
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomer-dependent reactivity in the solid state: topochemical [4 + 4] vs. [4 + 2] cycloaddition reactions†
Anu
Lal
and
Kana M.
Sureshan
 *
*
School of Chemistry, Indian Institute of Science Education and Research, Thiruvananthapuram, Kerala 695551, India. E-mail: kms@iisertvm.ac.in
First published on 18th June 2025
Abstract
This study investigates the reactivity of constitutional isomers of acylated N-hydroxybenzotriazole (HOBt) in solid-state photocycloaddition reactions. To understand the difference in solid-state reactivity of the isomers, we have synthesized two isomeric N- and O-acyl derivatives of HOBt via acylation with anthracene-9-carboxylic acid. Both O-anthr-9-oyl oxy-benzotriazole 1 and its N-acyl isomer 2 crystallized with the face-to-face arrangement of their anthracene units, with a separation of 3.8 Å between them, satisfying Schmidt's criteria, suggesting that they can undergo a topochemical [4 + 4] photocycloaddition reaction. Upon irradiation using blue light (456 nm), the isomer 2 underwent a quantitative [4 + 4] cycloaddition reaction yielding the dimer D2. However, isomer 1 did not undergo [4 + 4] cycloaddition; instead, it underwent a photochemical [4 + 2] cycloaddition with molecular oxygen, eventually yielding anthraquinone (AQ). The dimer D2 underwent a retro-cycloaddition reaction to revert back to the isomer 2. While isomers give the same product in solution-state reactions, our study establishes that isomers can react differently in the solid state and can give different products.
Introduction
N-Hydroxybenzotriazole (HOBt) exists in two tautomeric forms: hydroxy and azoxy forms.1 One of the main uses of HOBt is to activate the carboxylic acid in peptide coupling. As HOBt is an ambident nucleophile, its acylation gives two isomeric products; the O-acylated kinetic product and the N-acylated thermodynamic product.2 Depending on the polarity of the solvent, the nature of the acyl group, and reaction conditions, the ratio of these two isomeric products can vary.2 Furthermore, they can interconvert in solution via acyl migration to reach and maintain an equilibrium, with their ratio dependent on the nature of the solvent.2 The existence of two acylated forms is inconsequential for their use in acylation, such as peptide coupling, as both forms can act as acyl donors. Furthermore, as and when the most reactive form reacts, the dynamic interconversion sets in to maintain the equilibrium (replenish the reactive form) and to react further. Thus, in solution, there will be no apparent difference in reactivity between the two forms. However, the two forms can exhibit different reactivities in the solid state. For instance, the two isomeric acyl derivatives can behave differently in their topochemical reaction. To test this hypothesis, we have acylated HOBt with anthracene-9-carboxylic acid, as anthracene derivatives are known to undergo a light-induced topochemical [4 + 4]-cycloaddition reaction.3–18 Here, we report a remarkable difference in reactivity of two acylated HOBt derivatives in the solid state: [4 + 2]-cycloaddition of the O-acyl derivative and [4 + 4]-cycloaddition of the N-acyl derivative.Results and discussion
Acylation of HOBt with anthracene-9-carboxylic acid yielded O-acyl derivative 1 and N-acyl derivative 2 (Scheme 1 and ESI†). We obtained yellow-colored needle-like single crystals of isomer 1 (Fig. 1a) by slow evaporation of its solution in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of chloroform and toluene. Similarly, plate-like single crystals of isomer 2 were obtained (Fig. 1d) by slow evaporation of its solution in a 1:4 (v/v) mixture of ethyl acetate and petroleum ether. We determined their crystal structures by single-crystal X-ray diffraction (SCXRD) analysis.
:1 (v/v) mixture of chloroform and toluene. Similarly, plate-like single crystals of isomer 2 were obtained (Fig. 1d) by slow evaporation of its solution in a 1:4 (v/v) mixture of ethyl acetate and petroleum ether. We determined their crystal structures by single-crystal X-ray diffraction (SCXRD) analysis.
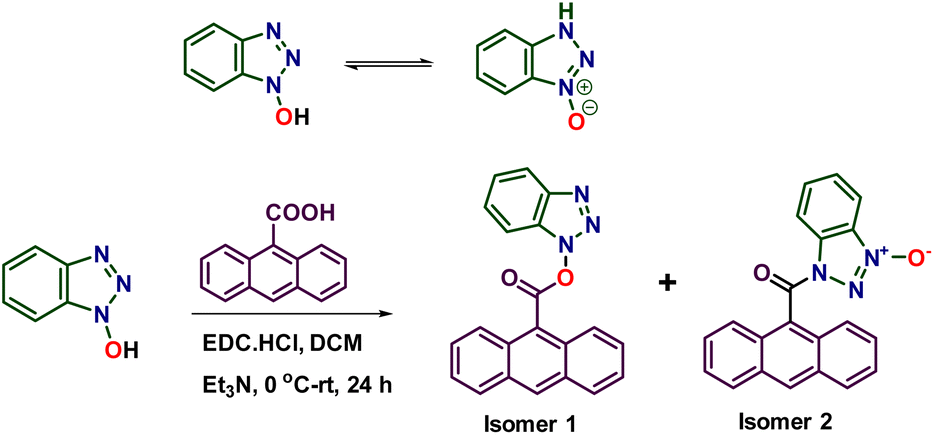 | ||
| Scheme 1 Synthesis of isomeric acyl derivatives of HOBt. | ||
 | ||
| Fig. 1 (a) Chemical structure and microscopic image of isomer 1. (b) Crystal packing along the crystallographic ‘bc’ axes showing C–H⋯N and C–H⋯O hydrogen bonding interactions. (c) The distance between the reacting groups is shown. The bond-forming carbons are highlighted in the ball and stick model. (d) Chemical structure and microscopic image of isomer 2. (e) The conformation stabilization via weak intramolecular interactions. (f) Crystal packing along the ‘a’ axis. (g) The appropriate distance between the reacting groups to undergo [4 + 4] cycloaddition reaction is shown. C–H⋯O (green), C–H⋯N (orange), anion⋯π (pink), and dipolar interaction between N⋯O (light blue) are shown. | ||
Isomer 1 crystallized in the P21/c space group (Fig. S1 and Table S1, ESI†) with a single molecule in the asymmetric unit. The plane of anthracene is almost parallel to the plane of benzotriazole (dihedral angle 16°). This conformation is stabilized by an intramolecular C–H⋯O hydrogen bond (Fig. 1b). An intermolecular C–H⋯O H-bond and π⋯π stacking between aromatic units stack the molecules parallelly along the ‘a’ axis (Fig. S1†). Weak C–H⋯N and C–H⋯O hydrogen bonds connect the molecules along the crystallographic ‘b’ and ‘c’ axes (Fig. 1b). The face-to-face packing of anthracene units places the reactive carbon ends 3.8 Å apart (Fig. 1c).
Isomer 2 crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with a single molecule in the asymmetric unit (Fig. S1 and Table S1, ESI†). The benzotriazole ring is almost perpendicular (dihedral angle 70°) to the anthracene ring. This conformation is stabilized by a weak intramolecular C–H⋯N and two intramolecular C–H⋯O hydrogen bonds (Fig. 1e).
space group with a single molecule in the asymmetric unit (Fig. S1 and Table S1, ESI†). The benzotriazole ring is almost perpendicular (dihedral angle 70°) to the anthracene ring. This conformation is stabilized by a weak intramolecular C–H⋯N and two intramolecular C–H⋯O hydrogen bonds (Fig. 1e).
Along the ‘a’ axis, molecules form a head-to-head dimer via attractive interaction between the dipoles of the triazoloxy units (O⋯N 3.0 Å), and such centrosymmetric dimers are further stabilized by anion⋯π interaction (O⋯C 3.2 Å) (Fig. 1f). These centrosymmetric dimers translate along the ‘a’ axis via π⋯π stacking between the phenyl rings (3.4 Å) of benzotriazole units and a C–H⋯O hydrogen bond, forming a columnar arrangement (Fig. S1†). Such dimer columns translate along one of the ‘bc’ diagonals via C–H⋯O hydrogen bonds (H20⋯O1: 2.5 Å; O2⋯H5: 2.7 Å). Along the other ‘bc’ diagonal, the dimeric columns translate via face-to-face π⋯π stacking (3.4 Å) between the anthracene units (Fig. S1†). This face-to-face arrangement makes a spacing of 3.8 Å between the reactive carbons in this case, which is optimal for a solid-state [4 + 4]-photocycloaddition reaction (Fig. 1g). Isomer 1 shows a melting point of 168 °C, whereas isomer 2 does not melt but decomposes at temperatures above 195 °C. The isomers 1 and 2 crystals were stable at room temperature if kept in the dark. However, when isomer 1 was exposed to room light for a prolonged time (a week or more) under ambient conditions, the sample began to change its color from yellow to an orange-brownish shade. But, isomer 2 did not change color when stored at ambient temperature under light. The Kubelka–Munk (K–M) diffuse reflectance spectrum of crystalline isomer 1 displayed two bands, one with a peak maximum of 303 nm and another with a broad structured band at 325–472 nm (Fig. S2†). The spectrum of isomer 2 exhibited a broad band at 200–500 nm (Fig. S2†). To assess the reactivity towards the [4 + 4] cycloaddition reaction, single crystals of both isomers were placed in a photoreactor and exposed to 456 nm light. Isomer 1 showed a noticeable color shift from bright yellow to light brown after 30 minutes of irradiation (Fig. 2a). After 24 hours of irradiation, the color of isomer 1 changed to orange-brown, and finally, it turned to dark brown after 96 hours of irradiation. The blue light exposure caused isomer 2 to change its color from dark yellow to light yellow after half an hour. After three hours of light exposure, the color of isomer 2 changed to wheat color, and it remained the same thereafter (Fig. 2b).
 | ||
| Fig. 2 (a) Images showing the color change in the isomer 1 crystals from light yellow to dark brown during irradiation. (b) Images of the isomer 2 crystals showing a color change from yellow to wheat color during irradiation. (c) Time-dependent 1H NMR spectra of isomer 1 during irradiation. (d) Expected and observed products of irradiation of isomer 1. | ||
To monitor the solid-state reaction systematically, we relied on time-dependent 1H NMR spectroscopy. 40 mg of crystals of 1 was irradiated, and small fractions were withdrawn at different times of irradiation and their 1H NMR spectra were recorded after dissolving in CDCl3. 1H NMR experiments conducted over six days showed the gradual disappearance of signals due to isomer 1 and the emergence of new signals due to the product (Fig. 2c). After 172 h of irradiation, the reaction reached a stagnant state with no further advancement, and even at this stage, traces of isomer 1 were present. We noticed, from the second day onwards, the emergence and gradual growth of tiny crystals on the wall of the vial. The 1H NMR spectrum of these newly formed crystals matched with new signals that emerged during the reaction (Fig. S3†), suggesting a sublimation of the product and its deposition on the walls. SCXRD analysis of one of the crystals deposited on the walls of the vial revealed that the product is surprisingly simple anthraquinone (AQ) rather than the expected [4 + 4]-cycloaddition reaction product, dimer D1 (Fig. 2d). The steric crowding in view of the head-to-head arrangement may be hindering their topochemical [4 + 4]-cycloaddition reaction.
The formation of AQ involves oxidation at 9,10-positions. Photochemical [4 + 2]-cycloaddition of anthracene and its derivatives with molecular oxygen to form anthracene-9,10-endoperoxide and its conversion to anthraquinone is well known.19–23 Also, molecular oxygen is known to participate in insertion reactions in crystals.24,25 Salzillo et al. reported the photo-induced topochemical conversion of 9,10-dinitro-anthracene to AQ. Though a definite mechanism was not proposed for this reaction, [4 + 2]-cycloaddition with oxygen followed by the homolytic cleavage of the endoperoxide and the subsequent elimination of NO2 is a probable mechanism.26 In a similar way, photooxygenation via [4 + 2]-cycloaddition reaction of isomer 1 would yield the endoperoxide EP. Light-induced homolytic cleavage of the endoperoxide bond followed by rearrangement would produce AQ, as shown in Fig. 3. Though rare, light-induced [4 + 2]-cycloaddition reactions are known in the literature.27 When crystals of isomer 1 were irradiated under an argon atmosphere, no reaction was observed (Fig. 4a). This confirms the involvement of molecular oxygen present in the air in the reaction of isomer 1 in the solid state.
 | ||
| Fig. 3 Plausible mechanism for the formation of anthraquinone (AQ) from isomer 1. | ||
 | ||
| Fig. 4 (a) 1H NMR spectra of isomer 1 crystals and irradiated crystals in blue light in an argon atmosphere for 60 h. (b) Time-dependent 1H NMR spectra of isomer 2. (c) MALDI-TOF spectrum of dimer D2. (d) Chemical structure of dimer D2. (e and f) Time-dependent PXRD spectra of isomers 1 (e) and 2 (f). | ||
Time-dependent 1H NMR spectral analysis revealed that isomer 2 starts reacting within 5 minutes of irradiation and converts smoothly and fully to a product within six hours of exposure to blue light (Fig. 4b). This study revealed that isomer 2 is more reactive than isomer 1. However, the crystals got disintegrated during the reaction and were not suitable for SCXRD analysis. We determined the structure of the product using various 1D and 2D-NMR techniques. In the product, all of the aromatic peaks were shifted significantly. The sharp singlet peak corresponding to the proton at the 10th position of anthracene completely disappeared after the irradiation, and shifted upfield from 8.8 ppm to 6 ppm, suggesting the dearomatization of the middle ring of anthracene. The COSY spectrum of the product could assign different protons as in D2 (Fig. 4d and S23†). With the help of 13C NMR (Fig. S21†), DEPT-135 (Fig. S22†) and HMQC (Fig. S24†), we could assign various carbon signals. Additionally, the correlation of H10 with C4 and C5 in the HMBC spectrum (Fig. S25†), confirmed the structure of the cycloadduct, D2 (Fig. 4d). Additionally, matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectral analysis (Fig. 4c) gave the molecular mass corresponding to the dimer, further confirming the structure of the product as dimer D2 (Fig. 4d).
We performed a time-dependent PXRD analysis of the reaction mixture to understand the nature of the reaction. The PXRD spectra of irradiated isomer 1 showed sharp peaks at every stage of the reaction, suggesting the crystalline nature of the reactant and product. The emergence of new reflection peaks and the disappearance of some of the peaks due to isomer 1 could be seen. The PXRD spectrum at later stages of the reaction shows peaks due to both unreacted isomer 1 and the product. As the product is sublimed and crystallized, we cannot classify it as a SCSC reaction. However, the crystalline phase of a minute amount of unreacted 1, even at the stage when the reaction reached a stagnant state, clearly suggests that the reaction happens in the crystalline state (Fig. 4e). The irradiated isomer 2 also maintained crystallinity throughout the reaction. This was evident from time-dependent PXRD spectra of the irradiated sample, which showed sharp peaks at every stage of the reaction and a smooth growth of the product phase at the expense of the reactant phase (Fig. 4f). Thus, isomer 2 exhibited a crystal-to-crystal transformation upon irradiation. Photo-dimers of anthracene derivatives are known to undergo retro-cycloaddition under thermal conditions.5,8,28–31 To understand the thermal retro-cycloaddition of dimer D2, we employed differential scanning calorimetry (DSC) analysis. The DSC analysis of isomer 2 showed a single exothermic peak with an onset temperature of 196 °C. This exothermic process corresponds to thermal decomposition, as the thermogravimetric analysis showed decomposition at this temperature. Partially reacted isomer 2 (sample irradiated for 2 h) showed two exothermic peaks at onset temperatures of 160 °C and 200 °C (Fig. 5a). The new exothermic peak could be due to a retro-cycloaddition reaction; cycloreversion of anthracene dimers is known to be an exothermic process.28 The DSC thermogram of the pure dimer D2 also displayed both exothermic peaks; while the intensity of the former exothermic peak increased, that of the exothermic peak at 198 °C remained unchanged. The TGA of dimer D2 was similar to the TGA of isomer 2, showing decomposition at 198 °C (Fig. S4†). These observations suggest that the dimer D2 undergoes retro-cycloaddition at 160 °C to form the isomer 2, which, upon further heating, undergoes decomposition at 198 °C.
 | ||
| Fig. 5 (a) DSC profile comparison of isomer 2, 2 h irradiated sample, and dimer D2 (6 h irradiated sample). (b) PXRD diffractogram of isomer 2, and the heated D2 (at 160 °C for 5 min) showing the crystal-to-crystal retrocycloaddition. (c) 1H NMR spectra showing three cycles of reversible cycloaddition–retrocycloaddition of isomer 2 showing repeatability. (d) PXRD profiles of isomer 2 and dimer D2 in three cycles showing the repeatability and the crystal-to-crystal reversible cycloaddition–retrocycloaddition. | ||
When a powdered sample of dimer D2 was heated at 160 °C for five minutes, it changed its color to mustard yellow, similar to the color of 2. The 1H NMR spectrum of this heated sample confirmed that the dimer D2 has completely reversed to the isomer 2 (Fig. S5†). PXRD profile of the dimer D2 heated at 160 °C for 5 minutes showed sharp peaks suggesting that the retro-cycloaddition is also topochemically controlled. The PXRD profile of this heated sample was indistinguishable from the PXRD profile of the freshly crystallized isomer 2 (Fig. 5b), suggesting that the retro-cycloaddition leads to the same crystal form as isomer 2. Irradiation of the heated sample yielded dimer D2, suggesting that photoinduced [4 + 4]-cycloaddition and heat-induced retro-cycloaddition are reversible crystal-to-crystal processes. We investigated the repeatability of cycloaddition–retrocycloaddition sequences by iterative irradiation-heating cycles and monitoring by 1H NMR spectroscopy (Fig. 5c) and powder X-ray diffractometry (Fig. 5d). This suggested that the reaction is reversible for several cycles, and the cyclic transformations are crystal-to-crystal processes. Thus, isomer 2 is a ‘two-state switchable crystal’ whose chemical state can be reversibly altered between two distinct states, similar to a ‘1’ and a ‘0’ in binary code, using appropriate stimuli (heat and light) and hence may find application in memory storage devices.
Having established the difference in solid-state reactivity of isomers 1 and 2, we were curious to know their solution-state reactivities. Irradiation of a solution of 1 in dichloromethane (DCM) under an oxygen atmosphere yielded a complex mixture of products containing AQ as the major product (Fig. S6†). Similarly, irradiation of a solution of isomer 2 in the presence of oxygen also yielded a complex mixture of products with anthraquinone as the major product, as in the case of isomer 1 (Fig. S7†). This clearly suggests that the solution-state reactivities of isomers are identical, as expected in view of their isomeric interconversion. Furthermore, solutions of pure isomer 1 and isomer 2 equilibrated to a mixture of the two, as expected (Fig. S8†). It is to be noted that under an argon atmosphere, isomer 1 gave a cycloadduct whereas isomer 2 gave a mixture of products (Fig. S9 and S26–S31†), suggesting that the dissolved oxygen enhances the equilibration.
Conclusions
In conclusion, reactants that exist as easily dynamically interconvertible structural isomers, usually do not yield different products originating from these isomers, when they participate in a conventional solution-state reaction. We tested our hypothesis that such isomers can behave differently in solid-state reactions by studying the light-induced topochemical cycloaddition reactions of isomeric N- and O-anthr-9-oyl oxy-benzotriazoles. Though both isomers had molecular arrangement, in their crystals, for their [4 + 4] cycloaddition, they followed different reaction pathways. While the N-acyl derivative underwent [4 + 4]-cycloaddition as expected, the O-acyl derivative underwent a photochemical [4 + 2]-cycloaddition with molecular oxygen, yielding an endoperoxide, which eventually yielded anthraquinone. This study demonstrates that topochemical reactions can be exploited to access structurally diverse products from a reactant that shows isomerism.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for isomer 1 and isomer 2 have been deposited at the CCDC under accession numbers 2422069 and 2422070,† respectively, and can be obtained from https://www.ccdc.cam.ac.uk/.Author contributions
KMS conceived the idea and designed the experiments. AL executed the project. KMS and AL analyzed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Veera Reddy Yatham for providing access to the irradiation facilities for carrying out the reactions. KMS thanks ANRF and MoE-STARS for research grants (CRG/2022/568, JCB/2023/0039, and STARS-2/2023-0222). AL thanks DST Inspire for a research fellowship.References
- S. Lee and T. R. Hoye, Org. Lett., 2024, 26, 5713–5718 CrossRef CAS PubMed
.
- B. D. Brink, J. R. DeFrancisco, J. A. Hillner and B. R. Linton, J. Org. Chem., 2011, 76, 5258–5263 CrossRef CAS PubMed
- E. Heller and G. M. J. Schmidt, Isr. J. Chem., 1971, 9, 449–462 CrossRef CAS
- N. Juneja, G. C. George and K. M. Hutchins, Angew. Chem., Int. Ed., 2025, 64, e202415567 CrossRef CAS PubMed
- C. Sun, J. J. Oppenheim, G. Skorupskii, L. Yang and M. Dincă, Chem, 2022, 8, 3215–3224 CAS
- M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. Van De Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779–784 CrossRef CAS PubMed
- W. Li, T. J. Gately, D. Kitagawa, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2024, 146, 32757–32765 CrossRef CAS PubMed
- X. Huang, Y. Xu, K. Fan, S. Bao, M. Kurmoo and L. Zheng, Angew. Chem., Int. Ed., 2018, 57, 8577–8581 CrossRef CAS PubMed
- Y.-S. Chen, C.-H. Wang, Y.-H. Hu, C.-Y. D. Lu and J.-S. Yang, J. Am. Chem. Soc., 2023, 145, 6024–6028 CrossRef CAS PubMed
- Q. Yu, M. Li, J. Gao, P. Xu, Q. Chen, D. Xing, J. Yan, M. J. Zaworotko, J. Xu, Y. Chen, P. Cheng and Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 18634–18640 CrossRef CAS PubMed
- T. Hughes, G. P. Simon and K. Saito, ACS Appl. Mater. Interfaces, 2019, 11, 19429–19443 CrossRef CAS PubMed
- C. Kuo, L. Hsu, Y. Chen, K. Goto, S. Maity, Y. Liu, S. Peng, K. V. Kong, T. Shinmyozu and J. Yang, Chem.–Eur. J., 2020, 26, 11511–11521 CrossRef CAS PubMed
- L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2011, 133, 12569–12575 CrossRef CAS PubMed
- K. Lam, R. J. Dillon, A. Carreras, T. Nishiuchi, T. Kubo, R. O. Al-Kaysi, D. Casanova and C. J. Bardeen, Phys. Chem. Chem. Phys., 2025, 27, 5305–5316 RSC
- R. Bhola, P. Payamyar, D. J. Murray, B. Kumar, A. J. Teator, M. U. Schmidt, S. M. Hammer, A. Saha, J. Sakamoto, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2013, 135, 14134–14141 CrossRef CAS PubMed
- X.-D. Huang, G.-H. Wen, S.-S. Bao, J.-G. Jia and L.-M. Zheng, Chem. Sci., 2021, 12, 929–937 RSC
- L.-Y. Hsu, S. Maity, Y. Matsunaga, Y.-F. Hsu, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Chem. Sci., 2018, 9, 8990–9001 RSC
- J.-F. Chen, D.-P. Gong, J. Wen, H. Ma and D.-K. Cao, Chem. Sci., 2016, 7, 451–456 RSC
- C. K. Das and N. S. Das, J. Chem. Technol. Biotechnol., 1982, 32, 643–649 CrossRef CAS
- D. Jiang, P. Niu, C. Tang, M. Zhang, W. Meng, P. Gao, L. Jin, A. Su, G. Zhang and L. Xu, Mol. Catal., 2024, 552, 113661 CrossRef CAS
- A. Putta, A. G. Sykes and H. Sun, J. Fluorine Chem., 2020, 235, 109548 CrossRef CAS
- S. Martins, J. P. S. Farinha, C. Baleizão and M. N. Berberan-Santos, Chem. Commun., 2014, 50, 3317 RSC
- J. Zhu, J. Zou, J. Zhang, Y. Sun, X. Dong and Q. Zhang, J. Mater. Chem. B, 2019, 7, 3303–3309 RSC
- T. Nakano, O. Nakagawa, T. Yade and Y. Okamoto, Macromolecules, 2003, 36, 1433–1435 CrossRef CAS
- T. Itoh, S. Nomura, M. Ohtake, T. Yoshida, T. Uno, M. Kubo, A. Kajiwara, K. Sada and M. Miyata, Macromolecules, 2004, 37, 8230–8238 CrossRef CAS
- T. Salzillo, I. Bilotti, R. G. Della Valle, E. Venuti and A. Brillante, J. Am. Chem. Soc., 2012, 134, 17671–17679 CrossRef CAS PubMed
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed
- G. C. George and K. M. Hutchins, Chem.–Eur. J., 2023, 29, e202302482 CrossRef CAS PubMed
- M. Servalli, N. Trapp and A. D. Schlüter, Chem.–Eur. J., 2018, 24, 15003–15012 CrossRef CAS PubMed
- P. Giri, A. Panda and M. K. Panda, Chem.–Eur. J., 2024, 30, e202303836 CrossRef CAS PubMed
- C. J. Easley, F. Tong, X. Dong, R. O. Al-Kaysi and C. J. Bardeen, Chem. Sci., 2020, 11, 9852–9862 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2422069 and 2422070. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02327k |
| This journal is © The Royal Society of Chemistry 2025 |