
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical-mediated proton transfer enables hydroxyl radical formation in charge-delocalized water†
Ruijuan
Zhao‡
a,
Qiuyue
Zhang‡
b,
Na
Yang
c,
Lei
Li
 a,
Zhenyu
Li
*b and
Chunhua
Cui
*a
a,
Zhenyu
Li
*b and
Chunhua
Cui
*a
aMolecular Electrochemistry Laboratory, Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 611731, China. E-mail: chunhua.cui@uestc.edu.cn
bKey Laboratory of Precision and Intelligent Chemistry, University of Science and Technology of China, Hefei 230026, China. E-mail: zyli@ustc.edu.cn
cSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu, 610054, China
First published on 23rd May 2025
Abstract
The Grotthuss mechanism has long defined our understanding of proton transfer in water, where protons migrate through hydrogen bond networks via structural diffusion. However, whether radical intermediates participate in this process remains unresolved. Herein, we demonstrate a radical-mediated proton transfer mechanism in acidic aqueous solutions. Using electron paramagnetic resonance spectroscopy, we detect hydroxyl radicals (˙OH) and water radical cations (H2O˙+), while isotope-labeled high-resolution mass spectrometry confirms the presence of crown ether H2O˙+ complexes in HCl solutions. Quantum simulations reveal that excess charge is delocalized over neighboring water molecules rather than localized on the hydronium ion (H3O+), forming positively charged water clusters. We thus reveal that the proton transfer proceeds through ˙OH⋯H+···H2O intermediates under ambient conditions. Our findings extend beyond the Grotthuss framework, proposing a mechanism driven by hydrogen bond imbalance and charge delocalization that spontaneously generates ˙OH in acidic environments. This work advances the understanding of proton transfer in water and has implications for acid-induced reactions, electrochemical processes, and degradation mechanisms in energy technologies.
Introduction
Proton transfer in aqueous solution is a fundamental process central to acid–base chemistry, biochemistry, and energy conversion technologies.1,2 The Grotthuss mechanism, which describes proton migration through hydrogen bond networks via structural diffusion, has long been the prevailing framework for understanding this process.2–6 This mechanism involves the interconversion of two limiting structures: the Zundel cation (H5O2+) and the eigen (H9O4+) cation6–9 (yet a recent report described that both clusters do not represent limiting configurations and distinct thermodynamic states10), where proton transfer occurs through the concerted breaking and reformation of hydrogen bonds. However, this classical model assumes that proton transfer proceeds without charge delocalization between water molecules, an assumption that may not hold under certain conditions.Theoretical studies suggest that localizing excess charge on the hydronium ion (H3O+) is energetically unfavorable, requiring 16–49 kcal mol−1.11–14 This raises the possibility that charge delocalization over neighboring water molecules may play a critical role in proton transfer dynamics.14 Despite this insight, the potential involvement of radical intermediates, such as hydroxyl radicals (˙OH) and water radical cations (H2O˙+), in proton transfer remains largely unexplored. The generation of these radicals typically requires high-energy processes, such as radiolysis15,16 or interfacial charge transfer,17–21 making their spontaneous formation in bulk water under ambient conditions seem unlikely.
In this study, we investigate proton transfer in acidic aqueous solutions using a combination of experimental techniques and quantum simulations. Through electron paramagnetic resonance (EPR) spectroscopy, we detect the formation of ˙OH and H2O˙+ radicals in HCl solutions with pH below 4.0. Isotope-labeled high-resolution mass spectrometry (HRMS) further confirms the presence of crown ether-H2O˙+ complexes, providing direct evidence for radical intermediates in proton transfer. Quantum simulations reveal that the excess charge is delocalized over neighboring water molecules rather than localized on the H3O+ ion, leading to the formation of positively charged water clusters. We thus uncover that the proton transfer proceeds via a ˙OH⋯H+···H2O intermediate under ambient conditions, enabling a radical-mediated proton transfer pathway.
This mechanism advances our fundamental understanding of proton transfer in water and has significant implications for acid corrosion of catalysts, such as Cu for acidic electrochemical CO2 reduction and oxidation-degradation of proton-exchange membrane in fuel cells. By revealing the role of radical intermediates in proton transfer, this work opens new avenues for research in catalysis, energy conversion, and environmental chemistry.
