
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulating room-temperature phosphorescence by electron–phonon coupling†
Liangwei
Ma‡
,
Muyu
Cong‡
,
Siyu
Sun
 and
Xiang
Ma
*
and
Xiang
Ma
*
Key Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: maxiang@ecust.edu.cn
First published on 4th April 2025
Abstract
Designing and optimizing efficient organic room-temperature phosphorescent (RTP) materials remains a captivating yet challenging endeavour due to the inherent difficulties in generating and stabilizing triplet excitons. Here, we report a suite of highly efficient phosphors characterized by near-unity intersystem crossing (ISC) yields. Surprisingly, upon doping these dyes into a polyvinyl alcohol matrix, their phosphorescence quantum yields (ΦP) spanned a wide range from 2.7% to 69.6%, governed by the position of the methyl substituent. Theoretical calculations and experimental results indicate that the variation in phosphorescence efficiency is primarily due to the strong electron–phonon coupling caused by the positional variation of the methyl substituents, rather than common factors such as ISC or energy levels. These findings provide a new insight into the design of high-performance organic RTP dyes.
In recent years, organic room-temperature phosphorescent (RTP) materials have garnered huge research interest due to their unique photophysical properties, such as long lifetimes and large Stokes shifts, which make them promising for applications in displays, bioimaging, anticounterfeiting, encryption, and electroluminescence.1–9 However, the construction of highly efficient organic RTP materials remains a significant challenge. The difficulties in constructing these materials stem from three main reasons: (1) spin-forbidden intersystem crossing (ISC) process: ISC is a spin-forbidden process, making it difficult to efficiently generate triplet excited states through photoexcitation. (2) Slow radiative transition rates: the radiative transition rates of triplet states are relatively slow (∼100–106 s−1), comparable to non-radiative relaxation processes caused by molecular vibration, rotation, or collision. This makes the triplet excited states highly susceptible to non-radiative deactivation. (3) Oxygen quenching: triplet excited states are easily quenched by oxygen through energy transfer processes.6,10–19 To address these challenges and achieve highly efficient RTP materials, various strategies have been proposed: (1) designing specific molecular structures (e.g., twisted molecular structures) or introducing heteroatoms, heavy atoms, or functional groups such as carbonyl groups into the molecular framework to enhance spin–orbit coupling (SOC) and promote ISC.1,20–30 (2) Utilizing polymer matrices, crystallization, or macrocycles to create a rigid microenvironment for phosphors that restricts the molecular vibration, rotation, and collision, which result in non-radiative transitions, and isolates dyes from oxygen effectively.29,31–40 Generally, molecular rotation and intermolecular collisions can be significantly suppressed or even eliminated in the solid state or in a rigid microenvironment.41 However, molecular vibrations are much more difficult to effectively suppress. This can be evidenced by the fact that fine structures in luminescence spectra are often only observable at very low temperatures and enhancement of phosphorescence at lower temperatures.10,15,40,42–44 Molecular vibrations are an intrinsic property related to electron–phonon coupling (EPC). Strong EPC typically implies strong molecular vibrational capability, which may lead to intense non-radiative transitions. Previous studies have already demonstrated that EPC is an important factor affecting the performance of organic optoelectronic materials.45–47 However, in the research on RTP materials, which are more susceptible to non-radiative transitions, little attention was paid to this extremely important factor.48 In current research, the general approach is to use a rigid microenvironment as an external means to suppress molecular vibrations. Theoretically, the ability of a rigid microenvironment is limited, with a specific threshold. When the intrinsic EPC is too strong, the molecular vibration can no longer be effectively suppressed, severely limiting the construction of highly efficient RTP dyes. Therefore, in the molecular design of phosphorescent dyes, EPC must be considered alongside SOC, electron configurations, and energy levels.
In this work, we systematically demonstrate the significant impact of EPC on the performance of phosphorescent dyes. As the position of the substituents changes, the ISC efficiency of these methyl-substituted thiochroman-4-one derivatives (MTOs, Scheme 1) approaches 100%, with the ISC rate constant (kISC) gradually increasing. In contrast, the phosphorescence quantum yield (ΦP) decreases from 69.6% (MTO-1@PVA) to 2.7% (MTO-4@PVA) with the increase of kISC. Temperature-dependent emission spectra show that materials with lower ΦP are more temperature-sensitive, indicating stronger EPC. Theoretical simulations reveal that MTO-4 has a Huang–Rhys factor of 23.06 at 82 cm−1, while the Huang–Rhys factors of the other three compounds do not exceed 1.0, indicating that MTO-4 has significantly stronger EPC. Combined with the temperature-dependent experiments, it is determined that the significant decrease in phosphorescence efficiency is primarily due to the excessively strong EPC in MTO-4, which causes molecular vibrations to exceed the threshold that the polyvinyl alcohol (PVA) matrix can effectively suppress. This work highlights the critical role of EPC in the design of efficient RTP materials and provides new insights into the mechanisms governing phosphorescence performance.
 | ||
| Scheme 1 Chemical structure of target compounds and the simplified Jablonski diagram for the transition process from the lowest excited state in aromatic compounds. | ||
The comprehensive synthetic protocols for MTO compounds are elaborately outlined in the ESI.† Their chemical structures were meticulously authenticated through 1H NMR, 13C NMR, and HRMS analyses. Upon dissolution in dichloromethane (DCM), the four dyes displayed strikingly analogous absorption spectra (Fig. 1a), with their maximum absorption peaks consistently centred around 350 nm. The subtle shifts in the maximum emission wavelengths can be attributed to the subtle perturbations induced by the varying methyl substituent positions on the energy levels of the frontier orbitals. This observation is consistent with the fact that the methyl group lacks π-orbitals to conjugate with the frontier orbitals of thiochroman-4-one. However, a marked disparity was observed in the fluorescence intensities among the dyes as the methyl substituent position varied (Fig. 1a). The fluorescence quantum yield (ΦF) was found to be 1.19% in DCM when the methyl group occupied the 6-substituent position (Table S1†), whereas it was virtually quenched (ΦF = 0.25%) when situated at the 5-substituent position. Furthermore, substantial differences in the fluorescence lifetimes of these compounds were discernible (Fig. 1b and S1†), indicative of substantial variations in the relaxation dynamics of the lowest singlet excited state (S1).
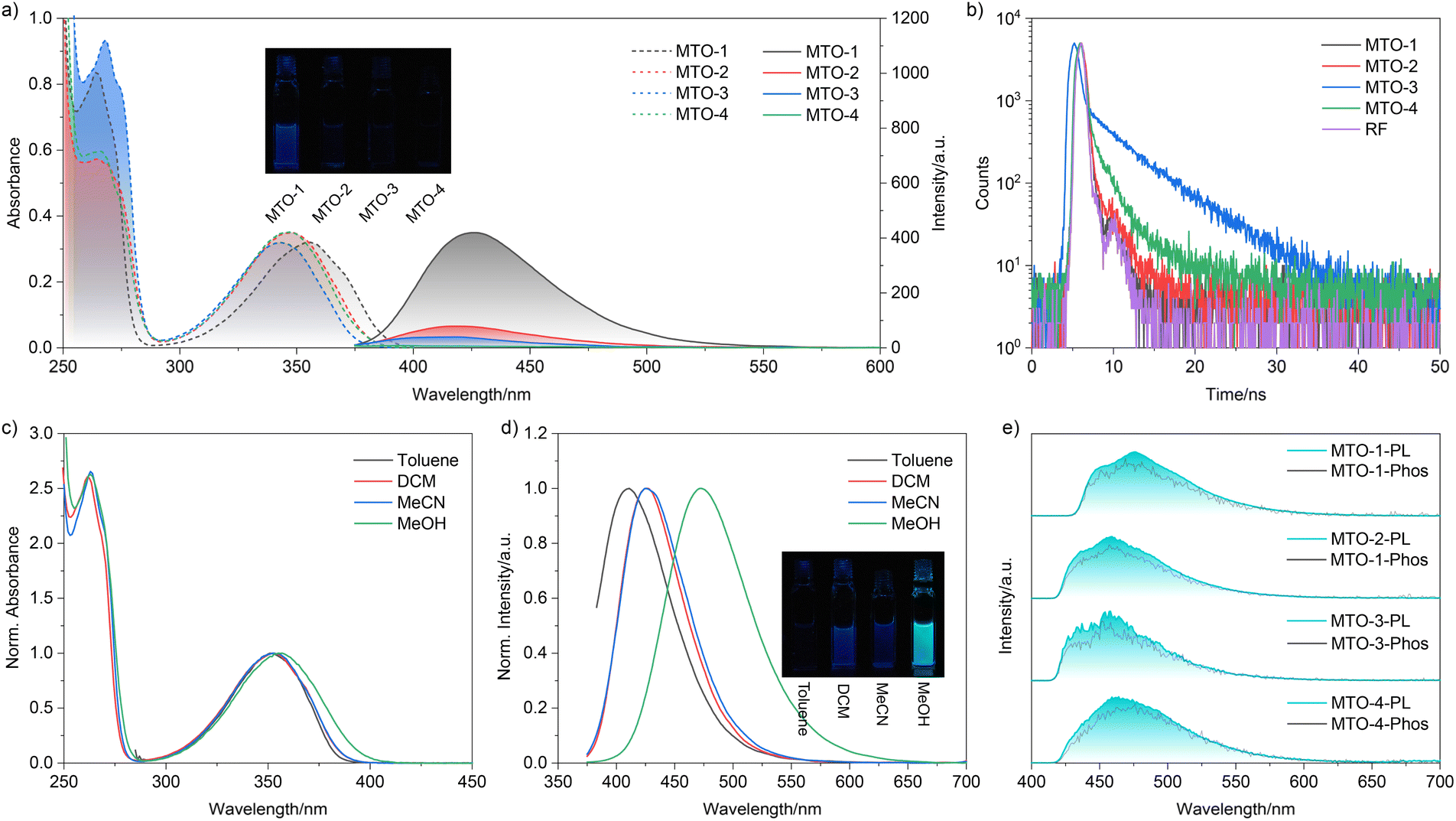 | ||
| Fig. 1 a) Absorption and emission spectra of MTOs in DCM; (b) lifetime spectra of MTOs in DCM; absorption (c) and emission (d) spectra of MTO-1 in different solvents; (e) steady-state and phosphorescence spectra of MTOs in MTHF at 77 K. | ||
To unravel the unusual impact of the methyl group on the photophysical properties, density functional theory (DFT) and time-dependent DFT (TDDFT) calculations were conducted using the G09 program with the M062x/aug-cc-pVTZ level of theory.39,49 As depicted in Table S2,† the S0 → S1 transition for these compounds predominantly involved HOMO/HOMO-2 → LUMO transitions. Despite their similar molecular frameworks (Fig. S2†), the contribution from the HOMO → LUMO transition, which characterizes a π → π* transition, progressively dwindled from 20.1% to 0% as the methyl substituent position varied. Notably, the HOMO-2 → LUMO transition, constituting the dominant contribution to the S0 → S1 transition, represents a typical n → π* transition. Thus, the S0 → S1 transition for these compounds is classified as a forbidden transition due to the non-conservation of orbital angular momentum. Correspondingly, with the decline in the HOMO → LUMO transition contribution, the f of these compounds plummeted from 0.0165 to 0.0002 (Table S2†). Conversely, the S0 → S2 transition for these compounds was primarily driven by the HOMO → LUMO transition, representing a spin-allowed process. Consequently, the S0 → S1 transition is designated as a dark state.50 Hence, the maximum absorption peak for these compounds is attributed to the S0 → S2 transition. The shoulder peak observed at 370 nm in the absorption spectrum of MTO-1 is assigned to the S0 → S1 transition due to its weak yet non-negligible f value.
