
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid and precise synthesis of acrylic polymers driven by visible light†
Changhoon Yu‡
a,
Jong-Kwon Ha‡ b,
Mincheol Parkc,
Jungwook Leea,
Jinho Choia,
Boyoung Y. Parkc,
Cyrille Boyer*d,
Seung Kyu Min*b and
Min Sang Kwon*a
b,
Mincheol Parkc,
Jungwook Leea,
Jinho Choia,
Boyoung Y. Parkc,
Cyrille Boyer*d,
Seung Kyu Min*b and
Min Sang Kwon*a
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: minsang@snu.ac.kr
bDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: skmin@unist.ac.kr
cDepartment of Biomedical and Pharmaceutical Sciences, Kyung Hee University, Seoul 02447, Republic of Korea
dCluster for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, and Australian Centre for Nanomedicine, School of Chemical Engineering, UNSW Australia, Sydney, NSW 2052, Australia. E-mail: cboyer@unsw.edu.au
First published on 2nd June 2025
Abstract
Rapid and precise acrylic polymer synthesis is essential for applications in drug delivery, programmable materials, and biosensors. However, achieving both speed and precision remains challenging, as reaction acceleration is typically coupled with increased radical concentration, leading to a trade-off between polymerization rate and molecular control. Photoiniferter RAFT polymerization, a catalyst-free, visible light-driven method, offers exceptional control but lacks a detailed mechanistic understanding of C–S bond photolysis. Here, we resolve this speed-control trade-off by leveraging a key photophysical feature of thiocarbonylthio compounds: C–S bond cleavage proceeds via an S1/S0 conical intersection (CI), enabling ultrafast, non-radiative relaxation and clean photolytic decomposition with minimal side reactions. Although quantum yield is low (0.3–0.5%), this mechanism inherently limits radical accumulation, even at elevated temperatures. As a result, propagation can be thermally accelerated without increasing termination, preserving excellent control. Coupled with flow chemistry, this strategy achieves 90% monomer conversion in 20 minutes with narrow dispersity (Đ = 1.02) and minimal dead chains (<2%). This work offers a scalable, energy-efficient route to precision polymers and advances the mechanistic understanding of controlled radical processes for next-generation materials.
Introduction
The rapid synthesis of well-defined acrylic polymers is essential for advancing a wide range of applications,1 including precision drug delivery systems,2 programmable materials,3 functional membranes,4 and biosensors.5 These technologies rely on precise control over polymer architectures—such as multi-block copolymers, and star or brush structures—to achieve their desired functions.6 However, synthesizing complex acrylic-based polymers with controlled architectures is typically slow, as it requires low radical concentrations, limiting scalability. Consequently, achieving both rapid polymerization and precise structural control remains a central challenge in radical-based polymer synthesis.7,8Reversible addition–fragmentation chain transfer (RAFT) polymerization, a widely used reversible-deactivation radical polymerization (RDRP), offers excellent control over polymer architecture by utilizing thiocarbonylthio (TCT) compounds as chain transfer agents (CTAs).9–14 Renowned for its versatility, RAFT is compatible with a broad range of acrylic monomers and solvents.9,10 However, achieving both high reaction rates and precise control remains a challenge. Increasing the reaction temperature accelerates polymerization by enhancing initiator decomposition and propagation rates. Yet, this also elevates radical concentrations, leading to more frequent bimolecular termination events and a corresponding decline in chain transfer efficiency.15 As a result, under elevated temperatures or high initiator concentrations, conventional RAFT polymerization can yield significant dead chain formation—often exceeding 20% within an hour—and broader molecular weight distributions.16,17 These issues underscore a fundamental limitation in reconciling polymerization speed with architectural precision.
Photoiniferter RAFT polymerization offers a potential solution to this dilemma. First introduced by Otsu et al. in 1982,18,19 this method eliminates the need for external initiators by utilizing a TCT-based compound that acts as an initiator, transfer agent, and reversible terminator (iniferter) upon C–S bond photolysis (Fig. 1a). Through the coexistence of reversible deactivation and degenerative chain transfer processes, photoiniferter RAFT polymerization demonstrates enhanced livingness relative to conventional RAFT polymerization.20,21 This advancement appears to be substantiated by recent studies under carefully controlled conditions, suggesting its potential for achieving superior control in polymerization processes. Interest in this method was reignited in 2015 by Qiao et al.22 and Boyer et al.23 demonstrated its potential under visible light irradiation, leading to remarkable advancements such as the synthesis of ultra-high molecular weight polymers,24–27 novel sequence tunability,28–32 and dispersed media applications33,34—outcomes unattainable with conventional RAFT polymerization. Additionally, our group recently demonstrated that poly(methyl acrylate) (PMA) synthesized through photoiniferter RAFT polymerization showed minimal dead chain formation (<2%) and a near-Poisson distribution (Đ = 1.01),35,36 achieving a level of control that surpasses conventional RDRP methods.