Results and discussion
pH-dependent formation of hydroxyl radicals (˙OH)
To investigate the potential formation of radical species in aqueous solutions, we first employed EPR to detect radicals in pure water. To enhance the stability and detectability of transient radicals, 100 mM 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was used as a spin-trapping agent.22,23 As expected, no EPR signals were detected in pure water, indicating the absence of detectable radicals under neutral conditions. To eliminate potential interference from oxygen-derived radicals, we used HCl (rather than oxygenated acids) to adjust the solution pH.24–26 Remarkably, upon decreasing the pH from neutral to 1.0, a distinct EPR signal corresponding to the DMPO-OH adduct (AN = AH = 1.49 mT)22 emerged at pH values below 4.0, with the signal intensity reaching its maximum at pH 1.5 (Fig. 1A). | ||
| Fig. 1 Detection of ˙OH in acidic aqueous solutions. (A) EPR spectra of DMPO-OH adducts at various pH values (neutral to 1.0) at 25 °C. The pH was adjusted using HCl solutions. (B) Overlay of EPR spectra for DMPO-17OH and DMPO-16OH adducts in H217O (90% 17O abundance). (C) High-resolution mass spectra of DMPO-OH adducts without and with H218O at pH 1.5. (D) Temperature-dependent EPR spectra of DMPO-OH adducts from 0 to 50 °C, measured using an EPR temperature controller. All experiments were performed in Ar-purged solutions containing 100 mM DMPO as a spin-trapping agent. | ||
To confirm the identity of the detected radical as ˙OH, we utilized an alternative spin-trapping agent, 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO).22 The BMPO-OH adducts, characterized by two distinct hyperfine splitting patterns (BMPO-OH-I: AN = 1.41 mT, AH = 1.54 mT; BMPO-OH-II: AN = 1.41 mT, AH = 1.28 mT),23,27 were observed (Fig. S1†), providing further evidence for the presence of ˙OH radicals.
To determine the origin of the oxygen atoms in the ˙OH radicals, we performed isotope-labeling experiments using H217O (90% 17O abundance). As shown in Fig. 1B, the EPR spectrum revealed a mixture of DMPO-17OH and DMPO-16OH adducts (AN = AH = 1.49 mT; AO17 = 0.46 mT),28 confirming that the ˙OH radicals are derived from water molecules. This finding was further corroborated by 18O isotope-labeling experiments using H218O, analyzed via HRMS. Peaks at m/z 130.08 and 132.09 were assigned to DMPO-16OH and DMPO-18OH, respectively (Fig. 1C), providing unambiguous evidence that ˙OH radicals originate from water. Additionally, the addition of 50 mM vitamin C, a known ˙OH scavenger,29 completely quenched the DMPO-OH signal (Fig. S2†), further validating the presence of ˙OH radicals.
To explore the excitation source responsible for ˙OH generation, we investigated the temperature dependence of the DMPO-OH signal. By varying the temperatures from 0 to 50 °C using an EPR temperature controller, we observed that the DMPO-OH signal was detectable even at 0 °C and increased in intensity with rising temperature, reaching a significant concentration at ambient temperature (25 °C) (Fig. 1D). This result indicates that thermal energy at room temperature is sufficient to drive the formation of ˙OH radicals in acidic solutions, highlighting an unrecognized implicit condition for radical generation.
The oxidizing reactions of ˙OH
The high reactivity of ˙OH radicals, evidenced by their strong oxidation potential (˙OH/OH−of ∼1.9 V),30 was further demonstrated through a series of oxidation reactions. For instance, ˙OH can oxidize Cu0 to Cu2+ (0.34 V), I− to I3− (0.54 V), and SO32− to SO3˙− (0.73 V).21,31–35 Contrary to the conventional understanding that HCl solutions are non-oxidizing due to the low electrode potential of H+/H2 (0 V) and the non-oxidizing nature of Cl−, we observed significant oxidation of high-purity Cu foil immersed in Ar pre-purged HCl solutions (pH 1.0–1.5). Optical microscopy and scanning electron microscopy (SEM) revealed pronounced surface roughening of the Cu foil at low pH (Fig. 2A), while X-ray fluorescence (XRF) analysis confirmed the increased dissolution of Cu2+ (Fig. S3†), The formation of Cu–Cl species was further supported by UV-vis spectroscopy, which showed enhanced absorption at lower pH values (Fig. 2B), consistent with the trend of DMPO-OH intensity (Fig. 1A). | ||
| Fig. 2 The oxidizing capability of ˙OH radicals in acidic solutions. (A) pH-dependent oxidation of Cu foil observed by optical microscopy (50×) and SEM. (B) UV-vis spectra of dissolved Cu2+ ions in solutions with varying pH. A 50 mM CuCl2 solution was used as a control. (C) UV-vis spectra of I3− formed by the oxidation of 20 mM I− by ˙OH radicals at different pH values. The oxidation of 20 mM I− to I3− by 0.05 mM H2O2 as a control. (D) EPR spectra of DMPO-SO3− adducts formed by the oxidation of 50 mM K2SO3 at different pH values. | ||
The oxidation of I− to I3− by ˙OH was monitored using UV-vis spectroscopy,36 with 20 mM I− solutions serving as the substrate. Two characteristic absorption bands at 284 and 350 nm, corresponding to I3− formation, were observed in acidic solutions (pH < 4.0) but were absent at higher pH values (Fig. 2C and standard spectra in Fig. S4†). Similarly, the oxidation of SO32− to SO3˙− was confirmed by the detection of the DMPO-SO3− adduct (AN = 1.47 mT; AH = 1.60 mT),22 with significantly higher intensity at pH 1.5 compared to pH 4.0 (Fig. 2D).
The ambient formation of ˙OH radicals in an acidic aqueous solution offers significant potential for synthetic applications, as reported in interface-rich aqueous systems.37–39 For instance, ˙OH can oxidize HCOO− to CO2˙− radicals, as evidenced by the EPR signal of the DMPO-CO2− adduct (AN = 1.56 mT; AH = 1.87 mT).23,40,41 This signal was further confirmed by HRMS (Fig. 3A and S5†), demonstrating the robust oxidizing capability of ˙OH under acidic conditions. Additionally, the well-documented reaction between CH3OH and ˙OH to form the ˙CH2OH radical was verified using DMPO as a spin-trapping agent (Fig. 3B). We also investigated the oxidation of benzoic acid to phenol, a classic organic transformation. HRMS revealed the formation of phenol in acidic solutions (Fig. 3C), further underscoring the synthetic utility of ˙OH radicals. These results collectively highlight the strong oxidizing capability of ˙OH radicals generated in acidic aqueous solutions.
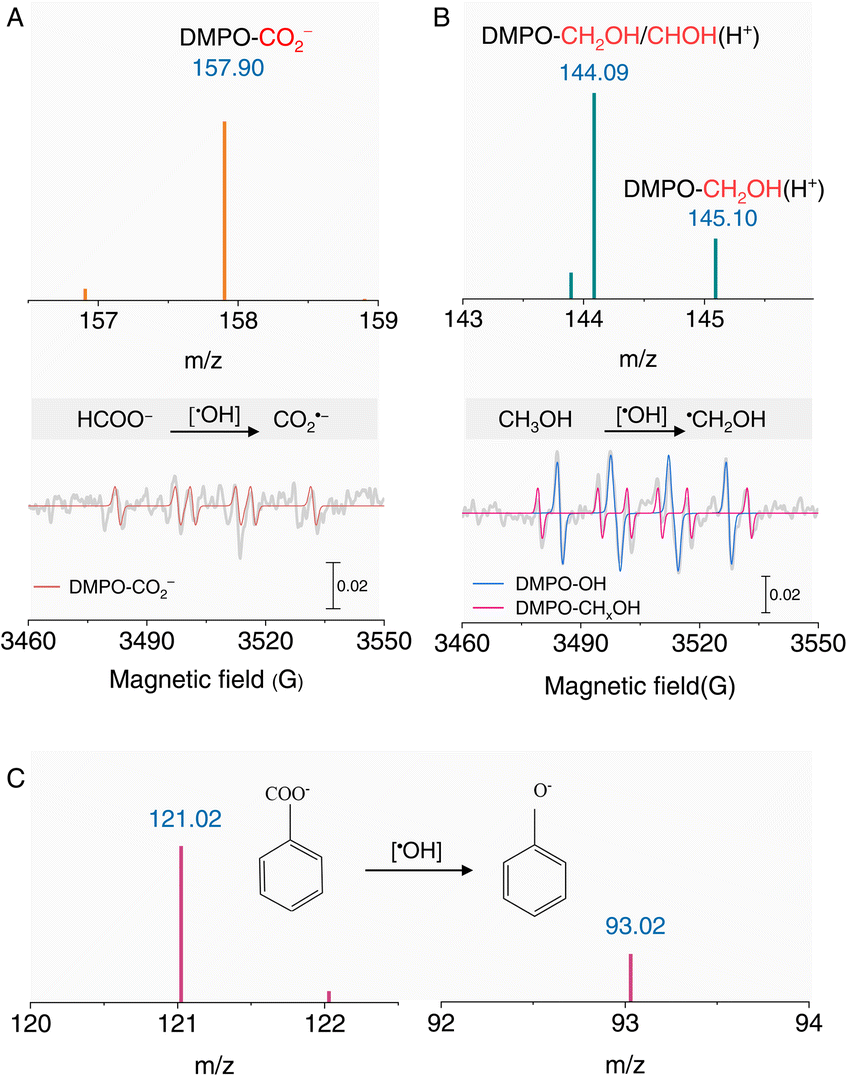 | ||
| Fig. 3 Synthetic applications of ˙OH radicals in acidic solutions. (A) Negative ion mode mass spectrum (top) and EPR spectrum (bottom) for the oxidation of HCOO− to CO2˙− by ˙OH radicals. (B) Positive ion mode mass spectrum (top) and EPR spectrum (bottom) for the conversion of CH3OH to ˙CH2OH radicals. DMPO-CHxOH adducts represent carbon-centered radicals derived from CH3OH. (C) Negative ion mode mass spectra for the oxidation of benzoic acid to phenol by ˙OH radicals. All experiments were performed at pH 1.5. | ||
A fundamental question arose: why can ˙OH radicals spontaneously generate in acidic aqueous solutions? While interfacial charge transfer has been shown to enable the formation of H2O2via the recombination of surface OH−-derived ˙OH radicals,42–44 this mechanism is unlikely to account for our observations. In highly acidic solutions, the concentration of OH− is negligible, making it improbable that ˙OH radicals originate from OH−. So, how should we understand the formation mechanism of ˙OH highlighted in this finding?
Determination of the water radical cation (H2O˙+) coordinated with 18-crown-6
Having established that ˙OH radicals originate from water molecules through isotope-labeled experiments (Fig. 1B and C), we hypothesize that the radical-mediated proton transfer proceeds via the reaction: H2O˙+ + H2O → ˙OH + H3O+. However, direct detection of the transient H2O˙+ species at pH 1.5 using EPR spectroscopy proved challenging.23 To overcome this limitation, we employed a complexation strategy using 18-crown-6 (18C6) to stabilize H2O˙+, thereby increasing its concentration and lifetime for EPR measurements.41 As shown in Fig. 4A, the stepwise addition of 18C6 led to a significant increase in the EPR intensity of both DMPO-OH adducts and DMPO-H2O+ species (AN = 1.58 mT; AH = 2.25 mT). The DMPO-OH signal arises from the classic spin-trapping mechanism, while the DMPO-H2O+ signal is attributed to the epoxidation of DMPO by the highly oxidizing H2O˙+ (with a redox potential exceeding 3 V).23 The correlation between increasing H2O˙+ concentration and enhanced ˙OH formation further supports the proposed mechanism, where H2O˙+ serves as the precursor for ˙OH radicals. | ||
| Fig. 4 Detection of H2O˙+ radicals stabilized by 18-crown-6 (18C6). (A) EPR spectra of DMPO-H2O+ and DMPO-OH adducts with increasing 18C6 concentration in Ar-purged HCl solution at pH 1.5. Higher 18C6 concentrations enhance the DMPO-H2O+ signal and increase the intensity of the DMPO-OH signal. (B–D) High-resolution mass spectra of 18C6 complexes in H2O, H218O, and D2O solutions, respectively. Isotope-labeled peaks are highlighted in color. | ||
To unequivocally confirm the formation of the 18C6-H2O˙+ complex, we conducted HRMS combined with isotope labeling. In 18C6-containing H2O, a peak at m/z 282.20 was observed, corresponding to the 18C6-H2O+ complex (Fig. 4B). This peak is distinct from the well-documented 18C6-H3O+ complex at m/z 283.20.45 To further validate the assignment, we introduced H218O into the solution, which resulted in isotope mass shifts to m/z 284.20 and 285.20, assigned to 18C6-H218O˙+ and 18C6-H318O+, respectively (Fig. 4C). Additionally, experiments using D2O revealed a series of peaks at m/z 282.20, 283.20, 284.21, 285.21, and 286.21, corresponding to 18C6-H2O˙+, 18C6-H3O+/18C6-HDO+, 18C6-D2O˙+, 18C6-HD2O+, and 18C6-D3O+, respectively (Fig. 4D). These isotope-labeled HRMS results provide unambiguous evidence for the formation of the 18C6-H2O˙+ complex, corroborating the EPR findings and confirming the stabilization of H2O˙+ by 18C6.