The S1 → S0 transition mirrored the trends observed for the S0 → S1 transition (Table S2†). For MTO-1, the S1 → S0 transition was primarily governed by the HOMO → LUMO transition (95.4%), with a minor contribution from the HOMO-2 → LUMO transition (2.2%). However, the contribution from the HOMO → LUMO transition significantly declined to 24.1% for MTO-2, further dwindling to 18.5% and 8.6% for MTO-3 and MTO-4, respectively. Concurrently, the HOMO-2 → LUMO transition contribution escalated to 62.3%, 67.6%, and 76.9% for MTO-2, MTO-3, and MTO-4. As a result, the fluorescence transition for these compounds transformed into a forbidden process as the methyl substituent position varied. Comparable phenomena were also evident in different solvents. The consistent maximum absorption and emission wavelengths of MTO-1 across various aprotic solvents with differing polarities supported the π → π* nature of the transition (Fig. 1c). An anomalous redshift in both absorption and emission spectra was observed for these compounds in protic solvents, accompanied by a marked increase in emission intensity (Fig. 1d). Correspondingly, the ΦF increased to 6.67% in methanol. This abnormal enhancement and redshift were attributed to hydrogen bonding interactions between the solvent and the carbonyl group of MTOs.51
Previous studies have established thiochroman-4-one derivatives as promising phosphorescent dyes with high ISC efficiency.18,52,53 Hence, the photophysical properties of these dyes were further investigated in 2-methyltetrahydrofuran at 77 K. As depicted in Fig. 1e, a broad emission band spanning 420–620 nm, adorned with a fine structure, was discernible in the steady-state spectra. Remarkably, the phosphorescence spectra of these compounds closely resembled their corresponding steady-state emission spectra. Most intriguingly, no appreciable fluorescence emission was detected at 77 K, strongly suggesting an efficient ISC process for these compounds.
Subsequently, these compounds were embedded in a PVA matrix, known to effectively suppress non-radiative transitions and isolate oxygen,15,54 thus enabling the fabrication of RTP materials. Powder X-ray diffraction analysis of these films (Fig. S3†) revealed an absence of discernible diffraction peaks, signifying their amorphous nature. Analogous to the behaviour of the dyes in solution, the maximum absorption peaks for these films were found around 350 nm (Fig. 2a). The steady-state emission spectra of the films displayed structureless emission bands spanning 380–700 nm (Fig. 2b). Specifically, MTO-1@PVA showcased a maximum emission wavelength at 485 nm, accompanied by a shoulder peak at approximately 420 nm. Notably, the shoulder peak vanished in the phosphorescence emission spectrum, implying that it corresponded to the weak fluorescence emission of MTO-1@PVA, while the dominant emission peak at 485 nm was attributed to the phosphorescence emission. This attribution was further substantiated by lifetime measurements performed at different temperatures (Fig. S4†). The phosphorescence lifetime of MTO-1@PVA was measured to be 21.92 ms at 297 K (Fig. 2c). Its photoluminescence quantum yield (ΦPL) was calculated to be 69.6% (Fig. S5† and Table 1). The excellent agreement between the excitation and absorption spectra (Fig. 2a) confirmed the origin of phosphorescence from MTO-1. Similarly, MTO-2@PVA displayed a much weaker shoulder peak around 420 nm. In contrast, MTO-3@PVA displayed a structureless emission peak at 460 nm, which was confidently assigned to the phosphorescence emission. The ΦPL values for MTO-2@PVA and MTO-3@PVA were determined to be 47.7% and 48.8%, respectively. Thus, it can be concluded that MTO-1@PVA, MTO-2@PVA, and MTO-3@PVA are essentially pure phosphorescent materials, a rarity among metal-free substances. However, MTO-4@PVA exhibited a distinct behaviour. As evident in Fig. 2b, the maximum emission wavelength of MTO-4@PVA was positioned at 435 nm in the steady-state emission spectrum, while its phosphorescence emission wavelength was identified as 470 nm, implying that the phosphorescence emission of MTO-4 was significantly weaker than its fluorescence emission within the PVA matrix. Correspondingly, the ΦPL of MTO-4@PVA was measured to be a mere 2.7%. The phosphorescence lifetime for MTO-4@PVA was found to be 12.0 ms. The fluorescence lifetimes of all films under scrutiny ranged from 1.44 to 2.47 ns (Fig. 2d). It is particularly noteworthy that MTO-1@PVA and MTO-4@PVA exhibited comparable decay patterns and lifetimes (2.34 and 2.47 ns, respectively). Meanwhile, the lifetime spectra and parameters of MTO-2@PVA closely resembled those of MTO-3@PVA.
 | ||
| Fig. 2 (a) Absorption and phosphorescence excitation spectra of MTOs in the PVA matrix; (b) steady-state (solid line) and phosphorescence spectra (dashed line) of MTOs in the PVA matrix; phosphorescence (c) and fluorescence (d) lifetime spectra of MTOs in the PVA matrix; (e) electron–hole distribution and energy levels of different excited states. | ||
| λ Abs/nm | λ F/nm | λ P/nm | τ F/ns | τ P/ms | Φ PL/% | Φ ISC/% | k ISC/s−1 | k P/s−1 | k nr/s−1 | |
|---|---|---|---|---|---|---|---|---|---|---|
a
 , where kS is the deactivation rate of the singlet excited state, and kT is the generation rate of the triplet excited state. kS and kT are determined according to the global fitting results of TA spectra.
b , where kS is the deactivation rate of the singlet excited state, and kT is the generation rate of the triplet excited state. kS and kT are determined according to the global fitting results of TA spectra.
b
 .
c .
c
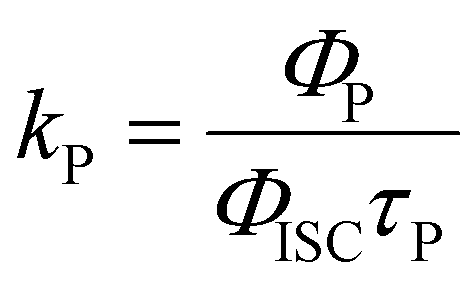 .
d .
d
 ; n.d. – too weak to record. ; n.d. – too weak to record.
|
||||||||||
| MTO-1@PVA | 356 | 423 | 488 | 2.34 | 21.92 | 69.6 | 99.999 | 1.50 × 1010 | 31.75 | 13.87 |
| MTO-2@PVA | 349 | n.d. | 470 | 1.46 | 10.28 | 47.7 | 99.995 | 1.75 × 1010 | 46.40 | 50.88 |
| MTO-3@PVA | 345 | n.d. | 464 | 1.44 | 11.77 | 48.8 | 99.996 | 2.05 × 1010 | 41.46 | 43.50 |
| MTO-4@PVA | 347 | n.d. | 468 | 2.47 | 12.00 | 2.7 | 99.995 | 2.55 × 1010 | 2.25 | 81.08 |
To delve deeper into the influence of the methyl group substituent position on the ISC process, TDDFT and ORCA calculations were also performed.18,23 As previously discussed, the S1 → S0 transition in MTOs progressively transitioned from a π → π* to an n → π* character with the change in the methyl group position. However, no conspicuous alteration in the frontier orbital distribution was observed (Fig. S2 and Table S2†). The energy levels of T1 and T2 exhibited slight fluctuations with the varying substituent positions (Table S2†). Notably, T2 represented the highest triplet state, often being either close to or lower in energy than the S1 state. Hence, S1 → T1 and S1 → T2 transitions were identified as plausible ISC pathways for generating triplet excitons. Fig. 2e reveals that the hole in the S0 → T1 transition of MTOs is primarily localized on the sulfur atom and benzene rings, corresponding to π orbitals, while the electron is primarily distributed over the benzene and carbonyl moieties, associated with π* orbitals. Although the electron distribution in the S0 → T2 transition mirrors that of the S0 → T1 transition, the hole is primarily contributed by the n orbital of the carbonyl moiety. Consequently, the S1 → T1 ISC process evolved from a forbidden to an allowed transition as the substituent position changed, according to El-Sayed's rule. Conversely, the S1 → T2 ISC process transformed from an allowed to a forbidden transition with the variation in the substituent position. ORCA calculations further substantiated the significant impact of the methyl substituent position on the SOC, which is intricately linked to the ISC process.
Femtosecond TA spectra of MTOs in the PVA matrix were recorded and are presented in Fig. 3 and S6.† No ground state bleach (GSB) signal was observable due to their absorptions lying outside our detection wavelength range, nor was any stimulated emission (SE) signal detected. A distinct excited state absorption (ESA) signal was promptly detected for MTO-1@PVA post-excitation (Fig. 3a). The initially formed ESA band featured a peak around 620 nm with a shoulder at approximately 525 nm (Fig. 3b). Within the first 200 ps, it evolved into a long-lived ESA band with peaks at 470 nm and 610 nm. Analogous TA spectra were also evident for MTO-2@PVA and MTO-3@PVA (Fig. S6a and b†). Remarkably, MTO-4@PVA displayed a markedly different TA spectrum compared to the other three films (Fig. S6c†). A distinct ESA signal spanning 400–600 nm, with peaks at 405 nm and 485 nm, was observed. The initial ESA band transformed into a long-lived ESA band spanning 400–600 nm, centred at 450 nm, within the first 100 ps (Fig. S6f†). Decay-associated difference spectra (DADS) for MTO-1@PVA were extracted via global analysis of the femtosecond TA data (Fig. S7†). The first decay-associated difference spectrum was attributed to the absorption of the singlet state (Fig. S7,† black line), displaying a broad signal in the 410–700 nm range with peaks around 520 nm and 620 nm, consistent with species present in the first 300 ps. The subsequent DADS was assigned to the absorption of the triplet state (Fig. S7,† red line), given its slow generation and decay within the 7 ns detection window. This spectrum exhibited a broad signal in the 410–700 nm range, with peaks around 450 nm and 615 nm. Comparable DADS were also discernible for the remaining three films (Fig. S6†). The decay kinetics curves of the singlet excited state and the generation kinetics of the triplet state of MTOs in the PVA matrix are compiled in Fig. 3c. Notably, the decay rate of the singlet excited state showed a pronounced increase from MTO-1 to MTO-4. The near-symmetric kinetics curves of decay and generation implied a highly efficient ISC process. The ISC yields (ΦISC) of MTOs were estimated to be >99.9% based on the kinetic parameters (Fig. 3d).55 The kISC for MTO-1 was calculated to be 1.50 × 1010 s−1, increasing to 1.75 × 1010 s−1 and 2.05 × 1010 s−1 for MTO-2 and MTO-3, respectively. MTO-4 displayed the highest kISC, reaching up to 2.55 × 1010 s−1. However, the ΦP of MTOs in the PVA matrix decreased from 69.6% to 2.7% as kISC increased. The TA data indicate that the ISC in the MTO series of compounds is highly efficient. Therefore, the phosphorescence quenching observed in MTO-4@PVA is unlikely to be due to issues with the generation of triplet excitons. Instead, strong non-radiative transitions are likely the primary cause.
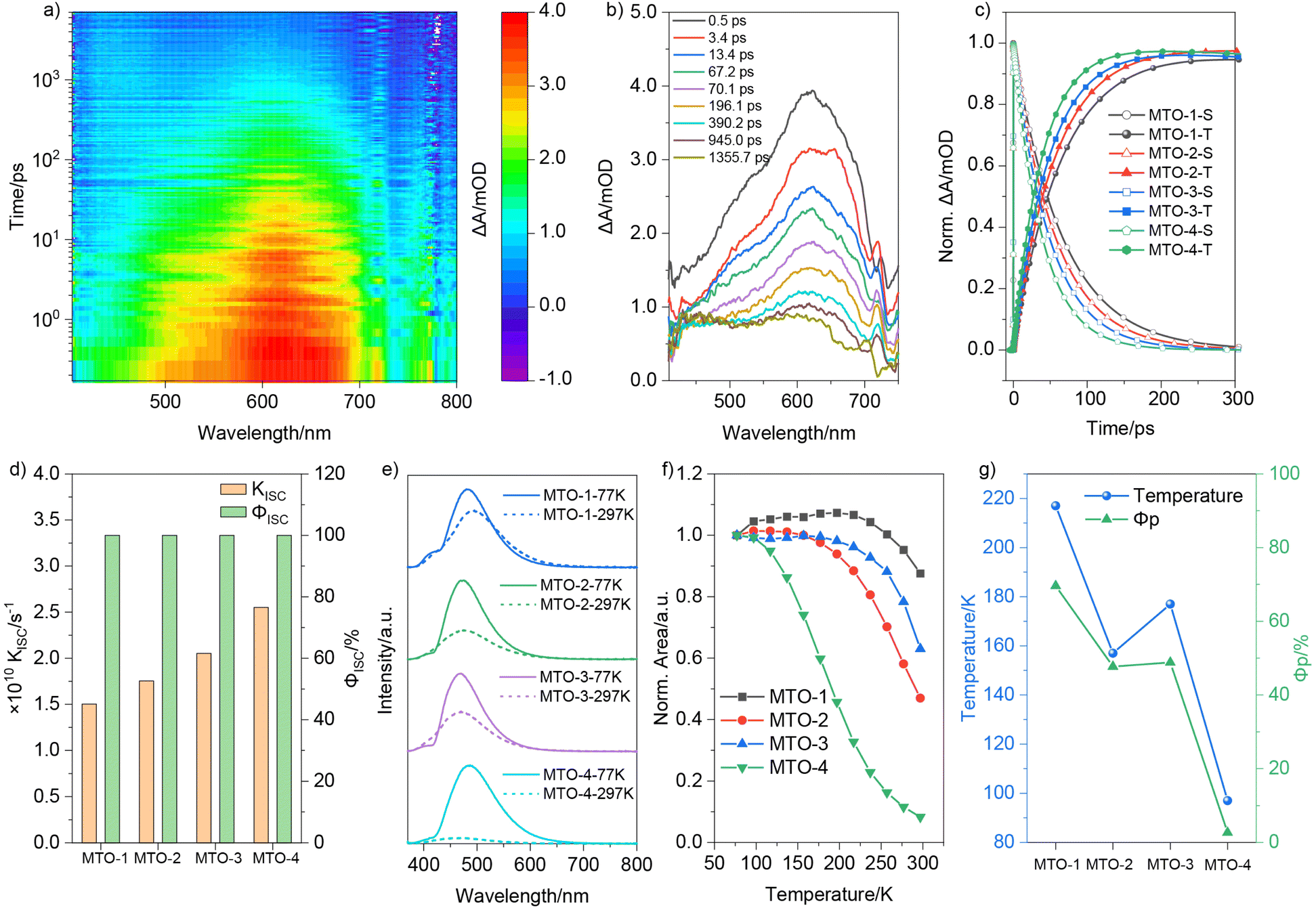 | ||
| Fig. 3 (a) Femtosecond broadband TA spectra of MTO-1 in PVA under 320 nm excitation; (b) TA spectra of MTO-1 at different pump-probe delay times; (c) normalized singlet and triplet state kinetics curves of MTOs in the PVA matrix; (d) the calculated ΦISC and kISC of MTOs according to DADS data; (e) steady-state spectra of MTOs in the PVA matrix at 77 K and 297 K; (f) normalized emission spectra area of MTOs in the PVA matrix at different temperatures; (g) transition temperature points and ΦP of MTOs in the PVA matrix. | ||
To exclude the possibility of oxygen-induced non-radiative transitions in the PVA matrix, the emission spectra of MTO-4@PVA were initially measured under different atmospheres. As depicted in Fig. S8,† no noticeable change in phosphorescence intensity was observed when the film atmosphere was switched from air to argon. The phosphorescence intensity of MTO-4@PVA significantly intensified in the steady-state spectrum upon decreasing the temperature from 297 K to 77 K (Fig. 3e). Specifically, the area of the emission peak grew by a factor of 12.1 with decreasing temperature. In comparison, MTO-1@PVA, MTO-2@PVA, and MTO-3@PVA exhibited increases of 1.1, 2.1, and 1.6 times, respectively. A pronounced elongation in the phosphorescence lifetime was also observed as the temperature dropped from 297 K to 77 K. The emission spectrum areas of MTOs in the PVA matrix at various temperatures were meticulously analysed (Fig. S9†) and plotted in Fig. 3f. The area of MTO-1@PVA showed a marginal increase when the temperature increased from 77 K to 97 K. A distinct decline, signifying the onset of triplet exciton quenching by molecular thermal motion, ensued once the temperature exceeded 217 K. Similar trends were observed for the remaining three films. The areas of MTO-2@PVA, MTO-3@PVA, and MTO-4@PVA started to diminish when the temperature surpassed 157 K, 177 K, and 97 K, respectively. Notably, MTO-4@PVA displayed a much steeper decrease rate compared to the other three films. Intriguingly, the transition temperature points followed the same trend as ΦP (Fig. 3g). The lower transition temperature points implied that triplet excitons in these films were more susceptible to quenching by molecular thermal vibration, hinting at a large knr.
To further understand the non-radiative pathways and the underlying mechanisms responsible for the phosphorescence quenching, we further used the DFT calculations to quantitatively assess the vibrational freedom of these compounds at the B3LYP/def2SVP level. The reorganization energy versus different normal vibrational modes is computed to evaluate the feasibility of a phosphor in its T1 state to decay via nonradiative decay (Fig. 4a). Compared to MTO-1, MTO-2 shows a slight increase in vibration at a low-frequency mode (377 cm−1), whereas MTO-3 primarily exhibits a slight increase in high-frequency vibration modes. The subtle changes in the vibrational modes of MTO-2 and MTO-3 may explain the decrease in their ΦP. MTO-4 has a remarkably high reorganization energy of 1891.3 cm−1 at a vibrational mode of 82 cm−1. Additionally, the reorganization energies at 565 cm−1 and 1192 cm−1 are both greater than 500 cm−1. The significant increase in vibrational modes may explain the strong non-radiative transitions in MTO-4. Further analysis of the root mean square deviation (RMSD) of this series of compounds revealed that, with the change in the position of the substituents, the difference between the optimal configurations of the T1 state and the S0 state gradually increases (Fig. 4b). Specifically, MTO-1 has the smallest RMSD value of 0.08, whereas MTO-4 has the largest, at 0.33—an increase of about four times. The large RMSD value of MTO-4 indicates stronger molecular vibrational behavior, providing more opportunities for non-radiative relaxation. We further calculated the Huang–Rhys factors of this series of compounds to characterize their EPC. As shown in Fig. 4c, the Huang–Rhys factors of MTO-1, MTO-2, and MTO-3 for various vibrational modes do not exceed 1. However, MTO-4 has four vibrational modes with Huang–Rhys factors exceeding 1.0, including a staggering 23.06 at 82 cm−1. Such a high Huang–Rhys factor indicates strong EPC in MTO-4. Therefore, despite having similar electronic structures and energy levels in the S0 and T1 states compared to the other three compounds, the T1 state of MTO-4 is highly susceptible to non-radiative deactivation.
