
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA path to perpetual chemical synthesis via photocatalytic cofactor regeneration
Vanshika
Jain
 * and
Pramod P.
Pillai
*
* and
Pramod P.
Pillai
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER), Dr Homi Bhabha Road, Pune, 411 008, India. E-mail: jain.vanshika@students.iiserpune.ac.in; pramod.pillai@iiserpune.ac.in
First published on 23rd May 2025
Abstract
Harnessing the power of the Sun for perpetual chemical synthesis is one of the most sustainable ways to reduce the carbon footprint in the chemical industry. In this regard, the natural photosynthetic machinery offers key insights into the sustainable production of chemical entities in a ceaseless manner. The natural process of photosynthesis couples light harvesting to produce cofactor molecules, which then participate in enzyme-driven dark cycles for continuous biocatalytic transformations. At the core of photosynthetic machinery is the constant regeneration and consumption of cofactors, which sustain the metabolic cycles continuously. Consequently, coupling the unique powers of photocatalysis and biocatalysis through cofactor shuttling emerges as an excellent opportunity for the ceaseless production of fine chemicals. The present Perspective highlights the design principles for integrating photocatalytically regenerated cofactors with natural enzymatic cycles for various chemical transformations. Further, we examine the existing limitations of the integrated system and highlight the efforts to alleviate them. Finally, we highlight the possibilities of incorporating ideas from different research fields, from material science to synthetic biology to organometallic chemistry, to develop robust cofactor-dependent photobiocatalytic systems for the perpetual synthesis of chemicals.
 Vanshika Jain | Vanshika Jain is a PhD student in Prof. Pramod P. Pillai's group at the Indian Institute of Science Education and Research (IISER) Pune. Her research interests include exploring light–matter interaction at the nanoscale, colloidal synthesis, surface chemistry, photocatalysis, and photobiocatalysis. Prior joining IISER Pune, she obtained her Masters in Chemistry from Indian Institute of Technology Chennai, India. |
 Pramod P. Pillai | Pramod P. Pillai is a Professor in the Department of Chemistry at the Indian Institute of Science Education and Research (IISER), Pune, India. His group's research is focused on controlling the interplay of forces to improve and impart newer properties at the nanoscale. Some of the properties of interest include light harvesting, catalysis, and self-assembly. He obtained his PhD in Chemistry from the National Institute for Interdisciplinary Science and Technology (NIIST), Trivandrum, India. Prior to joining IISER Pune, he was a postdoctoral fellow at Northwestern University, Evanston, IL, USA, and an Alexander von Humboldt postdoctoral fellow at Technische Universität, Dortmund, Germany. |
Introduction
Attaining sustainability in chemical synthesis and processing is a significant step towards achieving the UN Sustainable Development Goals of Responsible Consumption and Production and Climate Action. Utilizing surplus energy supplied by the Sun is considered the most sustainable and affordable solution to reach these goals. Consequently, the development and implementation of various solar harvesting technologies are of great interest to the scientific community.1–3 The origin of the attractive design principles for modern research in solar energy harvesting can be linked to the process of natural photosynthesis.1–5 In general, the photosynthetic process can be decoupled into two broad parts: the light cycle and the dark cycle (Fig. 1a).6,7 The light cycle is associated with the catalytic oxidation of H2O to produce O2 while simultaneously storing protons in the form of reduced nicotinamide cofactor, NADPH, through a tandem action of multiple components.6–9 The reduced nicotinamide cofactor then enters the Calvin cycle (dark cycle) for the perpetual fixation of CO2 into glucose.10,11 This transformation of photonic energy into chemical bonds is one of the pillars of modern solar energy research aiming at sustainable chemical production in a ceaseless and continuous manner. Thorough research over the years reveals the guarded secret behind the perpetual synthesis of biomolecules in natural photosynthesis, which is the uninterrupted regeneration and consumption of NAD(P)+ and NAD(P)H cofactors in the light and dark cycles, respectively (Fig. 1a).11–13 Mimicking this photosynthetic process with artificial light-harvesting components is highly desirable for perpetual chemical synthesis in a sustainable way. As it is clear, devising artificial photosynthetic systems will require ceaseless production of redox cofactors such as NAD(P)+/NAD(P)H, which will act as a shuttle between different catalytic components. Additionally, NAD(P)+/NAD(P)H redox cofactor is ubiquitous in metabolic pathways and is utilized by ∼80% of oxidoreductase enzymes for a wide range of biocatalytic reactions.14–19 This gives us the opportunity to combine artificial light-harvesting centers with natural catalytic machinery, i.e., enzymes, to explore and innovate new pathways for solar to chemical fuel production via photobiocatalysis. | ||
| Fig. 1 Mimicking natural photosynthesis with artificial light-harvesting components for the perpetual synthesis of chemicals. Schematic representation of light and dark cycles in (a) natural photosynthesis and (b) semi-artificial photosynthesis using a photocatalyst and enzyme. In natural photosynthesis, NADPH is regenerated in the light cycle, which then participates in the dark cycle (Calvin cycle) for the ceaseless production of sugars from CO2. In semi-artificial photosynthesis, a photocatalyst is employed to regenerate NAD(P)H, which is consumed by oxidoreductase enzymes in the dark cycle. A continuous regeneration and consumption of NAD(P)H in light and dark cycles, respectively, will allow the perpetual synthesis of fine chemicals in semi-artificial systems. | ||
The first step towards developing the photobiocatalytic system is to design an appropriate photocatalyst for cofactor regeneration (Fig. 1b). Extensive research over the years led to a library of photocatalysts for nicotinamide cofactor regeneration, starting from molecular systems (organic dyes and inorganic complexes), semiconductor oxides, quantum dots, plasmonic nanoparticles, 2-D materials, carbon nanostructures, and so on.15,20–34 In this Perspective, our aim is not to discuss each class of photocatalyst for cofactor regeneration, as a large number of reviews in this direction already exist.15,19,35–38 Rather, the current Perspective will focus on providing a wholesome picture of the semi-artificial mimics in perpetual chemical synthesis, the associated challenges, and possible ways to overcome these challenges. First, we will discuss proof-of-concept photobiocatalytic systems, where continuous and ceaseless production of chemicals can be achieved by combining light and dark cycles. Subsequently, we will discuss the challenges associated with both light and dark cycles in state-of-the-art photobiocatalytic systems. Finally, we will expand our focus to provide possible solutions to address the existing challenges.
Regeneration of nicotinamide adenine dinucleotide cofactors
Redox cofactors are ubiquitous in biological systems and are pivotal in diverse biological processes driven by oxidoreductase enzymes.10,14 A majority of oxidoreductase enzymes are dependent on nicotinamide adenine dinucleotide (NAD+/NADH) or its phosphorylated form (NADP+/NADPH).14,15 The redox ability of NAD+/NADH or NADP+/NADPH couple stems from the ability of the nicotinamide ring to accept/donate two electrons and a proton (a hydride ion equivalent) (Fig. 2a).39 This transfer or addition of hydride ion equivalent is highly regioselective and occurs exclusively at the C-4 position of the nicotinamide ring in the biological systems (Fig. 2a). A redox potential of −0.32 V vs. NHE makes these NADH or NADPH cofactors moderately strong reducing agents. Consequently, NADH or NADPH is involved in numerous enzymatic reactions such as reduction of carbonyl groups, carboxylic acids, unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, C–N bonds, nitro groups, peroxide, etc.10,40 Once the substrates are reduced by respective enzymes, NADH or NADPH oxidizes to NAD+ or NADP+, respectively, which gets regenerated in the subsequent light and biocatalytic cycles in a stoichiometric manner.
C bonds, C–N bonds, nitro groups, peroxide, etc.10,40 Once the substrates are reduced by respective enzymes, NADH or NADPH oxidizes to NAD+ or NADP+, respectively, which gets regenerated in the subsequent light and biocatalytic cycles in a stoichiometric manner.