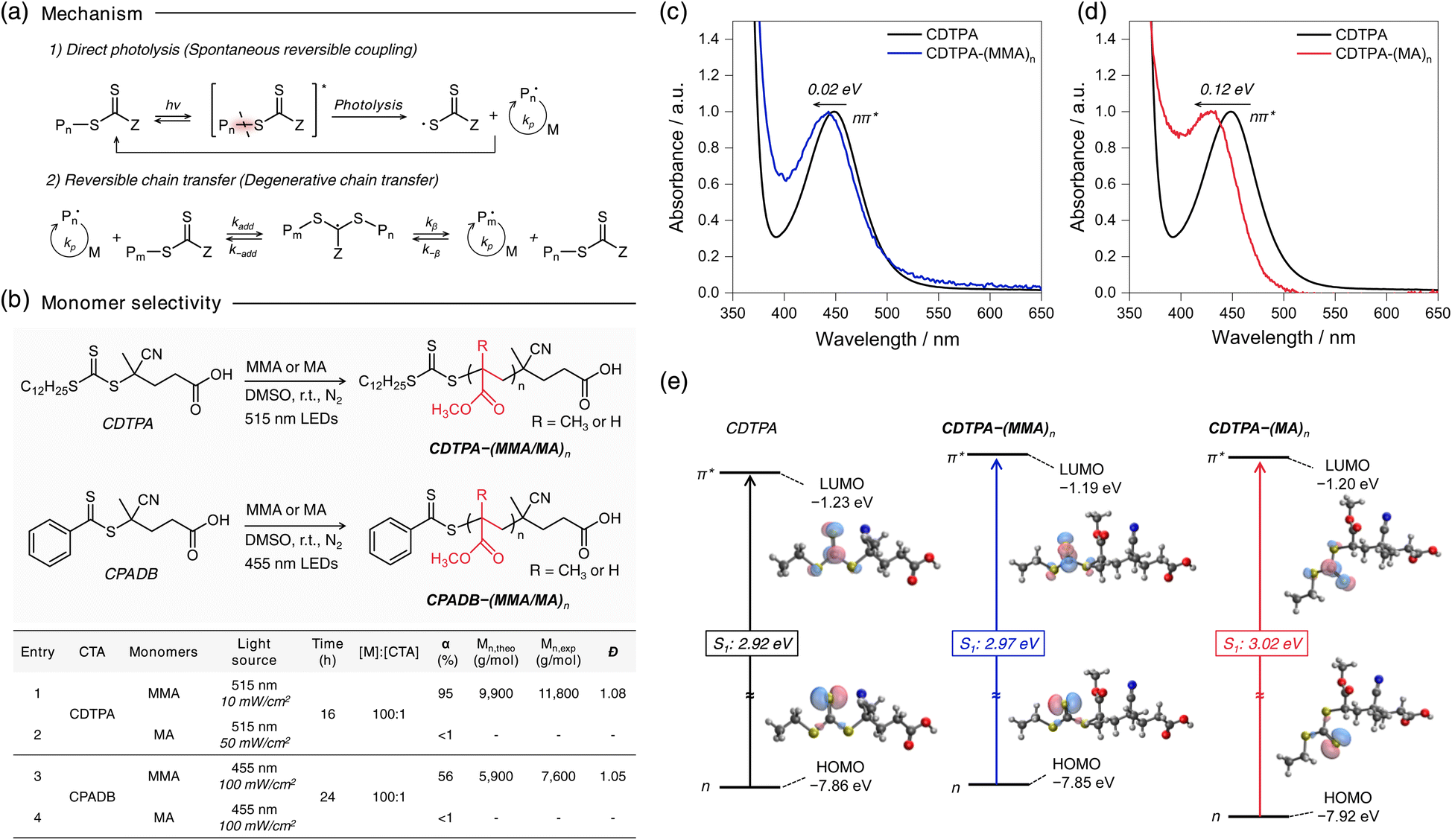 | ||
| Fig. 1 (a) Proposed mechanism of photoiniferter polymerization. Direct light absorption by the TCT moiety excites the agent, followed by the homolysis of the C–S bond to produce an active initiating/propagating and persistent TCT radical. Upon adding the TCT-based molecules, the former radical species participate in the degenerative transfer pathway of RAFT. The TCT radical deactivates the growing radical species in a reversible way, as seen in ATRP. (b) Results of photoiniferter polymerizations of methyl methacrylate (MMA) and methyl acrylate (MA) in the presence of CDTPA under different temperatures and luminous intensities. (c) UV-Vis graph of 200 μM CDTPA and CDTPA–(MMA)n solution in DMSO. (d) UV-Vis graph of 200 μM CDTPA and CDTPA–(MA)n solution in DMSO. (e) Calculated HOMO–LUMO gap and S1 value of CDTPA, CDTPA–(MMA)n and CDTPA–(MA)n. | ||
Despite these advances, achieving both rapid polymerization and precise control in photoiniferter RAFT polymerization remains rare, with accelerated reactions limited to specific solvent-induced conditions. Such cases, where monomer propagation rates (kp) are enhanced without an increase in radical concentration, have been observed almost exclusively in systems involving hydrophilic monomers like N,N-dimethylacrylamide in aqueous or ionic liquid environments.25,37 However, these instances represent isolated exceptions rather than a broadly applicable strategy. Alternative approaches, such as using acidic conditions38 or high-intensity irradiation,39 introduce additional complications, including increased susceptibility to TCT hydrolysis and degradation of TCT groups,40 ultimately compromising polymer livingness. The scarcity of effective, broadly applicable strategies highlights the urgent need for a universal approach.
In this study, we present a strategy that leverages the distinct mechanistic features of photoinitiated RAFT polymerization to achieve both rapid synthesis and precise architectural control. Our approach is guided by key mechanistic insights from quantum mechanical calculations, which reveal that the photolytic C–S bond cleavage in thiocarbonylthio (TCT) compounds proceeds via an S1/S0 conical intersection (CI) pathway. This CI enables a fast, efficient, and radiationless transition from the excited to the ground state, resulting in clean photolytic decomposition with minimal side reactions—albeit with low quantum yields (0.3–0.5%, as reported by the Falvey group);41,42 The photostability of nucleobases in biological systems is attributed to CIs, which enable rapid non-radiative relaxation and prevent side-inducing photochemical reactions. Crucially, this mechanism allows for the radical concentration generated through C–S bond dissociation to remain low, even at elevated temperatures, when light intensity is slightly reduced. As a result, the propagation rate can be significantly enhanced without increasing the likelihood of bimolecular termination. To further optimize the system, we integrated flow chemistry, which mitigates viscosity-related limitations and enhances scalability. This combined strategy enabled the efficient synthesis of well-defined acrylic polymers, achieving up to 90% monomer conversion within just 20 minutes while maintaining a narrow dispersity (Đ = 1.02) and minimal amount of dead chains (<2%).
