
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrosslinking 1,4-polybutadiene via allylic amination: a new strategy for deconstructable rubbers†
Mercie N. Hodges ,
Ana Paula Kitos Vasconcelos,
Laura J. Reed and
Matthew R. Golder*
,
Ana Paula Kitos Vasconcelos,
Laura J. Reed and
Matthew R. Golder*
Department of Chemistry and Molecular Engineering & Science Institute. University of Washington, Seattle, WA 98195, USA. E-mail: goldermr@uw.edu
First published on 19th May 2025
Abstract
As post-consumer rubbers (e.g., car tires) continue to accumulate in landfills and the environment, there is an increasing need for more reprocessible materials. Traditionally, devulcanization of rubbers requires excessive energy and releases toxic byproducts. Accordingly, downcycling (e.g., crumb rubber for asphalt or turf) is the major avenue for end-of-life thermoset elastomers. To enable alternative recycling pathways, herein we propose a two-step procedure to crosslink polybutadiene (PBD) as a substitute for vulcanization, resulting in deconstructable soft materials. First, we utilize the established C–H allylic amination of PBD to access thermoplastic elastomer pre-polymers functionalized with electrophilic hexafluoroisopropyl sulfamate (PBD-HFIPS). Then, PBD-HFIPS alcoholysis with diol crosslinkers yields thermoset specimens with tunable thermal, rheological, and mechanical properties dependent on crosslinker identity and density. Finally, treating these thermosets with a nucleophile cleaves sulfamate crosslinks and regenerates the thermoplastic with no characterizable differences from virgin PBD.
Introduction
Synthetic rubbers (e.g., polybutadiene, PBD; styrene-butadiene, SBR; nitrile-butadiene, NBR; polyisoprene, PI; ethylene propylene diene monomer, EPDM) are currently produced at approximately 30 million tons per year and remain essential to everyday life.1 In addition to vulcanized gaskets, hoses, membranes, seals, and shoe soles, many of these synthetic rubber feedstocks contribute to over 1 billion end-of-life tires globally per year. However, declining market demand for post-consumer rubber products, such as reclaimed tires, has highlighted collective knowledge gaps in how we process end-of-life thermoset elastomers. Traditionally, rubbers are irreversibly crosslinked via S8 vulcanization to generate polymer networks that are difficult to devulcanize (Fig. 1A) due to the strength of carbon-sulfur and sulfur–sulfur bonds.2–4 To improve upon the current state of the art, modulation of thermoplastic and thermoset elastomer backbone chemistry is imperative to create tunable, deconstructable rubbers. Post-polymerization modification (PPM)5 directly impacts bulk properties as demonstrated in recent examples employing commodity polymer feedstocks,6,7 such as polystyrene,8–10 polyacrylates,11,12 and polyolefins.13–17 For synthetic elastomers, however, few general strategies exist prior to work from our group;18,19 those that do focus on approaches that sacrifice the polymer microstructure (i.e., repeating 1,4-diene microstructure in polybutadiene) through alkene saturation or transposition.20 These issues persist when expanding elastomer PPM to thermosetting materials. For instance, Xin synthesized physically crosslinked ionomers by reacting butyl rubber with maleic anhydride followed by treatment with potassium hydroxide.21 In orthogonal work, Gandini used thiol–ene chemistry to functionalize PBD with furan; Diels–Alder and retro-Diels–Alder reactions with maleimide crosslinkers form and break the network, respectively (Fig. 1B).22 Notably, these emblematic examples sacrifice the PBD backbone microstructure that controls bulk thermomechanical properties.23,24 On the other hand, selenium-catalyzed C–H allylic amination25–27 of polyolefins (e.g., PBD, polynorbornene)18,19 introduces broad functionality into the polymer backbone while maintaining the original microstructure.28 With this paradigm in mind, we envisioned a novel two-step crosslinking approach that would maintain the parent alkene microstructure and enable chemical circularity. First, allylic amination of PBD with an activated sulfamate, such as hexafluoroisopropyl sulfamate (HFIPS),29 would enable solution-state characterization and analysis of a pre-polymer poised for subsequent crosslinking. Then, alcoholysis using a diol crosslinker would generate thermosets whose properties could be directly tuned by pre-polymer amination density and/or nucleophile identity. Lastly, nucleophilic cleavage of the sulfamate crosslinkers would induce network dissolution and regeneration of polydiene thermoplastics. Herein, we apply this workflow (Fig. 1C) to access degradable crosslinked PBDs with tunable mechanical properties by synthesizing Reprocessable Elastomers via Alcoholysis to Crosslink Thermoplastic Sulfamates (REACTS).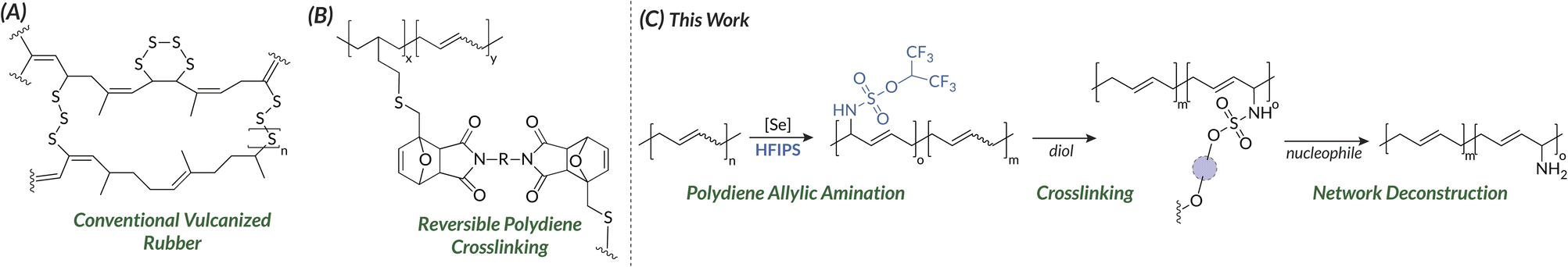 | ||
| Fig. 1 (A) Structure of vulcanized rubber with S8 linkages. (B) Previous examples of reprocessable crosslinked elastomers. (C) This work: reprocessable elastomers via alcoholysis to crosslink thermoplastic sulfamates (REACTS). | ||
Results and discussion
To establish the viability of REACTS, we first investigated the nucleophilic dimerization of a small molecule model following allylic amination with HFIPS. First, 4-phenyl-1-butene (1)25 was aminated with HFIPS using 20 mol% SePCy3 to afford 2 in 74% yield (Scheme 1). Then, 2 was treated with ethylene glycol to yield dimer 3,29 in a process akin to polymer crosslinking. Following unequivocal confirmation of 3 by 1H NMR spectroscopy and mass spectrometry (Fig. S3 and S7†), our next focus was to ensure that HFIPS substitution still occurred on polymeric substrates. To address this question, commercial PBD (Mn = 7 kDa, 87% cis–trans, 13% 1,2-vinyl branched) was aminated with HFIPS at a low target density based on stoichiometry (Scheme 2). Importantly, as we demonstrated in our previous work, amination of PBD does not alter the backbone microstructure (i.e., cis–trans ratio remains unchanged).18 Following purification by washing the crude polymer with MeOH and dialysis against acetone, the amination density was determined to be 6 mol% using 19F NMR spectroscopy (internal standard = 4,4-difluorobenzophenone). Following amination and purification, PBD-HFIPS-6 was treated with 4-fluorophenethyl alcohol (Scheme 2) to yield 4 (90% yield); as it was previously determined that sulfamoylation of alcohols proceeded more efficiently with weaker bases, we exclusively utilized pyridine as a base in these studies,29 and screened tetrahydrofuran (THF) and 1,2-dichloroethane (DCE) as co-solvents. Ultimately, we found that THF was a superior co-solvent to DCE as it maintained reagent solubility, an important consideration for implementing REACTS (Scheme 3). As expected, formation of 4 was observable via 19F NMR spectroscopy; disappearance of the diagnostic PBD-HFIPS-6 chemical resonance at −73.0 ppm and generation of a new resonance at −116.7 ppm (4) (Fig. S17†) was observed. Once we established that alcoholysis of 3 would occur with difunctionalized nucleophiles and that PBD-HFIPS-6 was competent for alcohol sulfamoylation, we then turned efforts to implementing REACTS. Briefly, we envisioned REACTS proceeding via initial pre-polymer formation by Se-catalyzed amination of PBD, followed by crosslinking using an appropriate diol. Hence, PBD (Mn = 7 kDa, 87% cis–trans, 13% 1,2-vinyl branched) was first aminated at two grafting densities, ca. 3 and 6 mol%, as quantified by 1H and 19F NMR spectroscopy (Fig. S9, S16, eqn S(1)–S(3)†). To initially assess crosslinking potential using REACTS, PBD-HFIPS-X and ethylene glycol (EG, X/2 equiv.) were dissolved in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pyridine (1:1 mixture, 1.5× by mass) and heated to 62 °C (Scheme 3). Higher density PBD-EG-6 gelled within 24 hours, while lower density PBD-EG-3 gelled within 48 hours. Importantly, samples prepared in the absence of either PBD-HFIPS or ethylene glycol remain fully soluble (Table S2†). Similar synthetic procedures were implemented for diethylene glycol (PBD-DEG-X) and triethylene glycol (PBD-TEG-X) crosslinked samples. Equilibrium swelling ratios for the collective samples were determined gravimetrically by swelling in THF for 40 hours (Table S4†). Differences in target crosslink density were apparent as PBD-R-3 samples had higher equilibrium swelling ratios (i.e., looser network structure) than those of PBD-R-6 samples. Additionally, while the difference in swelling ratios was not statistically significant between PBD-EG-6 and PBD-DEG-6, or between PBD-EG-3 and PBD-DEG-3, there was a significant difference between PBD-EG-X and PBD-TEG-X (p < 0.05), with the latter less swellable than the former (p < 0.01) (Table S5†). Overall, a longer crosslinker (i.e., TEG vs. EG) in PBD-R-X leads to a tighter network structure, possibly through increased hydrogen bonding when using higher order glycols.
:pyridine (1:1 mixture, 1.5× by mass) and heated to 62 °C (Scheme 3). Higher density PBD-EG-6 gelled within 24 hours, while lower density PBD-EG-3 gelled within 48 hours. Importantly, samples prepared in the absence of either PBD-HFIPS or ethylene glycol remain fully soluble (Table S2†). Similar synthetic procedures were implemented for diethylene glycol (PBD-DEG-X) and triethylene glycol (PBD-TEG-X) crosslinked samples. Equilibrium swelling ratios for the collective samples were determined gravimetrically by swelling in THF for 40 hours (Table S4†). Differences in target crosslink density were apparent as PBD-R-3 samples had higher equilibrium swelling ratios (i.e., looser network structure) than those of PBD-R-6 samples. Additionally, while the difference in swelling ratios was not statistically significant between PBD-EG-6 and PBD-DEG-6, or between PBD-EG-3 and PBD-DEG-3, there was a significant difference between PBD-EG-X and PBD-TEG-X (p < 0.05), with the latter less swellable than the former (p < 0.01) (Table S5†). Overall, a longer crosslinker (i.e., TEG vs. EG) in PBD-R-X leads to a tighter network structure, possibly through increased hydrogen bonding when using higher order glycols.
 | ||
| Scheme 1 Amination of 4-phenyl-1-butene (1) with HFIPS (2), followed by dimerization with ethylene glycol (3). | ||
 | ||
| Scheme 2 Amination of PBD with HFIPS followed by sulfamoylation of fluorophenethyl alcohol (4). | ||
 | ||
| Scheme 3 Generation of REACTS thermosets (PBD-XG-X) by alcoholysis of PBD-HFIPS pre-polymers using glycol crosslinkers. X = 3 mol% or 6 mol%. | ||
Oscillatory rheology showed that REACTS specimens behave as viscoelastic solids (Fig. 2A). As anticipated, frequency sweep experiments showed storage (G′) moduli above loss (G′′) moduli without any crossover points. Differences in crosslink density between PBD-TEG-6 and PBD-TEG-3 are revealed in a significantly higher plateau modulus for the former; affine network theory (i.e., G ∝ Mc)30–32 suggests ca. 4-fold reduction in molar mass between crosslinks for PBD-TEG-6. For comparison, photochemical thiol–ene crosslinked PBD samples were synthesized at similar target crosslinking densities (PBD-Thiol-X).33 These PBD-Thiol-X specimens approximate the random crosslinking (i.e., destruction of polymer microstructure) afforded from S8 vulcanization.34–44 The same trend was observed in these specimens, with the higher crosslink density sample resulting in a higher plateau storage modulus.
 | ||
| Fig. 2 (A) Frequency sweep of PBD-Thiol-X and PBD-TEG-X crosslinked specimens. (B) Compression analysis of representative PBD-TEG-X samples compared to PBD-Thiol-X. | ||
We then carried out compression testing analysis on emblematic PBD-TEG-X specimens (Fig. 2B and S40–S42†); these samples were the easiest to mold across all the PBD-R-X series, thereby serving as ideal representative formulations to probe mechanical properties of REACTS materials. As anticipated, we see that both PBD-TEG-3 and PBD-Thiol-3 are softer than PBD-TEG-6 and PBD-Thiol-8. Qualitatively, relative to PBD-TEG-6, PBD-Thiol-8 is stiffer with lower stress and strain at break while PBD-TEG-3 is softer with a higher strain at break than PBD-Thiol-3 (Fig. S37–S39†). Overall, despite PBD-TEG-X following the same trend as PBD-Thiol-X (i.e. lower density crosslinking leading to softer materials with lower strain at break), only the REACTS samples have the prospect for facile deconstruction to soluble byproducts (vide infra).
Next, the thermal stability of all PBD-R-6 and PBD-R-3 networks were analyzed using thermal gravimetric analysis (TGA). These materials showed a two-step decomposition curve, characteristic of aminated polydienes reported by Boydston19 and our group,18 with sulfamate groups thermally decomposing prior to the decomposition of the PBD backbone. Using 10% mass loss as the metric for REACTS specimen decomposition (Td,10%), we observed that PBD-R-6 samples had higher decomposition temperatures compared to their PBD-R-3 counterparts. We also observed that as crosslinker length increased (i.e., EG to DEG to TEG) decomposition temperature decreased, spanning PBD-TEG-3 (Td,10% = 194 °C) to PBD-EG-6 (Td,10% = 282 °C) (Fig. S27–S32†). The ability to tune the thermal stability of REACTS materials is distinct from PBD-Thiol-X samples that decompose at similar temperatures independent of crosslink density (Td,10% = 395–401 °C) (Fig. S25–S26†).
To demonstrate reprocessability of the REACTS platform, samples were decrosslinked using phenol under basic conditions (Table 1)45 to afford soluble polymer (Fig. 3A) and diphenyl glycol products alongside potassium sulfite. Isolated yields following removal of the small molecule byproduct were good to moderate (36–76%). As confirmed by 1H NMR spectroscopy, network deconstruction affords amine-functionalized PBD that is nearly indistinguishable from virgin PBD used to originally construct the networks (Fig. 3B and C).
|
|||
|---|---|---|---|
| Specimen | Time (h) | Temperature (°C) | Isolated yield |
| PBD-EG-3 | 24 | 45 | 57% |
| PBD-DEG-3 | 16 | 45 | 54% |
| PBD-DEG-6 | 48 | 60 | 76% |
| PBD-TEG-3 | 20 | 45 | 36% |

 | ||
| Fig. 3 (A) sample images of REACTS sample PBD-DEG-3 before and after decrosslinking. 1H NMR spectra of (B) PBD prior to REACTS crosslinking and (C) following network deconstruction of REACTS sample PBD-DEG-3. Scale bar = 1 cm. | ||
This process, which could be observed visually (Fig. S47†), proceeds from a swollen network to soluble products over ca. 16–48 hours; the exact conditions (i.e., time and temperature) for the selected REACTS samples empirically depended on crosslinker density and identity. Ostensibly, the low HFIPS density translates to an indetectable amount of free amine in the resultant PBD product by 1H NMR spectroscopy. As 1H NMR spectroscopy is not suitably sensitive to demonstrate the presence of the free amines, the PBD-NH2-3 material following thermoset deconstruction was fluorinated via an amidation reaction with trifluoracetic anhydride. This method generated a 19F NMR handle which could be used to characterize the amount of free amine on the parent PBD backbone (Fig. S12 and S18†); the amount was calculated to ca. 4% of monomers aminated after decrosslinking PBD-R-3, which is comparable to the ca. 3% of monomers aminated in PBD-HFIPS-3. Further, this claim is corroborated by nearly identical analytical gel permeation chromatography (GPC) retention times (Fig. S1†) between virgin PBD and the thermoplastic material recovered after decrosslinking. Importantly, neither component of the two-step crosslinking protocol nor the deconstruction procedure impacts polydiene backbone length or microstructure. Finally, to further exhibit the reprocessability of REACTS specimens, decrosslinked material described above (i.e., PBD-NH2-3) was resubjected to another round of amination with HFIPS (Fig. S13 and S19†). The resulting (PBD-NH2-3)-HFIPS-6 was then re-crosslinked with TEG; the same reactivity and curing properties as seen in the virgin materials were observed via vial inversion tests (Fig. S49†).
Conclusions
In conclusion, we have demonstrated the utility of post-polymerization C–H allylic amination to access deconstructable thermoset elastomers. Using the REACTS platform, a two-step procedure was developed that retains the original polydiene microstructure during generation of electrophilic sulfamate-tagged pre-polymers. Subsequent crosslinking via sulfamoylation with a variety of diols allows for tuning of mechanical properties as a function of crosslinker identity and density. Lastly, treatment of REACTS samples with an exogenous nucleophile effects thermoset deconstruction and regeneration of soluble aminated46,47 thermoplastic elastomers that can be reprocessed using a subsequent REACTS cycle. It is envisioned that this technology will be broadly applicable in the crosslinking of other unsaturated thermoplastics and ultimately represents a novel strategy to reversibly construct and deconstruct polymer networks in a nearly traceless fashion.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. N. H. and M. R. G. conceived of the idea. M. N. H. and L. R. conducted synthetic experiments and analysed physical properties. A. P. K. V. performed rheological and mechanical testing experiments. M. N. H. and M. R. G. wrote the manuscript; all authors discussed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by generous start-up funds from the University of Washington. M. N. H. acknowledges the University of Washington Clean Energy Institute for a graduate research fellowship. The authors thank Prof. Dianne Xiao for use of thermogravimetric analysis instrumentation. This material is based in part upon work supported by the state of Washington through the University of Washington Clean Energy Institute. NMR spectroscopy resources are supported under NIH S10 OD030224-01A1. The authors acknowledge Prof. Forrest Michael for helpful discussions.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- C. Fuschi, H. Pu, M. Macdonell, K. Picel, M. Negri and J. Chen, ACS Sustain. Chem. Eng., 2022, 10, 14074–14091 CrossRef CAS.
- E. Markl and M. Lackner, Materials, 2020, 13, 1246 CrossRef CAS PubMed.
- X. Lim, Nature, 2021, 593, 22–25 CrossRef CAS PubMed.
- K. A. Günay, P. Theato and H. A. Klok, J. Polym. Sci., Part A:Polym. Chem., 2013, 51, 1–28 CrossRef.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- J. B. Williamson, S. E. Lewis, R. R. Johnson, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef CAS PubMed.
- S. E. Lewis, B. E. Wilhelmy and F. A. Leibfarth, Chem. Sci., 2019, 10, 6270–6277 RSC.
- S. E. Lewis, B. E. Wilhelmy and F. A. Leibfarth, Polym. Chem., 2020, 11, 4914–4919 RSC.
- M. E. Skala, S. M. Zeitler and M. R. Golder, Chem. Sci., 2024, 15, 10900–10907 RSC.
- C. P. Easterling, T. Kubo, Z. M. Orr, G. E. Fanucci and B. S. Sumerlin, Chem. Sci., 2017, 8, 7705–7709 RSC.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS.
- P. T. Chazovachii, M. J. Somers, M. T. Robo, D. I. Collias, M. I. James, E. N. G. Marsh, P. M. Zimmerman, J. F. Alfaro and A. J. McNeil, Nat. Commun., 2021, 12, 4524 CrossRef CAS PubMed.
- N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267–275 RSC.
- C. Tian, H. Feng, Y. Qiu, G. Zhang, T. Tan and L. Zhang, Polym. Chem., 2022, 13, 5368–5379 RSC.
- J. X. Shi, N. R. Ciccia, S. Pal, D. D. Kim, J. N. Brunn, C. Lizandara-Pueyo, M. Ernst, A. M. Haydl, P. B. Messersmith, B. A. Helms and J. F. Hartwig, J. Am. Chem. Soc., 2023, 145, 21527–21537 CrossRef CAS PubMed.
- N. R. Ciccia, J. X. Shi, S. Pal, M. Hua, K. G. Malollari, C. Lizandara-Pueyo, E. Risto, M. Ernst, B. A. Helms, P. B. Messersmith and J. F. Hartwig, Science, 2023, 381, 1433–1440 CrossRef CAS PubMed.
- M. N. Hodges, M. J. Elardo, J. Seo, A. F. Dohoda, F. E. Michael and M. R. Golder, Angew. Chem., Int. Ed., 2023, 62, e202303115 CrossRef CAS PubMed.
- S. R. Gitter, W. P. Teh, X. Yang, A. F. Dohoda, F. E. Michael and A. J. Boydston, Angew. Chem., Int. Ed., 2023, 62, e202303174 CrossRef CAS PubMed.
- Y. Ren, T. P. Lodge and M. A. Hillmyer, J. Am. Chem. Soc., 1998, 120, 6830–6831 CrossRef CAS.
- L. Li, S. Zhao, C. Chen and Z. Xin, J. Polym. Res., 2015, 22, 1–6 CrossRef.
- E. Trovatti, T. M. Lacerda, A. J. F. Carvalho and A. Gandini, Adv. Mater., 2015, 27, 2242–2245 CrossRef CAS PubMed.
- E. M. Lloyd and S. L. Craig, Macromolecules, 2024, 57, 10349–10357 CrossRef CAS.
- A. Forens, K. Roos, C. Dire, B. Gadenne and S. Carlotti, Polymer, 2018, 153, 103–122 CrossRef CAS.
- W. P. Teh, D. C. Obenschain, B. M. Black and F. E. Michael, J. Am. Chem. Soc., 2020, 142, 16716–16722 CrossRef CAS PubMed.
- T. P. Maloney, J. L. Berman and F. E. Michael, Angew. Chem., Int. Ed., 2022, 61, e202210109 CrossRef CAS PubMed.
- A. F. Dohoda, V. L. Zottarelli, D. Hajikedir and F. E. Michael, Org. Lett., 2024, 26, 10213–10217 CrossRef CAS PubMed.
- S. E. Towell, M. Ratushnyy, L. S. Cooke, G. M. Lewis and A. V. Zhukhovitskiy, Nature, 2025, 640, 384–389 CrossRef CAS PubMed.
- M. A. Sguazzin, J. W. Johnson and J. Magolan, Org. Lett., 2021, 23, 3373–3378 CrossRef CAS PubMed.
- L. R. G. Treloar, Rep. Prog. Phys., 1973, 36, 755–826 CrossRef CAS.
- P. J. Flory, Polym. J., 1985, 17, 1–12 CrossRef CAS.
- C. W. H. Rajawasam, O. J. Dodo, M. A. S. N. Weerasinghe, I. O. Raji, S. V. Wanasinghe, D. Konkolewicz and N. De Alwis Watuthanthrige, Polym. Chem., 2024, 15, 219–247 RSC.
- M. J. Warner, J. P. Lassa, H. Narcross, A. Commisso, K. Ghosh, M. Romero, J. M. Schwartz, A. C. Engler, P. A. Kohl, S. C. Leguizamon and B. H. Jones, ACS Sustain. Chem. Eng., 2023, 11, 14538–14548 CrossRef CAS.
- S. Yang, Y. Wang, F. Wang, K. Zhang, X. Lv, H. Teng, R. Zheng, F. Luo and Q. Xing, eXPRESS Polym. Lett., 2025, 19, 94–106 CrossRef CAS.
- S. Basak and K. A. Cavicchi, J. Polym. Sci., 2025, 63, 486–492 CrossRef CAS.
- T. Alamfard, T. Lorenz and C. Breitkopf, Polymers, 2023, 15, 2058 CrossRef CAS PubMed.
- A. F. Behbahani, A. Rissanou, G. Kritikos, M. Doxastakis, C. Burkhart, P. Polińska and V. A. Harmandaris, Macromolecules, 2020, 53, 6173–6189 CrossRef CAS.
- P. C. Prabhu, S. Mohanty and V. K. Gupta, Rubber Chem. Technol., 2021, 94, 410–431 CrossRef CAS.
- A. Vasilev, T. Lorenz and C. Breitkopf, Polymers, 2021, 13, 315 CrossRef CAS PubMed.
- Z. H. Tang, H. Zeng, S. Q. Wei, S. W. Wu and B. C. Guo, Chin. J. Polym. Sci., 2021, 39, 1337–1344 CrossRef CAS.
- J. A. Herman, M. E. Seazzu, L. G. Hughes, D. R. Wheeler, C. M. Washburn and B. H. Jones, ACS Appl. Polym. Mater., 2019, 1, 2177–2188 CrossRef CAS.
- N. Ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef CAS.
- M. Saito, N. L. Yamada, K. Ito and H. Yokoyama, Macromolecules, 2023, 56, 4000–4011 CrossRef CAS.
- P. Berto, J. Mehats, A.-L. Wirotius, S. Grelier and F. Peruch, Macromolecules, 2022, 55, 4557–4567 CrossRef CAS.
- E. G. Burke and J. M. Schomaker, J. Org. Chem., 2017, 82, 9038–9046 CrossRef CAS PubMed.
- S. S. Scott, B. Kaur, C. H. M. Zheng, P. Brant, D. J. Gilmour and L. L. Schafer, J. Am. Chem. Soc., 2023, 145, 22871–22877 CrossRef CAS PubMed.
- N. Kuanr, I. Stoševski, D. P. Wilkinson and L. L. Schafer, ACS Appl. Energy Mater., 2024, 7, 3433–3442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, solution-state characterization, bulk properties testing. See DOI: https://doi.org/10.1039/d5sc02522b |
| This journal is © The Royal Society of Chemistry 2025 |