 | ||
| Fig. 2 Molecular structures of (a) nicotinamide adenine dinucleotide cofactors and (b) products of artificial regeneration of NAD(P)+. | ||
Artificially, these cofactors can be regenerated in numerous ways, which include chemical, homogeneous/heterogeneous catalytic, electrocatalytic, photocatalytic, or photoelectrocatalytic methods.15,20,41–53 Chemical regeneration of reduced cofactors is a non-catalytic pathway that involves the participation of reducing agents such as Na2S2O4, NaBH4, and dihydropyridine to reduce NAD(P)+.20,41–43 However, the high redox potential of these chemical agents often causes enzyme deactivation.44 Along with this, the large feedstock and high waste generation limit the use of this method for enzyme-coupled photocatalytic reactions.20,41–43 Electrochemical methods employ electrical energy as the redox currency to regenerate nicotinamide cofactors and have been reported to occur via three different strategies: direct electron transfer, indirect electron transfer, and indirect enzyme-coupled catalytic reduction.20,46,47 The direct regeneration methods lead to the reduction of NAD(P)+ directly on the electrode surface in two steps, first by forming NAD(P)˙ radical, followed by a second electron transfer step to anion and ultimately abstracting the proton to yield NAD(P)H.46,47 The indirect cofactor regeneration involves the use of additional electron mediators, which act as a shuttle between the electrode and NAD(P)+ to transfer two electrons in a single step. Viologen derivatives, neutral red, Co(III) complexes, Rh(III) complexes, and 5,5′-dithiobis(2-nitrobenzoic acid) are some of the commonly used electron mediators in the electrochemical regeneration of nicotinamide cofactors.46 Another strategy is to couple an electrochemical redox system comprising an appropriate electrode and an electron mediator with enzymes to regenerate cofactor molecules.47–49 From the library of enzymes, lipoamide dehydrogenase, diaphorase, and ferredoxin-NADP-reductase have been extensively used in enzyme-coupled electrochemical systems.47–49 Another commonly used strategy to regenerate NAD(P)H cofactors is by catalytic hydrogenation over supported metal catalysts such as Pt, Pd, Rh, Ru, and Ni in the presence of H2.20,52,53 In the context of mimicking the natural photosynthetic process of cofactor regeneration, the utilization of solar energy for chemical bond formation in the presence of appropriate photocatalysts offers one of the most sustainable ways to regenerate nicotinamide cofactors in artificial systems. A library of photocatalysts exists in literature, starting from molecular photoredox catalysts, bulk semiconductors, plasmonic metal nanoparticles, quantum dots, 2-D materials, etc., for photocatalytic NAD(P)H regeneration.20–38 And in this Perspective, we will restrict all the discussion to photocatalytic methods of cofactor regeneration, the key developments, and challenges associated.
Now, an important point to be noted here is the necessity of regioselective regeneration for 1,4-NAD(P)H in the light cycle, as it is the only enzymatically active isomer that can participate in the dark cycle for enzymatic biocatalytic reactions.19,20,24–26 However, there is a loss in selectivity for NAD(P)H regeneration when artificial methods are employed.20,54 While hydride (2e− and H+) transfer is the preferred path for cofactor regeneration in natural systems, artificial regeneration of NAD(P)H may proceed via a single electron and proton transfer pathway as well.55–57 The monoelectronic reduction of NAD(P)+ requires −0.94 V vs. NHE to form a highly reactive NAD(P)˙ radical, which can either abstract a H-atom to yield NAD(P)H or undergo a dimerization process to form NAD(P)2 dimers. Even though the reducing power of the monoelectronic reduction product is higher than the dielectronic reduction product (−0.94 V vs. −0.32 V vs. NHE for single electron reduction and two electron reduction, respectively),57 the rapid deactivation in aqueous medium makes this path a less probable one. Irrespective of the reaction mechanism, which in fact is highly debatable, the artificial regeneration of cofactors proceeds with the formation of 1,2-NAD(P)H and 1,6-NAD(P)H, both of which are enzymatically inactive (Fig. 2b) apart from the desired enzymatically active 1,4-NAD(P)H.20,54 This is one of the major challenges associated with the artificial regeneration of cofactors, which arises because of the non-specific interaction of the nicotinamide ring with the light-harvesting moiety.
In natural photosynthesis, a complex network of interactions (hydrogen-bonding, electrostatics, and hydrophobic) within the ferredoxin-NADP+ reductase enables the site-specific addition of hydride ion to NADP+ to form 1,4-NADPH.58 To achieve this selectivity in artificial systems, additional electron mediators or co-catalysts are usually employed in the light cycle. A versatile class of electron mediators includes Rh-based organometallic complexes such as [Cp*Rh(bpy)(H2O)]2+, [Cp*Rh(Phen)(H2O)]2+, [Cp*Rh(5,5′-CH2OH-bpy)(H2O)]2+, [Cp*Rh(5,5′-CH3-bpy)(H2O)]2+, [Cp*Rh(4,4′-OCH3-bpy)(H2O)]2+, etc.59–61 The Rh metal center serves as the active site for regioselective regeneration of NAD(P)H by the selective interaction of the amide group of NAD(P)+.59 Such a specific interaction enables hydride transfer at the C-4 position of the nicotinamide ring, yielding 1,4-NAD(P)H, and hence, Rh-based electron mediators act as ferredoxin-NAD(P)+ reductase enzyme mimics. As a result, Rh-based electron mediators dominate the area of artificial regeneration of nicotinamide cofactors.14,20–26
For photocatalytic regeneration of nicotinamide cofactors, it is equally important to extract the photoexcited holes from the photocatalyst for ceaseless chemical synthesis. Along with extracting holes from the photocatalyst, electron donors or hole scavengers also serve as buffering agents in maintaining the pH of the reaction mixture. It has been established that both oxidized and reduced forms of nicotinamide cofactors (NAD(P)+ and NAD(P)H) are susceptible to decomposition in extreme pH conditions.62,63 Specifically, reduced cofactors undergo hydration and anomerization under acidic conditions, whereas hydrolysis of nicotinamide-ribose bond takes place in oxidized cofactors under basic conditions.62,63 Moreover, the phosphorylated reduced cofactor (NAD(P)H) is more prone to decomposition than NADH. As a result, most of the photocatalytic cofactor regeneration reactions are optimized between pH 6 and 8 to minimize the decomposition of both cofactors.63,64 The pH buffering activity of triethanolamine (TEOA),65 along with its appropriate oxidation potential (−1.04 V vs. NHE) with respect to the majority of the photocatalysts, results in its wide use as the hole scavenger/electron donor in photocatalytic systems.15 Additionally, TEOA is also found to exhibit a rather interesting role in cofactor regeneration. Grzelczak and coworkers offered a new insight into the role of TEOA in cofactor regeneration wherein glycolaldehyde, an oxidation product of TEOA, reduces NAD+ in the light-independent cycle.66 Not only does TEOA serve as a chemical feedstock for NADH regeneration, but it also ensures a high pH to maintain the reducing power of glycolaldehyde. Besides TEOA, ascorbic acid and ethylenediaminetetraacetic acid (EDTA) have also been employed as hole scavengers and electron donors in photocatalytic cofactor regeneration.67–69 It must be mentioned that all the hole scavengers undergo irreversible oxidation that results in the accumulation of their oxidation products.70 In this context, water emerges as an ideal hole scavenger and electron donor, as no byproducts are accumulated. However, the sluggish kinetics of the water oxidation reaction due to high oxidation potential (1.23 V vs. SHE) limits the use of water in photocatalytic reactions.15
Continuous and ceaseless chemical production in semi-artificial photocatalytic systems
A key point that researchers often miss out on is that natural systems adapt and evolve not to maximize efficiency but rather to achieve ceaseless and sustained functions.71,72 While solar-to-chemical energy conversion using artificial light-harvesting systems or photocatalysts has achieved much higher efficiencies than plants,73 the operations are merely proof-of-concept and unpractical for sustained chemical production. In this section, we will discuss how coupling photocatalysts with natural catalytic factories in the form of enzymes enables the perpetual synthesis of fine chemicals via the constant regeneration and consumption of nicotinamide cofactors. The diversity of catalytic reactions that are possible is equivalent to the pool of NAD(P)+/NAD(P)H-dependent enzymes known in nature.14 Consequently, we will highlight distinctly different class of biocatalytic reactions carried out under semi-artificial photocatalytic systems.The initial era of research in the area of photobiocatalysis focused on the efficient regeneration of NAD(P)H cofactor, and enzymatic synthesis merely served as proof of the formation of biologically active currency – the 1,4-NAD(P)H.21–26 However, sustainable and continual synthesis by coupling light and dark cycles has been shown only recently.74–79 For instance, Liu and coworkers achieved sustained formation of L-lactate from pyruvate by coupling carbon nitride mesoporous spheres photocatalyst (light cycle) and L-lactate dehydrogenase enzyme (dark cycle) (Fig. 3a).74 Here, carbon nitride mesoporous spheres (CNMS) were chosen as the photocatalyst as they combine the excellent light-to-charge pair conversion of carbon nitride and the ability of structured mesoporous sphere architecture to enhance light intensity onto the active centers. Within 30 min of light illumination, CNMS results in 100% NADH yield, whereas bulk graphitic carbon nitride (g-C3N4) and mesoporous carbon nitride (mpg-C3N4) resulted in only 10% and 70% NADH yield under similar conditions, respectively (Fig. 3b). An excellent performance of CNMS was attributed to the improved light harvesting by inner reflection and field enhancement effect. The photoregenerated NADH was then coupled with L-lactate dehydrogenase to synthesize L-lactate from pyruvate (Fig. 3c). A stoichiometric conversion of pyruvate to L-lactate was achieved in 6 h under continuous light illumination, with an initial ratio of pyruvate to NAD+ being 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This essentially suggests the requirement of constant in situ regeneration of 1,4-NADH cofactor for the stoichiometric conversion of pyruvate (Fig. 3d). This work clearly shows a general pathway for synthesizing diverse chiral organic compounds using a cofactor-dependent photoenzymatic system.
:1. This essentially suggests the requirement of constant in situ regeneration of 1,4-NADH cofactor for the stoichiometric conversion of pyruvate (Fig. 3d). This work clearly shows a general pathway for synthesizing diverse chiral organic compounds using a cofactor-dependent photoenzymatic system.
 | ||
| Fig. 3 Coupling artificial light-harvesting components with biological enzymes for perpetual synthesis of value-added chemicals. (a) Schematics of continuous synthesis of L-lactate from pyruvate via NADH shuttling between photocatalytic and enzymatic cycles. The NADH regenerated by carbon nitride mesoporous spheres (CNMS) in the light cycle participates in the dark cycle driven by L-lactate dehydrogenase to convert pyruvate into L-lactate. (b) Plot showing the stoichiometric formation of L-lactate from pyruvate with an initial NAD+: pyruvate ratio of 1:5, enabled by continuous shuttling of NADH cofactors. Reprinted and adapted with permission from ref. 74. Copyright 2014 The Royal Society of Chemistry. | ||
In another example, our group coupled photocatalytic light and enzymatic dark cycles to produce butanol, a value-added fuel.77 Continuous production of butanol via the constant photoregeneration and consumption of NADH cofactor was powered by indium phosphide quantum dots (InP QDs) photocatalyst and alcohol dehydrogenase (ADH) enzyme (Fig. 4a).77 A strong electrostatic attraction between the oppositely charged InP QD photocatalyst and electron mediator ([Cp*Rh(bpy)(H2O)]2+) significantly enhanced the charge extraction and utilization processes under visible-light irradiation (Fig. 4a). This enabled the fast (∼30 min) and quantitative (>99%) photoregeneration of nicotinamide cofactors with a turn-over-frequency (TOF) of ∼1333 h−1. Such a fast photogeneration of cofactors is crucial for mimicking unidirectional flow of energy and electrons, as seen in natural Z-scheme. Once the NADH cofactor was regenerated, it was coupled with the ADH enzymatic dark cycle to produce butanol. Under sequential light (10 min) and dark (20 min) cycles, there was a continuous increase in the butanol formation (Fig. 4b). Interestingly, butanol was always formed in higher amounts in the light cycle than in the dark cycle, which can be attributed to the simultaneous consumption and regeneration of 1,4-NADH in the light cycle (Fig. 4b). Finally, under continuous light irradiation of the reaction mixture comprising InP QDs, ADH enzyme, NAD+, and butyraldehyde, the overall yield of butanol formation reached beyond the stochiometric limit of the initial NAD+ added (Fig. 4c). This clearly confirms the constant photoregeneration and consumption of the 1,4-NADH cofactors in multiple light and dark cycles, respectively, leading to the continuous biocatalytic synthesis of butanol, closely mimicking the simultaneous light and dark cycles in the natural thylakoid-stroma system.
 | ||
| Fig. 4 (a) Schematics showing the integration of a semi-artificial photosynthetic system based on InP QDs and ADH enzyme for butanol production. (b) Sequential light and dark cycles reveal the continuous production of butanol. The difference in slopes in the light and dark cycles corresponds to different rates of butanol formation in light and dark cycles, respectively. (c) Simultaneous light and dark cycles lead to butanol formation beyond the stoichiometric limit of initially added NAD+, confirming the in situ shuttling of NADH cofactor between multiple photocatalytic and enzymatic cycles. Adapted with permission from ref. 77. Copyright 2024 American Chemical Society. | ||
As cofactor shuttles between the light and dark cycles, the overall efficiency of the process is determined by a perfect balance between the kinetics of the two cycles. Often, there is a mismatch in the timescales of photocatalytic and enzymatic reactions.79 Therefore, the cofactor shuttling process must be optimized for the perpetual synthesis of chemical commodities. In this direction, Shi, Jiang, and coworkers developed an enzyme-photocoupled catalytic system (EPCS) based on a thylakoid membrane-inspired capsule (TMC) to maximize cofactor shuttling performance in light and dark cycles.80 A typical TMC comprised of protamine-induced titanium dioxide (p-TiO2) porous membrane, cadmium sulfide quantum dot photocatalyst (CdS QD), [Cp*Rh(bpy)H] (Rh*), and alcohol dehydrogenase (ADH) enzyme (Fig. 5a). Herein, CdS QDs and Rh* were electrostatically attached together and deposited on the inner surface of p-TiO2, and ADH was encapsulated in silica and immobilized on the outer p-TiO2 membrane. Such a system closely resembles natural photosynthetic machinery, as compartmentalization between photocatalytic and enzyme catalytic reactions can be achieved. The rate of propionaldehyde conversion was modulated by altering the distance between CdS QDs and ADH enzyme through control of the thickness of the p-TiO2 membrane (Fig. 5b). Interestingly, at a short distance of 73 nm, there was a negligible conversion, which was attributed to damaged capsular structure leading to loss of Rh*. However, with an increase in distance to 121 nm, the conversion rate reaches a maximum, and the reaction rate decreases with a further increase in the distance between the active sites (Fig. 5b). Moreover, the reaction rate was ∼4 times higher than the non-integrated system comprising of free ADH enzyme, confirming a rapid shuttling of cofactors between the photocatalytic and enzymatic reaction centers. An interesting parameter of shuttling number was introduced to represent the degree of the cofactor shuttling process by monitoring the conversion of propionaldehyde to propanol (Fig. 5c). Consequently, apparent shuttling number (ASN) and intrinsic shuttling number (ISN) were calculated to be 17.1 and 46.5, respectively, which were ∼8 times and ∼12 times higher than the non-integrated system with free ADH enzyme. A high value of ASN and ISN confirms the promoted cofactor shuttling in the integrated photobiocatalytic system.
 | ||
| Fig. 5 (a) Schematics of thylakoid membrane-inspired capsule (TMC) and cofactor shuttling module for propionaldehyde conversion. The spatial separation of photocatalytic and enzymatic centers was achieved through a porous titanium dioxide (p-TiO2) membrane. The photocatalytic unit comprising of CdS QD and Rh-mediator was immobilized on the inner membrane of p-TiO2, while silica-encapsulated ADH enzyme was immobilized on the outer membrane to carry out light-independent biocatalytic conversion of propionaldehyde. (b) Rate of propionaldehyde conversion in the integrated system as a function of the distance between photocatalytic and enzymatic reaction centers compared to the non-integrated system consisting of free ADH enzyme. (c) Apparent shuttling number (ASN) defining the degree of NAD+/NADH shuttling process as a function of distance between photocatalyst and enzyme active centers in the integrated system. A higher value of ASN for the integrated system confirms promoted cofactor shuttling in closely spaced catalytic centers. Adapted with permission from ref. 80. Copyright 2022 American Chemical Society. | ||
One step ahead in mimicking the natural photosynthetic route is wiring a series of enzymes in the light-independent cycle, as seen in the Calvin cycle.11 It is a perfect cascade and synergy between different enzymes that enable the continuous formation of complex macromolecules from simple building blocks such as CO2, N2, and H2O. Baeg and coworkers demonstrated the feasibility of coupling multiple NADH-dependent enzymes for multistep conversion of CO2 to methanol in the presence of graphene-coupled multi-anthraquinone-substituted porphyrin photocatalyst.81 As a proof-of-concept, formate dehydrogenase, formaldehyde dehydrogenase, and alcohol dehydrogenase were utilized to convert CO2 to formic acid, formaldehyde, and ultimately methanol in a sequential manner by respective enzymes. Later, Shi, Jiang, and coworkers improved the efficiency of these enzyme cascades for CO2 to methanol conversion by immobilizing the enzymes on protamine–titania microcapsules for enhanced cofactor shuttling.82 Both these examples show the compatibility of coupling different enzyme cascades wired through cofactor shuttling, which could possibly result in the production of valuable and synthetically challenging chemical commodities. However, sufficient attention has not been given to this direction, possibly because of the poor understanding of complex abiotic–biotic symbiosis and the challenges associated with the diffusion of substrates between different catalytic centers. Lessons from biological evolution will be useful for how an array of enzymes performs one-pot sequential reactions in a sustained way.
Challenges and perspectives
Engineering photocatalytic systems with electron mediators
It is quite evident that an additional electron mediator is a necessary component for the selective formation of 1,4-NAD(P)H and couple the light-independent enzymatic cycle with the light cycle. In most reports, an expensive Rh-based electron mediator is employed,14,20–26 and it is often freely diffusing in the reaction mixture (Fig. 6). As a result, precious Rh-M is lost in each cycle, impeding the practical applicability of cofactor regeneration.83,84 Alongside, the charge transfer from photocatalyst to Rh-M is influenced by the diffusion coefficient, which significantly affects the cofactor regeneration rate (see vide infra). Such an Rh-M diffusion-limited system poses an even more significant challenge of enzyme deactivation, ceasing the continuous formation of value-added products (Fig. 6).83,84 Freely diffusing Rh-M strongly interacts with enzymes by binding to cysteine, histidine, and tryptophan residues, leading to mutual inactivation.83,84 For instance, Shi, Jiang, and coworkers observed an enhanced aggregation between Rh-M and ADH enzymes within 2–4 min of light illumination.85 Under light illumination, the aqua ligand detaches due to a change in the electron density at the Rh center. Thus, the created vacant site is then available for enzyme residues to bind to the Rh center, leading to the inhibition of enzyme activity. Because of all these reasons, it becomes crucial to confine Rh-M spatially for enhanced cofactor shuttling and retention of enzyme activity. | ||
| Fig. 6 Comparison of photocatalytic systems integrated without and with Rh-M for cofactor regeneration. | ||
In this direction, Li, Farha, and coworkers integrated Cp*Rh(bpydc)Cl (Rh-M) into hierarchical mesoporous MOF, NU-1006 using solvent-assisted ligand incorporation.86 Here, NU-1006 is the photosensitizer comprised of water-stable zirconium-based MOF with carboxylate functionalized pyrene-based linkers. The OH/OH2 ligands in NU-1006 were replaced by carboxylate functionalized Rh-M, resulting in the immobilization of catalytic Rh sites with the photosensitizer (Fig. 7a). The mesoporous nature of MOF was used to encapsulate the formate dehydrogenase (FDH) enzyme for NADH-mediated CO2 reduction to formic acid. Combining Rh-M with the light-harvesting unit enhanced the charge transfer rate, as observed by the three times higher NADH regeneration rate than the physical mixture of MOF and Rh-M (Fig. 7b). Additionally, the encapsulation of FDH within the NU-1006 framework and proximity with the catalytic activity sites resulted in higher CO2 to formic acid conversion by surpassing the diffusion limitation of cofactor shuttling between the light and dark cycles.
 | ||
| Fig. 7 Integration of Rh-M with various light-harvesting components. (a) Schematic showing the photo-enzymatic conversion of CO2 to formic acid using Rh-M integrated and FDH encapsulated NU-1006 MOF. (b) NADH regeneration by Rh-M integrated NU-1006 MOF compared with the physical mixture of NU-1006 and freely diffusing Rh-M shows an enhanced charge transfer rate. Adapted with permission from ref. 86. Copyright 2020 American Chemical Society. (c) Schematic representation of core–shell MOF for integration of Rh-M with the light-harvesting component. (d) Plot showing the 1,4-NADH regeneration activity for multiple cycles, which confirms negligible loss of Rh-M from the surface of core–shell MOF. Adapted with permission from ref. 78. Copyright 2020 American Chemical Society. (e) Schematics of the photo-enzymatic system for methanol production with Rh-M embedded in a porous TiO2 over graphitic carbon nitride (GCN) to spatially separate it from the ADH enzyme. Such a strategy prevented aggregation-induced deactivation of the ADH enzyme. Adapted with permission from ref. 85. Copyright 2021 American Chemical Society. (f) Schematics showing the photoregeneration of 1,4-NADH with Rh-M modified CdS nanorods. The carboxylate groups of bipyridine moiety in Rh-M coordinate with surface Cd2+ and displace parent thiolate ligands. Adapted with permission from ref. 88. Copyright 2024 American Chemical Society. | ||
Along with enhancing the rate of cofactor shuttling, Shi, Jiang, and coworkers observed that attaching Rh-M with the photocatalyst helps preserve the cycling stability of Rh-M for multiple cycles.78 An interesting design of core–shell MOF to integrate photosensitizer and catalytic reaction centers, mimicking natural photosystem I (PSI), was reported here. Here, UiO-66-NH2 served as the photosensitizer core while catalytic Rh-M was integrated over UiO-67-bpydc shell (Fig. 7c). Upon light illumination, electrons are generated by the organic linker of the core (NH2-BDC) and are transferred to the coordinated Zr6O8 clusters. These Zr6O8 clusters act as mediators for the transfer of electrons from the photosensitizer to the Rh catalytic centers for the regeneration of NADH cofactor. The NADH regeneration rate of the integrated system was 2.08 times that of the Rh-M in homogeneous conditions, along with the retention of Rh-M catalytic activity up to three cycles (Fig. 7d). Interestingly, a synergy of electron transfer and electron utilization was achieved by optimizing the shell thickness and Rh-M loading on the core–shell MOF. Ultimately, efficient cofactor shuttling activity was reported for the continuous synthesis of amino acids using amino acid dehydrogenases.
More importantly, confining Rh-M can help alleviate issues related to enzyme deactivation. In the photobiocatalytic systems, enzymes are known to undergo irreversible inactivation via covalent or non-covalent pathways. For instance, the interaction of ADH enzyme with organometallic complex such as Rh-M results in aggregation of ADH by binding to cysteine, histidine, and tryptophan residues, deactivating the enzyme partially or even entirely. Shi, Jiang, and coworkers observed enhanced aggregation of ADH in the presence of Rh-M under light illumination.85 This challenge was overcome by embedding Rh-M in a TiO2 coating over the graphitic carbon nitride (GCN) photocatalyst (Fig. 7e).85 The size-sieving TiO2 coating also inhibited the direct interaction of ADH with GCN to protect ADH from oxidation by photogenerated holes. The Rh-M embedded photo-enzymatic system continuously produced methanol for at least three light–dark cycles of 60 min each. In contrast, the system comprising ADH, GCN, and free Rh-M was deactivated after the first light–dark cycle. Moreover, embedding Rh-M in proximity to GCN enhanced the continuous regeneration and consumption of NADH to produce methanol by at least three times.
At a glance, it may seem simple to attach Rh-M to the photocatalyst surface. However, it is not always trivial, considering the ever-expanding library of photocatalysts. For instance, it is synthetically challenging to design molecular photocatalysts integrated with Rh-M or achieve colloidally stable nanoparticles (NPs)/nanocrystals (NCs) with Rh-M tethered onto the surface.87 One way to attach Rh-M on the NP/NC surface is by synthesizing appropriate ligands bearing bipyridine headgroup, which can coordinate with the Rh center. However, along with synthesizing ligands, attaching the ligand of choice on the NP/NC surface is highly challenging as NP/NC can lose their colloidal stability and, consequently, their properties.87 Recently, Yang and coworkers successfully exchanged the parent thiolate ligands on CdS nanorods (NRs) with Rh-M containing carboxylic acid functionalized bipyridine unit (Fig. 7f).88 The two carboxylate groups of Rh-M coordinate with the surface Cd2+ ions either by attachment to unpassivated Cd2+ or by coordinate bonding to replace thiolate ligands (Fig. 7f). In-depth details about surface interactions were obtained through 2-D DOSY experiments, which revealed a 34% reduction in diffusion of Rh-M as compared to free Rh-M, clearly confirming the presence of Rh-M on the surface of CdS NRs. Interestingly, authors observed a reduction in NADH formation efficiency with the Rh-M modified CdS NRs compared to freely diffusing Rh-M. This was attributed to the reduced basicity of Rh-M due to the presence of electron-withdrawing carboxylic acid groups. This study clearly suggests the need for a rational design of surface ligands for NP/NC photocatalysts to achieve maximum cofactor regeneration activity and simultaneously overcome the issues related to freely diffusing Rh-M in the reaction mixture.
In an orthogonal direction, lessons must be taken from organometallic chemistry to develop a new class of electron mediators that enables selective regeneration of 1,4-NAD(P)H regioisomer. Over the past few decades, only the class of Rh-M with bipyridine ligands has been utilized in photocatalytic cofactor regeneration because of its excellent selectivity towards 1,4-NAD(P)H. Several other organometallic complexes have emerged in the last few years, albeit all belonging to the Ir family.89–92 For instance, Macchioni and coworkers reported half-sandwich Ir(III) catalysts [Cp*Ir(R-pysa)NO3] (pysa = k2-pyridine-2-sulfonamidate; R = H, 4-CF3, and 6-NH2) with excellent cofactor regeneration activity using phosphonic acid as a hydride source. With R = NH2, the TOF reached as high as 3731 h−1, approaching that of the formate dehydrogenase enzyme (Fig. 8a).89 The same group later reported another Ir-containing mediator, [Cp*Ir(R′-pica)Cl] (pica = R′-picolinamidate, R′ = H and Me), for electrochemical cofactor regeneration.90 Both the mediators exhibited intriguing selectivity, with 1,4-NADH and 1,6-NADH always in a 91:9 molar ratio (Fig. 8b). In-depth NMR studies revealed a rapid equilibration between the two isomers, thereby making it possible to reuse all the regenerated NADH when coupled with an NADH-dependent enzyme. So far, these Ir complexes have not been explored as regioselective catalysts or electron mediators for photocatalytic cofactor regeneration and hence open new opportunities in photobiocatalysis.
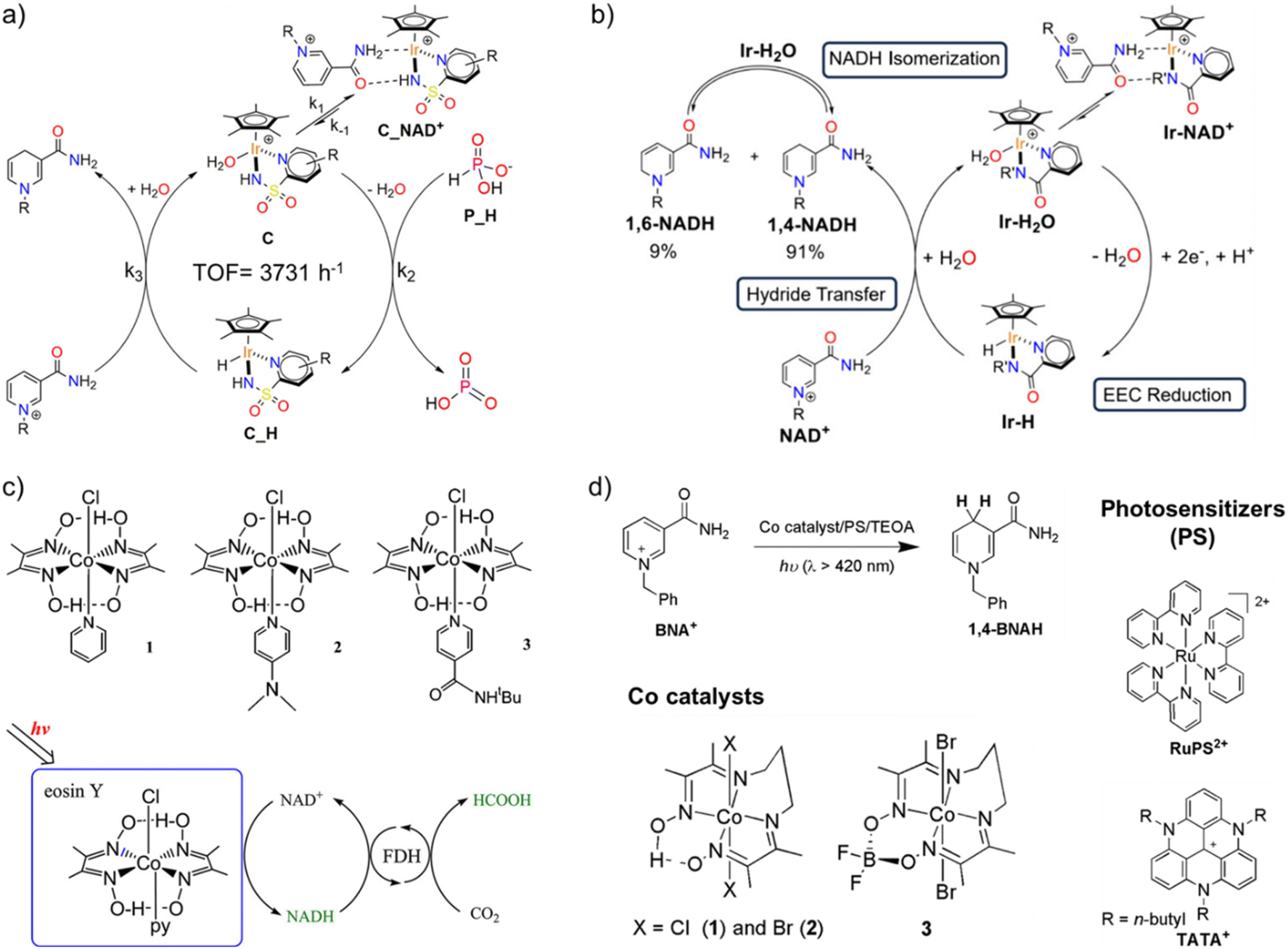 | ||
| Fig. 8 Various electron mediators for regioselective regeneration of 1,4-NADH. (a) Schematics showing 1,4-NADH regeneration using iridium-pyridine-2-sulfonamidate catalyst with phosphonic acid as the hydride source. Here, R = H, 4-CF3, or 6-NH2, out of which R = 6-NH2 yields the highest TOF of 3731 h−1 at 313 K. Adapted with permission from ref. 89. Copyright 2020 American Chemical Society. (b) Schematic showing the mechanism of NADH regeneration using [Cp*Ir(R′-pica)Cl] (pica = R′-picolinamidate, R′ = H and Me) electron mediator. A rapid equilibrium was established between 1,4-NADH and 1,6-NADH in 91:9 ratio. Adapted with permission from ref. 90. Copyright 2024 American Chemical Society. (c) Structures of earth-abundant cobaloximes complexes for 1,4-NADH regeneration in presence of eosin Y as the light-harvesting moiety. The regenerated 1,4-NADH was coupled to enzymatic dark cycle to reduce CO2 to formic acid. Adapted with permission from ref. 95. Copyright 2012 American Chemical Society. (d) Selective regeneration of 1,4-BNAH, a synthetic analogue of 1,4-NADH, in presence of cobalt diamine–dioxime complexes under visible light. Adapted with permission from ref. 97 Copyright 2020 The Royal Society of Chemistry. | ||
Despite the emergence of new electron mediators, the catalytic active center still remains the precious earth metals (Rh and Ir). However, biology's blueprint reveals the presence of earth-abundant metals, such as first-row (3d) transition metals and early second-row (4d) and third-row (5d) metals, at the catalytic centers of enzymes, which drive chemical transformations to sustain life.93,94 This leaves an obvious question: what restricts the use of earth-abundant metal sites in artificial systems, and what are the gaps in the fundamental understanding of how the natural system operates? Even though only a limited attempt has been made in this direction, all the results obtained are highly promising, which will allow us to move a step closer to understanding the natural biological processes as well. Inspired by the excellent H2 evolution capability of cobaloximes, Kim and coworkers employed three different cobaloximes complexes for hydride transfer to NAD+ to regenerate reduced cofactor molecules using eosin Y and TEOA as photosensitizer and sacrificial agent, respectively (Fig. 8c).95 The regeneration of reduced cofactor was only possible in the presence of cobalt catalysts, proving the crucial role of cobalt catalysts. The regenerated cofactor was then coupled with NADH-dependent formate dehydrogenase to reduce CO2 to formic acid, which confirmed that the cobalt catalysts were able to produce selective 1,4-NADH. A probable mechanism was proposed according to which photoexcited eosin Y reduces Co(III) centre to Co(I) via Co(II) intermediate. The Co(I) centre is then protonated to yield Co(III) hydride intermediate, which then transfers hydride to NAD+ at C-4 position to yield 1,4-NADH. Later, Nam, Fukuzumi, and coworkers employed one of these Co-catalysts to construct a full artificial photosynthetic system for regioselective NAD(P)H production with water as an electron and proton source.96 Herein, molecular models of PSI and PSII were combined to reduce NAD+ in presence of 9-mesityl-10-methylacridinium ion as the photocatalyst and cobaloximes (Co(III)(dmgH)2pyCl) as the catalyst. The regioselective formation of 1,4-NADH emanates from the formation of a kinetically favourable six-membered ring transition state that allows for preferential interaction of H-ligand with C-4 position of the nicotinamide ring, as seen in the case of Rh-based electron mediators. Along the same lines, Ko, Robert, and coworkers investigated different cobalt diimino-dioximes [Co(DO)(DOH)pnX2] (1 and 2; X = Cl and Br) and the BF2-bridged derivative [Co((DO)2BF2)pnBr2] catalysts for photocatalytic regeneration of NADH analogue i.e. 1-benzyl-1,4-dihydronicotinamide (1,4-BNAH) in presence of different photosensitisers such as [Ru(bpy)3]2+ (RuPS2+), 4,8,12-tri-n-butyl-4,8,12-triazatriangulenium hexafluorophosphate (TATA+), and xanthene-type dyes such as eosin Y and rose Bengal (Fig. 8d).97 The excellent regeneration of 1,4-BNAH in both aqueous and organic media calls for the use of earth-abundant Co-based catalysts with state-of-the-art photocatalysts for natural cofactor regeneration as well.
Another interesting aspect that is often overlooked is the significant contribution of dynamic conformational changes emanating from a complex network of non-covalent interactions in enzyme catalysis, which leads to the emergence of selectivity in biocatalytic reactions.10 With this thought, Yamauchi and coworkers utilized a Pt-modified TiO2 catalyst over a Ti mesh electrode for selective electrochemical regeneration of 1,4-NADH.98 Here, the specific interaction between the amide group of the nicotinamide ring and TiO2 surface and the excellent hydrogenation capability of Pt was the rationale for regioselective 1,4-NADH formation. In another example, Yang and coworkers explored strong metal–support interactions between Pt and TiO2 for selective 1,4-NADH regeneration.99 Detailed mechanistic insights revealed that NAD+ adsorbs over TiO2via carbonyl oxygen of the amide group of the nicotinamide fragment, and H2 adsorbed over Pt spills over the adsorbed NAD+ to form enol-NADH cation. Subsequent H transfer and keto–enol tautomerization result in the selective formation of 1,4-NADH. In another interesting approach, Bergamini and coworkers proposed a novel photocatalytic route to regenerate 1,4-NADH cofactors without the use of additional electron mediators.100 Here, an interesting and rather less explored approach of single electron transfer was used to regenerate cofactor molecules. Particularly, a luminescent metal complex mono(bipyridyl)-tetracyanoruthenate [Ru(bpy)(CN)4]2− was employed to attain a high monoelectronic reduction potential of −1.18 V vs. SCE for reducing NAD+ to NAD˙ radical. As NAD˙ radical undergoes rapid deactivation to form NAD2 dimers, a rapid proton donation is must to form NADH. Hence, L-cysteine (BDES–H = 360.3 kJ mol−1) was employed as the hydrogen atom donor and sacrificial electron donor to produce enzymatically active 1,4-NADH and outperformed widely used TEOA (BDEO–H = 441.0 kJ mol−1), which under similar conditions failed to produce the desired regioisomer. Despite these attempts, an obvious question still remains: can we design the light-harvesting components that preferentially interact with NAD(P)+ to regenerate regioselective 1,4-NAD(P)H without any additional electron mediator?
Compatibility and photostability of oxidoreductase enzymes and biological cofactors
While targeting the perpetual synthesis of fine chemicals via enzyme-coupled photocatalytic systems, a major challenge is associated with the inherent instability of the enzymes outside their natural environment. Apart from the deactivation by Rh-M, enzymes can irreversibly be deactivated by light irradiation, photogenerated holes, or reactive oxygen species (ROS) generated during photocatalysis.79,82,101–103 Therefore, it becomes crucial to protect enzymes from these deactivation species for the ceaseless production of chemicals. In natural photosynthetic system, there is a distinct compartmentalization of the light-driven NAD(P)H regeneration and dark Calvin cycle.104,105 The former takes place in the PSII and PSI embedded in the thylakoid membrane, while the latter takes place through a series of enzymes confined in the chloroplast stroma. The thylakoid membrane confines holes in the lumen, while the ROS scavenging system in chloroplast prevents damage to the biomolecules.104,105 Now, the challenge in replicating the natural thylakoid-stroma system is to achieve compartmentalization of light and dark cycles while maintaining the pristine cofactor shuttling activity.In an attempt to protect enzymes from the products of the light cycle, active and passive protection strategies have been proposed in the literature (Fig. 9a).106 In active passivation, a barrier is constructed to isolate the light-harvesting component, photogenerated holes, and ROS from the enzyme. In contrast, a protective shell is built around the enzyme to isolate it from the damage caused by the external environment in passive protection. As enzymes in the light-independent cycle must utilize NAD(P)H formed in the light cycle, the compartmentalization must allow unrestricted cofactor exchange to achieve the maximum potential of each catalytic cycle. Consequently, semi-permeable membranes with definite size-sieving abilities are often employed for the physical separation of photocatalytic and enzymatic cycles so that products of the light cycle, except the regenerated cofactor, do not interact with the enzyme.80,82,106–110 The overall rate of desired chemical transformation in the photobiocatalytic system will now depend on the rate of cofactor shuttling across the membrane, which in turn depends on the permeability and thickness of the membrane and the diffusion of the cofactor.
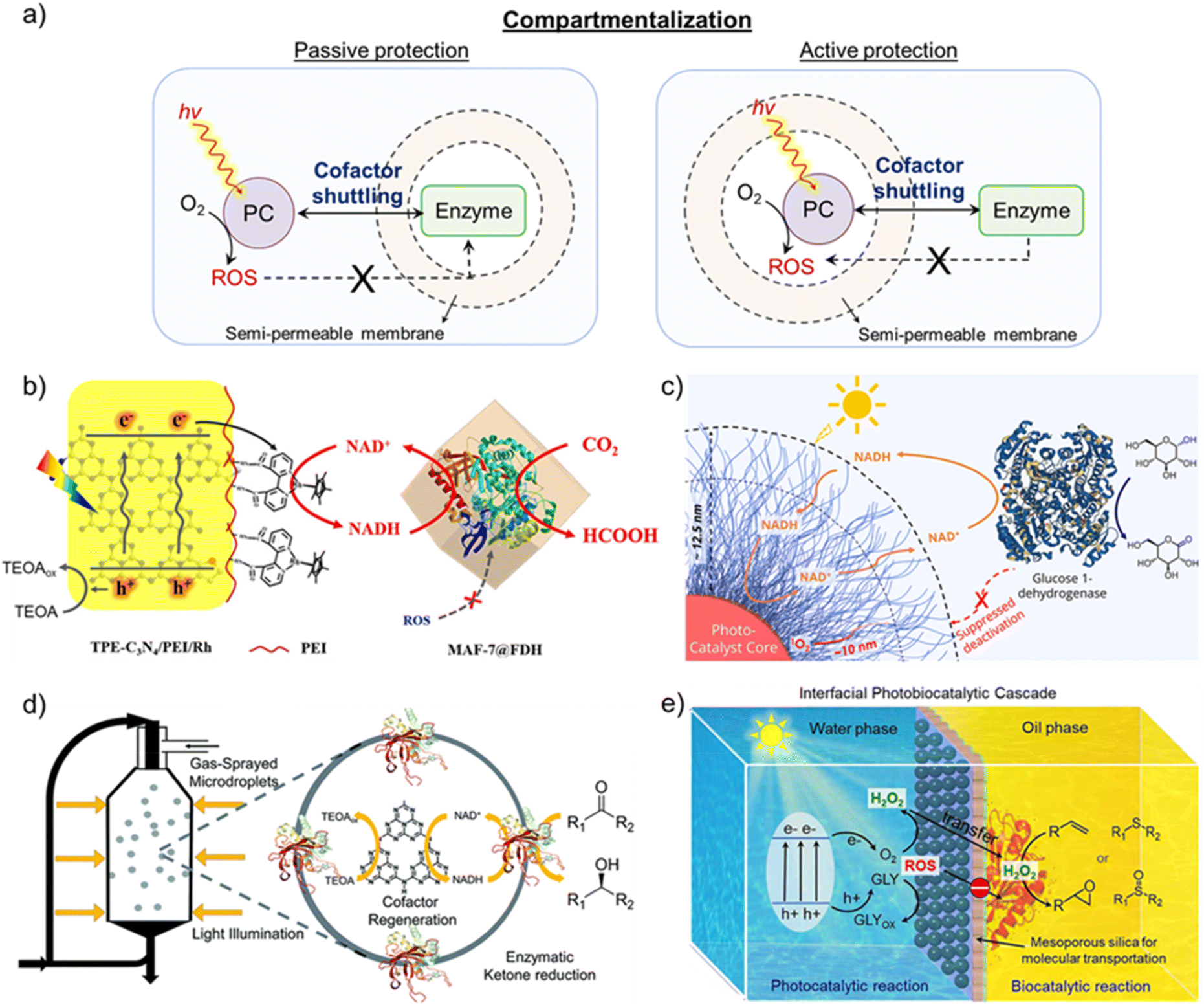 | ||
| Fig. 9 Compartmentalization of light and dark cycles. (a) Schematic representation of protection strategies to achieve compartmentalization in photobiocatalytic systems. Passive protection involves building a protective shell around the enzyme, whereas the light-harvesting component is isolated with a barrier in active passivation. (b) Schematics showing the functionally compartmental photocatalyst-enzyme hybrid system for formic acid production from CO2, wherein formate dehydrogenase enzyme was encapsulated inside the MAF-7 metal–organic framework to avoid its deactivation. Adapted with permission from ref. 107. Copyright 2020 American Chemical Society. (c) Schematic illustration of polymer micelle-vesicle core–shell structure coupled with glucose 1-dehydrogenase. The shell comprising a hydrophilic poly(ethylene glycol) layer acts as a shield between enzymes and ROS generated during the photoexcitation process to avoid enzyme deactivation from ROS. Adapted with permission from ref. 108. Copyright 2022 American Chemical Society. (d) A scheme showing the photoenzymatic system in microdroplets for producing chiral alcohols. A spatial arrangement of enzymes at the air–water interface and photocatalyst within the microdroplet leads to microscale compartmentalization. Adapted with permission from ref. 109. Copyright 2022 The Royal Society of Chemistry. (e) Schematic illustration of Pickering droplet interface to compartmentalize photocatalytic and biocatalytic cycles. The photocatalyst and enzymes are spatially isolated in different mediums of the Pickering emulsion, which restricts their direct interaction. The presence of mesoporous silica allows the selective transfer of reaction intermediates and blocks charge transfer to enzymes to prevent their deactivation. Adapted with permission from ref. 110. Copyright 2024 American Chemical Society. | ||
To avoid enzyme deactivation, Song and coworkers encapsulated formate dehydrogenase (FDH) into the MAF-7 metal–organic framework to separate it from the light-dependent cycle (Fig. 9b).107 MAF-7 is a class of zeolitic imidazole frameworks that possess excellent biocompatibility and special pore structure to encapsulate enzymes and prevent them from denaturing. The triazole linkers in MAF-7 are hydrophilic in nature, which helps maintain the conformation and stability of enzymes in confined spaces. A simple coprecipitation approach was utilized to encapsulate FDH, which allowed for the variation of FDH loading in MAF-7. Here, Rh-M was incorporated with thiophene-modified C3N4, which enabled photocatalytic regeneration of NADH. Upon integrating the photocatalytic center with FDH@MAF-7, formic acid production reached 16.75 mM after 9 h, which was 3.24 times higher than the homogenous counterpart. Even though encapsulation resulted in a minor decrease in FDH activity compared to free FDH, recyclability was achieved for up to 5 cycles (each cycle proceeded for 9 h) with 76% retention of formic acid yield. Such enhanced cyclic stability is clearly due to the compartmentalization of FDH, which protects it from oxidative denaturants formed in the light cycle. A slight decrease in encapsulated FDH activity (Km = 1.54 mM; kcat = 139.68 min−1) compared to free FDH (Km = 1.46 mM; kcat = 145.00 min−1) was attributed to the hindered diffusion of NADH and substrate across MAF-7.