Results & discussion
System of interest
The advent of photoiniferter RAFT polymerization has significantly advanced the precise control over polymer architectures. However, its applicability is limited by varying reactivity across different monomers, with the underlying mechanisms remaining unclear. Our preliminary investigation revealed distinct polymerization behaviors between methyl acrylate (MA) and methyl methacrylate (MMA), consistent with Johnson43 and Boyer's findings.23 Under 515 nm LED irradiation (25 °C, 10 mW cm−2) for 16 h, MMA polymerized efficiently in the presence of 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDTPA) (α = 99%) giving poly(methyl methacrylate) (PMMA), while MA showed minimal conversion (<1%) (Fig. 1b, entries 1 and 2). A similar trend was observed with 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPADB) under 455 nm LED (25 °C, 100 mW cm−2) for 24 h, where MMA reached 56% conversion, but MA remained unreacted (Fig. 1b, entries 3 and 4; Fig. S1†). These results suggest that monomer-selective polymerization stems from differences in C–S bond photolysis efficiency, implying MA exhibiting significantly lower bond dissociation QY than MMA.To further explore this distinct reactivity, we selected CDTPA, one of the most widely used iniferters in photoiniferter RAFT polymerization. 1H-NMR monitoring revealed that CDTPA was fully consumed within 2 hours, forming a single-unit monomer insertion with MA, as evidenced by peak shifts at 3.25–3.33 ppm (Fig. S2†). However, monomer conversion remained below 1%, even with continuous 515 nm LED irradiation at 50 mW cm−2 for an additional 14 hours. By contrast, MMA polymerization with CDTPA proceeded efficiently. To explore the differences further, UV-vis spectroscopy was employed to examine the absorption properties of CDTPA and its adducts, CDTPA–(MA/MMA)n, under polymerization conditions (Fig. 1c). The nπ* absorption band of CDTPA–(MA)n and CDTPA–(MMA)n showed slight blue shifts of 0.12 eV and 0.02 eV, respectively, relative to CDTPA, which aligned with computational predictions (Fig. 1d). Despite the reduced overlapped area with 515 nm LED emission spectrum—approximately four times lower than that of CDTPA (Fig. S3b†)—the nπ* band of CDTPA–(MA)n still aligns with the 515 nm LED light source. However, even under 16 hours of irradiation at an intensity five times stronger than that used for MMA (50 mW cm−2; Fig. 1b, entry 2), C–S bond dissociation does not occur in CDTPA–(MA)n.
Additional experiments conducted under further higher light intensities than previous irradiation conditions (250 mW cm−2) or elevated temperatures (60 °C) improved MA conversion to 62% and 30%, respectively (Table S1†). However, these results remained suboptimal compared to MMA polymerization under milder conditions. These observations suggest that (i) the photolysis of the C–S bond is intrinsically less efficient in CDTPA–(MA)n than in CDTPA–(MMA)n, and (ii) an activation barrier exists in the photolysis process, possibly associated with an S1/S0 conical intersection that limits efficient bond cleavage. This highlights the importance of optimizing reaction conditions to overcome the inefficiency of C–S bond photolysis across different monomers, further supporting the critical role of the CI in this mechanism.
Origin of C–S dissociation
Quantum chemical (QC) calculations were performed to fully understand the origin of the photoinduced C–S fission of CDTPA–(MA)n and CDTPA–(MMA)n. First, we evaluated the energies of the possible intermediates responsible for the C–S bond dissociation of CDTPA–MA and CDTPA–MMA (Fig. 2). As shown in Fig. 2a, the vertical S1 energies (i1 (S1,vert)) are far lower than those required for dissociation to produce a fragment in the S1 state , which lies at an exceptionally high energy level of 6.37 eV and 6.05 eV for the respective molecules. This energy difference indicates that the bond dissociation proceeds along the S0 PES rather than the S1 PES. Therefore, the molecules in the S1 state must experience internal conversion (IC) through an S1/S0 CI to reach a bond dissociation state on S0 (i3[R(D0) + Z(D0)]). Bond dissociation via triplet states is also a possibility; however, it is unlikely for CDTPA–MA and CDTPA–MMA, as the dissociation pathway is energetically unfavorable (0.57 eV and 0.29 eV, respectively; Fig. S4†). To enhance clarity and provide a concise overview of this process, we have schematically illustrated the C–S bond dissociation pathway in Scheme 1.
, which lies at an exceptionally high energy level of 6.37 eV and 6.05 eV for the respective molecules. This energy difference indicates that the bond dissociation proceeds along the S0 PES rather than the S1 PES. Therefore, the molecules in the S1 state must experience internal conversion (IC) through an S1/S0 CI to reach a bond dissociation state on S0 (i3[R(D0) + Z(D0)]). Bond dissociation via triplet states is also a possibility; however, it is unlikely for CDTPA–MA and CDTPA–MMA, as the dissociation pathway is energetically unfavorable (0.57 eV and 0.29 eV, respectively; Fig. S4†). To enhance clarity and provide a concise overview of this process, we have schematically illustrated the C–S bond dissociation pathway in Scheme 1.
 | ||
| Fig. 2 (a) Plausible photoinduced C–S bond dissociation pathway of CDTPA–MA and CDTPA–MMA. Plausible energy profiles for the photoinduced C–S bond dissociation of (b) CDTPA–MA (grey), CDTPA–MMA (red) and (c) CPADB–MA (grey), CPADB–MMA (blue). | ||
 | ||
| Scheme 1 Schematic representation of the photoinduced C–S bond photolysis pathway of CDTPA–MA and CDTPA–MMA. | ||
Subsequently, we investigated the minimum-energy CI structures for IC from S1 to S0, which act as a transition state of the C–S bond dissociation (Fig. 2b). In the ground state (S0), CDTPA–MMA showed a more linear structure than CDTPA–MA due to the steric hindrance of the methyl group of MMA. After Franck–Condon excitation (i1 (S1,vert)), the TCT moiety in both molecules started to rotate, and the molecules attained S1/S0 CI (iCI) with a transition to i3 [R(D0) + Z(D0)] through the i2 (S1,adia) state with structural relaxation.