 | ||
| Fig. 4 (a) Calculated reorganization energies of the T1 → S0 transition are plotted against the frequencies of MTOs. Representative high-intensity (>500 cm−1) vibration modes are shown as insets; (b) schematic distribution of RMSD T0 → S0 values; (c) Huang–Rhys factors of MTOs from T1 to S0. The insets show the most representative intramolecular vibration modes of MTO-4 in S0 and T1 states. | ||
Conclusions
In summary, a suite of efficient RTP materials was engineered by elaborate molecular designing. The S1 → S0 transition characteristics was effectively modulated between π → π* and n → π* transitions by altering the methyl group substituent position. All synthesized dyes displayed near-unity ΦISC values. The kISC increased gradually from MTO-1 to MTO-4 as the methyl group substituent position varied. However, contrary to expectations, the ΦP decreased from 69.6% to 2.7% alongside the rise in kISC. Theoretical calculations and experimental results indicate that the quenching of phosphorescence is primarily due to the strong EPC caused by the positional variation of the methyl substituents. This finding contrasts with common factors such as ISC or changes in energy levels, which are often considered in the context of phosphorescence quenching. The positional changes of the methyl groups significantly influence the vibrational modes and the interaction between electronic and vibrational states, leading to enhanced non-radiative relaxation and, consequently, the observed quenching of phosphorescence. These findings provide a new insight into the design of high-performance RTP dyes.Data availability
All the data are available in the ESI.†Author contributions
L. Ma, S. Sun, and X. Ma designed the materials and conceived the project. L. Ma conducted the molecule synthesis and characterization. M. Cong performed TA experiments. L. Ma and X. Ma wrote the manuscript. All authors engaged in discussions and conducted data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22125803, T2488302, 22020102006 and 22305080), Science and Technology Commission of Shanghai Municipality (Grant No. 24DX1400200), and the Fundamental Research Funds for the Central Universities.References
- Z. Wang, X. Cheng, Y. Xie, S. Liu, M. Dong, J. Zhao, F. Liang, Z. An and W. Huang, Recent Advances in Organic Room-Temperature Phosphorescence of Heteroatom (B/S/P)-Containing Chromophores, CCS Chem., 2023, 5, 292–309 CrossRef CAS.
- K. Jinnai, R. Kabe, Z. Lin and C. Adachi, Organic long-persistent luminescence stimulated by visible light in p-type systems based on organic photoredox catalyst dopants, Nat. Mater., 2022, 21, 338–344 CrossRef CAS PubMed.
- B. Ding, X. Ma and H. Tian, Recent Advances of Pure Organic Room Temperature Phosphorescence Based on Functional Polymers, Acc. Mater. Res., 2023, 4, 827–838 CrossRef CAS.
- L. Xu, H. Wei, G. Xie, B. Xu and J. Zhao, Ultralong MRTADF and Room-Temperature Phosphorescence Enabled Color-Tunable and High-Temperature Dual-Mode Organic Afterglow from Indolo[3,2-b]carbazole, Adv. Funct. Mater., 2024, 34, 2402428 CrossRef CAS.
- Y. Zhao, J. Yang, C. Liang, Z. Wang, Y. Zhang, G. Li, J. Qu, X. Wang, Y. Zhang, P. Sun, J. Shi, B. Tong, H. Y. Xie, Z. Cai and Y. Dong, Fused-Ring Pyrrole-Based Near-Infrared Emissive Organic RTP Material for Persistent Afterglow Bioimaging, Angew. Chem., Int. Ed., 2023, 63, e202317431 CrossRef PubMed.
- J. Yu, Z. Sun, H. Ma, C. Wang, W. Huang, Z. He, W. Wu, H. Hu, W. Zhao and W.-H. Zhu, Efficient Visible-Light-Activated Ultra-Long Room-Temperature Phosphorescence Triggered by Multi-Esterification, Angew. Chem., Int. Ed., 2023, 135, e202316647 CrossRef.
- Z. Xu, Z. Wang, W. Yao, Y. Gao, Y. Li, H. Shi, W. Huang and Z. An, Supercooled Liquids with Dynamic Room Temperature Phosphorescence Using Terminal Hydroxyl Engineering, Angew. Chem., Int. Ed., 2023, 62, e202301564 CrossRef CAS PubMed.
- Y. Li, Z. Huang, A. Shao, Z. Wu, Z. He, H. Tian and X. Ma, Aqueous up-conversion organic phosphorescence and tunable dual emission in a single-molecular emitter, Chem. Sci., 2025 10.1039/d4sc08330j.
- H. Sun, Y. Xiao, Y. He, X. Wei, J. Zou, Y. Luo, Y. Wu, J. Zhao, V. K.-M. Au and T. Yu, 3D printable organic room-temperature phosphorescent materials and printed real-time sensing and display devices, Chem. Sci., 2025, 16, 5299–5309 RSC.
- T. Wang, A. K. Gupta, S. Wu, A. M. Z. Slawin and E. Zysman-Colman, Conjugation-Modulated Excitonic Coupling Brightens Multiple Triplet Excited States, J. Am. Chem. Soc., 2023, 145, 1945–1954 CrossRef CAS PubMed.
- Q. Peng, H. Ma and Z. Shuai, Theory of Long-Lived Room-Temperature Phosphorescence in Organic Aggregates, Acc. Chem. Res., 2021, 54, 940–949 CrossRef CAS PubMed.
- Q. Meng, S. Gan, Q. Cheng, Z. Jiang, H. Zhu, G. Xie, R. Liu, S. Zhu and H. Zhu, Efficient tuning of nitrogen-doped carbon dots phosphorescence based on substrate regulation for multicolor and time-dependent anti-counterfeiting, Dyes Pigm., 2024, 224, 112002 CrossRef CAS.
- X. Ma, J. Wang and H. Tian, Assembling-Induced Emission: An Efficient Approach for Amorphous Metal-Free Organic Emitting Materials with Room-Temperature Phosphorescence, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- L. Ma and X. Ma, Recent advances in room-temperature phosphorescent materials by manipulating intermolecular interactions, Sci. China Chem., 2022, 66, 304–314 CrossRef.
- L. Gao, J. Huang, L. Qu, X. Chen, Y. Zhu, C. Li, Q. Tian, Y. Zhao and C. Yang, Stepwise taming of triplet excitons via multiple confinements in intrinsic polymers for long-lived room-temperature phosphorescence, Nat. Commun., 2023, 14, 7252 CrossRef CAS PubMed.
- L. Wei, F. Gao, C. He, Q. He, P. Jin, Y. Rong, T. Zhao, C. Yang and W. Wu, A new sensitization strategy for achieving organic RTP in aqueous solution: tunable RTP and UC emission in supramolecular TTA-UC systems, Sci. China Chem., 2023, 66, 3546–3554 CrossRef CAS.
- T. Wang, M. Liu, J. Mao, Y. Liang, L. Wang, D. Liu, T. Wang and W. Hu, Recent advances in long-persistent luminescence materials based on host–guest architecture, Chinese Chem. Lett., 2024, 35, 108385 CrossRef CAS.
- L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Highly Efficient Room-Temperature Phosphorescence Based on Single-Benzene Structure Molecules and Photoactivated Luminescence with Afterglow, Adv. Funct. Mater., 2021, 31, 2010659 CrossRef CAS.
- L. Ma, Y. Liu, X. Jin, T. Jiang, L. Zhou, Q. Wang, H. Tian and X. Ma, Triplet Exciplex Mediated Multi-Color Ultra-Long Afterglow Materials, Angew. Chem., Int. Ed., 2025, 64, e202500847 Search PubMed.
- M. A. El-Sayed, Spin-Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- J. Yang, M. Fang and Z. Li, Stimulus-Responsive Room Temperature Phosphorescence Materials: Internal Mechanism, Design Strategy, and Potential Application, Acc. Mater. Res., 2021, 2, 644–654 CrossRef CAS.
- Z. Chen, X. Chen, D. Ma, Z. Mao, J. Zhao and Z. Chi, Synergetic Conformational Regulations in Ground and Excited States for Realizing Stimulus-Responsive and Wide-Tuning Room-Temperature Phosphorescence, J. Am. Chem. Soc., 2023, 145, 16748–16759 CrossRef CAS PubMed.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, Efficient and Long-lived Room Temperature Organic Phosphorescence: Theoretical Descriptors for Molecular Designs, J. Am. Chem. Soc., 2018, 141, 1010–1015 CrossRef PubMed.
- M. Gao, Y. Tian, X. Li, Y. Gong, M. Fang, J. Yang and Z. Li, The Effect of Molecular Conformations and Simulated “Self-Doping” in Phenothiazine Derivatives on Room-Temperature Phosphorescence, Angew. Chem., Int. Ed., 2023, 62, e202214908 CrossRef CAS.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Molecular Engineering for Metal-Free Amorphous Materials with Room-Temperature Phosphorescence, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, 2009, ISBN: 978-1-891389-25-2 Search PubMed.
- C. A. M. Salla, G. Farias, M. Rouzieres, P. Dechambenoit, F. Durola, H. R. Bock, B. de Souza and I. H. Bechtold, Persistent solid-state phosphorescence and delayed fluorescence at room temperature from a twisted hydrocarbon, Angew. Chem., Int. Ed., 2019, 58, 6982–6986 CrossRef CAS PubMed.
- Z. Yuan, J. Wang, L. Chen, L. Zou, X. Gong and X. Ma, Methanol Dynamically Activated Room-Temperature Phosphorescence from a Twisted 4-Bromobiphenyl System, CCS Chem., 2020, 2, 158–167 CrossRef CAS.
- S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Aqueous Phase Phosphorescence: Ambient Triplet Harvesting of Purely Organic Phosphors via Supramolecular Scaffolding, Angew. Chem., Int. Ed., 2018, 57, 17115–17119 CrossRef CAS PubMed.
- Z. Xie, Z. Mao, H. Wang, Y. Xiao, X. Zhang, T. Yu, Z. An and W. Huang, Dual-channel mechano-phosphorescence: a combined locking effect with twisted molecular structures and robust interactions, Light Sci. Appl., 2024, 13, 85 CrossRef CAS PubMed.
- X. Ma, C. Xu, J. Wang and H. Tian, Amorphous Pure Organic Polymers for Heavy-Atom-Free Efficient Room-Temperature Phosphorescence Emission, Angew. Chem., Int. Ed., 2018, 57, 10854–10858 CrossRef CAS PubMed.
- Z. A. Yan, X. Lin, S. Sun, X. Ma and H. Tian, Activating Room-Temperature Phosphorescence of Organic Luminophores via External Heavy-Atom Effect and Rigidity of Ionic Polymer Matrix, Angew. Chem., Int. Ed., 2021, 60, 19735–19739 CrossRef CAS PubMed.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Boosting the Quantum Efficiency of Ultralong Organic Phosphorescence up to 52% via Intramolecular Halogen Bonding, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS.
- E. Hamzehpoor, C. Ruchlin, Y. Tao, C. H. Liu, H. M. Titi and D. F. Perepichka, Efficient room-temperature phosphorescence of covalent organic frameworks through covalent halogen doping, Nat. Chem., 2023, 15, 83–90 CrossRef CAS PubMed.
- C. Li, X. Li and Q. Wang, Supramolecular self-assembling strategy for constructing cucurbit[6]uril derivative-based amorphous pure organic room-temperature phosphorescence complex featuring extra-high efficiency, Chinese Chem. Lett., 2022, 33, 877–880 CrossRef CAS.
- J. Wang, Z. Huang, X. Ma and H. Tian, Visible-Light-Excited Room-Temperature Phosphorescence in Water by Cucurbit[8]uril-Mediated Supramolecular Assembly, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- X. K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Supramolecular Pins with Ultralong Efficient Phosphorescence, Adv. Mater., 2021, 33, 2007476 CrossRef CAS PubMed.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Confining isolated chromophores for highly efficient blue phosphorescence, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed.
- H. Wang, H. Ma, N. Gan, K. Qin, Z. Song, A. Lv, K. Wang, W. Ye, X. Yao, C. Zhou, X. Wang, Z. Zhou, S. Yang, L. Yang, C. Bo, H. Shi, F. Huo, G. Li, W. Huang and Z. An, Abnormal thermally-stimulated dynamic organic phosphorescence, Nat. Commun., 2024, 15, 2134 CrossRef CAS PubMed.
- G. Wang, X. Chen, Y. Zeng, X. Li, X. Wang and K. Zhang, Dual-Mechanism Design Strategy for High-Efficiency and Long-Lived Organic Afterglow Materials, J. Am. Chem. Soc., 2024, 146, 24871–24883 CrossRef CAS PubMed.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- B. Chen, W. Huang and G. Zhang, Observation of Chiral-selective room-temperature phosphorescence enhancement via chirality-dependent energy transfer, Nat. Commun., 2023, 14, 1514 CrossRef CAS PubMed.
- M. Molkenthin, E. Hupf and B. J. Nachtsheim, Dibenzyl isophthalates as versatile hosts in room temperature phosphorescence host–guest systems, Chem. Sci., 2025, 16, 2819–2829 RSC.
- B. Du, Y. Wu, X. Wang, H. Tian, S. Shao and L. Wang, Multi-resonance emitters with room-temperature phosphorescence in amorphous state and excited by visible light, Chem. Sci., 2024, 15, 19432–19442 RSC.
- J. Li, Y. Dong, R. Wei, G. Jiang, C. Yao, M. Lv, Y. Wu, S. H. Gardner, F. Zhang, M. Y. Lucero, J. Huang, H. Chen, G. Ge, J. Chan, J. Chen, H. Sun, X. Luo, X. Qian and Y. Yang, Stable, Bright, and Long-Fluorescence-Lifetime Dyes for Deep-Near-Infrared Bioimaging, J. Am. Chem. Soc., 2022, 144, 14351–14362 CrossRef CAS PubMed.
- Y. Jiang, Y. Li, F. Liu, W. Wang, W. Su, W. Liu, S. Liu, W. Zhang, J. Hou, S. Xu, Y. Yi and X. Zhu, Suppressing electron-phonon coupling in organic photovoltaics for high-efficiency power conversion, Nat. Commun., 2023, 14, 5079 CrossRef CAS PubMed.
- X. Gong, O. Voznyy, A. Jain, W. Liu, R. Sabatini, Z. Piontkowski, G. Walters, G. Bappi, S. Nokhrin, O. Bushuyev, M. Yuan, R. Comin, D. McCamant, S. O. Kelley and E. H. Sargent, Electron–phonon interaction in efficient perovskite blue emitters, Nat. Mater., 2018, 17, 550–556 CrossRef CAS PubMed.
- H. Zhang, S. Wu, Y. Liang, Z. Zhang, H. Wei, Q. Yang, P. Hu, C. Liu, Z. Yang, C. Zheng, G. Shi, Z. Chi and B. Xu, Enabling efficient and ultralong room-temperature phosphorescence from organic luminogens by locking the molecular conformation in polymer matrix, Chem. Eng. J., 2024, 497, 154949 CrossRef CAS.
- X. Zhu, Y. Jia, Y. Liu, J. Xu, H. He, S. Wang, Y. Shao, Y. Zhai and Y. Zhu, Enhancing Built-in Electric Fields via Molecular Symmetry Modulation in Supramolecular Photocatalysts for Highly Efficient Photocatalytic Hydrogen Evolution, Angew. Chem., Int. Ed., 2024, 63, e202405962 CrossRef CAS PubMed.
- J. V. Morris, U. Brühlmann, O. Serafimov and J. R. Huber, Excited State Behavior of Aromatic Amines in Solution: Dihydrophenazine Derivatives, Ber. Bunsenges. Phys. Chem., 1974, 78, 1348–1353 CrossRef CAS.
- L. Ma, B. Ding, Z. Yuan, X. Ma and H. Tian, Triboluminescence and Selective Hydrogen-Bond Responsiveness of Thiochromanone Derivative, ACS Mater. Lett., 2021, 3, 1300–1306 CrossRef CAS.
- L. Ma, Y. Liu, H. Tian and X. Ma, Switching Singlet Exciton to Triplet for Efficient Pure Organic Room-Temperature Phosphorescence by Rational Molecular Design, JACS Au, 2023, 3, 1835–1842 CrossRef CAS PubMed.
- Q. Xu, L. Ma, S. Sun and X. Ma, Achieving visible-light-excited organic room-temperature phosphorescence by manipulating p–π conjugation, J. Mater. Chem. C, 2021, 9, 14623–14627 RSC.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Ultralong room temperature phosphorescence from amorphous organic materials toward confidential information encryption and decryption, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- M. Lv, X. Lu, Y. Jiang, M. E. Sandoval-Salinas, D. Casanova, H. Sun, Z. Sun, J. Xu, Y. Yang and J. Chen, Near-Unity Triplet Generation Promoted via Spiro-Conjugation, Angew. Chem., Int. Ed., 2022, 61, e202113190 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02149a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |