
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolable monoatomic monovalent bismuth complexes with a redox non-innocent bis-silylenyl carborane ligand†
Jian
Xu
a,
Shenglai
Yao
 a,
Verònica
Postils
b,
Eduard
Matito
cd,
Christian
Lorent
e and
Matthias
Driess
*a
a,
Verònica
Postils
b,
Eduard
Matito
cd,
Christian
Lorent
e and
Matthias
Driess
*a
aMetalorganic and Inorganic Materials, Department of Chemistry, Technische Universität Berlin, 10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de
bTheoretical Chemistry Group, Molecular Chemistry, Materials and Catalysis Division (MOST), Institute of Condensed Matter and Nanosciences, Université Catholique de Louvain, Place Louis Pasteur 1, B-1348 Louvain-la-Neuve, Belgium
cDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain
dIkerbasque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Euskadi, Spain
ePhysical and Biophysical Chemistry, Department of Chemistry, Technische Universität Berlin, 10623 Berlin, Germany
First published on 19th May 2025
Abstract
Utilizing the chelating bis(silylenyl)carborane [SiII(closo-CB)SiII] (A, SiII = PhC(NtBu)2Si, CB = o-C2B10H10) ligand, a series of unprecedented bis(silylene)-stabilized monovalent bismuth complexes {[SiII(closo-CB)SiII]Bi}X (X = I, 1a; X = OTf, 1b), {[SiII(nido-CB)SiII]Bi} (2) and ({[SiII(nido-CB)SiII]Bi}K(thf)2)2 ([3K(thf)2]2) were synthesized, isolated and characterized. The electronic structures of the bismuth complexes are significantly influenced by the redox-active nature of the CB scaffold. Remarkably, a one-electron injection to 1b with KC8 does not furnish a Bi0 complex but reduction of the CB backbone giving rise to the neutral BiI radical complex 2. Notably, compound 1b can also undergo a two-electron reduction with two molar equiv. of potassium naphthalenide, resulting in the formation of the diamagnetic BiI anion complex 3 as a dimer bridged via two K(thf)2 cations. Density functional theory calculations reveal that upon reduction from 1a to 2, and 2 to 3, the added electron predominantly localizes within the carborane cage, with a marked preference for the carbon atoms, ruling out that these species exhibit characteristics of a molecular bismuth(0) electride.
Introduction
Bismuth, known since ancient times, is the heaviest stable element in the periodic table, possessing an extraordinarily long half-life of 1.9 × 1019 years.1,2 Despite its inherent stability, bismuth has been widely utilized in diverse research fields, including medical science, physical and materials sciences, and applied chemistry.3–7 In recent years, significant progress has been made in the study of low-valent bismuth compounds,8–10 highlighting their multiple accessible oxidation states and redox-active nature. These characteristics enable bismuth to participate in a variety of chemical transformations, presenting exciting opportunities for innovative catalytic applications.11–13 Furthermore, bismuth has emerged as a promising central ion in single-molecule magnets (SMMs) due to its strong spin–orbit coupling and stable multivalent states.14–16Monoatomic zero-valent complexes of Group 14 elements, known as tetrylones, adopt the general formula L: → E0 ←: L (E = C, Si, Ge, Sn, Pb; L = Lewis donor ligands).17–21 Mono-valent cationic bismuth species can be regarded as the heaviest isoelectronic analogs of tetrylones, that is plumbylones, possessing two lone pairs on the central atom.22,23 The first cationic BiI compound (I, Fig. 1)24 was synthesized by reducing BiCl3 in the presence of cyclic alkyl(amino)carbenes (cAACs). Subsequently, the bis(silylene)-supported BiI cation complex [{SiII(TBD)SiII}BiI][BArF4] II (ref. 25) (SiII = PhC(NtBu)2Si, TBD = 1,8,10,9-triazaboradecalin, ArF = 3,5-(CF3)2-C6H3) was prepared through a one-pot reaction between the bis(silylene) {SiII(TBD)SiII} ligand and [IPr → BiBr3] (IPr = 1,3-bis(2,6-diisopropylphenyl)-imidazole-2-ylidene), followed by treatment with Na[BArF4] in THF at −30 °C and reduction with two molar equiv. of potassium graphite (KC8). Recently, our group reported the bis(germylene)-supported cationic BiI complex III.26 Due to the redox non-innocent nature of the germylene moieties, the positive charge of the BiI cation migrates to one of the Ge atoms within the bis(germylene) ligand, resulting in the chelating germylium–germylene BiI complex III.
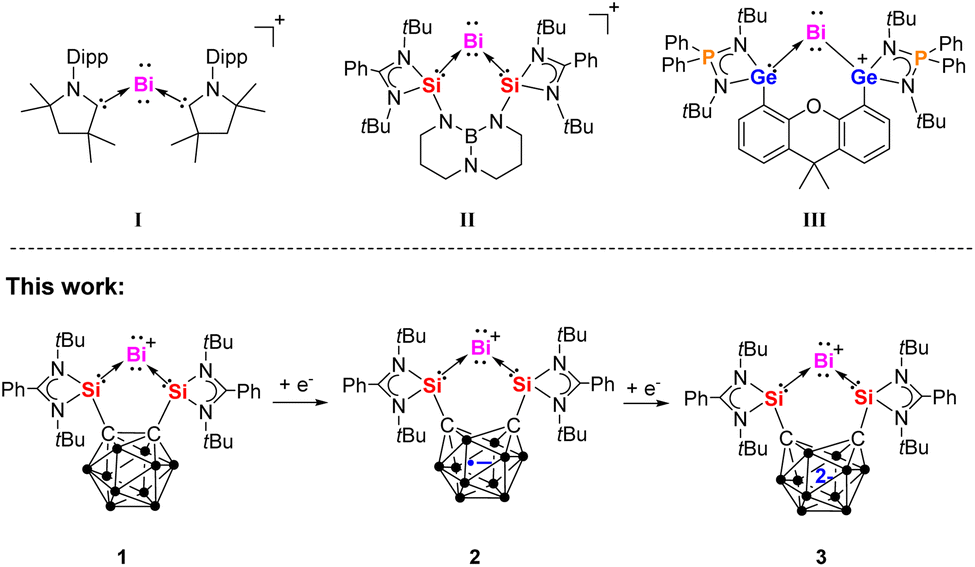 | ||
| Fig. 1 Known examples of cationic BiI complexes I–III and the BiI species 1–3 of this work bearing a redox non-innocent carborane cage. | ||
In recent years, our group has successfully designed and synthesized several chelating bis(NHSi) ligands (NHSi = N-heterocyclic silylene) featuring electronically and geometrically diverse spacers.27 These bis(NHSi) ligands have facilitated the isolation of various low-valent main-group compounds, including zero-valent group 14 and mono-valent group 15 complexes, which exhibit fascinating electronic structures and unique chemical reactivities.28–35 The bis(silylenyl)-o-carborane ligand [SiII(closo-CB)SiII] (A, SiII = PhC(NtBu)2Si, CB = o-C2B10H10)36 (Scheme 1), featuring a relatively short SiII⋯SiII distance of approximately 3.3 Å, was first reported by our group in 2016. The latter ligand acts as a strong chelating Lewis donor due to the silylene moieties and exhibits interesting redox non-innocence attributed to the carborane spacer. These properties have proven effective in stabilizing monoatomic Si0 and Ge0 complexes33,34 as well as containing the isoelectronic NI cation.35 Herein, we report the synthesis and characterization of the BiI cation complexes {[SiII(closo-CB)SiII]Bi}X (X = I, 1a; X = OTf, 1b) supported by the bis(silylenyl)-o-carborane A. Strikingly, the one-electron and two-electron reductions of 1b using KC8 and KC10H8, respectively, yield no Bi0 species but the neutral and anionic BiI complexes {[SiII(nido-CB)SiII]Bi} 2 and ({[SiII(nido-CB)SiII]Bi}K(thf)2)2 ([3K(thf)2]2), both featuring a nido-C2B10 core, yet in different reduced states. The electronic structures of this series of BiI complexes are further elucidated through Density Functional Theory (DFT) calculations.
 | ||
| Scheme 1 Synthesis of BiI cation complexes 1a and 1b from bis(silylenyl)-o-carborane A. | ||
Results and discussion
Synthesis of bis(silylene)-stabilized mono-valent single atom bismuth complexes
The dipotassium bis(silylenyl)-nido-dicarboranate precursor [{SiII(nido-CB)SiII}][K2(thf)4] (B),33 generated in situ from bis(silylenyl)-o-carborane (A) and two molar equivalents of KC8 in THF (Scheme 1), reacts with one equiv. of BiI3 at −30 °C to afford the BiI cation complex 1a. After workup, the desired BiI complex {[SiII(closo-CB)SiII]Bi}I (1a) was isolated as brown-yellow needle-shaped crystals in 57% yield. Subsequent treatment of 1a with one equiv. of AgOTf in dichloromethane (DCM) results in the replacement of the iodide counterion by OTf−, yielding the {[SiII(closo-CB)SiII]Bi}OTf complex 1b as an orange powder in 77% yield. The 29Si{1H} NMR spectra of 1a and 1b show singlets at δ = 68.7 and 66.9 ppm, respectively, both exhibiting significant downfield shifts relative to A (δ = 18.9 ppm).28The molecular structures of 1a and 1b were determined by single-crystal X-ray diffraction (scXRD) analysis. Both exhibit a discrete ionic structure with a similar five-membered C2Si2Bi ring in the cation, where the central BiI site is coordinated to two silicon atoms. The Si–Bi bond lengths range from 2.5774(6) to 2.5958(9) Å (Fig. 2), similar to those in the [{SiII(TBD)SiII}BiI][BArF4] complex II (Fig. 1, 2.557(1) and 2.561(8) Å).25 The Si–C bond lengths span from 1.923(2) to 1.937(4) Å, while the C1–C2 distances in 1a and 1b [1.691(5) and 1.692(3) Å] are very close to that in A (1.71 Å).36 Notably, the Si1–Bi1–Si2 angles of 79.28(3)° and 79.486(18)° in 1a and 1b are slightly more acute than in complex II [82.10(3)°].25
 | ||
| Fig. 2 Molecular structures of the cations in 1a and 1b. Thermal ellipsoids are drawn at the 50% probability level. H atoms, anionic moieties and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (deg.): 1a: Bi1–Si2 2.5958(9), Bi1–Si1 2.5940(10), C2–C1 1.691(5), Si2–C2 1.937(4), Si1–C1 1.937(4), Si1–Bi1–Si2 79.28(3), C1–Si1–Bi1 115.14(11), C2–Si2–Bi1 115.65(11). 1b: Bi1–Si2 2.5774(6), Bi1–Si1 2.5931(6), C2–C1 1.692(3), Si1–C1 1.923(2), Si2–C2 1.925(2), Si1–Bi1–Si2 79.486(18), C1–Si1–Bi1 115.25(7), C2–Si2–Bi1 115.53(7). | ||
Notably, the cyclic voltammogram (CV) of 1b exhibits two quasi-reversible reduction processes at E1/2 = −1.36 V and −1.68 V vs. Fc/Fc+ (see ESI Fig. S11†). This prompted us to explore its chemical reduction. Upon mixing 1b with one equivalent of KC8 in THF at −30 °C, a deep-red solution formed, from which complex 2 was isolated in 43% yield (Scheme 2).
 | ||
| Scheme 2 Reversible redox reactions between 1b, 2 (with its resonance structure 2′) and [3K(thf)2]2. | ||
An scXRD analysis revealed that compound 2 crystallizes as a neutral BiI radical complex in the orthorhombic space group Cmcm. Its molecular structure features an open-cage nido-carborane backbone with a C⋯C distance of 2.268 Å (Fig. 3). The Si1–Bi1 bond length of 2.576(2) Å is comparable to those in complexes 1a and 1b (2.5774(6)–2.5958(9) Å). However, the Si1–C1 bond in complex 2 (1.851(8) Å) is significantly shorter than in 1a and 1b (1.923(2)–1.937(4) Å). Notably, the Si1–Bi1–Si1a bond angle in complex 2 (86.25(8)°) is substantially larger than in 1a and 1b (∼79°), likely due to the open-cage nature of the carborane backbone.
 | ||
| Fig. 3 Molecular structure of 2. Thermal ellipsoids are drawn at the 50% probability level. H atoms and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (deg.): C1–Si1 1.851(8), Si1–Bi1 2.576(2), C1⋯C1a 2.268, Si1–Bi1–Si1a 86.25(8), C1–Si1–Bi1 117.1(3). | ||
Compound 2 is paramagnetic and shows broad resonance peaks in solution 1H NMR spectra at room temperature (see ESI, Fig. S13†). Accordingly, the electron paramagnetic resonance (EPR) spectrum of 2 recorded at room temperature in THF (Fig. 4) exhibits an isotropic signal at g = 2.0229 (line width = 29.1 G). Though the band shape is very similar to the one of the known NI anionic carborane radical {[SiII(nido-CB)SiII]NI},35 the g-value is slightly higher than those typical for such organic radicals. Magnetic interactions with the nearby very heavy Bi nucleus could be the origin. An unpaired spin located at the Bi itself can be excluded based on the EPR spectrum at 10 K (see ESI, Fig. S14†), which lacks the characteristic broad and multiline features of a Bi-centred radical (Bi0).26 Overall, the observations indicate that the unpaired electron is localized in the carborane cage (see electronic structure discussion below).
 | ||
| Fig. 4 EPR spectrum of compound 2 in THF (top) recorded at 293 K and the corresponding simulation (bottom). The g-value of the radical species is 2.023 and the line width 29.1 G. | ||
To investigate the reversibility of the latter one-electron reduction, compound 2 was allowed to react with equimolar amount of AgOTf in THF at room temperature. Indeed, compound 1b was quantitatively regenerated after stirring for 10 minutes (Scheme 2). Given the carborane moiety of bis(NHSi) can store one or two electrons, we further explored the two-electron reduction of 1b. The reaction of 1b with two molar equiv. of potassium naphthalenide (KC10H8) in THF at −30 °C yielded complex ({[SiII(nido-CB)SiII]Bi}K(thf)2)2 ([3K(thf)2]2) as a brown powder in 51% yield (Scheme 2). Its 1H NMR spectrum in THF-d8 displays one singlet at δ = 1.29 ppm for the tert-butyl groups, while the 29Si{1H} NMR spectrum exhibits a singlet at δ = 51.8 ppm, significantly upfield-shifted compared to 1a (δ = 68.7 ppm) and 1b (δ = 66.9 ppm).
An scXRD analysis reveals that ([3K(thf)2]2) adopts a dimeric structure in the solid state, with two [K(thf)2]+ moieties acting as linkers via B–H⋯K⋯H–B interactions (Fig. 5). The Si–Bi bond lengths in ([3K(thf)2]2) (2.6266(13) and 2.6138(13) Å) are slightly longer than those in 1a and 1b (2.5774(6)–2.5958(9) Å) and in 2 (2.576(2) Å). Additionally, the Si–Bi1–Si bond angle in ([3K(thf)2]2) (90.27(4)°) is larger than in 1a and 1b (∼79°) and 2 (86.25(8)°). In line with that, the carborane backbone in ([3K(thf)2]2) features a C⋯C distance of 2.576 Å, notably longer than that in 2 (2.268 Å), indicating a further reduced carborane cage. Furthermore, the Si–C distances in ([3K(thf)2]2) (1.786(5) and 1.789(5) Å) are significantly shorter than those in 1a and 1b (1.923(2)–1.937(4) Å) and in 2 (1.851(8) Å). These structural features confirm that the open cage in ([3K(thf)2]2) corresponds to a nido-carborane anion. It should be mentioned that complex 2 can also be synthesized via a metathesis reaction between 1b and ([3K(thf)2]2) in THF at room temperature. In addition, complex ([3K(thf)2]2) can be obtained through the one-electron reduction of complex 2 with KC8 in THF (Scheme 2). This indirectly confirms the redox reversibility between the family members of this BiI series.
 | ||
| Fig. 5 Molecular structure of ([3K(thf)2]2). Thermal ellipsoids are drawn at the 50% probability level. H atoms and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (deg.): Bi1–Si1 2.6266(13), Bi1–Si2 2.6138(13), C1⋯C2 2.576, Si1–C1 1.786(5), Si2–C2 1.789(5), Si2–Bi1–Si1 90.27(4), C1–Si1–Bi1 116.52(16), C2–Si2–Bi1 115.93(16). | ||
The reactivity of 1b towards methyl trifluoromethanesulfonate (MeOTf) was also investigated to evaluate the nucleophilic character of BiI. Upon addition of MeOTf at 40 °C in DCM, the yellow solution of 1b gradually decolorized over 4 hours, yielding colorless {[SiII(closo-CB)SiII]BiMe} [OTf]2 (4) in 61% yield after workup (Scheme 3). The 1H NMR spectrum of 4 in DCM-d2 displays a singlet at δ = 2.47 ppm for the methyl group,37 downfield-shifted compared to free BiMe3 (δ = 1.11 ppm).37a The 29Si{1H} NMR spectrum exhibits a singlet at δ = 62.0 ppm, significantly upfield-shifted compared to 1a (δ = 68.7 ppm) and 1b (δ = 66.9 ppm) but downfield-shifted relative to ([3K(thf)2]2) (δ = 51.8 ppm). The 19F NMR spectrum shows a singlet at δ = −78.7 ppm. Notably, treatment of 4 with PMe3 in DCM at room temperature quantitatively regenerated 1b immediately along with the formation of PMe4OTf.
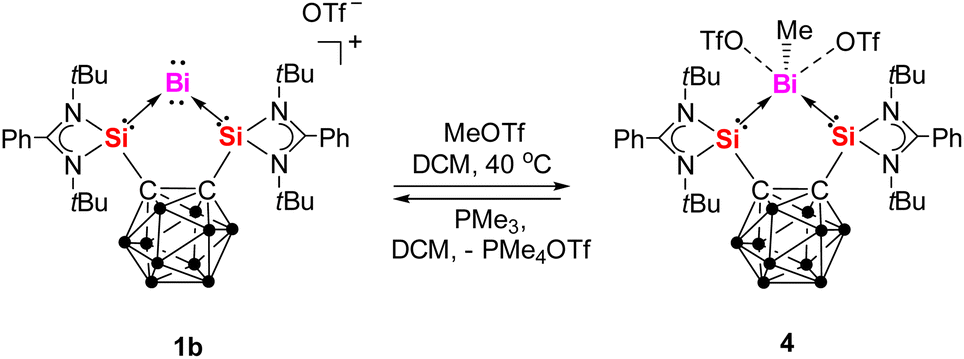 | ||
| Scheme 3 Reaction of 1b with MeOTf and the reverse reaction with PMe3. | ||
An scXRD analysis revealed that 4 crystallizes in the monoclinic space group P21/n. The dication in 4 features a five-membered C2Si2Bi ring, with a methyl group and two triflate anions coordinated to the bismuth atom (Fig. 6). The bismuth center thus adopts a distorted tetragonal pyramidal geometry (τ5 = 0.25),37b with the methyl group occupying the apical position, suggesting the presence of a lone electron pair opposite the tetragonal plane. The Si–Bi bond lengths in 4 (2.7003(17) and 2.7160(19) Å) are significantly longer than those observed in complexes 1–3 (1: 2.5774(6)–2.5958(9) Å; 2: 2.576(2) Å; 3: 2.6266(13) and 2.6138(13) Å). The Bi1–C3 bond length of 2.278(9) Å in 4 is comparable to that in the [BiMe]2+ complexes derived from II (2.300 Å)25 and III (2.247(7) Å).26 Notably, the shortest Bi⋯O interaction between the bismuth center and the two triflate anions in 4 is 3.025 Å, indicating weak coordination between the bismuth center and the triflate anions.
 | ||
| Fig. 6 Molecular structure of 4. Thermal ellipsoids are drawn at the 50% probability level. H atoms and solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (deg.): Bi1–Si2 2.7003(17), Bi1–Si1 2.7160(19), Bi1–C3 2.278(9), C2–C1 1.717(9), Si1–C1 1.929(7), Si2–C2 1.917(6), Si1–Bi1–Si2 80.44(5), C1–Si1–Bi1 112.1(2), C2–Si2–Bi1 112.24(19), C3–Bi1–Si1 96.5(2), C3–Bi1–Si2 94.7(3). | ||
Computational analysis
To elucidate the electronic structures of these monovalent bismuth complexes, Density Functional Theory (DFT) calculations were performed using the CAM-B3LYP/def2-TZVPD level in Gaussian 16 (see ESI† for details).38Table 1 compiles the HOMO–LUMO gaps of these species, confirming the predominantly single-determinant character of their wavefunctions and thus supporting the use of Kohn–Sham DFT as an appropriate method. Moreover, to discard large electron delocalization errors, we have compared CAM-B3LYP and B3LYP values, the latter of which are included in the ESI for comparison (see ESI, Table S11 and Fig. S34†). The experimental scXRD data were used for the analysis, although several optimized structures—which show good agreement with the experimental ones—are also included in the ESI for comparison (see Fig. S35 and S36†). Table 1 includes the ionization potential of these species, which serves as an indication of their stability with respect to the loss of one electron, and the first hyperpolarizabilities, β. The first hyperpolarizability increases from 1a to 2, and 2 to 3, reaching relatively large values which are compatible with delocalized charge distributions. In order to assess the changes in the electron distribution upon reduction, we have also employed the quantum theory of atoms in molecules (QTAIM)39 partition to compute atomic charges and delocalization indices (DI), which are covalent bond orders.40,41 We also run various tests to discard the electride character of 2 and 3.42,43 We carried out a complete topological analysis of the electron density for 1a, 2, and 3, identified the critical points according to QTAIM, and examined the regions with negative values of the Laplacian of the electron density. No features indicative of an isolated electron—such as a non-nuclear attractor or regions of localized electron density revealed by Laplacian analysis, which are characteristic of electrides—were identified (see Section D4 in the ESI†). Conversely, the first hyperpolarizabilities of these compounds (see Table 1) are consistent with the presence of labile and loosely bound electrons, albeit not isolated or localized as expected in electrides.The skeleton of 1a, 2, and 3 is divided into four parts: the Bi center, the two amidinato silylene units, and the carborane cage. The amidinato silylene units barely change upon reduction from 1a to 2, and 2 to 3, and their data is only included in the ESI† for the sake of completion. Fig. 7 and Table 1 contain data concerning the electron distribution of the relevant parts of structures 1a, 2, and 3.
 | ||
| Fig. 7 Evolution of selected QTAIM atomic charges (in black) and delocalization indices (shaded and blue) of 1a, 2 and 3 structures upon reduction. | ||
We focus on the Bi center, the two carbon atoms in the carborane cage, and the rest of the cage (BH cage, hereafter). 1a exhibits a regular 2c–2e bond (DI = 1.01) between Bi and Si (which is essentially maintained after reduction to 2 and 3), while the bonds between Si and the C atoms in the carborane cage are partially covalent (DI = 0.49) with a high ionic component (Si holds a +2.06 charge, whereas C has a large negative charge of −1.75e). 1a also exhibits a C–C covalent bond (DI = 0.97) within the carborane cage.
Upon reduction of 1a to 2, the most significant change is the reduction of the C–C bonding interaction (DI = 0.38), which further decreases upon reduction to 3 (DI = 0.22). This C–C weakening is caused by the opening of the cage at the top, as reflected in the increase of the C–C bond distance from 1.69 Å in 1a to 2.27 Å in 2, and subsequently to 2.58 Å in 3. The cage opening is accompanied by a slight increase of the covalent bond orders between C and the neighboring B atoms (see ESI, Fig. S31†), and a slight increase of the Si–C bond strength, which is reflected by the higher covalent character (DI = 0.56 in 2, DI = 0.63 in 3). This electron reorganization is also reflected in the picture of the Laplacian of the electron density given in Fig. S32 and S33,† and the number of electrons localized in the C atoms (see ESI, Fig. S31†).
Hence, upon reduction from 1a to 2, the extra electron mostly localizes in the carborane cage, especially in the carbon atoms, as the BH cage actually loses some electron density upon reduction. The Bi atomic charge also increases by 0.4e. Based on the calculated partial charge distribution, resonance structure 2′ (Scheme 2) is proposed for compound 2, in which the negative charges are delocalized over the bismuth atom and the carborane cage, while the two positive charges are distributed over the two silicon atoms. Upon reduction from 2 to 3, 0.26 additional electrons localize in each C atom, whereas the other half electron is split between the Bi, which has now −0.45 electrons, and the BH cage, which restores the +2 charge it had in 1a.
The analysis of the molecular orbitals aligns well with the conclusions drawn thus far (see Fig. 8). The SOMO of the BiI compound 2 shows a large fraction of the electron density of the unpaired electron on the C atoms of the carborane (Fig. 8). This stabilized σ* orbital is reminiscent of the NI radical {[SiII(closo-CB)SiII]NI} supported by the same bis(silylenyl) carborane ligand35 and the diphenyl-o-carborane system reported by Adillon et al.44
 | ||
| Fig. 8 Molecular orbital diagrams for selected orbitals of complexes 1a (*without considering the I− counterion), 2, and 3. The front tert-butyl groups of the silylenyl units are removed for clarity. Molecular orbitals isovalue of 0.050 e Å–3. | ||
Conclusion
In summary, we have successfully synthesized and characterized a series of bis(silylene)-stabilized monovalent single atom bismuth complexes supported by a chelating bis(silylenyl)-o-carborane ligand. The redox-active nature of the carborane scaffold plays a crucial role in modulating the electronic structures of these BiI complexes. The one-electron reduction of the cationic complex 1b with KC8 afforded the neutral radical complex 2, while its two-electron reduction with KC10H8 led to the formation of the anionic dimeric complex 3. These transformations highlight the remarkable redox flexibility of the bis(silylenyl)carborane framework in stabilizing monovalent bismuth. Furthermore, the reversibility of the redox processes, as demonstrated by the quantitative regeneration of 1b upon oxidation of 2 with AgOTf, underscores the dynamic redox behavior of these complexes. X-ray crystallographic analyses and spectroscopic data reveal significant structural variations among 1–3, particularly in the Bi–Si bonding and carborane core geometry. DFT calculations revealed that upon reduction from 1a to 2, the added electron predominantly localizes within the carborane cage, with a marked preference for the carbon atoms. This localization weakens the central C–C bond. A similar trend is observed in the subsequent reduction from 2 to 3, where the additional electron again accumulates in the carborane framework. Further analysis rules out the possibility that these species exhibit the characteristics of molecular electrides. Overall, this work expands the understanding of bismuth redox chemistry and the role of cooperative carborane-based silylene ligands in stabilizing low-valent main-group species. The unique electronic properties of these robust BiI complexes are expected to pave the way to new types of Bi-based redox catalysts.Data availability
All experimental and computational data associated with this work are available in the ESI.†Author contributions
Jian Xu carried out the synthetic experiments and analyzed the experimental data. Shenglai Yao assisted in the XRD refinement of the compounds and edited the manuscript. Christian Lorent collected the EPR data. Verònica Postils performed the DFT calculations. Verònica Postils and Eduard Matito analyzed the electronic structure of the compounds. Matthias Driess supervised the work and edited the manuscript. The manuscript was written through the contribution of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by DFG (German Research Foundation) under Germanýs Excellence Strategy – EXC 2008-390540038 – UniSysCat and DR-226/25-1. We particularly thank Paula Nixdorf for the assistance in the XRD measurements and Christian Teutloff from the FU Berlin for helpful discussion regarding the EPR data. Grants PID2022-140666NB-C21 funded by MCIN/AEI/10.13039/501100011033 and “FEDER Una manera de hacer Europa”, and the grants funded by the Gobierno Vasco (IT1584-22, and PIBA_2023_1_0055) are acknowledged. V.P. also thanks WBI (Wallonie-Bruxelles International) for her IN.WBI Excellence Grant. Calculations were performed on the computing facilities at the DIPC.Notes and references
- R. Mohan, Nat. Chem., 2010, 2, 336 CrossRef CAS PubMed.
- P. De Marcillac, N. Coron, G. Dambier, J. Leblanc and J. P. Moalic, Nature, 2003, 422, 876–878 CrossRef CAS PubMed.
- N. Yang and H. Sun, Coord. Chem. Rev., 2007, 251, 2354–2366 CrossRef CAS.
- C. Stewart, K. Konstantinov, S. McKinnon, S. Guatelli, M. Lerch, A. Rosenfeld, M. Tehei and S. Corde, Phys. Med., 2016, 32, 1444–1452 CrossRef.
- X. Huang, S. Huang, P. Biswas and R. Mishra, J. Phys. Chem. C, 2016, 120, 28924–28932 CrossRef CAS.
- P. Sprenger, W. Kleist and J. D. Grunwaldt, ACS Catal., 2017, 7, 5628–5642 CrossRef CAS.
- V. K. Srivastava, M. Maiti, G. C. Basak and R. V. Jasra, J. Chem. Sci., 2014, 126, 415–427 CrossRef CAS.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- M. Mato, F. Wang and J. Cornella, Adv. Synth. Catal., 2024, 366, 740–744 CrossRef CAS.
- C. Lichtenberg, Chem. Commun., 2021, 57, 4483–4495 RSC.
- M. Mato, D. Spinnato, M. Leutzsch, H. W. Moon, E. J. Reijerse and J. Cornella, Nat. Chem., 2023, 15, 1138–1145 CrossRef CAS PubMed.
- T. Tsuruta, D. Spinnato, H. W. Moon, M. Leutzsch and J. Cornella, J. Am. Chem. Soc., 2023, 145, 25538–25544 CrossRef CAS PubMed.
- S. Ni, D. Spinnato and J. Cornella, J. Am. Chem. Soc., 2024, 146, 22140–22144 CrossRef CAS PubMed.
- P. Zhang, F. Benner, N. F. Chilton and S. Demir, Chem, 2022, 8, 717–730 CAS.
- E. R. Pugliese, F. Benner and S. Demir, Chem.–Eur. J., 2023, 29, e202302687 CrossRef CAS PubMed.
- P. Zhang, R. Nabi, J. K. Staab, N. F. Chilton and S. Demir, J. Am. Chem. Soc., 2023, 145, 9152–9163 CrossRef CAS PubMed.
- G. Frenking, M. Hermann, D. M. Andrada and N. Holzmann, Chem. Soc. Rev., 2016, 45, 1129–1144 RSC.
- S. Yao, Y. Xiong and M. Driess, Acc. Chem. Res., 2017, 50, 2026–2037 CrossRef CAS PubMed.
- P. K. Majhi and T. Sasamori, Chem.–Eur. J., 2018, 24, 9441–9455 CrossRef CAS PubMed.
- S. Yao, Y. Xiong, A. Saddington and M. Driess, Chem. Commun., 2021, 57, 10139–10153 RSC.
- S. Yao, A. Saddington, Y. Xiong and M. Driess, Acc. Chem. Res., 2023, 56, 475–488 CrossRef CAS PubMed.
- W. Petz and G. Frenking, Adv. Inorg. Chem., 2022, 79, 247–299 CrossRef.
- R. Deb, P. Balakrishna and M. Majumdar, Chem.–Asian J., 2022, 17, e202101133 CrossRef CAS PubMed.
- M. M. Siddiqui, S. K. Sarkar, M. Nazish, M. Morganti, C. Köhler, J. Cai, L. Zhao, R. Herbst-Irmer, D. Stalke, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2021, 143, 1301–1306 CrossRef CAS PubMed.
- X. Wang, B. Lei, Z. Zhang, M. Chen, H. Rong, H. Song, L. Zhao and Z. Mo, Nat. Commun., 2023, 14, 2968 CrossRef CAS PubMed.
- J. Xu, S. Pan, S. Yao, C. Lorent, C. Teutloff, Z. Zhang, J. Fan, A. Molino, K. B. Krause, J. Schmidt, R. Bittl, C. Limberg, L. Zhao, G. Frenking and M. Driess, J. Am. Chem. Soc., 2024, 146, 6025–6036 CrossRef CAS PubMed.
- A. Saddington, S. Yao and M. Driess, Adv. Inorg. Chem., 2023, 82, 119–156 CrossRef CAS.
- Y. P. Zhou, M. Karni, S. Yao, Y. Apeloig and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 15096–15099 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, A. Kaushansky, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 12916–12927 CrossRef CAS PubMed.
- Y. Wang, M. Karni, S. Yao, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 1655–1664 CrossRef CAS PubMed.
- J. Xu, C. Dai, S. Yao, J. Zhu and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202114073 CrossRef CAS PubMed.
- J. Xu, S. Pan, S. Yao, G. Frenking and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202209442 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 14864–14868 CrossRef CAS PubMed.
- S. Yao, T. Szilvási, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess, Angew. Chem., Int. Ed., 2020, 59, 22043–22047 CrossRef CAS PubMed.
- Y. P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef CAS PubMed.
- (a) J. Ramler, F. Fantuzzi, F. Geist, A. Hanft, H. Braunschweig, B. Engels and C. Lichtenberg, Angew. Chem., Int. Ed., 2021, 60, 24388–24394 CrossRef CAS; (b) A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev C01, 2016 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1999, 103, 304–314 CrossRef CAS.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- V. Postils, M. Garcia-Borràs, M. Solà, J. M. Luis and E. Matito, Chem. Commun., 2015, 51, 4865–4868 RSC.
- S. P. Sitkiewicz, E. Ramos-Cordoba, J. M. Luis and E. Matito, J. Phys. Chem. A, 2021, 125, 4819–4826 CrossRef CAS PubMed.
- E. H. Adillon and J. C. Peters, J. Am. Chem. Soc., 2024, 146, 30204–30211 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2428091–2428095. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02644j |
| This journal is © The Royal Society of Chemistry 2025 |