The CI energies were higher by 0.23 eV and 0.24 eV in CDTPA–MA and CDTPA–MMA, respectively, than those of i2 (S1,adia). These results suggest that the CI acts as an activation barrier in the photolysis process and behaves as a transition state in the thermal process, which is in good agreement with the experimental results (discussed in the below section). While activation barriers of almost the same energy were observed in CDTPA–MA and CDTPA–MMA, the process from CI to the bond dissociation state i3 [R(D0) + Z(D0)] was found to be very different in CDTPA–MA and CDTPA–MMA, due to the large energy stabilization of tertiary radical species derived from CDPTA–MMA.40 In CDTPA–MMA, the energy of i3 [R(D0) + Z(D0)] was significantly lower than that of iCI, making the dissociation process one of the most favorable pathways. However, in CDTPA–MA, because the bond dissociation state energy was similar to that of the CI (iCI → i0 (S0)), the C–S bond dissociation was less competitive than the other nonradiative decay channels from the CI, leading to a considerably low bond-dissociation QY.
To further explore the C–S bond dissociation of dithiobenzoate-type iniferters, we also conducted QC calculation on CPADB–MA and CPADB–MMA (Fig. 2c and S5†). Similar to the CDTPA case, the CI energies for CPADB–MA and CPADB–MMA were found to be higher than their respective i2 (S1,adia) states by 0.29 eV and 0.36 eV, respectively, acting as activation barriers. However, the i3 [R(D0) + Z(D0)] state of CPADB–MA was significantly higher in energy compared to its CI, making bond dissociation impossible. This high-energy CI state of CPADB–MA is the fundamental reason for the widely reported acrylate–CPADB mismatch,23,40 as it results in a near-zero C–S bond photolysis QY. In contrast, the bond dissociation process for CPADB–MMA, though feasible, was inefficient, with only 0.1 eV of stabilization. These findings align closely with our preliminary experimental results, as illustrated in Fig. 1b, and underscore the critical role of the relative energy positions of the i2 (S1,adia), iCI, and i3 [R(D0) + Z(D0)] states in determining the efficiency of C–S bond photolysis.
Optimization of the polymerization of MA
Building on the mechanistic insights into C–S bond photolysis, we investigated the polymerization behavior of MA by selecting a 455 nm LED as a new light source (Table 1). This choice was motivated by the significantly higher light absorption of CDTPA−(MA)n, which shows approximately 10 times greater overlap with the 455 nm LED emission compared to 515 nm LED emission due to its molar extinction coefficient (∼22 M−1 cm−1) at 455 nm, while exhibiting almost no absorption at 515 nm (Fig. S3b†). As our previous report,35 PMA polymerized under 455 nm LEDs (25 °C, 100 mW cm−2) led to a very high conversion of 95% in 16 h and demonstrated excellent controllability (Đ = 1.01) (Table 2 – entry 1). In this study, all Đ and MW values were determined using size exclusion chromatography (SEC) coupled with multi-angle light scattering (MALS), refractive index (RI) and UV detectors, resulting SEC curves combined of RI (black), MALS (red), and UV at 235 nm (green) and 310 nm (blue) (see ESI† for detailed SEC setups). Furthermore, the characteristic signals of the trithiocarbonate (TTC) functionality were clearly observed in the 1H-NMR spectrum, with no end-group decomposition signal, indicating high end-group fidelity (Fig. S6†).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Light source | Temp (°C) | Time (h) | [M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CTA]:[I] :[CTA]:[I] |
αb (%) | Mn,theoc (g mol−1) | Mn,expd (g mol−1) | Đd |
| a [MA]/[CDTPA] = 100/1, [MA] = 5.45 M in DMSO, 455 nm LED as a light source under various temperature for photoiniferter RAFT polymerzation. [MA]/[CDTPA]/[AIBN] = 100/1/0.1, [MA] = 5.45 M in DMSO, 80 °C for conventional RAFT polymerzation.b Conversion (%) is calculated by 1H-NMR comparing the ratio of monomer and polymer peaks (Fig. S6).c Mn,theo is calculated using the following equation: Mn,theo = [MA]0/[CDTPA]0 × MWM × α + MWCDTPA, where [MA]0, [CDTPA]0, MWM, α, and MWCDTPA correspond to MA and CDTPA concentration, molar mass of MA, monomer conversion, and molar mass of CDTPA.d Mn,exp and Đ are determined by SEC (see ESI for setup). | ||||||||
| 1 | 455 nm | r.t. | 16 | 100:1:0 |
95 | 8600 | 9100 | 1.01 |
| 100 mW cm−2 | ||||||||
| 2 | — | 80 | 4 | 100:1:0.1 (AIBN) |
97 | 8800 | 7100 | 1.07 |
| 3 | 455 nm | r.t. | 12 | 100:1:0 |
94 | 8500 | 8800 | 1.01 |
| 200 mW cm−2 | ||||||||
| 4 | 455 nm | 60 | 3 | 100:1:0 |
93 | 8400 | 8000 | 1.01 |
| 100 mW cm−2 | ||||||||
| 5 | 455 nm | 60 | 2 | 100:1:0 |
97 | 8800 | 10300 |
1.02 |
| 200 mW cm−2 | ||||||||
| 6 | 455 nm | 80 | 2 | 100:1:0 |
91 | 8200 | 7200 | 1.02 |
| 100 mW cm−2 | ||||||||
| 7 | 455 nm | 80 | 1.5 | 100:1:0 |
95 | 8600 | 8400 | 1.03 |
| 200 mW cm−2 | ||||||||
| 8 | 455 nm | 80 | 5 | 100:1:0 |
90 | 8100 | 7200 | 1.02 |
| 30 mW cm−2 | ||||||||
 |
||||||||

|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Light source | Temp (°C) | Time (h) | [M]:[CTA] |
αb (%) | Mn,theoc (g mol−1) | Mn,expd (g mol−1) | Đd |
| a [MMA]/[CPADB] = 100/1, [MMA] = 4.65 M in DMSO, 455 nm LED as a light source under various temperature for photoiniferter RAFT polymerzation.b Conversion (%) is calculated by 1H-NMR comparing the ratio of monomer and polymer peaks.c Mn,theo is calculated using the following equation: Mn,theo = [MMA]0/[CPADB]0 × MWM × α + MWCPADB, where [MMA]0, [CPADB]0, MWM, α, and MWCPADB correspond to MMA and CPADB concentration, molar mass of MA, monomer conversion, and molar mass of CPADB.d Mn,exp and Đ are determined by SEC. | ||||||||
| 1 | 455 nm | 60 | 12 | 100:1 |
90 | 9300 | 10500 |
1.06 |
| 100 mW cm−2 | ||||||||
| 2 | 455 nm | 80 | 8 | 100:1 |
80 | 8300 | 8800 | 1.04 |
| 100 mW cm−2 | ||||||||
| 3 | 455 nm | 80 | 12 | 100:1 |
90 | 9300 | 10400 |
1.03 |
| 50 mW cm−2 | ||||||||

To quantify the dead chain portions of the as-prepared PMA, high-performance liquid chromatography (HPLC) was performed with four detectors (Fig. S7†). The polymers eluted in two distinct peaks, as indicated by the MALS signal. These peaks displayed a clear difference in the UV detector at 310 nm due to the absorption by the TTC group. The major fraction, labeled “fraction 2,” which accounted for 98.3%, exhibited absorption at 310 nm. In contrast, the minor fraction, labeled “fraction 1” (1.7%), did not show this absorption. Both fractions were collected and reanalyzed by SEC (Fig. S7a†). The results indicated that the major fraction displayed a slightly narrower peak compared to the as-prepared PMA (Đ = 1.01) and was identified as the living chain due to the presence of the TTC group. On the other hand, “fraction 1” exhibited a broad bimodal peak (Đ = 1.27), which, due to the absence of a TTC moiety, was attributed to the dead chains that terminated spontaneously through coupling and disproportionation during polymerization.44
Conversely, conventional RAFT polymerization using azobisisobutyronitrile (AIBN) as an initiator (control experiment) comparable conversion but exhibited broader dispersity (α = 97% and Đ = 1.07) (Fig. S8†), resulting in a significant (15%) proportion of dead chains (Fig. S7b†). This high amount of dead polymers was attributed to the formation of exogenous radicals generated by the thermal decomposition of AIBN, resulting in the formation of new chains. In contrast, photoiniferter RAFT polymerization operates without external initiators and enables precise control of radical concentration via inefficient C–S bond photolysis QY. These results confirm that, under optimized conditions, photoiniferter RAFT polymerization of MA predominantly yields living chains with a near-Poisson distribution, underscoring the synthetic advantage of the photoiniferter RAFT method in achieving both rapid polymerization and excellent architecture control.
Origin of high controllability and monomer scope expanding
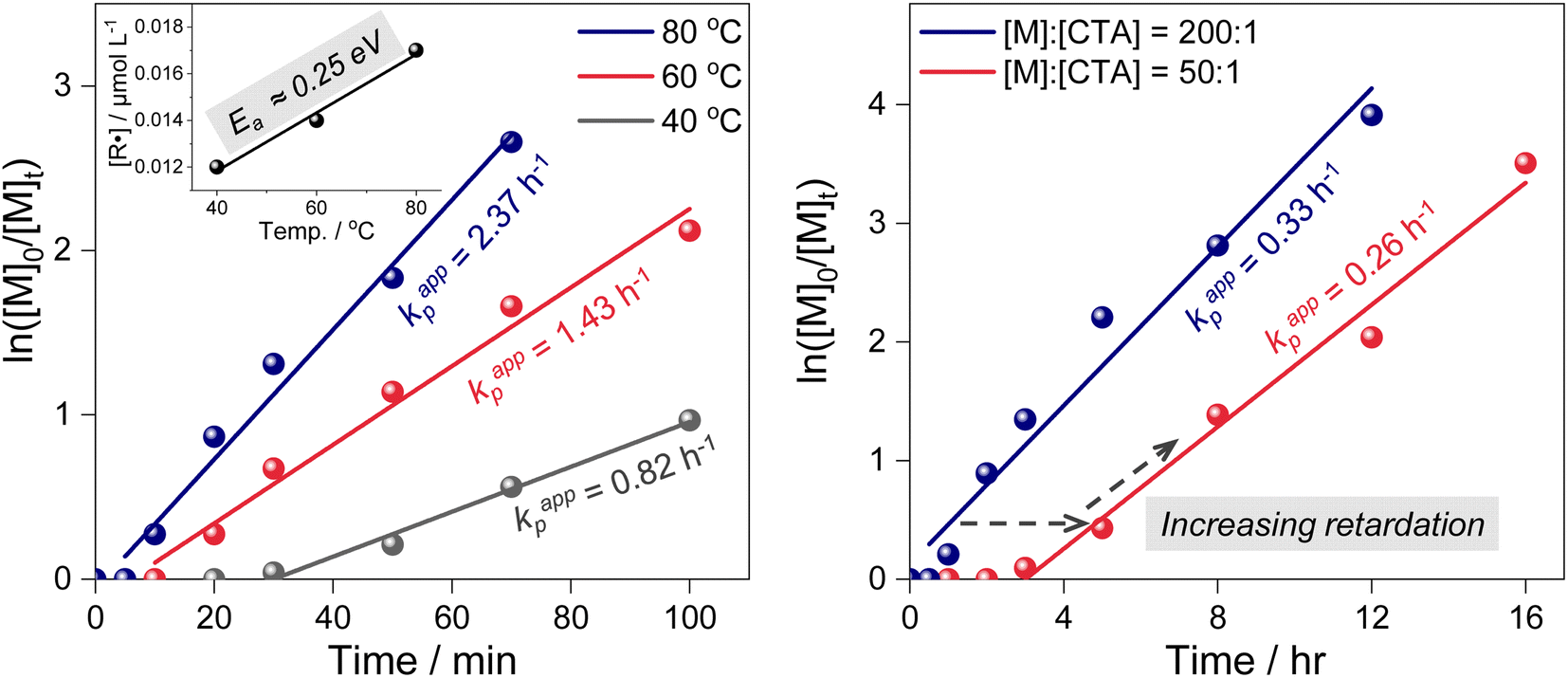
To investigate the origin of the high controllability, photoiniferter RAFT polymerizations were performed under 455 nm irradiation at a high light intensity (200 mW cm−2) and/or at elevated temperatures (60 °C and 80 °C). The conditions yielded PMAs with a broader dispersity (Table 1, entries 3–7), suggesting that TCT photolysis occurred faster at a higher light intensity. At excessively higher intensities (300–400 mW cm−2), photolysis accelerates further, but the resulting increase in radical concentration compromises control (Table S2†).36 Additionally, elevated temperatures facilitated the overcoming of the activation barrier, increasing the chance of accessing the CI and accelerating TCT photolysis. Despite the increased photolysis rate, the inherently low QY (0.3–0.5%) governed by the CI limits radical accumulation. This effectively suppresses the steady-state radical concentration [R˙], which in turn minimizes the bimolecular termination rate Rt = kt[R˙]2 while still sustaining efficient chain propagation.Kinetics experiments conducted at different temperatures indeed support this argument (Table 1, bottom left). The apparent rate of propagation (kp,app) was determined from the kinetics plots, and the radical concentration ([R˙]) was estimated as follows: [R˙] ∼ kp,app/kp, where kp is the rate of propagation.44 An evident increase in [R˙] was observed for polymerization at a higher temperature, clearly confirming the existence of an activation barrier in TCT photolysis that can be overcome by thermal energy. To further evaluate the activation energy of the photolysis process, we analyzed the temperature-dependent variation in [R˙]. The resulting activation energy was approximately 24.4 kJ mol−1 (∼0.25 eV), closely matching the value obtained from quantum chemical calculations (0.24 eV, vide supra). The corresponding Arrhenius plot is presented in Fig. S9.†”. Surely, the CI is expected to play a crucial role in providing an activation barrier and a nonradiative pathway to the ground state, resulting in inefficient C–S bond dissociation and, ultimately, lower radical concentrations (Fig. 2 and Scheme 1). Finally, we investigated the occurrence of chain transfer processes. As the CDTPA concentration increased (i.e., at lower DP targets), pronounced retardation and an extended inhibition period were observed (Table 1, bottom right). Under these conditions, photolysis efficiency and total radical concentration were confirmed to remain effectively constant under same light irradiation intensities (Fig. S10†), indicating that the observed retardation originates from the increased formation of RAFT intermediate adducts. These intermediates transiently trap radicals, reducing the concentration of free propagating species. This behavior reflects a typical chain transfer process, which promotes homogeneous chain growth and ensures high chain-end fidelity.45
Considering these mechanistic features, the polymerization conditions were further optimized. Since the kp increases with temperature, its activation energy for MA is approximately 16–18 kJ mol−1,46,47 while the rate of termination (kt) has a lower activation energy of around 4–7 kJ mol−1.48 This disparity makes kp more temperature-dependent, resulting in a higher kp/kt ratio at elevated temperatures. Consequently, the reaction time can be significantly reduced without compromising control by increasing the temperature while lowering the light intensity to keep the radical concentration low. Notably, under these optimized conditions—high temperature with reduced light intensity—the polymerization achieved a level of controllability in just 5 hours comparable to that observed under room temperature conditions (Table 1, entry 8).
The optimized conditions (i.e., with CDTPA under 455 nm LED irradiation of 100 mW cm−2 at 25 °C) provided excellent control over the polymerization at high conversion for a wide range of acrylic monomers such as hydrophobic, hydrophilic, zwitterionic, fluorine (Fig. S11†), and solvents (Table S3†). Due to the excellent controllability and broad monomer scope of the proposed polymerization method, polyacrylates with varying compositions, MWs, and architectures were prepared.
Optimization of the polymerization of MMA
We then expanded our studies to the polymerization of MMA. The photoiniferter RAFT polymerization of MMA provided PMMA with a broader MW distribution (Đ = 1.08) at a similar monomer conversion to PMA (α = 99%; Fig. 1b, entry 1). As for the case of MA, [R•] was also evaluated. Even under 515 nm LED irradiation with a lower luminous intensity (10 mW cm−2 at 25 °C), an order of magnitude higher value was obtained (Fig. S12†). This suggests that the observed controllability can be attributed to the high concentration of propagating radical species produced by the more efficient C–S bond dissociation than CDTPA–MA. To improve the controllability for the polymerization of MMA, we changed the iniferter to CPADB (Table 2). This is because CPADB–MMA exhibits an unfavorable energy structure for C–S dissociation similar to that of CDTPA–MA (Fig. 2). Polymerization was also performed at a higher temperature to increase the kp of MMA. Since MMA, like MA, has a higher activation energy for kp compared to kt, it benefits from improved controllability at elevated temperatures.47 Indeed, applying this strategy under reduced light intensity resulted in a significant enhancement in conversion with narrower dispersity (α = 90% and Đ = 1.03), further validating our approach (Table 2, entry 3). Additionally, given the numerous variations in light intensity and temperature in this study, we have summarized all polymerization results in Table S4† to provide a clear overview of the observed polymerization trends under these conditions. The applicability of this process beyond acrylate monomers was explored using styrene as a representative example (Table S5†), under the same optimized conditions established for MA (455 nm LED irradiation at 100 mW cm−2, 25 °C). Unlike acrylic monomers, photoiniferter RAFT polymerization of styrene required significantly longer reaction times (>72 h) and exhibited reduced controllability (Đ > 1.19) when using trithiocarbonate (TTC)-based iniferters, while no polymerization was observed with CPADB. This behavior is likely attributed to the fundamentally different CI structure of the styrene–CTA adduct, as well as distinct RAFT equilibrium dynamics. Further optimization of reaction parameters is currently underway and remains an active area of investigation.Polymerization of MA under flow reactor
Based on these findings, we have attained a comprehensive understanding of C–S bond photolysis, enabling us to synthesize highly precise polyacrylates and polymethacrylates within a short timeframe. To further enhance these results, we transitioned from batch reactors to flow reactors for photoiniferter RAFT polymerization. This transition not only significantly reduces light intensity loss due to the shorter light path length but also markedly improves reaction speed and homogeneity (Table 3).49,50 Maintaining the reaction temperature at 80 °C and employing 265 mW cm−2 of 450 nm LEDs irradiation for the photoiniferter RAFT polymerization of MA, we achieved a 94% conversion in 50 minutes, albeit with a somewhat broad dispersity (Đ = 1.06; Table 3, entry 1). The high dispersity observed was attributed to poor mixing within the flow reactors, as evidenced by the low Reynolds number (Re ∼0.38, see ESI†) calculated under our experimental conditions, which indicated laminar flow.51 Under such conditions, the residence time distribution (RTD) broadens, leading to higher dispersity and lower conversion due to mixing being restricted to molecular diffusion.52–54 To address these challenges, we implemented a strategy of incorporating SiO2 glass beads into the reactor tubing. This approach enhanced mixing by promoting turbulence flow and improving overall controllability.55
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reactor volume (mL) | SiO2 bead size (mm) | Time (min) | αb (%) | Mn,theoc (g mol−1) | Mn,expd (g mol−1) | Đd |
| a [MA]/[CDTPA] = 100/1, [MA] = 5.45 M in DMSO, 450 nm LED as a light source under 80 °C for photoiniferter RAFT polymerzation under flow reactor.b Conversion (%) is calculated by 1H-NMR comparing the ratio of monomer and polymer peaks.c Mn,theo is calculated using the same equation described in Table 1.d Mn,exp and Đ are determined by SEC. | |||||||
| 1 | 9.0 | — | 50 | 94 | 8500 | 11400 |
1.06 |
| 2 | 6.4 | 0.2 | 30 | 94 | 8500 | 9500 | 1.04 |
| 3 | 6.4 | 0.2 | 20 | 89 | 8100 | 9300 | 1.02 |
| 4 | 6.8 | 0.3 | 20 | 89 | 8100 | 8800 | 1.03 |
| 5 | 7.5 | 0.4 | 20 | 85 | 7700 | 8500 | 1.03 |
| 6 | 8.0 | 0.5 | 20 | 86 | 7800 | 8400 | 1.02 |
| 7 | 8.4 | 0.75 | 20 | 85 | 7700 | 8100 | 1.02 |
 |
|||||||

As anticipated, using 0.2 mm SiO2 beads, we achieved comparable conversion in just 30 minutes with a slight reduction in dispersity (α = 94%, Đ = 1.04; Table 3, entry 2). Notably, reducing the reaction time to 20 minutes achieved similar conversion with even narrower dispersity (α = 89%, Đ = 1.02; Table 3, entry 3 and bottom right), indicating that continued C–S bond photolysis during the end of time frame polymerization, when most monomers are already consumed, likely influences dispersity over time; in fact, further thorough studies are currently underway. Screening different bead sizes consistently yielded high monomer conversion (>85%) resulting in PMAs with narrow dispersity. Furthermore, a good agreement between Mn,theo and Mn,SEC was observed, demonstrating the successful polymerization of well-defined PMA in a remarkably short period using a flow reactor (Table 3, entries 4–7). For qualitative comparison with previously reported photoiniferter RAFT polymerizations of acrylic monomers under flow conditions, key results from this study and relevant literature are summarized in Table S6.†49,56 Furthermore, under these optimized conditions, we also examined the effects of increasing monomer concentration and targeting higher molecular weights (Table S7†). These adjustments significantly increased the viscosity of the reaction medium, resulting in broadened RTD and diminished mixing efficiency, consistent with observations reported by Leibfarth and co-workers.54 Consequently, polymerization control was adversely affected.
Conclusion
This study establishes a fundamental understanding of photoiniferter RAFT polymerization by elucidating the C–S bond photolysis mechanism via S1/S0 conical intersection-mediated pathways. Leveraging these insights, we have developed a broadly applicable strategy that enables rapid and precise acrylic polymer synthesis, independent of specific solvent constraints. By optimizing reaction conditions—balancing reduced light intensity with elevated temperature—and incorporating flow chemistry, we achieved highly efficient polymerization, attaining 90% monomer conversion in just 20 minutes with exceptional control over dispersity (Đ = 1.02). This method offers a scalable, catalyst-free platform for high-precision polymer manufacturing. These advancements overcome critical challenges in radical polymerization, presenting an energy-efficient and environmentally sustainable alternative. Beyond refining the theoretical framework of light-driven polymerization, this work lays a foundation for future advancements in sustainable materials, high-performance macromolecular engineering, and next-generation polymeric systems.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. S. K. and C. Y. conceived the project. M. S. K. supervised the project with support of C. B. C. Y. designed and performed most of the experiments. J.-K. H. and S. K. M. conducted QC calculations and analyzed the results. M. P. and B.-Y. P. performed flow photopolymerization and analyzed obtained results. J. L. and J. C. helped with the photoiniferter RAFT reproducibility tests and SEC measurements. C. Y. and M. S. K. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Samsung Research Funding & Incubation Center of Samsung Electronics under project number SRFC-MA2101-05 and National Research Foundation of Korea (NRF) funded by the Korea government (MSIT) (RS-2024-00354184). C. B is the recipient of an Australian Research Council Australian Laureate Fellowship (Project No. FL220100016) funded by the Australian Government. The authors are thankful to Daewhan Kim (Department of Materials Science and Engineering, Seoul National University) for his work on photoiniferter RAFT polymerization of styrene during the revision.References
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- A. Walther, Adv. Mater., 2020, 32, 1905111 Search PubMed.
- X. Feng, J. Zhu, J. Jin, Y. Wang, Y. Zhang and B. Van der Bruggen, Prog. Mater. Sci., 2024, 144, 101285 CrossRef CAS.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035–3097 RSC.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Adv. Sci., 2020, 7, 2001656 CrossRef CAS PubMed.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- M. Semsarilar and V. Abetz, Macromol. Chem. Phys., 2020, 222, 2000311 CrossRef.
