
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium-mediated nucleophilic aromatic substitution of hydrogen in benzene†
Stanislav Melnikov a,
Donghun Hwangbc,
Philip Gabberta,
Bohyun Parkbc,
Martin Lutzd,
Mu-Hyun Baik*cb and
Daniël L. J. Broere*a
a,
Donghun Hwangbc,
Philip Gabberta,
Bohyun Parkbc,
Martin Lutzd,
Mu-Hyun Baik*cb and
Daniël L. J. Broere*a
aOrganic Chemistry and Catalysis, Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: d.l.j.broere@uu.nl
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: mbaik2805@kaist.ac.kr
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
dStructural Biochemistry, Bijvoet Centre for Biomolecular Research, Faculty of Science, Utrecht University, Universiteitsweg 99, Utrecht, The Netherlands
First published on 16th June 2025
Abstract
The direct functionalization of unactivated hydrocarbons remains a significant challenge in modern chemistry. In this study, we demonstrate that a simple ruthenium complex featuring a chelating tBuPN ligand can mediate the nucleophilic aromatic substitution of hydrogen (SNArH) in benzene. Key intermediates were kinetically trapped in low-temperature NMR experiments, providing crucial insights into the reaction mechanism. These findings are further supported by isotopic labeling and comprehensive DFT studies. The data shows that the substitution proceeds via an unprecedented mechanism, involving reversible rear-side nucleophilic addition of the exogenous nucleophile to the ruthenium-bound benzene, followed by an intramolecular hydride migration facilitated by deprotonation of tBuPN ligand. The broad range of nucleophiles amenable to this reaction, including classical non-nucleophilic bases, showcases the versatility of this reaction and makes it a promising candidate for further developments in the area of SNArH.
Introduction
Nucleophilic aromatic substitution (SNAr) reactions are widely employed in both academic and industrial settings to structurally modify aromatic ring scaffolds.1–5 While the electron-rich π-system in aromatic molecules typically makes them react as nucleophiles in substitution reactions,6–8 several strategies have been developed to render them susceptible to nucleophilic attack.1,9–12 The most common strategy involves functionalization of the aromatic ring with electron-withdrawing groups (EWGs), which reduce the electron density on the arene. This facilitates nucleophilic addition at the position bearing a leaving group, such as F or Cl, leading to the formation of a σX-adduct (Fig. 1a), also known as the Jackson–Meisenheimer complex. For benzene derivatives, this step is generally rate-determining due to the loss of aromaticity, which is restored upon subsequent extrusion of the leaving group.1,9 Although the addition of nucleophiles is kinetically more facile in positions occupied by hydrogen to give a σH-adduct (Fig. 1b), the nucleophilic aromatic substitution of hydrogen (SNArH) is prohibited by unfavorable elimination of a hydride anion.1,2 Nevertheless, formal SNArH reactions have been realized in a two-step procedure involving a separate oxidation of the pre-formed σH-adduct or through incorporation of a leaving group in the nucleophile.12–15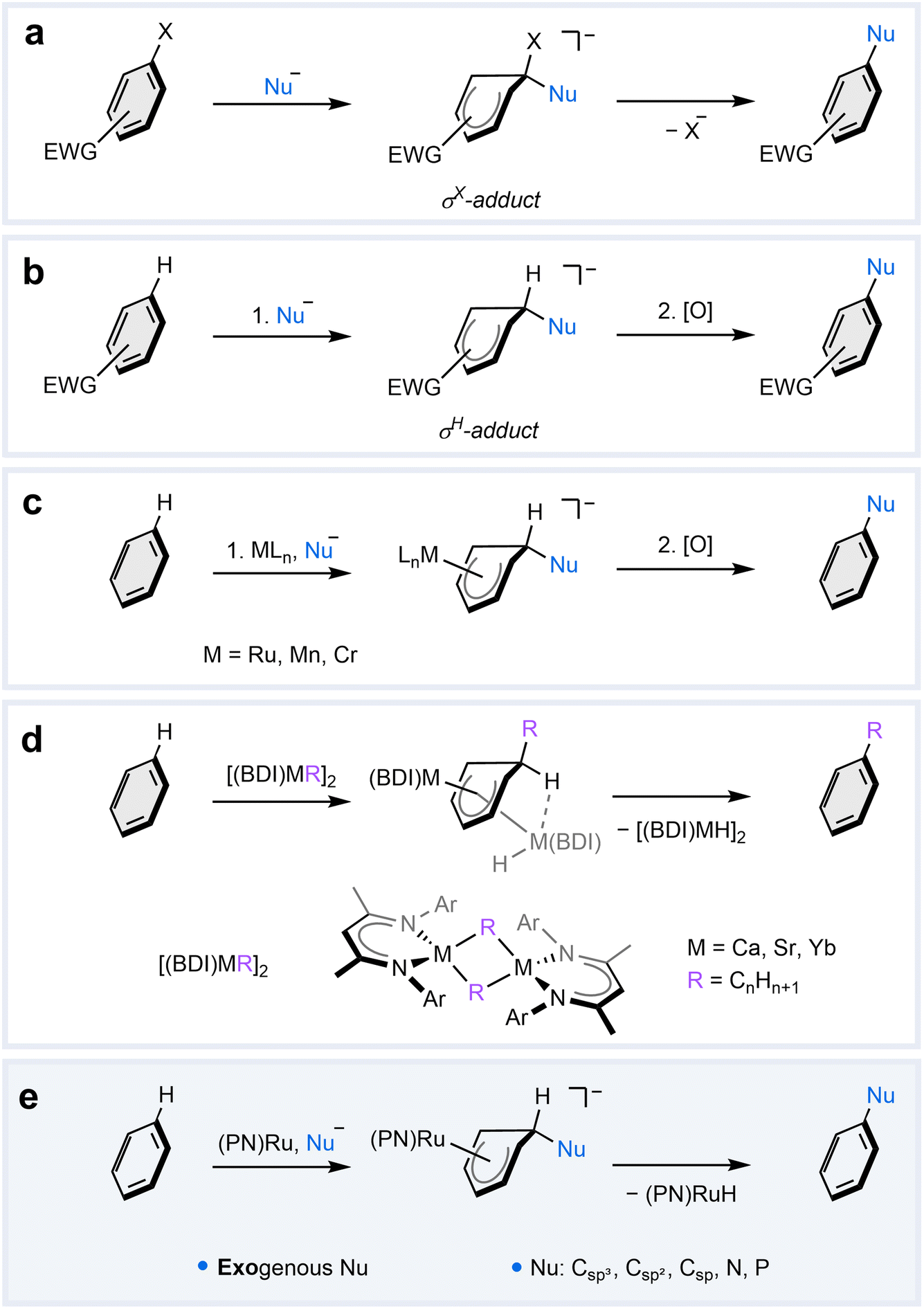 | ||
| Fig. 1 Strategies for nucleophilic aromatic substitution. (a) SNAr in electron-deficient arenes through initial formation of a σX-adduct followed by elimination of a leaving group (X). (b) Nucleophilic addition to electron-deficient arenes to give a σH-adduct, which can be oxidized in a subsequent reaction to realize a formal SNArH. (c) Nucleophilic addition to in transition metal arene complexes resulting in the formation of metal-stabilized σH-adducts, which can be oxidized to mediate a formal SNArH reaction. (d) SNArH alkylation of benzene with β-diketiminate (BDI) complexes of Ca,29 Sr,30 and Yb,31 which employ endogenous nucleophiles; no oxidation step is required. (e) This work: metal-mediated SNArH in benzene with a readily accessible tBuPN–Ru complex and various types of exogenous nucleophiles; no oxidation step is required. | ||
An alternative strategy that circumvents the functionalization of the aromatic ring with EWGs involves the coordination of arenes to transition metal complexes. Nucleophilic addition to the metal-bound arene yields stable cyclohexadienyl complexes, which are considered metal-stabilized analogs of the σH-adducts. These reactions have been extensively investigated, particularly with Cr, Mn, and Ru complexes.16–28 However, to realize the substitution reaction, this strategy still requires subsequent oxidation of the metal-stabilized σH-complex (Fig. 1c) or a conventional leaving group on the arene.
The first example of a metal-mediated SNArH reaction that requires neither EWGs nor leaving groups on the arene was reported in 2017 by Hill, Maron, and co-workers. They found that dimeric β-diketiminate (BDI) supported organocalcium nucleophiles are capable of the direct nucleophilic alkylation of benzene.29 Subsequently, similar reactivity has been demonstrated by BDI-supported strontium30 and ytterbium31 complexes. Recent computational studies on the Ca and Sr complexes suggest that the mechanism involves an intramolecular front-side nucleophilic addition of the alkyl to the metal-coordinated arene (Fig. 1d).32 Subsequently, a second metal complex performs a rear-side hydride abstraction in an intermolecular manner. Although this process allows for oxidant-free SNArH, it has thus far been limited to alkylation due to the required pre-formation of endogenous metal-bound nucleophiles. Consequently, the potential for other types of nucleophiles in metal-mediated SNArH reactions remains unclear.
Herein, we report a new metal-mediated pathway for SNArH in benzene with exogenous nucleophiles (Fig. 1e). This distinct reactivity is enabled by a simple ruthenium complex, which not only activates the arene towards nucleophilic attack but also mediates a unique intramolecular hydride transfer that completes the SNArH reaction. In this article, we elucidate the underlying mechanism of this unusual reaction through a combination of isotopic labeling, kinetic trapping of key intermediates, and computational studies. Finally, we demonstrate the versatility of this reaction, highlighting its compatibility with a range of exogenous nucleophiles.
Results and discussion
Synthesis and characterization
While exploring Milstein-like aromatization–dearomatization cooperativity33 with complex [(tBuPN)RuCl(C6H6)]PF6 (1, where tBuPN is 2-((di-tert-butylphosphaneyl)methyl)pyridine), we discovered that the addition of 1 equiv. of KN(TMS)2 to 1 in THF at room temperature results in the formation of a mixture of two inseparable species in a 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 ratio (Fig. 2a). Analysis of the mixture by 1H nuclear magnetic resonance (NMR) spectroscopy in THF-d8 revealed that the major species featured all the characteristic resonances of the anticipated deprotonated compound [(tBuPN*)RuCl(C6H6)K(THF)n]PF6 (2-K) (see ESI Section S1† for characterization). Surprisingly, the second species featured a doublet at δ = −7.77 ppm and two doublets of doublets at δ = 3.63 and 3.26 ppm, all integrating to 1H. These distinct resonances are characteristic of the presence of a hydride and diastereotopic methylene hydrogens in a non-deprotonated form of the tBuPN ligand, respectively. Additionally, this new species lacked a coordinated benzene molecule; instead, five separated multiplets were found in the 1H spectrum in the range from δ = 6.27 to 4.64 ppm. Based on the spectroscopic data we assign this species to [(tBuPN)RuH(PhN(TMS)2)]PF6 (3, Fig. 2a). Intrigued by the formation of this unusual species, which we hypothesized to be the result of SNArH at the coordinated benzene, we undertook a more detailed investigation into this surprising reactivity.
:30 ratio (Fig. 2a). Analysis of the mixture by 1H nuclear magnetic resonance (NMR) spectroscopy in THF-d8 revealed that the major species featured all the characteristic resonances of the anticipated deprotonated compound [(tBuPN*)RuCl(C6H6)K(THF)n]PF6 (2-K) (see ESI Section S1† for characterization). Surprisingly, the second species featured a doublet at δ = −7.77 ppm and two doublets of doublets at δ = 3.63 and 3.26 ppm, all integrating to 1H. These distinct resonances are characteristic of the presence of a hydride and diastereotopic methylene hydrogens in a non-deprotonated form of the tBuPN ligand, respectively. Additionally, this new species lacked a coordinated benzene molecule; instead, five separated multiplets were found in the 1H spectrum in the range from δ = 6.27 to 4.64 ppm. Based on the spectroscopic data we assign this species to [(tBuPN)RuH(PhN(TMS)2)]PF6 (3, Fig. 2a). Intrigued by the formation of this unusual species, which we hypothesized to be the result of SNArH at the coordinated benzene, we undertook a more detailed investigation into this surprising reactivity.
 | ||
| Fig. 2 Substitution of a hydride at a coordinated benzene with KN(TMS)2. (a) The reaction between 1 and KN(TMS)2. The addition of 1 equiv. of the base leads to the mixture of 2-K and 3 (70:30), whereas 2 equiv. yield compound 4. (b) Solid-state molecular structure of 4 (ellipsoids drawn at the 30% probability level). The hydrogen atoms, except the hydride, are omitted for clarity. Selected bonds (Å) and angles (°): Ru(1)–PhN(TMS)centroid2 = 1.7821(7), Ru(1)–N(1) = 2.0961(11), Ru(1)–P(1) = 2.3325(3), Ru(1)–C(15) = 2.4275(13), Ru(1)–C(16) = 2.3775(14), Ru(1)–C(17) = 2.2091(14), Ru(1)–C(18) = 2.2055(14), Ru(1)–C(19) = 2.1776(14), Ru(1)–C(20) = 2.2409(13), C(1)–C(2) = 1.382(2), C(2)–N(1) = 1.3910(18), C(2)–C(3) = 1.4415(19), C(3)–C(4) = 1.359(2), C(4)–C(5) = 1.407(2), C(5)–C(6) = 1.370(2), N(1)–C(6) = 1.3555(18), N(1)–Ru(1)–P(1) = 81.75(3), N(1)–Ru(1)–PhN(TMS)centroid2 = 131.28(4), P(1)–Ru(1)–PhN(TMS)centroid2 = 140.17(3). | ||
Adding complex 1 to 2 equiv. of KN(TMS)2 in THF at room temperature results in an immediate color change from yellow to brown. From this mixture (tBuPN*)RuH(PhN(TMS)2) (4) was isolated in 97% yield (Fig. 2a). The 1H NMR spectrum of 4 in C6D6 shows a doublet at δ = −7.76 ppm and a doublet at δ = 3.49 ppm, which both integrate to 1H and are characteristic of a hydride and a methine linker of a dearomatized tBuPN ligand (tBuPN*), respectively. Similar to 3, the 1H NMR spectrum of 4 also displays five magnetically coupled multiplets (all integrating to 1H) between δ = 5.11 and 4.14 ppm and a large singlet at 0.25 ppm, which integrates to 18H. From this data, we concluded that complex 4 is the deprotonated analog of 3. This is supported by the X-ray crystal structure determination (Fig. 2b, ESI Section S6.2†) of brown single crystals of 4 obtained from a concentrated pentane solution at −40 °C.
The solid-state structure of 4 revealed a piano-stool type complex in which ruthenium is bound to a silylated aniline, a tBuPN* ligand, and a hydride, which was located in difference Fourier map. The found interatomic distances in the tBuPN* ligand are characteristic of a dearomatized pyridine ring and a deprotonated methylene linker (Fig. 2b), which agrees with the NMR data. This anionic binding mode of the bidentate ligand enforces the small N(1)–C(2)–C(1)–P(1) dihedral angle of 2.10(19)°. The coordination sphere of the metal center is best described as a distorted piano-stool geometry with a very long Ru–anilinecentroid distance of 1.7821(7) Å (only 4% of reported η6-arene Ru structures have this distance longer according to the Cambridge Structural Database,34 see ESI Fig. S167–S169†). The ruthenium is bound unevenly to all the carbons of the aniline moiety, and a slight ring slippage is observed away from the N(TMS)2 side with respect to the coordinated metal (0.194 Å). It is noticeable that the Ru–C bonds are clustered into two groups. Two of the bonds are longer compared to the other four (Ru–C = 2.3775(14)–2.4275(13) Å for C(15)–C(16) vs. Ru–C = 2.1776(14)–2.2409(13) Å for C(17)–C(20), see ESI Tables S7 and S8†). This type of arene coordination can also be considered as ‘η4+η2’ and is associated with the trans-effect of the tertiary phosphine35,36 as well as steric repulsion emerged between PhN(TMS)2 fragment and tBuPN* ligand. This explains the relatively large separation of the aromatic aniline resonances in the 1H NMR spectrum.
Mechanistic investigations
The reactivity observed for complex 1 is particularly remarkable given that KN(TMS)2 is typically regarded as a non-nucleophilic base, while benzene is typically inert to nucleophilic aromatic substitution. Hence, we set out to elucidate the underlying mechanism of this reaction. To ascertain the origin of the hydride in 4, we synthesized an isotopically labeled analog, 4-D (Fig. 3). For that, benzene-d6 was reduced with lithium metal and ethylenediamine in Et2O/EtOD according to a modified birch-like procedure,37 which gave a mixture of partially deuterated cyclohexadienes. A subsequent reaction with ruthenium trichloride gave partially deuterated Ru2Cl4(benzene)2 (5), which was used to synthesize 1-D with 61% deuteration of the coordinated benzene. Reacting 1-D with two equiv. of KN(TMS)2 yielded compound 4-D with 57% and 56% deuteration for the coordinated aniline and hydride, respectively. The relative integration of the signals in 1H NMR spectra of compound 4-D shows an equal degree of deuteration between the hydridic and aromatic resonances, confirming that the hydride originates from the benzene (ESI Fig. S45 and S46†). It is noteworthy that no H/D scrambling with the tBuPN ligand was observed in the product, excluding arene C–H bond activation via metal–ligand cooperation.38,39 | ||
| Fig. 3 Isotopic labeling experiments. Synthetic route towards compound 4-D. The purple balls represent the positions where a significant amount of deuterium was detected by qNMR spectroscopy. The reaction conditions: (i) Li metal, ethylenediamine, EtOD in THF at 0 °C; (ii) RuCl3·xH2O in EtOD (reflux). For more details on isotopic labeling see ESI Section S1.† | ||
Based on the isotopic labeling experiments, two plausible mechanistic pathways were hypothesized for the substitution reaction: (i) deprotonation of tBuPN with subsequent aromatic substitution of the hydride (Fig. 4a top) and (ii) aromatic substitution followed by deprotonation of the ligand (Fig. 4a bottom). To shed light on the operational mechanism, a series of low-temperature NMR experiments were performed involving the reaction between 1 and KN(TMS)2. After the addition of only 1 equiv. of KN(TMS)2 to 1 at −78 °C in THF-d8, 1H NMR analysis at −60 °C showed conversion to a single major species that neither contained a hydride nor a symmetrical benzene. The proton signals corresponding to the former aromatic ring of the benzene were found as six equally integrating multiplets in a wide range of δ = 5.10 to 2.76 ppm. Based on extensive 2D NMR analysis, we assign this compound as intermediate Int1 (Fig. 4b, ESI Fig. S66–S71†), which contains a η5-cyclohexadienyl ligand formed by nucleophilic addition of N(TMS)2− to the coordinated benzene, resembling the Jackson–Meisenheimer complex. The extremely upfield-shifted proton signal at δ = 2.76 ppm corresponds to the allylic proton of the cyclohexadienyl fragment. Surprisingly, the methylene linker of the complex is intact despite the addition of such a potent base as KN(TMS)2. Slow warming up of the reaction mixture to RT results in the decomposition of Int1 into a mixture of species that converted to predominantly complex 2-K after 16 hours at RT (ESI Fig. S72†). This shows that the nucleophilic addition that forms Int1 is a kinetically driven process, contrarily to the deprotonation of the ligand that leads to 2-K, which is a more thermodynamically favorable pathway. Moreover, it also shows that the nucleophilic addition is reversible (Fig. 4b).
 | ||
| Fig. 4 Plausible mechanisms and mechanistic investigations of the SNArH reaction with KN(TMS)2. (a) Plausible mechanistic pathway involving first deprotonation of the methylene linker of the tBuPN ligand followed by nucleophilic aromatic substitution of a hydride (top path) and a pathway wherein the SNArH precedes the deprotonation of the tBuPN ligand (bottom path). (b) Mechanistic NMR experiments at RT (i) and −60 °C (ii) featuring the structures of two kinetically-trapped metal-stabilized Jackson–Meisenheimer intermediates Int1 and Int2. | ||
1H NMR analysis at −60 °C of a mixture resulting from the addition of 2 equiv. of KN(TMS)2 to 1 in THF-d8 at −78 °C allowed us to observe a new species Int2. Similar to the analogous experiment with 1 equiv. of KN(TMS)2 that gave Int1, no resonances in the hydridic region were detected, and seven equally integrating resonances within δ = 5.28 and 3.10 ppm were found. Together with 1H–31P HMBC analysis, a broad signal at δ = 3.25 ppm was assigned as a methine proton, suggesting deprotonation of the tBuPN ligand (Fig. 4b, ESI Fig. S73–S78†). The other six signals were magnetically coupled, as shown by 1H–1H TOCSY NMR analysis, and were assigned as η5-cyclohexadienyl protons. From this data, we conclude that compound Int2 is another metal-stabilized Jackson–Meisenheimer intermediate, which forms upon the deprotonation of Int1. Warming up the reaction mixture to RT resulted in a clean formation of compound 4 already at −20 °C, supporting our findings regarding the nature of Int2 (ESI Fig. S79–S82†). The fact that compound 4 forms cleanly only upon warming up demonstrates that the hydride migration is the rate-determining step of the reaction from 1 to 4.
As noted above, nucleophilic addition to transition metal complexes has been well established since the 1960s. However, metal-stabilized Jackson–Meisenheimer intermediates are typically very stable and do not undergo hydride migration to the metal center. To gain insight into the role of the PN ligand in this unusual reactivity, we conducted a comparative study using a series of alternative ligands (see ESI, Section S2.3†). Specifically, we synthesized five analogous complexes featuring bidentate ligands with varying electronic and steric properties: 2-((di-tert-butylphosphaneyl)oxy)pyridine (PON), N-ethyl-N-(pyridin-2-ylmethyl)ethanamine (NN), 4,4′-di-tert-butyl-2,2′-bipyridine (tBu-bpy), 1,10-phenanthroline (phen), N1,N1,N2,N2-tetramethylethane-1,2-diamine (TMEDA). Remarkably, upon reacting them in an analogous manner to complex 1 with KN(TMS)2, only the reaction with [(PON)RuCl(C6H6)]PF6 led to the formation of the corresponding hydride complex [(PON)RuH(C6H5N(TMS)2)]PF6, which displayed similar spectral features as complex 3. The fact that the other complexes showed no reactivity toward nucleophilic aromatic substitution, underscores the critical role of ligand design in enabling SNArH reactivity. Furthermore, the successful hydride migration observed with the PON ligand demonstrates that the ligand deprotonation observed in 4 is not vital for the Ru-mediated SNArH.
Computational studies
To gain a deeper understanding of the mechanism of the SNArH reaction, density functional theory (DFT) calculations were performed using the ORCA 5.0.3 software at the B3LYP-D3(BJ)/def2-TZVPD/def2-ECP(Ru)//B3LYP-D3(BJ)/def2-SVP/def2-ECP(Ru) level of theory (see Methods for details). Given the complexity and potential inaccuracies associated with modeling the solvated structure of the complex associated with K+ and THF,40 the N(TMS)2− anion without its cationic partner was utilized to model the entire reaction. Both mechanistic scenarios depicted in Fig. 4a were analyzed – where nucleophilic addition to 1 (1-TS, Fig. 5) precedes deprotonation of the PN ligand or vice versa (1-TS′, ESI Fig. S123, see ESI Section S3 for more detail†). Consistent with the experimental results, the calculations show that the formation of metal-stabilized Jackson–Meisenheimer complex Int1 via 1-TS, featuring a barrier of 7.1 kcal mol−1, is more facile than the formation of deprotonated complex 2 via 1-TS′, which has a 3.0 kcal mol−1 higher barrier (for comparison of both pathways computed at RT see ESI Fig. S125†). The subsequent deprotonation of Int1 by a N(TMS)2− anion forms Int2-Cl with a barrier of 18.4 kcal mol−1. This deprotonation results in the dearomatization of the PN ligand, enhancing the electron-donating ability of the nitrogen in the pyridine moiety and rendering the metal center more electron-rich. As such, it facilitates Cl− dissociation to generate the required vacant site for hydride migration. Combined with the anionic nature of Int2-Cl, extrusion of Cl− to give Int2 is more favorable compared to an alternative pathway where Cl− extrusion occurs prior to PN ligand deprotonation (see ESI Fig. S124†). That said, our computations show that this alternative pathway is still feasible, and this is further underlined by the observation that the SNArH is also possible with the analogous complex featuring the PON ligand that cannot dearomatize. The final step that completes the SNArH reaction from Int2 to 4 traverses Int2-TS with a barrier of 14.5 kcal mol−1, and is best described as an intramolecular hydride migration. Aromatization of η5-cyclohexadienyl ligands has been proposed via indirect reductive elimination with metal hydrides41 and alkyls,42 but not via direct migration to the metal center. Notably, it was investigated for re-aromatization of a η5-cyclohexadienyl Rh complex, but calculations showed a far too high barrier of 49.9 kcal mol−1 for this to be feasible.43 Instead, a more favorable stepwise pathway involving ring slippage followed by allylic β-hydride elimination was found. This shows that the facile hydride transfer from Int2 to 4 features an exceptionally low barrier. Notably, based on the calculated reaction profile at 298.15 K, the barrier for the deprotonation step of Int1 via Int1-TS (ΔG‡1 = 18.4 kcal mol−1) is higher than that of the intramolecular hydride migration via Int2-TS (ΔG‡2 = 14.8 kcal mol−1), which seems inconsistent with the observation of Int2 in low-temperature NMR experiments. However, when calculated at the reaction temperature at which the experiment was performed (195.15 K), the barrier for the deprotonation decreases to 11.6 kcal mol−1, while the barrier for the intramolecular hydride migration remains nearly unaffected between the two temperatures. This drop in the barrier of Int1-TS can be attributed to the diminished entropic penalty for the bimolecular process at lower temperatures. As a result, at 195.15 K, the deprotonation barrier becomes lower than that of the hydride migration (see ESI Fig. S126† for the reaction profiles computed at 195.15 K). These results corroborate the experimental observation of Int2 at low temperatures and its selective conversion into 4 upon warming to RT.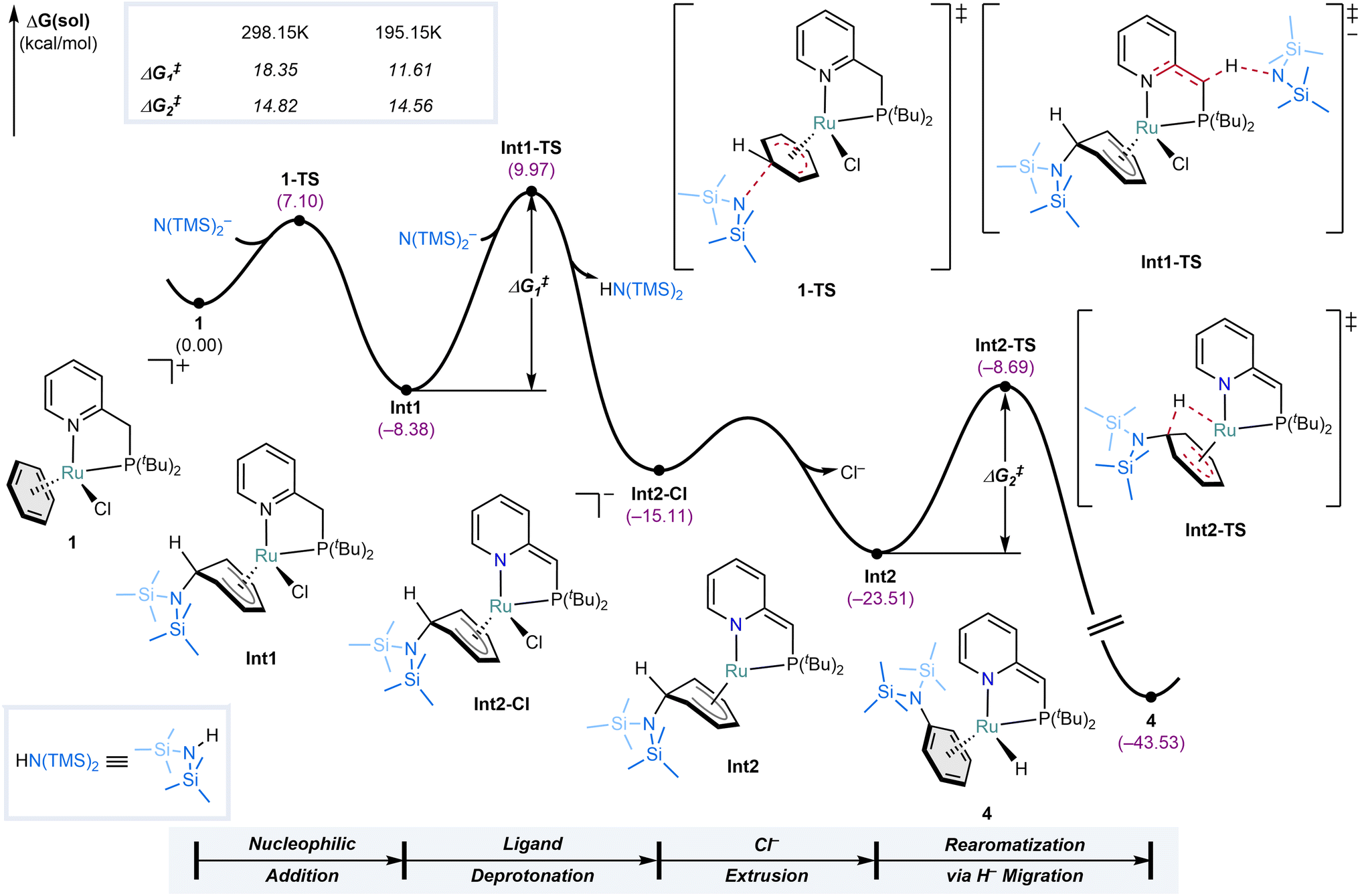 | ||
| Fig. 5 Computed reaction profile of the SNArH mediated by complex 1. Only the most facile pathway is shown. For details and other calculated energy profiles, see ESI Section S3.1.† | ||
Scope of the reaction
The state-of-the-art SNArH reactions using β-diketiminate complexes (Fig. 1d) are limited to intramolecular alkyl nucleophiles. Given that the reactivity described above involves an exogenous nucleophile that is commonly used as a non-nucleophilic bulky base, we investigated the scope of nucleophiles amenable to the Ru-mediated SNArH reaction. Motivated by both the need to obtain the metal-free substitution product and the difficulties in accurately determining spectroscopic yields with some nucleophiles used, we first developed a protocol that enables liberation of the silylated aniline in 4 (ESI Section S4.1†). Although it is known that arene exchange reactions on Ru are challenging, especially in neutral complexes featuring electron-rich arenes,44,45 we found that irradiation of a refluxing benzene solution of 4 with a 365 nm UV light for 72 h enables partial arene exchange to give “free” PhN(TMS)2 in a modest yield of 40% based on GC analysis (VI, Fig. 6). This confirms the strong binding energy between the ruthenium center and bis-silylated aniline (see ESI Section S3.3† for TD-DFT calculations). Although it is not optimal, we used this protocol to assess the scope of nucleophiles amenable to the SNArH reaction in 1. GC analysis of the reaction mixtures (after arene exchange) with C(sp3)-based nucleophiles n-butyl lithium (nBuLi) and benzyl potassium (BnK) showed the formation of the targeted butyl benzene (I) in 9% yield and diphenylmethane (II) in 31% yield. Similarly, a reaction with phenylmagnesium bromide as a C(sp2)-based nucleophile showed the formation of biphenyl (III) in 35% yield. A reaction with vinylmagnesium bromide to give styrene (IV) is incompatible with our protocol, given that the reaction product polymerizes under UV light.46 However, 1H NMR analysis of the reaction mixture after mixing 1 and vinyl magnesium bromide showed 21% formation of two hydride species at similar chemical shifts as those observed for complex 4 (ESI Fig. S145 and S146†). This suggests the formation of a similar complex as 4 but with coordinated styrene instead. Using different arene exchange protocols, triphenylphosphine (VI) and diphenylacetylene (VII) were generated by reaction with potassium diphenyl phosphide or lithium phenyl acetylide, respectively, albeit in only 6% yield. Although these non-optimized yields and protocols are far from practical, they demonstrate that the Ru-mediated SNArH reaction is compatible with a variety of nucleophiles. | ||
Fig. 6 SNArH on benzene mediated by complex 1 with various nucleophiles. I: n-butyllithium, II: benzyl potassium, III: phenylmagnesium bromide, IV: vinylmagnesium bromide (quantified based on the qNMR data of the corresponding complex), V: potassium diphenylphosphide (analyzed in the form of its oxide), VI: potassium bis(trimethylsilyl)amide, VII: lithium phenylacetylide (basic work-up was done instead of the UV irradiation). The yields were determined by GC analysis. * based on spectroscopic yield of the corresponding hydride complex; ** based on GC yield of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 following a reaction with H2O2; for more details, see ESI Section S4.2.† PPh3 following a reaction with H2O2; for more details, see ESI Section S4.2.† | ||
Conclusion
In conclusion, we have uncovered a new ruthenium-mediated pathway for nucleophilic aromatic substitution of hydrogen (SNArH) on benzene. Remarkably, this is the first example of SNArH on a transition metal arene complex using exogenous nucleophiles that do not require a leaving group on the arene nor subsequent oxidation steps. Our detailed mechanistic studies show that the reaction involves reversible rear-side addition of the nucleophile followed by an unprecedented intramolecular hydride migration from the metal-stabilized Jackson–Meisenheimer intermediate to the metal center. This last key step is facilitated by deprotonation of the tBuPN ligand, which makes the metal center more electron-rich, thereby promoting chloride dissociation and subsequent hydride migration. The reaction demonstrates broad versatility, accommodating diverse nucleophiles, including Csp3, Csp2, Csp, N, and P-based ones. We envision that this study opens up new avenues for developing new stoichiometric and catalytic SNArH reactions. To achieve this, our ongoing efforts are aimed at enabling efficient arene exchange and vacant site regeneration via hydride loss.Methods
Synthetic procedures
Note: see ESI Section S1† for synthetic procedures and characterization of the compounds presented in this study.Note: the product has moderate solubility only in DCM and MeCN. Washing with THF (note that as little as possible of THF should be used as 1 is partially soluble in THF) is required to get rid of byproducts of the reaction, the crystal structure of one of them – (tBuPN)2RuCl2– was fortuitously also obtained (see ESI Section S6.3†).
1H NMR (400 MHz, CD2Cl2, 298 K): δ = 9.24 (d, 3JH,H = 5.0 Hz, 1H), 7.87–7.80 (m, 1H), 7.44 (d, 3JH,H = 7.8 Hz, 1H), 7.39–7.34 (m, 1H), 6.11 (d, 3JH,P = 0.7 Hz, 6H), 3.89 (dd, 2JH,H = 16.4, 2JH,P = 8.7 Hz, 1H), 3.31 (dd, 2JH,H = 16.4, 2JH,P = 13.3 Hz, 1H), 1.57 (d, 3JH,P = 14.5 Hz, 9H), 1.21 (d, 3JH,P = 13.4 Hz, 9H).
13C{1H} NMR (101 MHz, CD2Cl2, 298 K): δ = 163.0 (d, 4JC,P = 3.1 Hz), 157.5 (s), 140.5 (d), 125.1 (s), 125.0 (s), 89.7 (d, 2JC,P = 2.4 Hz), 39.6 (d, 1JC,P = 2.3 Hz), 39.5 (d, 2JC,P = 3.1 Hz), 33.6 (d, 1JC,P = 23.7 Hz), 31.6 (d, 2JC,P = 2.3 Hz), 29.9 (d, 2JC,P = 2.7 Hz).
31P{1H} NMR (162 MHz, CD2Cl2, 298 K): δ = 90.8 (s, 1P), −144.4 (hept, 1JP,F = 710.8 Hz, 1P).
19F NMR (376 MHz, CD2Cl2, 298 K): δ = −72.7 (d, 1JF,P = 711.0 Hz, 6F).
Anal. calcd for C20H30ClNP2RuF6: C, 40.24; H, 5.07; N, 2.35. Found: C, 39.69; H, 5.04; N, 2.22.
ATR-IR (film, N2 flow): ν = 3090 (w), 2964 (m), 2924 (m), 2873 (w), 1607 (w), 1474 (m), 1441 (m), 1387 (w), 1373 (w), 1312 (w), 1269 (w), 1178 (w), 1024 (w), 876 (w), 835 (s), 776 (w), 734 (m), 702 (w), 621 (w), 557 (s), 493 (w), 460 (w) cm−1.
Note: the crude 1H NMR spectrum shows the formation of ∼50% species 2-K and ∼20% species 3 based on the relative integral values. For a cleaner synthesis route towards 2-K as well as an alternative route to the mixture of 2K and 3 see ESI Section S1.†
For 2-K:
1H NMR (400 MHz, THF-d8, 298 K): δ = 8.21 (ddd, J = 5.3, 1.6, 1.2 Hz, 1H), 7.62 (ddd, J = 7.9, 7.9, 1.5 Hz, 1H), 7.06 (dddd, J = 7.8, 5.3, 1.3, 1.3 Hz, 1H), 6.72 (dd, J = 7.9, 1.0 Hz, 1H), 5.82 (s, 6H), 3.33 (d, 2JH,P = 3.0 Hz, 1H), 1.36 (d, 3JH,P = 14.9 Hz, 9H), 1.08 (d, 3JH,P = 15.4 Hz, 9H).
31P{1H} NMR (162 MHz, THF-d8, 298 K): δ = 97.5 (s, 1P), −144.5 (hept, 1JP,F = 710.1 Hz, 1P).
For 3:
1H NMR (400 MHz, THF-d8, 298 K): δ = 8.86 (d, J = 5.4 Hz, 1H), 7.71 (t, J = 8.3 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.14 (dd, J = 7.6, 6.7 Hz, 1H), 6.27 (t, J = 6.2 Hz, 1H), 6.21 (d, J = 5.5 Hz, 1H), 5.69 (dd, J = 6.2, 1.5 Hz, 1H), 5.44–5.39 (m, 1H), 4.64 (dt, J = 5.9, 1.6 Hz, 1H), 3.64–3.56 (m, 1H, overlapped with a THF signal), 3.26 (dd, J = 17.2, 7.6 Hz, 1H), 1.34 (d, 3JH,P = 13.7 Hz, 9H), 1.25 (d, 3JH,P = 13.0 Hz, 9H), 0.30 (s, 18H), −7.77 (d, J = 42.5 Hz, 1H).
31P{1H} NMR (162 MHz, THF-d8, 298 K): δ = 111.8 (d*, 2JP,H = 11.2 Hz, 1P), −144.5 (hept, 1JP,F = 710.1 Hz, 1P).
*The doublet appears due to partial coupling with the hydride.
1H NMR (400 MHz, C6D6, 298 K): δ = 7.33 (dd, 3JH,H = 6.3, 4JH,H = 0.9 Hz, 1H), 6.53 (dddd, 3JH,H = 9.0, 3JH,H = 6.3, 5JH,P = 2.1, 4JH,H = 1.4 Hz, 1H), 6.41 (d, 3JH,H = 8.8 Hz, 1H), 5.38 (ddd, 3JH,H = 7.1, 3JH,H = 6.3, 4JH,H = 1.4 Hz, 1H), 5.11 (dd, 3JH,H = 6.2, 3JH,H = 5.9 Hz, 1H), 4.94 (dd, 3JH,H = 6.2, 3JH,H = 5.2 Hz, 1H), 4.77 (dd, 3JH,H = 6.0, 3JH,H = 4.9 Hz, 1H), 4.67 (dd, 3JH,H = 6.0, 3JH,H = 1.9 Hz, 1H), 4.14 (ddd, 3JH,H = 5.7, 4JH,H = 2.2, 4JH,H = 1.8 Hz, 1H), 3.49 (d, 2JH,P = 2.6 Hz, 1H), 1.31 (d, 3JH,P = 12.4 Hz, 9H), 1.27 (d, 3JH,P = 13.2 Hz, 9H), 0.25 (s, 18H), −7.76 (d, 2JH,P = 43.4 Hz, 1H).
13C{1H} NMR (101 MHz, C6D6, 298 K): δ = 170.9 (d, 2JC,P = 15.6 Hz), 154.4 (s), 130.8 (d, 4JC,P = 2.3 Hz), 130.7 (s), 115.2 (d, 3JC,P = 17.2 Hz), 101.4 (s), 92.4 (s), 90.6 (d, 2JC,P = 5.7 Hz), 83.4 (s), 77.4 (s), 74.1 (d, 2JC,P = 3.1 Hz), 62.3 (d, 1JC,P = 60.3 Hz), 38.3 (d, 1JC,P = 14.5 Hz), 36.2 (d, 1JC,P = 34.3 Hz), 31.1 (d, 2JC,P = 3.4 Hz), 30.2 (d, 2JC,P = 5.0 Hz), 3.3 (s).
31P{1H} NMR (162 MHz, C6D6, 298 K): δ = 98.8 (s).
ATR-IR (film, N2 flow): ν = 3045 (m), 2954 (s), 2864 (s), 2893 (m), 2034 (w, br), 1604 (s), 1535 (w), 1511 (w), 1488 (s), 1446 (s), 1381 (w), 1359 (w), 1358 (w), 1285 (m), 1253 (m), 1225 (m), 1205 (m), 1179 (w), 1146 (w), 1101 (w), 1017 (w), 1000 (m), 933 (m), 892 (s), 840 (m), 810 (m), 758 (w), 726 (w), 687 (w), 667 (w), 616 (w), 503 (w), 463 (w) cm−1.
Despite several attempts using spectroscopically pure samples, the reactive nature of 4 precluded obtaining a satisfactory elemental analysis.
X-ray crystal structure determination of 4
C26H47N2PRuSi2, Fw = 575.87, orange block, 0.41 × 0.40 × 0.17 mm3, monoclinic, P21/c (no. 14), a = 13.9298(4), b = 16.2626(4), c = 13.8275(4) Å, β = 112.534(2), V = 2893.25(15) Å3, Z = 4, Dx = 1.322 g cm−3, μ = 0.70 mm−1. The diffraction experiment was performed on a Bruker Kappa ApexII diffractometer with a sealed tube and Triumph monochromator (λ = 0.71073 Å) at a temperature of 150(2) K up to a resolution of (sinθ/λ)max = 0.65 Å−1. The crystal was broken into several fragments. Two orientation matrices were used for the intensity integration of the major fragments using the Eval15 software.53 Only the non-overlapping reflections were used for structure solution and refinement. A multi-scan absorption correction and scaling were performed with SADABS54 (correction range 0.68–0.75). A total of 41381 reflections were measured, 6635 reflections were unique (Rint = 0.021), and 6179 reflections were observed [I > 2σ(I)]. The structure was solved with Patterson superposition methods using SHELXT.55 Structure refinement was performed with SHELXL-2018 (ref. 56) on F2 of all reflections. Non-hydrogen atoms were refined freely with anisotropic displacement parameters. All hydrogen atoms were located in difference Fourier maps. Metal-bound hydrogen atom H1 and hydrogens H16–H20 of the coordinated phenyl group were refined freely with isotropic displacement parameters. All other hydrogen atoms were refined with a riding model. 329 parameters were refined with no restraints. R1/wR2 [I > 2σ(I)]: 0.0200/0.0507. R1/wR2 [all refl.]: 0.0217/0.0516. S = 1.045. Residual electron density between −0.42 and 0.49 e Å−3. Geometry calculations and checking for higher symmetry were performed with the PLATON program.57
Note: for further structural details see ESI Section S6.†
Computational details
All calculations were carried out using DFT58 as implemented in ORCA 5.0.359–61 with the B3LYP,62,63 including Grimme's D3 dispersion correction with Becke–Johnson damping.64–69 Geometry optimizations and analytical vibrational frequency calculations were carried out with the def2-SVP basis set70 with def2-ECP for Ru.71 For all optimized structures, the intermediates were confirmed with no imaginary vibrational frequency, while transition states showed a single imaginary frequency with a motion corresponding to the proper transitions. The solvated energies of optimized structures were re-evaluated by additional single-point calculations on each optimized geometry using the def2-TZVPD basis set.70 For all calculations, the RIJCOSX approximation72,73 was utilized with the auxiliary basis set def2/J.74 To model the solution environment for tetrahydrofuran, the solvation model based on density (SMD)75 was utilized with parameters that have been implemented in ORCA. TD-DFT calculations for modeling excited states were conducted as implemented in Q-Chem 5.4 software.76 Geometries from the optimized geometry with ORCA were utilized for the calculations of excited states. Single Excitation Configuration Interaction (CIS)77 and Tamm–Dancoff approximation78 were utilized to reduce the computation cost without damage to the quality of the results. The functional and basis set for the calculations of the excited state are identical to those for DFT calculations.
Data availability
All data can be found in the main text, Methods section, or the ESI Materials.† CCDC 2355856–2355858 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif. The spectroscopic files that support the findings of this study are openly available in the Yoda data repository at DOI: https://doi.org/10.24416/UU01-BYOFTG.Author contributions
S. M. and D. L. J. B. conceived the project. S. M. performed the experiments, including the synthesis, characterization, and analysis with support from P. G. D. H. and B. P. carried out computational calculations. M. L. performed crystallographic measurements, including the acquisition, solving, and interpretation of the crystal structures. D. L. J. B. supervised the project. The original draft was written by S. M. and was reviewed by M.-H. B. and D. L. J. B. with contributions by all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by the Netherlands Organization for Scientific Research (Project OCENW. M20.016 to D. L. J. B). We thank the Institute for Basic Science in Korea for financial support (IBS-R010-A1). The X-ray diffractometer has been financed by the Netherlands Organization for Scientific Research (NWO). We deeply thank Dr M.-E. Moret, Dr S. Tretiakov, E. Kounalis, R. Bienenmann, L. Killian, and L. Riddell for fruitful discussions and suggestions throughout the project.References
- F. Terrier, Modern nucleophilic aromatic substitution, John Wiley & Sons, Weinheim, 2013 Search PubMed.
- J. F. Bunnett and R. E. Zahler, Aromatic Nucleophilic Substitution Reactions, Chem. Rev., 1951, 49(2), 273–412 CrossRef CAS.
- N. E. Tay, W. Chen, A. Levens, V. A. Pistritto, Z. Huang, Z. Wu, Z. Li and D. A. Nicewicz, 19F− and 18F− arene deoxyfluorination via organic photoredox-catalysed polarity-reversed nucleophilic aromatic substitution, Nat. Catal., 2020, 3(9), 734–742 CrossRef CAS PubMed.
- C. N. Neumann, J. M. Hooker and T. Ritter, Concerted nucleophilic aromatic substitution with 19F− and 18F−, Nature, 2016, 534(7607), 369–373 CrossRef CAS PubMed.
- E. E. Kwan, Y. Zeng, H. A. Besser and E. N. Jacobsen, Concerted nucleophilic aromatic substitutions, Nat. Chem., 2018, 10(9), 917–923 CrossRef CAS PubMed.
- C. Friedel and J. M. Crafts, Sur une nouvelle méthode générale de synthèse d’hydrocarbures, d’acétones, etc, Compt. Rend., 1877, 84, 1392–1395 Search PubMed.
- G. A. Olah, Aromatic substitution. XXVIII. Mechanism of electrophilic aromatic substitutions, Acc. Chem. Res., 1971, 4(7), 240–248 CrossRef CAS.
- B. Galabov, D. Nalbantova, P. V. R. Schleyer and H. F. Schaefer, Electrophilic Aromatic Substitution: New Insights into an Old Class of Reactions, Acc. Chem. Res., 2016, 49(6), 1191–1199 CrossRef CAS PubMed.
- K. Błaziak, W. Danikiewicz and M. Mąkosza, How Does Nucleophilic Aromatic Substitution Really Proceed in Nitroarenes? Computational Prediction and Experimental Verification, J. Am. Chem. Soc., 2016, 138(23), 7276–7281 CrossRef PubMed.
- M. Mąkosza, Electrophilic and nucleophilic aromatic substitution: Analogous and complementary processes, Russ. Chem. Bull., 1996, 45(3), 491–504 CrossRef.
- M. Mąkosza, Electrophilic and Nucleophilic Aromatic Substitutions are Mechanistically Similar with Opposite Polarity, Chem.–Eur. J., 2020, 26(67), 15346–15353 CrossRef PubMed.
- M. Mąkosza, Nucleophilic substitution of hydrogen in electron-deficient arenes, a general process of great practical value, Chem. Soc. Rev., 2010, 39(8), 2855–2868 RSC.
- O. N. Chupakhin and V. N. Charushin, Recent advances in the field of nucleophilic aromatic substitution of hydrogen, Tetrahedron Lett., 2016, 57(25), 2665–2672 CrossRef CAS.
- M. Makosza and J. Winiarski, Vicarious nucleophilic substitution of hydrogen, Acc. Chem. Res., 1987, 20(8), 282–289 CrossRef CAS.
- M. Makosza and A. Kwast, Vicarious nucleophilic substitution of hydrogen. Mechanism and orientation, J. Phys. Org. Chem., 1998, 11(5), 341–349 CrossRef CAS.
- R. J. Card and W. S. Trahanovsky, Arene-metal complexes. 12. Reaction of (η6-benzene)tricarbonylchromium with n-butyllithium, J. Org. Chem., 1980, 45(13), 2555–2559 CrossRef CAS.
- M. F. Semmelhack, H. T. Hall Jr, R. Farina, M. Yoshifuji, G. Clark, T. Bargar, K. Hirotsu and J. Clardy, η5-Cyclohexadienyltricarbonylchromium(0) complexes from addition of carbon nucleophiles to η6-benzenetricarbonylchromium(0). Formation, chemical and spectroscopic features, and x-ray diffraction analysis, J. Am. Chem. Soc., 1979, 101(13), 3535–3544 CrossRef CAS.
- M. F. Semmelhack, G. R. Clark, J. L. Garcia, J. J. Harrison, Y. Thebtaranonth, W. Wulff and A. Yamashita, Addition of carbon nucleophiles to arene-chromium complexes, Tetrahedron, 1981, 37(23), 3957–3965 CrossRef CAS.
- F. Rose-Munch and E. Rose, cine and tele Nucleophilic Substitutions in (η6-Arene)tricarbonylchromium and Tricarbonyl(η5-cyclohexadienyl)manganese Complexes, Eur. J. Inorg. Chem., 2002, 2002(6), 1269–1283 CrossRef.
- M. Brookhart, A. R. Pinhas and A. Lukacs, Reaction of lithium dimethylcuprate with C6H6Mn(CO)3+. Observation of methyl group migration from manganese to the arene ring in C6H6(CO)2MnMe, Organometallics, 1982, 1(12), 1730–1731 CrossRef CAS.
- G. Winkhaus, L. Pratt and G. Wilkinson, 738. π-Cyclohexadienylmanganese tricarbonyl and related compounds, J. Chem. Soc., 1961, 3807–3813 RSC.
- G. A. M. Munro and P. L. Pauson, Alkyl and Acyl Dicarbonylarenemanganese Complexes: The Reduction of Metal-Bound Carbonyl Groups, Isr. J. Chem., 1976, 15(3–4), 258–261 CrossRef.
- R. C. Cambie, S. A. Coulson, L. G. Mackay, S. J. Janssen, P. S. Rutledge and P. D. Woodgate, Recations of cationic (η6-chloroarene) complexes of iron and ruthenium with some O-silyl and C-silyl compounds, J. Organomet. Chem., 1991, 409(3), 385–409 CrossRef CAS.
- A. J. Pearson and J. N. Heo, Approaches to the Fully Functionalized DEF Ring System of Ristocetin A via Highly Selective Ruthenium-Promoted SNAr Reaction, Org. Lett., 2000, 2(19), 2987–2990 CrossRef CAS PubMed.
- B. Therrien, Functionalised η6-arene ruthenium complexes, Coord. Chem. Rev., 2009, 253(3–4), 493–519 CrossRef CAS.
- H. Werner, R. Werner, C. Burschka and V. Aromaten(phosphan)metall-Komplexe, Zur Addition von Carbanionen an (Benzol)ruthenium(II)- und -osmium(II)-Komplexe. Kristall- und Molekülstruktur von (exo-6-n-C4H9-η5-C6H6)OsI(PMe3)2, Chem. Ber., 1984, 117(1), 152–160 CrossRef CAS.
- K. Chen, Y. Ma, Y. Lin, J. Y. Li and H. Shi, Ruthenium/η5-Phenoxo-Catalyzed Amination of Phenols with Amines, J. Am. Chem. Soc., 2024, 146(23), 15833–15842 CrossRef CAS PubMed.
- T. Schulte, Z. Wang, C. C. Li, A. Hamad, F. Waldbach, J. Pampel, R. Petzold, M. Leutzsch, F. Bahns and T. Ritter, Ruthenium Phenoxo Complexes: An Isolobal Ligand to Cp with Improved Properties, J. Am. Chem. Soc., 2024, 146(23), 15825–15832 CrossRef CAS PubMed.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Organocalcium-mediated nucleophilic alkylation of benzene, Science, 2017, 358(6367), 1168–1171 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Nucleophilic Aromatic Substitution at Benzene with Powerful Strontium Hydride and Alkyl Complexes, Angew. Chem., Int. Ed., 2019, 58(16), 5396–5401 CrossRef PubMed.
- G. M. Richardson, I. Douair, S. A. Cameron, J. Bracegirdle, R. A. Keyzers, M. S. Hill, L. Maron and M. D. Anker, Hydroarylation of olefins catalysed by a dimeric ytterbium (II) alkyl, Nat. Commun., 2021, 12(1), 3147 CrossRef CAS PubMed.
- Z. W. Qu, H. Zhu, R. Streubel and S. Grimme, Organo-Group 2 Metal-Mediated Nucleophilic Alkylation of Benzene: Crucial Role of Strong Cation−π Interaction, ACS Catal., 2023, 13(3), 1686–1692 CrossRef CAS.
- J. R. Khusnutdinova and D. Milstein, Metal–Ligand Cooperation, Angew. Chem., Int. Ed., 2015, 54(42), 12236–12273 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr. B, 2016, 72(2), 171–179 CrossRef CAS PubMed.
- D. Arquier, L. Vendier, K. Miqueu, J.-M. Sotiropoulos, S. Bastin and A. Igau, Crucial Role of the Amidine Moiety in Methylenamino Phosphine-Type Ligands for the Synthesis of Tethered η6-Arene-η1-P Ruthenium(II) Complexes: Experimental and Theoretical Studies, Organometallics, 2009, 28(17), 4945–4957 CrossRef CAS.
- M. A. Bennett, G. B. Robertson and A. K. Smith, Divalent ruthenium complexes containing non-planar hexahapto-benzene, J. Organomet. Chem., 1972, 43(2), C41–C43 CrossRef CAS.
- J. Burrows, S. Kamo and K. Koide, Scalable Birch reduction with lithium and ethylenediamine in tetrahydrofuran, Science, 2021, 374(6568), 741–746 CrossRef CAS PubMed.
- L. Schwartsburd, M. A. Iron, L. Konstantinovski, E. Ben-Ari and D. Milstein, A Dearomatized Anionic PNP Pincer Rhodium Complex: C–H and H–H Bond Activation by Metal–Ligand Cooperation and Inhibition by Dinitrogen, Organometallics, 2011, 30(10), 2721–2729 CrossRef CAS.
- E. Ben-Ari, G. Leitus, L. J. W. Shimon and D. Milstein, Metal−Ligand Cooperation in C−H and H2 Activation by an Electron-Rich PNP Ir(I) System: Facile Ligand Dearomatization−Aromatization as Key Steps, J. Am. Chem. Soc., 2006, 128(48), 15390–15391 CrossRef CAS PubMed.
- J. A. Spivey and D. B. Collum, Potassium Hexamethyldisilazide (KHMDS): Solvent-Dependent Solution Structures, J. Am. Chem. Soc., 2024, 146(26), 17827–17837 CrossRef CAS PubMed.
- R. H. Crabtree and C. P. Parnell, Arene and cyclohexadienyl complexes as intermediates in the selective catalytic dehydrogenation of cyclohexenes to arenes, Organometallics, 1985, 4(3), 519–523 CrossRef CAS.
- M. B. Fischer, E. J. James, T. J. McNeese, S. C. Nyburg, B. Posin, W. Wong-Ng and S. S. Wreford, Hydrogen-transfer reactions catalyzed by low-valent, tertiary phosphine complexes of zirconium. Molecular structure of hydrido(η5-cyclooctadienyl)bis[1,2-bis(dimethylphosphino)ethane]zirconium, J. Am. Chem. Soc., 1980, 102(15), 4941–4947 CrossRef CAS.
- W. Q. Wu, Y. Lin, Y. Li and H. Shi, Catalytic Dehydrogenative (3 + 2) Cycloaddition of Alkylbenzenes via π-Coordination, J. Am. Chem. Soc., 2023, 145(17), 9464–9470 CrossRef CAS PubMed.
- N. V. Shvydkiy and D. S. Perekalin, Reactions of arene replacement in transition metal complexes, Coord. Chem. Rev., 2020, 411, 213238 CrossRef CAS.
- M. G. Choi, T. C. Ho and R. J. Angelici, Arene Binding Affinities in [CpRu(η6-arene)]+ Complexes: Models for the Adsorption of Arenes on Hydrodesulfurization Catalysts, Organometallics, 2008, 27(6), 1098–1105 CrossRef CAS.
- O. F. Olaj, H. F. Kauffmann and J. W. Breitenbach, The Diels-Alder intermediate as a chain transfer agent in spontaneous styrene polymerization 1. New evidence from the kinetic analysis of photoinitiated polymerization, Makromol. Chem., 1976, 177(10), 3065–3071 CrossRef CAS.
- P. M. Fanara, S. N. MacMillan and D. C. Lacy, Planar-Locked Ru-PNN Catalysts in 1-Phenylethanol Dehydrogenation, Organometallics, 2020, 39(20), 3628–3644 CrossRef CAS.
- M. L. Scheuermann, K. A. Grice, M. J. Ruppel, M. Roselló-Merino, W. Kaminsky and K. I. Goldberg, Hemilability of P(X)N-type ligands (X = O, N–H): rollover cyclometalation and benzene C–H activation from (P(X)N)PtMe2 complexes, Dalton Trans., 2014, 43(31), 12018–12025 RSC.
- B. S. Kim, J. Jiménez, F. Gao and P. J. Walsh, Palladium-Catalyzed Benzylic C–H Arylation of Azaarylmethylamines, Org. Lett., 2015, 17(23), 5788–5791 CrossRef CAS PubMed.
- C. Keck, C. Maichle-Moessmer and H. F. Bettinger, Photo electron transfer induced desilylation of N, N-bis (trimethylsilyl) aminodibenzoborole to aminodibenzoborole, Chem. Commun., 2019, 55(52), 7470–7473 RSC.
- Y. Wang, W. X. Zhang, Z. Wang and Z. Xi, Procedure-controlled selective synthesis of 5-acyl-2-iminothiazolines and their selenium and tellurium derivatives by convergent tandem annulation, Angew. Chem., Int. Ed., 2011, 50(35), 8122–8126 CrossRef CAS PubMed.
- C. B. Van Beek, N. P. Van Leest, M. Lutz, S. D. De Vos, R. J. K. Gebbink, B. De Bruin and D. L. J. Broere, Combining metal–metal cooperativity, metal–ligand cooperativity and chemical non-innocence in diiron carbonyl complexes, Chem. Sci., 2022, 13(7), 2094–2104 RSC.
- A. M. Schreurs, X. Xian and L. M. Kroon-Batenburg, EVAL15: a diffraction data integration method based on ab initio predicted profiles, J. Appl. Crystallogr., 2010, 43(1), 70–82 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination, J. Appl. Crystallogr., 2015, 48(1), 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr. A, 2015, 71(1), 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71(Md), 3–8 Search PubMed.
- A. L. Spek, Structure validation in chemical crystallography, Biol. Crystallogr., 2009, 65(2), 148–155 CrossRef CAS PubMed.
- R. G. Parr, Density Functional Theory of Atoms and Molecules, in Horizons of Quantum Chemistry, ed. K. Fukui, B. Pullman, Springer Netherlands, Dordrecht, 1980, pp. 5–15, DOI:10.1007/978-94-009-9027-2_2.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2(1), 73–78 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, WIREs. Comput. Mol. Sci., 2018, 8(1), e1327 CrossRef.
- F. Neese, Software update: The ORCA program system—Version 5.0, WIREs. Comput. Mol. Sci., 2022, 12(5), e1606 CrossRef.
- A. D. Becke, A new mixing of Hartree-Fock and local density-functional theories, J. Chem. Phys., 1993, 98(2), 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37(2), 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- J. C. Slater and J. C. Phillips, Quantum Theory of Molecules and Solids Vol. 4: The Self-Consistent Field for Molecules and Solids, Phys. Today, 1974, 27, 49 CrossRef.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58(8), 1200–1211 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38(6), 3098–3100 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Energy-adjusted ab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77(2), 123–141 CrossRef CAS.
- F. Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix, J. Comput. Chem., 2003, 24(14), 1740–1747 CrossRef CAS PubMed.
- B. Helmich-Paris, B. de Souza, F. Neese and R. Izsák, An improved chain of spheres for exchange algorithm, J. Chem. Phys., 2021, 155(10), 104109 CrossRef CAS PubMed.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- E. Epifanovsky, A. T. Gilbert, X. Feng, J. Lee, Y. Mao and N. Mardirossian, et al., Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155(8), 084801 CrossRef CAS PubMed.
- J. B. Foresman, M. Head-Gordon, J. A. Pople and M. J. Frisch, Toward a systematic molecular orbital theory for excited states, J. Phys. Chem., 1992, 96(1), 135–149 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 314(3–4), 291–299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC [2355856–2355858]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02090e |
| This journal is © The Royal Society of Chemistry 2025 |