Electron quantum delocalization and the resultant proton transfer via H2O˙+
In contrast to pure water, the formation of H2O˙+ in an acidic solution is likely driven by hydrogen bond (HB) imbalance, which facilitates change delocalization in the presence of a dissociated H3O+–Cl pair.11 To study this phenomenon, we applied ab initio molecular dynamics simulation to evaluate the electronic charge transfer through the HB at 298.15 K (see Methods†).13 First, we calculated the average charge transferred to a water molecule in pure water (Fig. 5A), which was found to be approximately 0.010e, consistent with previously reported values (∼0.008e).46 This value is lower than that observed in the water dimer system (0.020e) due to the more symmetric hydrogen bonding network in liquid water. However, the introduction of H3O+ disrupts this symmetry, creating an imbalance in HB donors and acceptors and enabling charge delocalization. | ||
| Fig. 5 Ab initio molecular dynamics (AIMD) simulations. (A) Charge distribution in pure water. Charge distributions on neighboring H2O molecules (B) and in the second hydration shell (C) of the H3O+ core in HCl solutions. Snapshots of the H2O˙+-mediated proton transfer process: (D) initial state, (E) formation of H2O˙+ radicals, (F) generation of H3O+ and ˙OH radicals, and (G) subsequent Grotthuss proton transfer. The radical intermediates and the proton transfer pathways are highlighted by yellow circles and arrows, respectively. Spin density isosurfaces of 0.02 au−3 for (E–G) are shown to visualize radical formation. | ||
We next analyzed the charge distribution around the H3O+ core and its neighboring water molecules (Fig. 5B). The simulations revealed that the H3O+ induces a significant positive charge on its neighboring H2O molecules, increasing the likelihood of H2O˙+ formation. A slight redistribution of charge was also observed in the second hydration shell (Fig. 5C), indicating that the Grotthuss proton transfer facilitates dynamic electron density redistribution along the proton transport trajectory. This charge delocalization is reminiscent of the strong charge segregation observed at the oil/water or gas/water interfaces,17,20 but it is significantly more pronounced than the fleeting charge transfer (H2O+δ⋯H2O−δ) in pure water.47
To further explore the proton transfer process, we simulated a water box with an excess proton (Fig. 5D). Upon the formation of H2O˙+ radical intermediates (Fig. 5E), proton transfer occurred within 150 femtoseconds, consistent with the fastest known chemical process.15 This rapid transfer leads to the generation of detectable ˙OH radicals and new H3O+ ions (Fig. 5F, S6 and Movie S1†). The newly formed H3O+ ions can subsequently transfer protons via the classic Grotthuss mechanism (Fig. 5G), a process that is significantly slower than the radical-mediated proton transfer observed here.
Implications of radical-mediated proton transfer
Our experimental observations of H2O˙+ and ˙OH radical intermediates, combined with theoretical simulations of charge transfer in protonated water, provide strong evidence for a radical-mediated proton transfer mechanism. This mechanism, which involves the rapid formation and transfer of H2O˙+ and ˙OH radicals, offers an alternative explanation for the anomalously high proton mobility observed in acidic solutions. Given the efficiency of radical reactions compared to the classical “structural diffusion” mechanism, this radical-mediated pathway may play a significant role in proton transport under certain conditions.The charge delocalization observed in acidic water is a general phenomenon that can lead to the formation of water-derived radical species. This finding has broad implications for understanding the reactivity of the H3O+-containing aqueous system under ambient conditions. For instance, the presence of H3O+ ions can significantly enhance the chemical reactivity of water, particularly under external excitations, such as under radiolysis,15 electric field,48–51 or in microdroplets/aerosols,52,53 where proton accumulation at interfaces further promotes radical formation.54
The intrinsic formation of ˙OH in acidic solutions has important practical implications. For example, in electrocatalysis, the presence of ˙OH can lead to the corrosion of catalysts, such as Cu in electrochemical CO2 reduction under strongly acidic conditions.55 Similarly, the degradation of ionomers and carbon supports in proton-exchange membrane fuel cells (PEMFCs) may be exacerbated by ˙OH radicals, particularly at elevated operating temperatures (∼80 °C), where thermal excitation further promotes radical generation.56,57 These findings highlight the need to carefully consider the role of radical-mediated processes in the design and optimization of electrochemical systems.
Conclusions
Our study reveals a new proton transfer mechanism in acidic water, where the charge spreads across water molecules to form unstable intermediates (H2O˙+), leading to the spontaneous creation of highly reactive hydroxyl radicals (˙OH). This discovery challenges the traditional view that water is stable at room temperature and shows a deep connection between proton transfer and radical reactions. Understanding this mechanism helps explain real-world issues like material corrosion and fuel cell degradation while also guiding the design of better catalysts and energy technologies. This work reshapes our understanding of water's chemistry and opens new paths for innovation in chemistry, energy, and environmental science.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
C. C. conceived this study and led the project. R. Z. carried out the experiments. Q. Z. and Z. L. performed molecular dynamics simulations. N. Y. and L. L. implemented part measurements and data analysis. C. C. and R. Z. wrote the manuscript. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (22372027 and 22393913) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0450101).Notes and references
- Q. Wu, N. Yang, M. Xiao, W. Wang and C. Cui, Nat. Commun., 2024, 15, 9145 CrossRef CAS PubMed
.
- S. S. Xantheas, Nature, 2009, 457, 673–674 CrossRef CAS PubMed
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS
- C. T. Wolke, J. A. Fournier, L. C. Dzugan, M. R. Fagiani, T. T. Odbadrakh, H. Knorke, K. D. Jordan, A. B. McCoy, K. R. Asmis and M. A. Johnson, Science, 2016, 354, 1131–1135 CrossRef CAS PubMed
- T. C. Berkelbach, H.-S. Lee and M. E. Tuckerman, Phys. Rev. Lett., 2009, 103, 238302 CrossRef PubMed
- F. Dahms, B. P. Fingerhut, E. T. J. Nibbering, E. Pines and T. Elsaesser, Science, 2017, 357, 491–494 CrossRef CAS PubMed
- M. Schröder, F. Gatti, D. Lauvergnat, H.-D. Meyer and O. Vendrell, Nat. Commun., 2022, 13, 6170 CrossRef PubMed
- Y. Tian, J. N. Hong, D. Y. Cao, S. F. You, Y. Z. Song, B. W. Cheng, Z. C. Wang, D. Guan, X. M. Liu, Z. P. Zhao, X. Z. Li, L. M. Xu, J. Guo, J. Chen, E. G. Wang and Y. Jiang, Science, 2022, 377, 315–319 CrossRef CAS
- J. A. Fournier, W. B. Carpenter, N. H. C. Lewis and A. Tokmakoff, Nat. Chem., 2018, 10, 932–937 CrossRef CAS
- S. Di Pino, E. D. Donkor, V. M. Sánchez, A. Rodriguez, G. Cassone, D. Scherlis and A. Hassanali, J. Phys. Chem. B, 2023, 127, 9822–9832 CrossRef CAS PubMed
- J. M. J. Swanson and J. Simons, J. Phys. Chem. B, 2009, 113, 5149–5161 CrossRef CAS PubMed
- T. D. Kühne and R. Z. Khaliullin, Nat. Commun., 2013, 4, 1450 CrossRef PubMed
- C. Schran, O. Marsalek and T. E. Markland, Chem. Phys. Lett., 2017, 678, 289–295 CrossRef CAS
- M. Flór, D. M. Wilkins, M. de la Puente, D. Laage, G. Cassone, A. Hassanali and S. Roke, Science, 2024, eads4369 CrossRef PubMed
- Z. H. Loh, G. Doumy, C. Arnold, L. Kjellsson, S. H. Southworth, A. Al Haddad, Y. Kumagai, M. F. Tu, P. J. Ho, A. M. March, R. D. Schaller, M. S. B. Yusof, T. Debnath, M. Simon, R. Welsch, L. Inhester, K. Khalili, K. Nanda, A. I. Krylov, S. Moeller, G. Coslovich, J. Koralek, M. P. Minitti, W. F. Schlotter, J. E. Rubensson, R. Santra and L. Young, Science, 2020, 367, 179–182 CrossRef CAS PubMed
- M.-F. Lin, N. Singh, S. Liang, M. Mo, J. P. F. Nunes, K. Ledbetter, J. Yang, M. Kozina, S. Weathersby, X. Shen, A. A. Cordones, T. J. A. Wolf, C. D. Pemmaraju, M. Ihme and X. J. Wang, Science, 2021, 374, 92–95 CrossRef CAS PubMed
- S. Pullanchery, S. Kulik, B. Rehl, A. Hassanali and S. Roke, Science, 2021, 374, 1366–1370 CrossRef CAS PubMed
- S. Q. Lin, X. Y. Chen and Z. L. Wang, Chem. Rev., 2022, 122, 5209–5232 CrossRef CAS PubMed
- L. Q. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2022, 61, e202210765 CrossRef CAS
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2024, 63, e202400118 CrossRef CAS PubMed
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS
- L. Li, Q. Wu, S.-k. Xiang, S. Mu, R. Zhao, M. Xiao, C. Long, X. Zheng and C. Cui, J. Phys. Chem. Lett., 2023, 14, 9183–9191 CrossRef CAS PubMed
- F. N. Brünig, M. Rammler, E. M. Adams, M. Havenith and R. R. Netz, Nat. Commun., 2022, 13, 4210 CrossRef
- M. Thamer, L. De Marco, K. Ramasesha, A. Mandal and A. Tokmakoff, Science, 2015, 350, 78–82 CrossRef
- F. Xie, D. S. Tikhonov and M. Schnell, Science, 2024, 384, 1435–1440 CrossRef CAS PubMed
- A. Misak, V. Brezova, M. Chovanec, K. Luspai, M. J. Nasim, M. Grman, L. Tomasova, C. Jacob and K. Ondrias, Antioxidants, 2021, 10, 1286 CrossRef CAS
- L. Wang, Q. Li, Y. Fu, Z. Wang, H.-y. Zhu and M. Sillanpaa, ACS ES&T Water, 2023, 3, 532–541 Search PubMed
- P. Li, Z. Shen, W. Wang, Z. Ma, S. Bi, H. Sun and Y. Bu, Phys. Chem. Chem. Phys., 2010, 12, 5256–5267 RSC
- D. A. Armstrong, R. E. Huie, W. H. Koppenol, S. V. Lymar, G. Merényi, P. Neta, B. Ruscic, D. M. Stanbury, S. Steenken and P. Wardman, Pure Appl. Chem., 2015, 87, 1139–1150 CrossRef CAS
- H.-M. Hung, M.-N. Hsu and M. R. Hoffmann, Environ. Sci. Technol., 2018, 52, 9079–9086 CrossRef CAS PubMed
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed
- L. L. Perissinotti, M. A. Brusa and M. A. Grela, Langmuir, 2001, 17, 8422–8427 CrossRef CAS
- W. Deng, H. Zhao, F. Pan, X. Feng, B. Jung, A. Abdel-Wahab, B. Batchelor and Y. Li, Environ. Sci. Technol., 2017, 51, 13372–13379 CrossRef CAS PubMed
- D. Ma, B. Hu, W. Wu, X. Liu, J. Zai, C. Shu, T. Tadesse Tsega, L. Chen, X. Qian and T. L. Liu, Nat. Commun., 2019, 10, 3367 CrossRef PubMed
- X.-P. Zhang, Y.-N. Li, Y.-Y. Sun and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 18394–18398 CrossRef CAS PubMed
- Y. Meng, R. N. Zare and E. Gnanamani, J. Am. Chem. Soc., 2023, 145, 19202–19206 CrossRef CAS PubMed
- D. Xia, H. Zhang, Y. Ju, H.-b. Xie, L. Su, F. Ma, J. Jiang, J. Chen and J. S. Francisco, J. Am. Chem. Soc., 2024, 146, 11266–11271 CAS
- L. Zhao, X. Song, C. Gong, D. Zhang, R. Wang, R. N. Zare and X. Zhang, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2200991119 CrossRef CAS PubMed
- X. Zheng, Q. Wu, M. Xiao, L. Li, R. Zhao and C. Cui, Chem.–Eur. J., 2024, 30, e202303383 CrossRef CAS PubMed
- R. Zhao, L. Li, Q. Wu, W. Luo, Q. Zhang and C. Cui, Nat. Commun., 2024, 15, 8367 CrossRef PubMed
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 19294–19298 CrossRef CAS PubMed
- B. Chen, Y. Xia, R. He, H. Sang, W. Zhang, J. Li, L. Chen, P. Wang, S. Guo, Y. Yin, L. Hu, M. Song, Y. Liang, Y. Wang, G. Jiang and R. N. Zare, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2209056119 CrossRef CAS PubMed
- A. J. Colussi, J. Am. Chem. Soc., 2023, 145, 16315–16317 CrossRef CAS PubMed
- M. Bühl and G. Wipff, J. Am. Chem. Soc., 2002, 124, 4473–4480 CrossRef PubMed
- A. J. Lee and S. W. Rick, J. Chem. Phys., 2011, 134 Search PubMed
- D. Ben-Amotz, Science, 2022, 376, 800–801 CrossRef CAS PubMed
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS
- G. Cassone, J. Phys. Chem. Lett., 2020, 11, 8983–8988 CrossRef CAS PubMed
- Z. Futera and N. J. English, J. Chem. Phys., 2017, 147, 031102 CrossRef PubMed
- V. Conti Nibali, S. Maiti, F. Saija, M. Heyden and G. Cassone, J. Chem. Phys., 2023, 158 Search PubMed
- M. A. Mehrgardi, M. Mofidfar and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 7606–7609 CrossRef CAS
- K. Li, K. Gong, J. Liu, L. Ohnoutek, J. Ao, Y. Liu, X. Chen, G. Xu, X. Ruan, H. Cheng, J. Han, G. Sui, M. Ji, V. K. Valev and L. Zhang, Cell Rep. Phys. Sci., 2022, 3, 100917 CrossRef CAS
- H. Wei, E. P. Vejerano, W. Leng, Q. Huang, M. R. Willner, L. C. Marr and P. J. Vikesland, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 7272–7277 CrossRef CAS PubMed
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed
- H. Xie, X. Xie, G. Hu, V. Prabhakaran, S. Saha, L. Gonzalez-Lopez, A. H. Phakatkar, M. Hong, M. Wu, R. Shahbazian-Yassar, V. Ramani, M. I. Al-Sheikhly, D.-e. Jiang, Y. Shao and L. Hu, Nat. Energy, 2022, 7, 281–289 CrossRef CAS
- L. Gubler, S. M. Dockheer and W. H. Koppenol, J. Electrochem. Soc., 2011, 158, B755 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02206a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |