- A. Zharov and O. Nikolaeva, Russ. J. Phys. Chem. B, 2010, 4, 839–845 CrossRef.
- R. E. Banks, B. E. Smart and J. Tatlow, Organofluorine chemistry: principles and commercial applications, 1994 Search PubMed.
- M. A. Sidebottom, C. P. Junk, H. L. S. Salerno, H. E. Burch, G. S. Blackman and B. A. Krick, Macromolecules, 2018, 51, 9700–9709 CrossRef CAS.
- N. Sinha, H. Choi, M.-Y. Song, H.-J. Jang, Y.-H. Oh and K.-D. Song, J. Comput. Theor. Chem., 2023, 1225, 114159 CrossRef CAS.
- S. Xiao, Y. Chen, M. Tang, S. Tian, H. Xia, Y. Wang, J. Tang, Y. Li and X. Zhang, High Volt., 2024, 9, 509–517 CrossRef.
- O. Marrec, T. Billard, J.-P. Vors, S. Pazenok and B. R. Langlois, J. Fluorine Chem., 2010, 131, 200–207 CrossRef CAS.
- C. Bonnefoy, E. Chefdeville, A. Panosian, G. Hanquet, F. R. Leroux, F. Toulgoat and T. Billard, Chem.–Eur. J., 2021, 27, 15986–15991 CrossRef CAS PubMed.
- A. Tlili, F. Toulgoat and T. Billard, Angew Chem. Int. Ed. Engl., 2016, 55, 11726–11735 CrossRef CAS PubMed.
- E. Castagnetti and M. Schlosser, Chem.–Eur. J., 2002, 8, 799–804 CrossRef CAS.
- Q. W. Zhang and J. F. Hartwig, Chem. Commun., 2018, 54, 10124–10127 RSC.
- P. Golz, K. Shakeri, L. Maas, M. Balizs, A. Perez-Bitrian, H. D. Kemmler, M. Kleoff, P. Vossnacker, M. Christmann and S. Riedel, Chem.–Eur. J., 2024, 30, e202400861 CrossRef CAS.
- C.-P. Zhang and D. A. Vicic, Organometallics, 2012, 31, 7812–7815 CrossRef CAS.
- A. Turksoy, T. Scattolin, S. Bouayad-Gervais and F. Schoenebeck, Chem.–Eur. J., 2020, 26, 2183–2186 CrossRef CAS.
- C. M. Tovar, M. B. Blanco, I. Barnes, P. Wiesen and M. A. Teruel, Atmos. Environ., 2014, 88, 107–114 CrossRef CAS.
- N. O. Andrella, N. Xu, B. M. Gabidullin, C. Ehm and R. T. Baker, J. Am. Chem. Soc., 2019, 141, 11506–11521 CrossRef CAS.
- B. Ghaffari, L. L. T. N. Porto, N. Johnson, J. S. Ovens, C. Ehm and R. T. Baker, ACS Org. Inorg. Au., 2024, 4, 628–639 CrossRef CAS PubMed.
- K. I. Petko and A. A. Filatov, Chem. Heterocycl. Compd., 2021, 57, 666–671 CrossRef CAS.
- E. Schmitt, S. Bouvet, B. Pégot, A. Panossian, J.-P. Vors, S. Pazenok, E. Magnier and F. R. Leroux, Org. Lett., 2017, 19, 4960–4963 CrossRef CAS PubMed.
- M. Talavera and T. Braun, Chem.–Eur. J., 2021, 27, 11926–11934 CrossRef CAS.
- M. Talavera and T. Braun, Chem. Sci., 2022, 13, 1130–1135 RSC.
- A. L. Raza and T. Braun, Chem. Sci., 2015, 6, 4255–4260 RSC.
- A. L. Raza, M. F. Kuehnel, M. Talavera, M. Teltewskoi, M. Ahrens, P. Kläring, T. Braun and D. Lentz, J. Fluorine Chem., 2018, 214, 80–85 CrossRef CAS.
- M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew Chem. Int. Ed. Engl., 2010, 49, 3947–3951 CrossRef CAS PubMed.
- M. Talavera, C. N. von Hahmann, R. Muller, M. Ahrens, M. Kaupp and T. Braun, Angew Chem. Int. Ed. Engl., 2019, 58, 10688–10692 CrossRef CAS.
- M. A. Howells, R. D. Howells, N. C. Baenziger and D. J. Burton, J. Am. Chem. Soc., 1973, 95, 5366–5370 CrossRef CAS.
- D. J. Burton, S. Shin-ya and R. D. Howells, J. Fluorine Chem., 1980, 15, 543–546 CrossRef CAS.
- A. A. Facundo, A. Arévalo, G. Fundora-Galano, M. Flores-Álamo, E. Orgaz and J. J. García, New J. Chem., 2019, 43, 6897–6908 RSC.
- D. Noveski, T. Braun, M. Schulte, B. Neumann and H.-G. Stammler, Dalton Trans., 2003, 4075–4083 RSC.
- S. Wada and R. F. Jordan, Angew Chem. Int. Ed. Engl., 2017, 56, 1820–1824 CrossRef CAS.
- N. O. Andrella, K. Liu, B. Gabidullin, M. Vasiliu, D. A. Dixon and R. T. Baker, Organometallics, 2018, 37, 422–432 CrossRef CAS.
- N. Pfister, T. Braun, P. Wittwer and M. Ahrens, Z. Anorg. Allg. Chem., 2018, 644, 1064–1070 CrossRef CAS.
- Y. Zong and G. C. Tsui, Org. Lett., 2024, 26, 1261–1264 CrossRef CAS PubMed.
- A. Foris, Magn. Reson. Chem., 2004, 42, 534–555 CrossRef CAS PubMed.
- T. Ahrens, M. Teltewskoi, M. Ahrens, T. Braun and R. Laubenstein, Dalton Trans., 2016, 45, 17495–17507 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- U. Allwörden and G. V. Röschenthaler, Chem. Ztg., 1988, 112, 69–76 Search PubMed.
- S. Bonfante, C. Lorber, J. M. Lynam, A. Simonneau and J. M. Slattery, J. Am. Chem. Soc., 2024, 146, 11578 CrossRef CAS PubMed.
- M. H. Holthausen, M. Mehta and D. W. Stephan, Angew Chem. Int. Ed. Engl., 2014, 53, 6538–6541 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and analytics for all compounds and details for the DFT calculations. See DOI: https://doi.org/10.1039/d5sc02056e |
| This journal is © The Royal Society of Chemistry 2025 |