- T. Otsu and M. Yoshida, Makromol. Chem., Rapid Commun., 1982, 3, 127–132 CrossRef CAS.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- R. W. Hughes, M. E. Lott, R. A. Olson S and B. S. Sumerlin, Prog. Polym. Sci., 2024, 156, 101871 CrossRef CAS.
- M. Hartlieb, Photo-Iniferter RAFT Polymerization, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS PubMed.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, ACS Symp. Ser., 2015, 1187, 247–267 CrossRef CAS.
- S. Zhu, W. Kong, S. Lian, A. Shen, S. P. Armes and Z. An, Nat. Synth., 2025, 4, 15–30 CrossRef CAS.
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS.
- C. L. G. t. Davidson, M. E. Lott, L. Trachsel, A. J. Wong, R. A. Olson, D. I. Pedro, W. G. Sawyer and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 1224–1230 CrossRef PubMed.
- R. N. Carmean, M. B. Sims, C. A. Figg, P. J. Hurst, J. P. Patterson and B. S. Sumerlin, ACS Macro Lett., 2020, 9, 613–618 CrossRef CAS PubMed.
- Y. Xia, G. M. Scheutz, C. P. Easterling, J. Zhao and B. S. Sumerlin, Angew. Chem., Int. Ed., 2021, 60, 18537–18541 CrossRef CAS PubMed.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, Block Copolymer Sequence Inversion through Photoiniferter Polymerization, ACS Macro Lett., 2019, 8, 1461–1466 CrossRef CAS PubMed.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376–8383 CrossRef CAS PubMed.
- C. Fu, Z. Huang, C. J. Hawker, G. Moad, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 4637–4643 RSC.
- J. Gardiner, I. Martinez-Botella, J. Tsanaktsidis and G. Moad, Polym. Chem., 2016, 7, 481–492 RSC.
- J. I. Bowman, C. B. Eades, M. A. Vratsanos, N. C. Gianneschi and B. S. Sumerlin, Angew. Chem., Int. Ed., 2023, 62, e202309951 CrossRef CAS PubMed.
- Z. Wu, Z. He, Y. Zhou, T. Kou, K. Gong, F. Nan, T. T. Bezuneh, S. Han, C. Boyer and W. W. Yu, Angew. Chem., Int. Ed., 2025, 64, e202422975 CrossRef CAS PubMed.
- C. Yu, J. Choi, J. Lee, S. Lim, Y. Park, S. M. Jo, J. Ahn, S. Y. Kim, T. Chang, C. Boyer and M. S. Kwon, Adv. Mater., 2024, 36, 2403048 CrossRef CAS PubMed.
- J. Lee, Y. Kwon, C. Yu, D. Konkolewicz and M. S. Kwon, Polym. Chem., 2025, 16, 1798–1806 RSC.
- A. R. S. Santha Kumar, S. Allison-Logan, J. R. Finnegan, N. K. Singha, M. Ashokkumar and G. Qiao, ACS Macro Lett., 2023, 12, 1012–1018 CrossRef CAS PubMed.
- M. N. Antonopoulou, N. P. Truong, T. Egger, A. A. Kroeger, M. L. Coote and A. Anastasaki, Angew. Chem., Int. Ed., 2024, 64, e202420733 CrossRef PubMed.
- J. R. Lamb, K. P. Qin and J. A. Johnson, Polym. Chem., 2019, 10, 1585–1590 RSC.
- T. G. McKenzie, L. P. d. M. Costa, Q. Fu, D. E. Dunstan and G. G. Qiao, Polym. Chem., 2016, 7, 4246–4253 RSC.
- M. D. Thum, S. Wolf and D. E. Falvey, J. Phys. Chem. A, 2020, 124, 4211–4222 CrossRef CAS PubMed.
- D. Jadoun and M. Kowalewski, J. Phys. Chem. Lett., 2021, 12, 8103–8108 CrossRef CAS PubMed.
- J. R. Lamb, K. P. Qin and J. A. Johnson, Polym. Chem., 2019, 10, 1585–1590 RSC.
- K. Kim, J. Ahn, M. Park, H. Lee, Y. J. Kim, T. Chang, H. B. Jeon and H.-j. Paik, Macromolecules, 2019, 52, 7448–7455 CrossRef CAS.
- K. G. E. Bradford, L. M. Petit, R. Whitfield, A. Anastasaki, C. Barner-Kowollik and D. Konkolewicz, J. Am. Chem. Soc., 2021, 143, 17769–17777 CrossRef CAS PubMed.
- J. Van Herck, S. Harrisson, R. A. Hutchinson, G. T. Russell and T. Junkers, Polym. Chem., 2021, 12, 3688–3692 RSC.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191–254 CrossRef CAS.
- M. Buback and C. Kowollik, Macromolecules, 1998, 31, 3211–3215 CrossRef CAS.
- M. A. Bereś, B. Zhang, T. Junkers and S. Perrier, Kinetic investigation of photoiniferter-RAFT polymerization in continuous flow using inline NMR analysis, Polym. Chem., 2024, 15, 3166–3175 RSC.
- F. Lauterbach, M. Rubens, V. Abetz and T. Junkers, Angew. Chem., Int. Ed., 2018, 57, 14260–14264 CrossRef CAS PubMed.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- N. Corrigan, A. Almasri, W. Taillades, J. Xu and C. Boyer, Macromolecules, 2017, 50, 8438–8448 CrossRef CAS.
- N. Corrigan, R. Manahan, Z. T. Lew, J. Yeow, J. Xu and C. Boyer, Macromolecules, 2018, 51, 4553–4563 CrossRef CAS.
- M. H. Reis, T. P. Varner and F. A. Leibfarth, Macromolecules, 2019, 52, 3551–3557 CrossRef CAS.
- F. Zhong, Y. Zhou and M. Chen, Polym. Chem., 2019, 10, 4879–4886 RSC.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02594j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |