
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplification effect of side group regulation via imidazolate linkages of covalent organic frameworks for efficient oxygen reduction†
Mengyuan Chenab,
Zhiqiang Zhub,
Youxin Ji*a,
Xiangtao Kong c,
Diandian Han*b and
Lipeng Zhai*b
c,
Diandian Han*b and
Lipeng Zhai*b
aNational Engineering Research Center for Advanced Polymer Processing Technology, The Key Laboratory of Material Processing and Mold of Ministry of Education, College of Materials Science and Engineering, Zhengzhou University, Zhengzhou, 450002, P. R. China. E-mail: yxji@zzu.edu.cn
bDepartment Henan Key Laboratory of Functional Salt Materials, Center for Advanced Materials Research, Zhongyuan University of Technology, Zhengzhou 450007, P. R. China. E-mail: 6788@zut.edu.cn; zhailp@zut.edu.cn
cHenan Key Laboratory of New Optoelectronic Functional Materials, College of Chemistry and Chemical Engineering, Anyang Normal University, Anyang 455000, P. R. China
First published on 26th May 2025
Abstract
Metal-free covalent organic framework (COF) materials are promising catalysts for the oxygen reduction reaction (ORR). However, it is difficult to markedly modulate their catalytic performance by only changing the side groups because their partially conjugated linkages weaken the influence of the electronic and charge transfer properties along the frameworks. Here, we demonstrated the amplification effect of micro-changes in the side units of COFs by constructing imidazole linkages for the ORR, and four different electron effect groups (H, F, OMe, and OH) were introduced. Owing to the conjugated linkages, electronic effects can be transmitted to the active sites through the skeleton. Notably, the half-wave potential (E1/2) and mass activity of OH-COF were 0.80 V vs. RHE and 12.43 A g−1, respectively, which were 180 mV more positive and 11 times more than those of H-COF, indicating the superior catalytic activity of OH-COF. Theoretical calculations revealed that different side groups influenced the binding ability of the intermediates and thus contributed to the modulated catalytic properties. This work demonstrates that a small change in the pore surface leads to substantial variations in the electrocatalytic performance, offering valuable insights into the relationship between the structural design and performance of COFs as electrocatalysts.
Introduction
The oxygen reduction reaction (ORR) is essential for the cathodic reaction in fuel cells and metal–air batteries.1–8 Metal-free carbon-based catalysts have gained popularity for catalysing the ORR owing to their cheap and abundant sources, high activity, stability, and large specific surface area.9–11 Strategies such as defect engineering,12–14 heteroatom doping,15,16 and surface modification17–20 have been used to boost the electrocatalytic behaviour of metal-free carbon materials. However, the active sites of carbon-based materials cannot be precisely regulated, and the location and number of catalytically active sites are still a major challenge.21–26 Hence, the fabrication of metal-free catalysts with accurate and plentiful active sites is critical for the ORR.Covalent organic frameworks (COFs) represent a kind of crystalline porous material, fabricated by thermodynamic reversible polymerization of small organic molecular monomers with periodic topological structures.27–33 With the advantages of structural designability, functional diversity, high specific surface area, uniform pore distribution, and excellent stability, COFs are generally applied in gas storage and separation,34 catalysis,35–38 energy storage,39–42 and sensing.43 Meanwhile, the excellent charge transport capacity and predictable active sites of COFs make them possible to be employed as electrocatalysts for the ORR,44–49 oxygen evolution reaction (OER),50 hydrogen evolution reaction (HER),51,52 and carbon dioxide reduction reaction (CO2RR).53–55 Numerous studies have introduced metals into COFs to improve the catalytic performance of the catalyst.56,57 Single-atom metals can be integrated into the COF skeleton as catalytic sites, which are conducive to improving the electronic environment of the COF skeleton and increasing the active sites, thereby improving the catalytic activity of COFs. For instance, Kundu et al. constructed triazine-based COFs post-modified with metal ions such as Ni, Co, and Fe, which displayed superior performance for the OER and HER.58 Feng et al. synthesized a series of metalloporphyrin-based COFs exhibiting high catalytic activity for the ORR by introducing N-hybrid porphyrins into metal centers.59 However, the metal sites of metal-doped COFs are easily oxidized or inactivated and cannot exist stably, which limits their commercial application.60,61 Therefore, metal-free COFs have been considered potential candidates for the ORR.62–67 In 2020, Fang et al. first constructed metal-free COFs for the ORR.68 Subsequently, aiming to enhance the catalytic activity, selectivity, and stability, various building blocks with different kinds of atoms and different linkages have been developed.69–71 However, it is still difficult to modulate the catalytic performance by changing only the side chain groups of COFs. Because most of these catalytic COFs are linked by non-conjugated or semi-conjugated connection linkages, their corresponding π-electron delocalization/transferring abilities via linkages between monomers are weak, resulting in the interaction between the side groups and the catalytic sites being isolated.72–74
Herein, we sought to amplify the modulatory effects of the side groups by constructing imidazole-linked COFs. Unlike other linkages, imidazole linkages have excellent chemical stability and a conjugated π-electron system and provide fast electronic/charge transport paths.75 To modulate the catalytic properties of COFs, different substituent functional groups (–H, –F, –OMe, and –OH) were introduced along the pore walls of the imidazolate COFs (R-COFS, R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, F, OMe, OH) and constructed by multicomponent reactions (MCRs) (Scheme 1). The OH-COF, OMe-COF and F-COF electron donating/-withdrawing features exhibited higher ORR electrocatalytic activity than H-COF in alkaline electrolytes, demonstrating that the electronic effect of the substituent functional group can modulate the electronic structure and change the charge distribution throughout the COFs. In addition, OH-COF with a strong electron-donating effect group (–OH) exhibited superior ORR catalytic performance to OMe-COF with a moderate electron-donating effect group (–OMe) and F-COF with a strong electron-accepting effect group (–F). Therefore, groups with stronger electronic capabilities can generate stronger conjugation effects and increase the electron density of imidazole through the electron push–pull effect enhancing the ORR electrocatalytic activity.
H, F, OMe, OH) and constructed by multicomponent reactions (MCRs) (Scheme 1). The OH-COF, OMe-COF and F-COF electron donating/-withdrawing features exhibited higher ORR electrocatalytic activity than H-COF in alkaline electrolytes, demonstrating that the electronic effect of the substituent functional group can modulate the electronic structure and change the charge distribution throughout the COFs. In addition, OH-COF with a strong electron-donating effect group (–OH) exhibited superior ORR catalytic performance to OMe-COF with a moderate electron-donating effect group (–OMe) and F-COF with a strong electron-accepting effect group (–F). Therefore, groups with stronger electronic capabilities can generate stronger conjugation effects and increase the electron density of imidazole through the electron push–pull effect enhancing the ORR electrocatalytic activity.
 | ||
| Scheme 1 Design strategy of imidazole-linked COFs as electrocatalysts for the ORR. | ||
Results and discussion
A set of robust imidazole-linked COFs (H-COF, F-COF, OMe-COF, and OH-COF) with similar skeletons but different substituent functional groups (H, F, OMe, and OH) over the pore channels was synthesized using a one-pot three-component Debus–Radziszewski condensation reaction under solvothermal conditions (Fig. 1a).76,77 The H-COF was synthesized using 2,7-ditert-butylpyrene-4,5,8,10-tetraone (t-BuPyT), ammonium acetate (NH4OAc), and 1,3,5-tris (4-formylphenyl) benzene (TFPB) in a mixed solution of 1,4-dioxane/mesitylene (0.2 ml/0.8 ml) at 150 °C for 5 d. Similarly, F-COF, OMe-COF, and OH-COF were synthesized using 1,3,5-tris (3-fluoro-4-formylphenyl) benzene (TFFPB) or 5′-(4-formyl-3-methoxyphenyl)-3,3′′-5′-(4-formyl-3-methoxyphenyl)-3,3′′-dimethoxy-[1,1′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3′,1′′-terphenyl]-4,4′′-dicarbaldehyde (TOMeFPB) or 5′-(4-formyl-3-hydroxyphenyl)-3,3′′-dihydroxy-[1,1′:3′,1′′-terphenyl]-4,4′′-dicarbaldehyde (THOFPB), respectively, with t-BuPyT and NH4OAc in the mixed solution of 1,4-dioxane/mesitylene (0.5 ml/0.5 ml) at 150 °C for 5 d. The yields of the H-COF, F-COF, OMe-COF, and OH-COF were 81%, 80%, 85%, and 84%, respectively. As shown in the top views of these COFs (Fig. 1b), the substituent functional groups (F, H, OMe, and OH) were uniformly tethered to the pore walls.
:3′,1′′-terphenyl]-4,4′′-dicarbaldehyde (TOMeFPB) or 5′-(4-formyl-3-hydroxyphenyl)-3,3′′-dihydroxy-[1,1′:3′,1′′-terphenyl]-4,4′′-dicarbaldehyde (THOFPB), respectively, with t-BuPyT and NH4OAc in the mixed solution of 1,4-dioxane/mesitylene (0.5 ml/0.5 ml) at 150 °C for 5 d. The yields of the H-COF, F-COF, OMe-COF, and OH-COF were 81%, 80%, 85%, and 84%, respectively. As shown in the top views of these COFs (Fig. 1b), the substituent functional groups (F, H, OMe, and OH) were uniformly tethered to the pore walls.
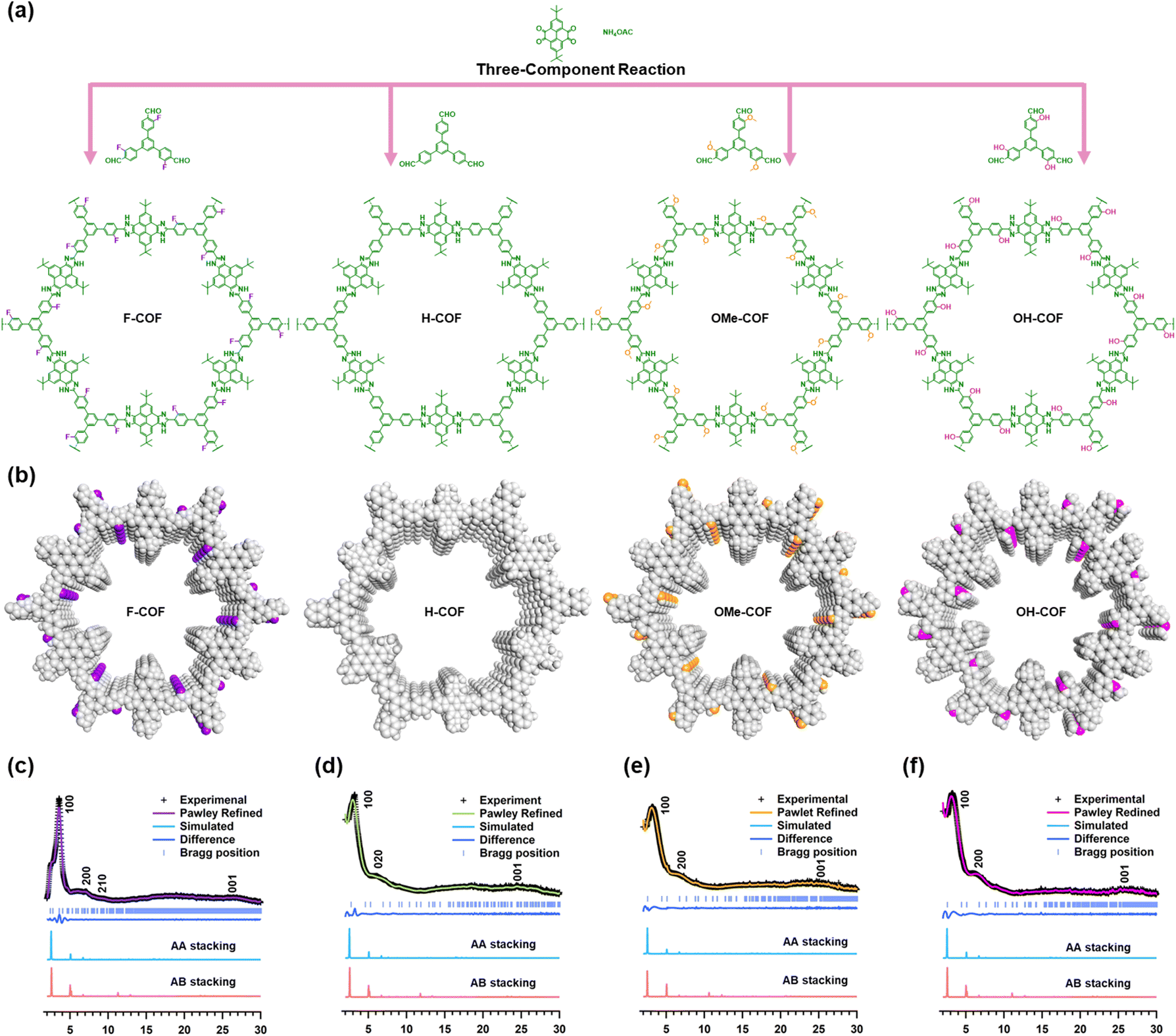 | ||
| Fig. 1 Synthesis and PXRD patterns of F-COF, H-COF, OMe-COF and OH-COF. (a) Schematic synthesis route of H-COF, F-COF, OMe-COF, and OH-COF. (b) Top view structures of the as-prepared COFs (purple, white, orange, and pink spheres represent F, H, OMe, and OH groups). PXRD patterns of (c) F-COF, (d) H-COF, (e) OMe-COF, and (f) OH-COF: the experimental curves, Pawley-refined patterns, the discrepancies between experimental and Pawley-refined PXRD profiles, and theoretical simulations based on AA stacking and AB stacking modes. | ||
X-ray powder diffraction (PXRD) was employed to investigate the crystalline structure of the R-COFs.78 The diffraction peaks of H-COF were observed at 3.18°, 6.13°, and 25.34°, corresponding to the 100, 020, and 001 crystal planes, respectively (Fig. 1c). Similarly, the diffraction peaks of the F-COF appeared at 3.58°, 6.36°, 7.18°, and 26.70°, corresponding to the 100, 200, 210, and 001 crystal planes, respectively (Fig. 1d). Three obvious diffraction peaks were observed at 3.28°, 6.80°, and 24.95°, corresponding to the 100, 200, and 001 crystal planes of OMe-COF (Fig. 1e). The OH-COF displayed peaks at 3.41°, 6.55°, and 26.01°, corresponding to the 100, 200, and 001 crystal planes, respectively (Fig. 1f). The density functional tight binding method was used to simulate the corresponding theoretical structures of the R-COFs. The comparison of the PXRD patterns derived from Pawley refinements with the experimental PXRD curves for the four synthesized COFs revealed minor discrepancies (P6, a = b = 40.22 Å, c = 5.39 Å, Rwp = 6.91% and Rp = 4.49% for H-COF; P6, a = b = 40.21 Å, c = 5.56 Å, Rwp = 6.79% and Rp = 4.51% for F-COF; P6, a = b = 40.15 Å, c = 5.48 Å, Rwp = 5.94% and Rp = 3.42% for OMe-COF; P6, a = b = 40.18 Å, c = 5.52 Å, Rwp = 4.36% and Rp = 3.24% for OH-COF) (Fig. 1c–f). In addition, the relative stacking energies of R-COFs reveal that the AA stacking is more stable than AB (H-COF: ΔE = −33.23 kcal mol−1; F-COF: ΔE = −40.88 kcal mol−1; OMe-COF: ΔE = −37.99 kcal mol−1; OH-COF: ΔE = −53.16 kcal mol−1), indicating that the theoretical simulation patterns from AA stacking mode were in good agreement with the experimental results (Fig. 1c–f).
N2 adsorption–desorption measurements were conducted to characterize the permanent porosity of the R-COFs. All four COFs exhibit characteristic type-I N2 adsorption isotherms, suggesting the presence of microporous structures (Fig. 2a). The Brunauer–Emmett–Teller method was used to obtain specific surface areas of H-COF, F-COF, OMe-COF and OH-COF, which were 467.89, 538.36, 628.64, and 664.57 m2 g−1, respectively. Employing nonlocal density functional theory to analyse the pore size allocation of R-COFs revealed that the prominent peak of these COFs was located at 1.57 nm (Fig. S1†), which aligns well with the theoretical values derived from the AA stacking mode.
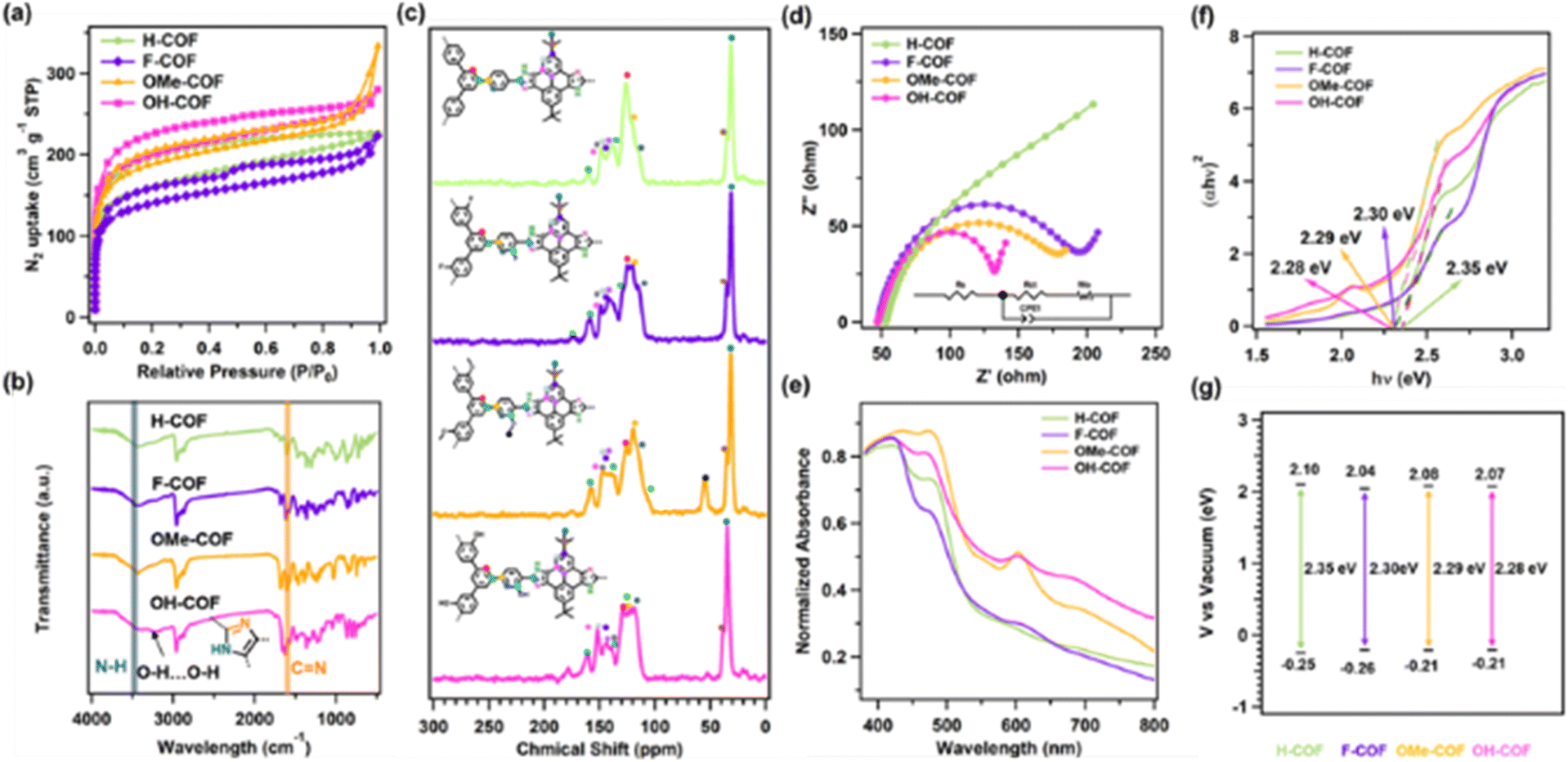 | ||
| Fig. 2 Structural characterization, EIS and UV/vis spectroscopy spectra of H-COF, F-COF, OMe-COF and OH-COF. (a) Nitrogen sorption isotherm curves. (b) FT-IR curves. (c) The 13C NMR spectra, (d) Nyquist plots of electrochemical impedance spectroscopy (EIS), (e) UV/Visible diffuse reflectance plots, (f) Tauc plots and (g) LUMO and HOMO levels of H-COF (green), F-COF (purple), OMe-COF (orange) and OH-COF (pink), respectively. | ||
The morphologies of the R-COFs were analyzed using field emission scanning electron microscopy (FE-SEM) and high-resolution transmission electron microscopy (HR-TEM). The FE-SEM images depicted aggregated regular spherical morphology of H-COF, F-COF, and OMe-COF with diameters of approximately 2.6, 8, and 1 μm (Fig. S2†), respectively. The OH-COF exhibited an aggregated irregular spherical morphology with a diameter of approximately 1.2 μm. In addition, specific elements were evenly distributed in the COF structures, as shown in the energy dispersive X-ray spectroscopy mapping images (Fig. S3–S6†). Notably, the decrease in the size of OH-COF relative to that of H-COF may be due to the influence of the intermolecular hydrogen bonds in OH-COF. The TEM images further illustrate the spherical morphology of the R-COFs (Fig. S7–S10†). The thermogravimetric results obtained under N2 and air atmospheres indicated that the thermal decomposition temperature of R-COFs reached 400 °C, demonstrating excellent stability of R-COFs (Fig. S11†).
The chemical structures and functional groups of the R-COFs were determined using Fourier transform infrared spectroscopy. All COFs showed distinct stretching vibration peaks near 1620 and 3450 cm−1, corresponding to the typical signal of the CN and N–H bonds of imidazole rings in the COF structures, respectively (Fig. 2b). Meanwhile, the almost complete disappearance of the H–CO and CO stretching vibration peaks indicated that the aldehyde and carbonyl groups reacted almost completely, further confirming the successful synthesis of the R-COFs (Fig. S12–S15†). Moreover, the OH-COF exhibited a broad stretching vibration peak corresponding to intermolecular hydrogen bonding (O–H) at 3210 cm−1 (Fig. S15†). In addition, solid-state nuclear magnetic resonance (13C NMR) spectroscopy revealed typical carbon atom resonance signals at 153, 127, and 119 ppm, further confirming the presence of imidazole moieties in the COFs' network (Fig. 2c).
The chemical states and molecular structures of the R-COFs were determined using X-ray photoelectron spectroscopy (Fig. S16–S19†). The N 1s spectrum of the COFs displays CN and N–H bond peaks, demonstrating the synthesis of imidazole. The surface wettability of the R-COFs was tested using contact-angle (CA) measurements. The CA values of H-COF, F-COF, OMe-COF, and OH-COF toward water were 119°, 136°, 125°, and 100°, respectively (Fig. S20†). The smallest CA of OH-COF among all the synthesized COFs, owing to the abundant hydrophilic OH groups, indicates that O2 diffusion to the active sites could be facilitated.
The charge transfer resistances of all the synthesized COFs were determined using electrochemical impedance spectroscopy. As shown in Fig. 2d, the semicircular radius of the Nyquist curve of OH-COF was smaller than those of H-COF, F-COF, and OMe-COF (Fig. 2d), indicating its lowest interfacial charge-transfer resistance. The electron transfer performance of the R-COFs was evaluated using UV/vis diffuse reflectance spectroscopy. The absorbance edges of the H-COF, F-COF, OMe-COF, and H-COF were approximately 550, 568, 575, and 596 nm, respectively (Fig. 2e). F-COF, OMe-COF, and OH-COF exhibited increased absorption wavelengths compared to H-COF. This red shift indicates a decrease in the electronic transition energy of the three COFs, suggesting that groups with electronic effects are beneficial for reducing the energy of electronic transitions and promoting electron transfer in the reaction. Interestingly, the pronounced red shift of OH-COF may be attributed to the hydrogen bonds between molecules expanding the π conjugation of the COF skeleton, changing the electronic distribution of the molecule and resulting in a reduction in electron transition energy, thereby accelerating electronic transfer. The optical band gaps of H-COF, F-COF, OMe-COF, and OH-COF calculated using Tauc plots were 2.35, 2.30, 2.29, and 2.28 eV (Fig. 2f). The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels were obtained using Mott–Schottky measurement (Fig. S21†). The LUMO levels of H-COF, F-COF, OMe-COF, and OH-COF were 2.1, 2.04, 2.08, and 2.07 eV, and the corresponding HOMO levels were −0.25, −0.26, −0.21, and −0.21 eV, respectively (Fig. 2g). Importantly, OMe-COF and OH-COF exhibit higher HOMO levels than H-COF and F-COF, which illustrates that COFs with electron-donating groups possess stronger reductive ability.
Notably, the E1/2 of OH-COF achieved a record value compared with that of the reported COF-based electrocatalysts (Fig. 3g). The LSV curves of R-COFs were obtained to evaluate the electrochemical reaction kinetics and material transport characteristics (Fig. 3h and S24†). By using the rotating-ring disk electrode (RRDE) test, the electron-transfer number and H2O2 productivity could be obtained. Compared with H-COF, F-COF and OMe-COF, the n of OH-COF (3.7) is the highest with the H2O2 productivity lower than 20%, demonstrating that the ORR catalysed by OH-COF is a typical four-electron process (Fig. 3i).
 | ||
| Fig. 3 (a) CV curves for OH-COF in oxygen saturated (solid curve) and saturated (dotted curve) solutions. (b) LSV plots at 1600 rpm of H-COF, F-COF, OMe-COF, OH-COF and commercial Pt/C in 0.1 M KOH solution (oxygen saturated). (c) Contrast of Eonset, E1/2 and electron transfer numbers of H-COF, F-COF, OMe-COF, and OH-COF. (d) Tafel slope and (e) Cdl values of H-COF, F-COF, OMe-COF, and OH-COF. (f) Comparisons of the TOFs and mass activities of H-COF, F-COF, OMe-COF, and OH-COF. (g) Comparisons of the onset and half-wave potential of the reported metal-free catalysts and OH-COF (this research). (h) LSV curves of OH-COF at various rotation speeds. (i) Electron transfer number and H2O2 productivity obtained from the RRDE measurement of H-COF, F-COF, OMe-COF, and OH-COF. | ||
Cyclic voltammetry tests were employed to assess the catalytic performance of the R-COFs in N2- or O2-saturated 0.1 M KOH solutions. F-COF, OMe-COF, and OH-COF all exhibited distinct reduction peaks with high current densities, whereas H-COF exhibited a wider reduction peak and lower current density under an O2 atmosphere. No peaks were observed under the N2 atmosphere, which clearly indicates that these COFs materials exhibited catalytic activity for the ORR (Fig. 3a and S22†). In particular, the OH-COF showed a higher peak potential for oxygen reduction (0.88 V vs. RHE) (Fig. S22†), indicating the higher catalytic activity of the OH-COF. To further explore the ORR performances of R-COFs with different electronic effect groups, linear sweep voltammetry (LSV) curves were first obtained in a 0.1 M KOH solution with rotation set to 1600 rpm. As shown in Fig. 3b, the half-wave potential (E1/2) of OH-COF reached 0.80 V vs. RHE, demonstrating superior ORR activity compared to H-COF (0.62 V), F-COF (0.63 V), and OMe-COF (0.70 V) (Fig. 3b and c. The half-wave potential of OH-COF (0.80 V vs. RHE) is only 40 mV lower than that of commercial Pt/C (0.84 V vs. RHE), indicating that OH-COF exhibits high activity close to that of platinum-based catalysts. In addition, imine-linked PDA-OMe-COF was synthesized as a comparative sample to determine the effect of linkage on ORR performance (Fig. S23–S25†). As shown in Fig. S26,† the E1/2 of PDA-OMe-COF is 0.65 V vs. RHE, which is smaller than that of OMe-COF (0.70 V vs. RHE), indicating that the imidazole linkage is more conducive to amplifying the ORR activity by side substituents than the imine linkage. The onset potential (Eonset) of OH-COF (0.89 V) was higher than those of H-COF (0.79 V), F-COF (0.81 V), and OMe-COF (0.84 V). The Tafel slopes of H-COF, F-COF, OMe-COF, and OH-COF calculated from the LSV curves were 107.5, 80.9, 70.9, and 53.7 mV dec−2 (Fig. 3d). OH-COF showed the smallest Tafel slope among the four COFs, indicating that OH-COF had the fastest ORR kinetics. The electrochemically active surface areas (ECSAs) of R-COFs were evaluated using electrochemical double-layer capacitance (Cdl) (Fig. 3e and S27†). The Cdl values of H-COF, F-COF, OMe-COF, and OH-COF were 6.14, 4.07, 2.12, and 1.84 mF cm−2, respectively (Fig. 3e). The largest Cdl of OH-COF proves the fastest reaction kinetics in the process of the ORR catalysed by OH-COF. The turnover frequency (TOF) and mass activity indicate the fundamental activities of the COF catalysts. As presented in Fig. 3f, the TOF and mass activity of OH-COF are 0.024 s−1 and 12.43 A g−1, respectively, with the aldehyde monomer in the sample as the active site. These values surpass those of H-COF (0.0019 s−1 and 1.13 A g−1), F-COF (0.0044 s−1 and 2.25 A g−1), and OMe-COF (0.013 s−1 and 6.33 A g−1). Therefore, the highest active site utilization efficiency was achieved for OH-COF owing to the strong electron-donating ability of the –OH group, which boosted the electron density of the active site, making it more susceptible to proton attack and reduction.
Notably, the E1/2 of OH-COF achieved a record value compared with that of reported COF-based electrocatalysts (Fig. 3g). The LSV curves of the R-COFs were obtained to evaluate the electrochemical reaction kinetics and material transport characteristics (Fig. 3h and S28†). Using a rotating-ring disk electrode test, the electron transfer number and H2O2 productivity were obtained. Compared to the H-COF, F-COF, and OMe-COF, the n value of OH-COF (∼3.7) was the highest with the H2O2 productivity lower than 20%, demonstrating that the ORR catalyzed by OH-COF is a typical four-electron process (Fig. 3i).
Density functional theory calculations were conducted to explore the mechanism of the ORR catalyzed by R-COFs. The HOMO and LUMO of H-COF are localized on the imidazole ring and phenyl unit, respectively, which display typical D-A properties, whereas in OH-COF and OMe-COF, the HOMO extends to the hydroxyl and methoxy regions, indicating strong electron-donating abilities that facilitate charge transfer during the ORR. In F-COF, the LUMO also extends to the F atom, suggesting that the F atom has a strong electron-accepting ability, enhancing the reactivity of F-COF (Fig. S29†). To directly visualize the electronic properties of –F, –OMe, and –OH and the impact of –F, –OMe, and –OH on the ORR process, we selected the main atoms, such as –F, –OMe, –OH, and imidazole rings, as active sites for theoretical calculations.
The catalytic sites of the H-COF, F-COF, OMe-COF, and OH-COF were determined using the Bader charge distribution map (Fig. S30†). The most probable catalytic centers are indicated by red arrows in Fig. 4a, identified using the most negatively charged sites. In addition, density of states calculations showed that OH-COF exhibited the smallest band gap around the Fermi level compared to F-COF, H-COF, and OMe-COF, suggesting outstanding semiconductor properties of OH-COF (Fig. 4b). As shown in Fig. S31–S34†, the charge distribution difference illustrations of these COFs clearly indicate that the corresponding electron transfer was enhanced over the conjugated frameworks, particularly for OH-COF. Three intermediate states (*OOH, *O, and *OH) exist in the oxygen reduction (Fig. S35–S37† and 4c). The corresponding free energy variations (ΔG) of each ORR step process catalyzed by each COF were calculated at U = 1.23 (Fig. 4d). According to these results, the transformation of O2 into *OOH was the rate-determining step in the oxygen reduction. As illustrated in Fig. 4e, the ΔG values of the rate-determining step were computed to be 1.71, 1.54, 1.48, and 1.45 eV for H-COF, F-COF, OMe-COF, and OH-COF, respectively. Notably, OH-COF exhibited the lowest ΔG value, suggesting that the intermediate *OOH was most readily formed during the ORR process of OH-COF, thereby establishing OH-COF as the most catalytically active among all COFs.
 | ||
| Fig. 4 Analysis of the ORR mechanism. (a) The most likely catalytic sites in the COFs. (b) Density of states (DOS) of H-COF, F-COF, OMe-COF, and OH-COF. (c) The ORR process structure diagram of OH-COF. (d) Comparison of the free energy changes for these COF catalysts. (e) The corresponding ΔG values for the rate determining step of these COF catalysts. | ||
As shown in Fig. 5a, a zinc–air battery (ZAB) was prepared using OH-COF to evaluate its practical application based on its superior ORR performance. The ZAB exhibited an open-circuit voltage of 1.465 V (Fig. 5b). As shown in Fig. 5c and d, owing to its favourable mass transfer characteristics, a maximum power density of 119.5 mW cm−2 was observed in the ZAB. Moreover, the OH-COF electrode exhibited a stable cycle life, maintaining its performance over 200 h during continuous discharge at an applied current density of 5.0 mA cm−2 (Fig. 5e). At a current density of 20 mA cm−2, the ZAB was continuously discharged for 24 h, and the calculated specific capacity reached 854 mA h g−1, indicating its impressive battery efficiency (Fig. 5f) and reinforcing the viability of utilizing COFs as effective catalysts in battery environments. In addition, the stability of OH-COF assessed at 0.7 V vs. RHE reached 20 h with negligible current density loss (only 0.5%), proving the high stability of OH-COF in an alkaline solution (Fig. 5g). One ZAB composed of OH-COF was capable of illuminating an LED panel displaying “ZZU” (Fig. 5g, inset), demonstrating that the OH-COF catalyst has substantial potential for application in the field of ZABs.
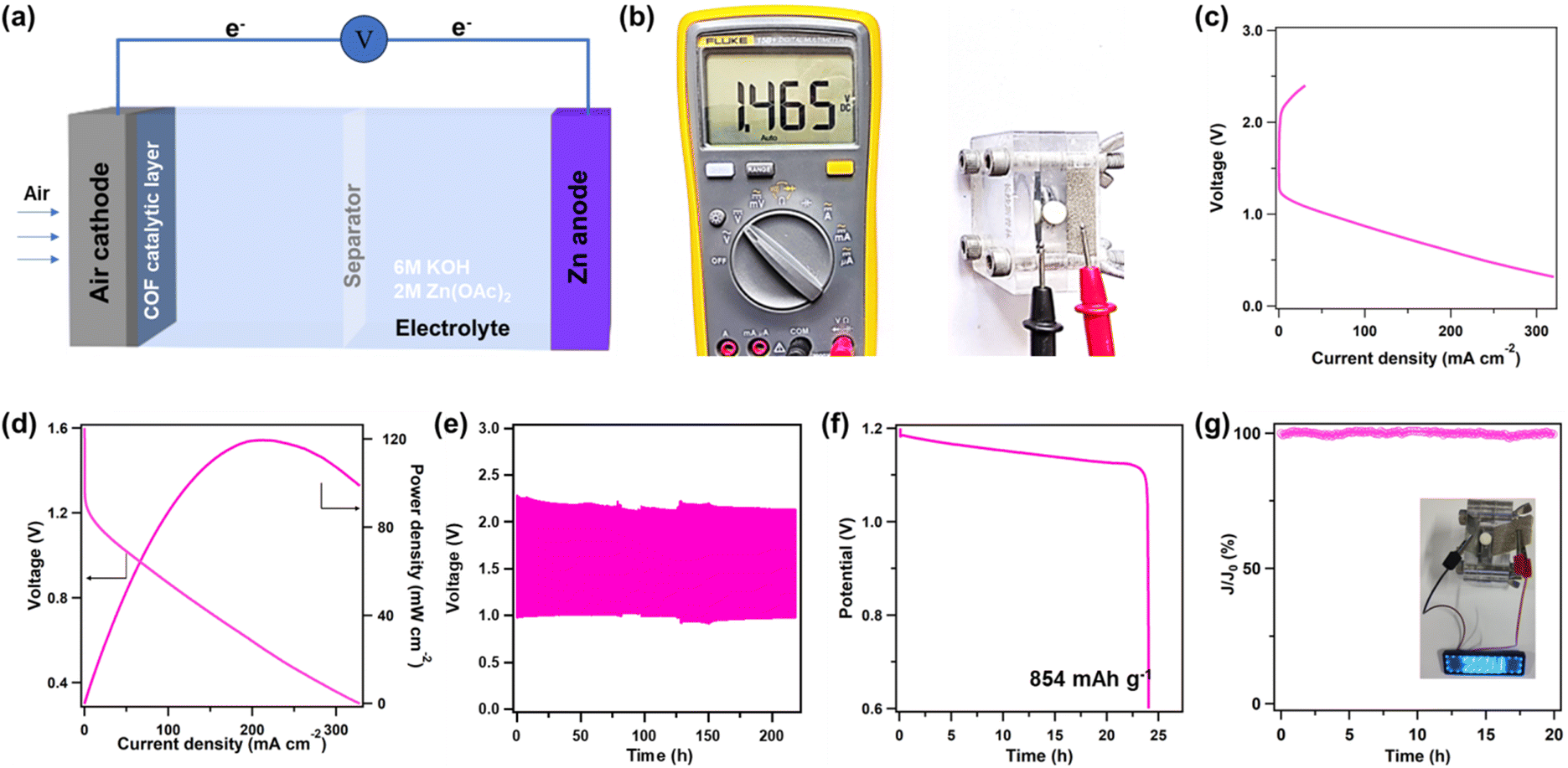 | ||
| Fig. 5 Capability of a zinc–air battery assembled using OH-COF. (a) Schematic of the ZAB. (b) Open circuit voltage. (c) Charge–discharge voltage profile. (d) Power density plots. (e) The durability of the ZAB prepared with OH-COF tested at 5 mA cm−2. (f) Discharge profiles at a fixed current density of 20 mA cm−2. (g) Long-time stability test of OH-COF (inset: an image of a “ZZU” LED panel driven by the ZAB). | ||
Conclusions
We developed four R-COFs containing different electron-effect groups (–H, –F, –OMe, and –OH) via a one-pot three-component Debus–Radziszewski condensation reaction. The corresponding surface groups with different electronic effects can interact with π-conjugated skeletons to promote electron transfer within the COF skeleton. Therefore, by adopting a pore-surface engineering strategy, charge transfer within the entire COF framework can be precisely regulated through the surface functional groups. These COFs were subsequently employed as metal-free ORR electrocatalysts. Notably, the E1/2 of OH-COF was 0.80 V (vs. RHE), exhibiting a record ORR activity compared to previously reported metal-free COF catalysts. In view of the exploration and comprehensive understanding of the influence of the electronic effects of pore wall groups on the charge density of the active sites, the pore surface modification strategy adopted in this study offers experimental and theoretical principles for the construction and improvement of the electrocatalytic performance of metal-free COFs.Data availability
Data available on request from the authors.Author contributions
Y. Ji and L. Zhai conceived the project, designed experiments, and provided funding. M. Chen, D. Han, and Z. Zhu conducted the experiments. X. Kong performed computational calculations. M. Chen, D. Han, and L. Zhai wrote the manuscript and all authors revised the manuscript. All the authors contributed to the data interpretation, discussion, and manuscript revision. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52403288), the Natural Science Foundation of Henan Province (242300421073), the Program for Science & Technology Innovation Talents in Universities of Henan Province (23HASTIT015), the Henan Provincial Science and Technology R&D Program Joint Fund (235200810071), and Central Plains Elite Young Top-notch Talents.Notes and references
- M. K. Debe, Nature, 2022, 486, 43–51 CrossRef PubMed.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- Q. Shi, Q. Liu, Y. Ma, Z. Fang, Z. Liang, G. Shao, B. Tang, W. Yang, L. Qin and X. Fang, Adv. Energy Mater., 2020, 10, 1903854 CrossRef CAS.
- P. Yu, L. Wang, F. Sun, Y. Xie, X. Liu, J. Ma, X. Wang, C. Tian, J. Li and H. Fu, Adv. Mater., 2019, 31, 1901666 CrossRef PubMed.
- C. Zhao, Bo. Li, J. Liu, J. Huang and Q. Zhang, Chin. Chem. Lett., 2019, 30, 911–914 CrossRef CAS.
- Y. Lian, W. Yang, C. Zhang, H. Sun, Z. Deng, W. Xu, L. Song, Z. Ouyang, Z. Wang, J. Guo and Y. Peng, Angew. Chem., Int. Ed., 2020, 59, 286–294 CrossRef CAS PubMed.
- B. Chi, L. Zhang, X. Yang, Y. Zeng, Y. Deng, M. Liu, J. Huo, C. Li, X. Zhang, X. Shi, Y. Shao, L. Gu, L. Zheng, Z. Cui, S. Liao and G. Wu, ACS Catal., 2023, 13, 4221–4230 CrossRef CAS.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- A. A. Gewirth, J. A. Varnell and A. M. Diascro, Chem. Rev., 2018, 118, 2313–2339 CrossRef CAS PubMed.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- L. Zhang, J. M. Theresa Agatha Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS PubMed.
- J. Guo, C.-Y. Lin, Z. Xia and Z. Xiang, Angew. Chem., Int. Ed., 2018, 57, 12567–12572 CrossRef CAS PubMed.
- S. Zhang, W. Xia, Q. Yang, Y. V. Kaneti, X. Xu, S. M. Alshehri, T. Ahamad, M. S. A. Hossain, J. Na, J. Tang and Y. Yamauchi, Chem.–Eur. J., 2019, 396, 25154 Search PubMed.
- Y. Wang, J. Wu, S. Tang, J. Yang, C. Ye, J. Chen, Y. Lei and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202219191 CrossRef CAS PubMed.
- J. Diao, Y. Qiu, S. Liu, W. Wang, K. Chen, H. Li, W. Yuan, Y. Qu and X. Guo, Adv. Mater., 2020, 32, 1905679 CrossRef CAS PubMed.
- B. Gui, X. Liu, Y. Cheng, Y. Zhang, P. Chen, M. He, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2022, 134, e202113852 CrossRef.
- M. Jahan, Q. Bao and K. Loh, J. Am. Chem. Soc., 2012, 134, 6707–6713 CrossRef CAS PubMed.
- H. Qin, Y. Wang, X. Duan, H. Lei, X. Zhang, H. Zheng, W. Zhang and R. Cao, J. Energy Chem., 2021, 53, 77–81 CrossRef CAS.
- A. Aljabour, H. Awada, L. Song, H. Sun, S. Offenthaler, F. Yari, M. Bechmann, M. C. Scharber and W. Schöfberger, Angew. Chem., Int. Ed., 2023, 62, e202302208 CrossRef CAS PubMed.
- X. Han, W. Zhang, X. Ma, Ch. Zhong, N. Zhao, W. Hu and Y. Deng, Adv. Mater., 2019, 31, 1808281 CrossRef PubMed.
- X. Lyu, Y. Jia, X. Mao, D. Li, G. Li, L. Zhuang, X. Wang, D. Yang, Q. Wang, A. Du and X. Yao, Adv. Mater., 2020, 32, 2003493 CrossRef CAS PubMed.
- J. Durst, C. Simon, F. Hasché and H. Gasteiger, J. Electrochem. Soc., 2014, 162, F190 CrossRef.
- Z. Liao, Y. Wang, Q. Wang, Y. Cheng and Z. Xiang, Appl. Catal. B Environ., 2019, 243, 204–211 CrossRef CAS.
- Q. Feng, S. Zhao, D. He, S. Tian, L. Gu, X. Wen, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 2773–2776 CrossRef CAS PubMed.
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- Y. Zhao, W. Dai, Y. Peng, Z. Niu, Q. Sun, C. Shan, H. Yang, G. Verma, L. Wojtas, D. Yuan, Z. Zhang, H. Dong, X. Zhang, B. Zhang, Y. Feng and S. Ma, Angew. Chem., Int. Ed., 2020, 59, 4354–4359 CrossRef CAS PubMed.
- T. Ma, Y. Zhou, C. S. Diercks, J. Kwon, F. Gándara, H. Lyu, N. Hanikel, P. Pena-Sánchez, Y. Liu, N. J. Diercks, R. O. Ritchie, D. M. Proserpio, O. Terasaki and O. M. Yaghi, Nat., Synth., 2023, 2, 286–295 CrossRef CAS.
- X. Guan, F. Chen, S. Qiu and Q. Fang, Angew. Chem., Int. Ed., 2023, 62, e202213203 CrossRef CAS PubMed.
- F. Jin, T. Wang, H. Zheng, E. Lin, Y. Zheng, L. Hao, T. Wang, Y. Chen, P. Cheng, K. Yu and Z. Zhang, J. Am. Chem. Soc., 2023, 145, 6507–6515 CrossRef CAS PubMed.
- Y. Zhu, P. Shao, L. Hu, C. Sun, J. Li, X. Feng and B. Wang, J. Am. Chem. Soc., 2021, 143, 7897–7902 CrossRef CAS PubMed.
- Y. Ying, S. Bo Peh, H. Yang, Z. Yang and D. Zhao, Adv. Mater., 2022, 34, 2104946 CrossRef CAS.
- M. Zhen, T. Zhou, W. Weng, J. Unruangsri, K. Hu, W. Yang, C. Wang, K. A. I. Zhang and J. Guo, Angew. Chem., Int. Ed., 2021, 60, 9642–9649 CrossRef PubMed.
- J. M. Seo, H. J. Noh, J. P. Jeon, H. Kim, G. F. Han, S. K. Kwak, H. Y. Jeong, L. Wang, F. Li and J. Baek, J. Am. Chem. Soc., 2022, 144, 19973–19980 CrossRef CAS PubMed.
- D. Wu, Q. Xu, J. Qian, X. Li and Y. Sun, Chem.–Eur. J., 2019, 25, 3105–3111 CrossRef CAS PubMed.
- H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, A. Kravchenko, T. Liu, Y. Yang, Y. Fang, L. Wang, J. Guan, I. Furó, M. S. G. Ahlquist and L. Sun, Nat. Catal., 2022, 5, 414–429 CrossRef CAS.
- J. Xu, Y. He, S. Bi, M. Wang, P. Yang, D. Wu, J. Wang and F. Zhang, Chem. Int. Ed., 2019, 131, 12193–12197 Search PubMed.
- Y. Xu, P. Cai, K. Chen, Q. Chen, Z. Wen and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202215584 CrossRef CAS PubMed.
- Y. Yue, H. Li, H. Chen and N. Huang, J. Am. Chem. Soc., 2022, 144, 2873–2878 CrossRef CAS PubMed.
- Z. Guo, Y. Zhang, Y. Dong, J. Li, S. Li, P. Shao, X. Feng and B. Wang, J. Am. Chem. Soc., 2019, 141, 1923–1927 CrossRef CAS PubMed.
- M. Liu, Y. Chen, X. Huang, L. Dong, M. Lu, C. Guo, D. Yuan, Y. Chen, G. Xu, S. Li and Y. Lan, Chem. Int. Ed., 2022, 61, e202115308 CrossRef.
- Q. Zuo, G. Cheng and W. Luo, Dalton Trans., 2017, 46, 9344–9348 RSC.
- S. Bhunia, A. Pena-Duarte, H. Li, H. Li, M. F. Sanad, P. Saha, M. A. Addicoat, K. Sasaki, T. A. Strom, M. J. Yacaman, C. R. Cabrera, R. Seshadri, S. Bhattacharya, J.-L. Bredas and L. Echegoyen, ACS Nano, 2023, 17, 3492–3505 CrossRef CAS PubMed.
- X. Yu, Y. Ma, C. Li, X. Guan, Q. Fang and S. Qiu, Chem. Res. Chin. Univ., 2022, 38, 167–172 CrossRef CAS.
- J. Yue, Y. Wang, X. Wu, P. Yang, Y. Ma, X. Liu and B. Tang, Chem. Commun., 2021, 57, 12619–12622 RSC.
- Z. Mei, G. Zhao, C. Xia, S. Cai, Q. Jing, X. Sheng, H. Wang, X. Zou, L. Wang, H. Guo and B. Xia, Angew. Chem., Int. Ed., 2023, 62, e202303871 CrossRef CAS PubMed.
- H. Chen, Q. Li, W. Yan, Z. Gu and J. Zhang, Chem.–Eur. J., 2020, 401, 126149 CAS.
- M. Liu, S. Liu, C. Cui, Q. Miao, Y. He, X. Li, Q. Xu and G. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202213522 CrossRef CAS PubMed.
- Y. Ma, Y. Fu, W. Jiang, Y. Wu, C. Liu, G. Che and Q. Fang, J. Mater. Chem. A, 2022, 10, 10092 RSC.
- X. Sun, Y. Hu, Y. Fu, J. Yang, D. Song, B. Li, W. Xu and N. Wang, Small, 2024, 20, 2305978 CrossRef CAS PubMed.
- C. He, D. Si, Y. Huang and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202212507 CrossRef.
- Y. Zhang, L. Dong, S. Li, X. Huang, J. Chang, J. Wang, J. Zhou, S. Li and Y. Lan, Nat. Commun., 2021, 12, 6390 CrossRef CAS PubMed.
- J. Yi, D. Si, R. Xie, Q. Yin, M. Zhang, Q. Wu, G. Chai, Y. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 17108–17114 CrossRef CAS PubMed.
- Z. Fu, H. Zhuo, X. Liu, W. Li, H. Song, Z. Shi, L. Feng, T. Jin, W. Chen and Y. Chen, ACS Appl. Energy Mater., 2025, 8, 1051–1059 CrossRef CAS.
- S.-W. Ke, W. Li, Y. Gu, J. Su, Y. Liu, S. Yuan, J.-L. Zuo, J. Ma and P. He, Sci. Adv., 2023, 9, eadf2398 CrossRef CAS PubMed.
- M. Punniyamoorthy, S. Singha Roy, M. Kathiresan and S. Kundu, ACS Appl. Energy Mater., 2024, 7, 4111–4120 CrossRef CAS.
- P. Shao, Z. Ren, B. Zhao, X. Wang, J. Li, J. Xie, B. Wang and X. Feng, J. Am. Chem. Soc., 2025, 147, 8769–8777 CrossRef CAS PubMed.
- V. Hasija, S. Patial, P. Raizada, A. Aslam Parwaz Khan, A. M. Asiri, Q. Van Le, V.-H. Nguyen and P. Singh, Coord. Chem. Rev., 2022, 452, 214298 CrossRef CAS.
- H. Zhang, M. Zhu, O. G. Schmidt, S. Chen and K. Zhang, Adv. Energy Sustainability Res., 2021, 2, 2000090 CrossRef CAS.
- Z. You, B. Wang, Z. Zhao, Q. Zhang, W. Song, C. Zhang, X. Long and Y. Xia, Adv. Mater., 2023, 35, 2209129 CrossRef CAS PubMed.
- S. Royuela, E. Martinez-Periñán, M. P. Arrieta, J. I. Martinez, M. M. Ramos, F. Zamora, E. Lorenzo and J. L. Segura, Chem. Commun., 2020, 56, 1267–1270 RSC.
- Q. Xu, Y. Tang, X. Zhang, Y. Oshima, Q. Chen and D. Jiang, Adv. Mater., 2018, 30, 1706330 CrossRef PubMed.
- R. Bao, Z. Xiang, Z. Qiao, Y. Yang, Y. Zhang, D. Cao and S. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216751 CrossRef CAS PubMed.
- M. Martinez-Fernández, E. Martinez-Periñán, S. Royuela, J. I. Martinez, F. Zamora, E. Lorenzo and J. L. Segura, Appl. Metamater., 2022, 26, 101384 Search PubMed.
- X. Yan, B. Wang, L. Ren, X. Long and D. Yang, Angew. Chem., Int. Ed., 2022, 134, e202209583 CrossRef.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104 CrossRef CAS PubMed.
- M. O. Cichocka, Z. Liang, D. Feng, S. Back, S. Siahrostami, X. Wang, L. Samperisi, Y. Sun, H. Xu, N. Hedin, H. Zheng, X. Zou, H. Zhou and Z. Huang, J. Am. Chem. Soc., 2020, 142, 15386–15395 CrossRef CAS PubMed.
- P. Garcia-Arroyo, E. Martinez-Perinan, J. J. Cabrera-Trujillo, E. Salagre, E. G. Michel, J. I. Martinez, E. Lorenzo and J. L. Segura, Nano Res., 2022, 15, 3907–3912 CrossRef CAS.
- S. An, X. Li, S. Shang, T. Xu, S. Yang, C. Cui, C. Peng, H. Liu, Q. Xu, Z. Jiang and J. Hu, Angew. Chem., Int. Ed., 2023, 62, e202218742 CrossRef CAS PubMed.
- G. Cai, L. Zeng, L. He, S. Sun, Y. Tong and J. Zhang, Chem. - Asian J., 2020, 15, 1963–1969 CrossRef CAS PubMed.
- Q. Wang, C. Wang, K. Zheng, B. Wang, Z. Wang, C. Zhang and X. Long, Angew. Chem., Int. Ed., 2024, 63, e202320037 CrossRef CAS PubMed.
- X. Li, S. Yang, M. Liu, X. Yang, Q. Xu, G. Zeng and Z. Jian, Angew. Chem., Int. Ed., 2023, 62, e202304356 CrossRef CAS PubMed.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- P. Wang, S. Ding, Z. Zhang, Z. Wang and W. Wang, J. Am. Chem. Soc., 2018, 41, 18004–1800 Search PubMed.
- Z. Zhang, P. Wang, Y. Sun, T. Yang, S. Ding and W. Wang, J. Am. Chem. Soc., 2024, 146, 822–4829 Search PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 61, 48–52 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02145f |
| This journal is © The Royal Society of Chemistry 2025 |