While Song and coworkers provided passive protection in the above-discussed work, Chen, Li, and coworkers chose the active protection strategy to compartmentalize light and dark cycles.108 Here, polymer micelle-vesicle core–shell structures composed of a hydrophobic polymer core and hydrophilic polymer shell were constructed to avoid the deactivation of the enzyme (Fig. 9c).108 The hydrophobic polymer core was designed as a photocatalytic polymer where a metal-free organic dye was attached to the polymer backbone, whereas hydrophobic cores formed the nanoreactors (Fig. 9c). In particular, amphiphilic block polymers were prepared using poly(ethylene) glycol as the hydrophilic block and triphenylamine–thiophene-based dye (TTMN) integrated with polymethacrylate as the hydrophobic block. These polymers were self-assembled to form micelle-vesicle core–shell structures, which, in the presence of glucose–dehydrogenase, continuously oxidized glucose to gluconolactone. The hydrophilic shell ensured free diffusion of cofactors, while enzymes, being larger in size, could not diffuse into the shell. An appropriate shell thickness corresponding to the diffusion length of ROS ensured the trapping and deactivation of ROS before its interaction with the enzyme. The optimized design of the micelle-vesicle core–shell structure enabled 10 consecutive photobiocatalytic cycles with 70% preservation of the enzyme activity after the last cycle, along with 85% of the photocatalytic activity.
In an interesting approach by Zare, Ge, and coworkers, microdroplets were used to compartmentalize an enzyme-coupled photocatalytic system.109 The microdroplet reactor was prepared by gas-spraying the reaction mixture comprising graphitic carbon nitride (GCN) as photocatalyst, triethanolamine (TEOA) as hole scavenger, Rh-M, and Sporobolomyces salmonicolor carbonyl reductase (SsCR) for production of chiral alcohols (Fig. 9d). In the microdroplet reactor, SsCR was exclusively distributed at the air–water interface, while GCN was uniformly distributed within the microdroplet, leading to compartmentalization at the microscale. Such spatial compartmentalization avoided enzyme inactivation from photogenerated holes, as was evident from the retention of 50% enzymatic activity even after 5 h of light illumination. In contrast, only <10% of the enzyme activity was retained after 1 h of light illumination when the reaction was performed in bulk solution state. Alongside providing protection to the enzymes, microdroplets enabled higher reaction rates owing to the regionalized distribution of regenerated NADH and enzyme.
In addition to microdroplets, Pickering droplet interfaces have recently emerged for compartmentalization.110–113 Typically, Pickering emulsion droplets are oil-in-water droplets stabilized by solid particles at the interface.110,111 As a result, different catalysts (here, photocatalyst and enzyme) can be compartmentalized in distinct mediums at the droplet interface. Zhou, Yang, and coworkers regulated photobiocatalytic reactions with Pickering droplets for enzymatic alkene epoxidation and thioether oxidation via photocatalytically generated H2O2 (Fig. 9e).110 Gold nanoparticle loaded-ultrathin graphitic carbon nitride nanosheets (Au/g-C3N4-NSs) were used as the photocatalyst to produce H2O2. To avoid interaction with the photocatalyst, enzymes were immobilized on the hydrophobic surface of Janus mesoporous silica nanosheets (JMSNs), which were functionalized with the aminopropyl and octyl groups on each side. A mixture of enzyme/JMSNs and Au/g-C3N4-NSs was used to form Pickering emulsion under vigorous stirring conditions. While hydrophobic enzyme/JMSNs assembled in the inner interfacial layer of oil drops, Au/g-C3N4-NSs were positioned in the outer interfacial layer. Thus, a spatial arrangement of photocatalysts and enzymes could be achieved with nanochannels of JMSNs connecting the two. Furthermore, the insulating nature of silica prevented the transfer of photoexcited charge carriers, which can deactivate enzymes and only allow selective transportation of reaction intermediates. Pickering droplet reactors showed a 2.0–5.8-fold rate enhancement compared to bulk systems, along with additional merits of enzyme protection from light illumination, oxidative inactivation, and overcoming diffusion limitation of reaction intermediates.
From the above discussion, it is clear that optimal shuttling of cofactors and compatibility between the photocatalytic and enzyme catalytic cycles must be ensured for continuous and ceaseless chemical synthesis. Precise compartmentalization of the light-dependent and -independent cycles offers a way to achieve the same. Despite witnessing several compartmentalization strategies, all of them are at their preliminary stage and require significant improvement, as enzymes would still lose their activity. Further elucidation of enzyme deactivation mechanisms in photobiocatalytic systems is required to devise advanced protection strategies. An aid from biomimetic materials research may afford an alternative to construct nano/micro-sized compartments, as seen in the natural chloroplast.114
It is quite intuitive that enzymes are prone to denature when exposed to external conditions. It is clear from the above discussion that the deactivation pathways become more prominent when coupled with a photocatalytic system. As enzymes are the best-known catalysts sustaining life on Earth, we must take lessons from their structure and function to design efficient and robust synthetic analogs. For instance, an NAD(P)H-dependent artificial transfer hydrogenase (ATHase) based on biotinylated [Cp*Ir(Biot-p-L)Cl] complex was reported by Ward and coworkers, which was further coupled for multi-enzymatic cascade transformations under physiological conditions (Fig. 10a and b).115 While enzymatic NAD(P)H regeneration was employed after its consumption by ATHase for imine reduction, in principle, a photocatalyst can easily be used for NAD(P)H regeneration, thereby achieving a complete mimic of the natural photosynthetic system. So far, the introduction and testing of such synthetic analogs have not been carried out, and hence, they hold potential avenues for ceaseless chemical transformations.
 | ||
| Fig. 10 (a) Structure of artificial transfer hydrogenase (ATHase) and its further assembly inside streptavidin. (b) Coupling of biotinylated-ATHase with alcohol dehydrogenase enzyme (ADH) for conversion of linear aminoalcohol into a cyclic amine via an imine. Adapted with permission from ref. 115. Copyright 2016 American Chemical Society. (c) Schematics of metal-ion catalyzed reduction of keto-acid in the presence of NADH without needing an oxidoreductase enzyme. Here, metal ions such as Al3+/Fe2+/3+ mimic the role of enzymes by pre-organizing and activating the substrates. Adapted with permission from ref. 117. Copyright 2024 Elsevier Inc. (d) Structure of nicotinamide cofactors, oxidized (left) and reduced (right). In artificial cofactor, β-D-ribose oxygen(X) is replaced by methylene group to form 2,3-dihydroxy cyclopentane ring yielding carba-NADP/H. (e) Thermal decomposition studies clearly show the increased lifetime of artificial carbon-NADP/H at elevated temperatures compared to NADP/H. Adapted with permission from ref. 119. Copyright 2021 Wiley VCH. | ||
It is interesting to note that NAD(P)H was a central molecule in early metabolism and had been known before the existence of enzymes.116 The question arises: can prebiotic chemistry help identify non-enzymatic routes to perform biocatalytic transformations? In an interesting study, Mayer and Moran reported that certain metal ions can enable nonenzymatic hydride transfer between NADH and keto acid in a stereoselective manner.117 A catalytic amount of Al3+/Fe2+/3+ could replace the enzyme's function to reduce keto acid in the presence of NADH by pre-organizing the interaction of keto acid with NADH for stereoselective transformation (Fig. 10c). Utilization of such non-enzymatic catalysts will surely alleviate issues related to enzyme degradation and, at the same time, help gain an understanding of the origins of life.
Similar to enzymes, the regenerated NAD(P)H cofactor can also undergo oxidation by its interaction with photogenerated holes and ROS.118 Along with the back oxidation to NAD(P)+, NAD(P)H can oxidize to intermediate radicals such as NADH+˙, NAD˙, or fragment into ADP-ribose and nicotinamide or undergo hydrolysis and ring opening.118 This is one of the challenges in photobiocatalysis involving shuttling cofactors, which is often neglected while devising a complete system. This can be addressed by developing modified and robust cofactors that have similar compatibility with enzymes to natural cofactors. One such modified cofactor is carba-NAD(P)+/NAD(P)H, where the α-D-ribose oxygen atom is replaced by a methylene group, resulting in a 2,3-dihydroxy cyclopentane ring (Fig. 10d).119 Along with enhanced thermal stability and lower decomposition rate in the acidic medium (Fig. 10e), carba-NAD(P)H was accepted by most NAD(P)H-dependent oxidoreductase enzymes. This report demonstrates that cofactor engineering can be of special interest for photobiocatalytic conversion in harsh conditions; however, it has not been tested so far.
Stability of photocatalysts under continuous illumination
In the context of continuous production of chemicals through semi-artificial photoenzymatic systems, the stability of light-harvesting components is as crucial as the stability of enzymes and biological cofactor molecules. In general, the long-term stability of the photocatalyst under continuous light illumination is a major concern, as most of the photocatalysts tend to deactivate after a few hours of operation.120–122 However, this particular aspect is seldom studied and reported in the literature. Depending on the nature of the photocatalyst, different deactivation pathways exist that are believed to hamper their continuous operation. For instance, eosin Y (EY), one of the widely used photoredox catalysts, is known to undergo degradation in both oxic and anoxic media when excited at its absorption maximum.120 In the presence of O2, the photoexcited EY (triplet state) gets quenched, leading to the formation of reactive oxygen species, which then attack the conjugated system to cause discoloration/bleaching. On the other hand, different debromination processes take place under anoxic conditions that result in single and double debrominated products. Another widely used photosensitizers belong to the family of ruthenium trisbipyridine complexes, which also exhibit long-term instability under light irradiation.121,122 This stems from the instability of the reduced and oxidized forms of the Ru complexes as they undergo ligand substitution by solvent molecules or anions present in the reaction mixture.121,122 In contrast to the molecular photocatalysts, semiconductor-based photocatalysts suffer from more complex and multiple deactivation pathways, which include the accumulation of charge carriers leading to photocorrosion, surface poisoning by reaction intermediates, structural changes, defect formation, etc.123–125The strategies developed for enhancing the stability of photocatalysts will also align well to improve the stability of photobiocatalytic systems. One of the ways to mitigate the instability issue is by applying a protective layer.123,126,127 An ideal protective layer must exhibit good charge transfer properties and not hamper light absorption by the photocatalyst. In a report by Lewis and coworkers, NiOx was sputtered over InP photoanodes to provide stabilization against photocorrosion and led to stable water oxidation activity for over 48 h in alkaline medium.126 Improving the adhesion between the photocatalyst and protective layers may also help in enhancing the photostability for long-term applications.123,127,128 For instance, Shin and coworkers inserted reduced graphene oxide between Pt catalysts and CdS layer over Cu(In, Ga)Se2 photocathode to increase the stability of photocathode from 2.5 h to more than 7 h for hydrogen evolution under continuous light irradiation.128 Another interesting way to enhance the stability is by precise engineering of the electronic levels in the photocatalyst.129,130 Yu and coworkers demonstrated that Ag doped CdS QDs can enhance the stability by creating an acceptor energy level near the valence band of CdS.129 This newly formed acceptor energy level can abstract the holes. On the other hand, the photoexcited electrons are transferred from the conduction band of CdS to Ag deposited on its surface. Another promising way to improve the stability is by fine-tuning the photocatalyst interaction with the reactants and intermediates, so as to avoid surface poisoning.87 In this direction, our group demonstrated that using mixed-charged gold nanoparticles (AuNPs) comprising both positively and negatively charged ligands helps in regulating the electrostatic interactions between the AuNP photocatalyst and the reactants.131 This, in turn, provides a balanced adsorption–desorption of the reactant/intermediates and enhances the colloidal stability of the photocatalyst, thereby imparting recyclability. Nevertheless, all these strategies are currently limited to laboratory-scale applications but hold potential to construct large-scale photocatalytic platforms. This strongly suggests the need for sincere efforts in designing appropriate photocatalysts, which requires dedicated research in addressing the fundamental challenges associated with photocatalysis in general.
Scale-up
Even though enzymes exhibit nearly 100% selectivity with high reaction rates, their isolation requires an extensive purification process. As a result, the scalability of the photobiocatalytic systems can be limited to the laboratory scale, hence making them impractical for commercial use. Keeping in mind these challenges, Reisner and coworkers fabricated a 50 cm2 semi-artificial system prototype (medium-scale prototype device) consisting of carbon nitride and formate dehydrogenase enzyme for outdoor testing of CO2 to formate transformation.132 This has been reported to be the largest integrated semiartificial solar fuel system to date and was continuously operational for three days under natural sunlight. This report calls for such ambitious research directions in taking photobiocatalysis to a practical stage. Moreover, the fundamentals involved in enzymatic photobiocatalytic systems will be crucial for systems involving whole cells, which are projected to reduce processing costs. The use of whole cells will also direct the utilization of complex metabolic pathways,133–135 resulting in complex biomolecule production that is unattainable by photo-enzymatic systems.In another direction, the growing interest in developing flow systems will enable the continuous production of fuels in photoenzymatic systems.136,137 Grzelczak and coworkers performed cofactor regeneration (light cycle) under flow conditions, while the dark reactions utilizing the regenerated cofactor for nanoparticle synthesis were performed in batch.136 The flow photoreactor consisted of a column filled with AuPd/agarose composite, which was coupled to an online UV-vis spectrophotometer for real-time monitoring of the NADH regenerated. As a proof-of-concept, three complete light and dark cycles were performed by separating the nanoparticles formed in each dark cycle and supplying the permeate back to the flow photoreactor. Advancing in this direction, Bergamini and coworkers designed a fully automated microfluidic system controlling all the steps required for cofactor regeneration, right from the uptake of reactants to their mixing, irradiation of the reaction mixture, and NADH collection.137 Using this, NADH regeneration was carried out for multiple cycles without any external intervention. To expedite the optimization, reproducibility, and scale-up processes in photoredox catalysis, Noël's group developed RoboChem, an automated robotic platform for photocatalytic organic synthesis.138 The use of Bayesian optimization algorithms allows for spanning across diverse reaction variables. The robotic platform is highly modular and holds immense promise in the digitalization of photobiocatalytic platforms as well.
Conclusions and outlook
The rise of photobiocatalysis in the past two decades unfurled exciting directions to enter. With the natural photosynthetic Z-scheme serving as a quintessential photobiocatalytic platform, semi-artificial mimics involving artificial light harvesters and natural enzymes enable the one-pot production of molecules and materials. The coupling between the light and dark cycles via cofactor shuttling has emerged as a promising strategy for ceaseless and sustainable chemical synthesis, expanding into diverse, challenging reactions, including enantioselective synthesis, amino acid synthesis, etc. This Perspective builds on this direction by first summarizing the recent developments in semi-artificial photoenzymatic systems achieving a continuous production of chemicals through shuttling of NAD(P)+/NAD(P)H cofactors. A seemingly trivial task of coupling light and dark cycles for attaining perpetual chemical synthesis, is, in fact, highly challenging. Here, we outline key roadblocks in engineering multiple component systems exhibiting perfect synergy between the photocatalyst and enzymes, fast regeneration and shuttling of cofactors, and robust overall performance.Through this Perspective, it is clear that interfacing enzymes with artificial light-harvesting materials enables bypassing the limitations of individual components and offers a way to combine their strengths to perform catalytic transformations at their best. The presence of a vast enzyme inventory offers excellent flexibility in devising photobiocatalytic systems. There lies a huge untouched list of NAD(P)H-dependent oxidoreductase enzymes that could transform synthetic chemical manufacturing. Next, to take these semi-artificial photoenzymatic systems to newer heights, it is crucial to develop enzyme cascades to match the complexity of natural systems. Often, a single enzyme is coupled with the photoregenerated cofactor, yielding only simpler molecules in the end. A judicious choice of enzymes will enable a unidirectional flow of electron and substrate flux towards complex and value-added molecules. Additionally, it is noticed that most research revolves around carbon-based cycles such as C–C bond formation, C–H activation, CO2 reduction, etc. Alongside this, parallel research must focus on developing photobiocatalytic systems that mimic nitrogen and phosphorous cycles, which are essential life-building blocks. This may require the involvement of cofactors other than the nicotinamide ones, such as flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). Thus, opportunities lie in developing flavin-based photoenzymatic systems, which remain less explored.
Developing cofactor-dependent photobiocatalytic systems involves amalgamating diverse areas of material science, photocatalysis, electrochemistry, enzymology, microbiology, synthetic biology, etc. With joint efforts, the expedition of photobiocatalysis involving enzymes will definitely open new pathways in solar energy transduction for perpetual chemical synthesis in a sustainable way.
Data availability
No new data were generated or analyzed as part of this perspective.Author contributions
All authors contributed to the discussion, writing, and provided feedback on the manuscript. P. P. P. conceived and coordinated the Perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Soumendu Roy, Dr Indra Narayan Chakraborty, Mr Ankit Dhankhar, and Ms Namitha Deepak for their contributions to establishing the area of photobiocatalysis in our research group. The authors acknowledge the financial support from DST-SERB India Grant CRG/2023/001711. V. J. thanks MoE/PMRF for the fellowship.References
- G. Ciamician, The photochemistry of the future, Science, 1912, 36, 385–394 CrossRef CAS.
- D. G. Nocera, The artificial leaf, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS.
- N. S. Lewis, Research opportunities to advance solar energy utilization, Science, 2016, 351, aad1920 CrossRef PubMed.
- Q. Wang, C. Pornrungroj, S. Linley and E. Reisner, Strategies to improve light utilization in solar fuel synthesis, Nat. Energy, 2022, 7, 13–24 CrossRef.
- J. Lv, J. Xie, A. G. A. Mohamed, X. Zhang, Y. Feng, L. Jiao, E. Zhou, D. Yuan and Y. Wang, Solar utilization beyond photosynthesis, Nat. Rev. Chem., 2023, 7, 91–105 CrossRef PubMed.
- J. Barber, Photosynthetic energy conversion: Natural and artificial, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- G. Govindjee, D. Shevela and L. O. Björn, Evolution of the Z-scheme of photosynthesis: A perspective, Photosynth. Res., 2017, 133, 5–15 CrossRef PubMed.
- R. Hill and F. Bendall, Function of the two cytochrome components in chloroplasts: A working hypothesis, Nature, 1960, 186, 136–137 CrossRef CAS.
- L. N. M. Duysens, J. Amesz and B. M. Kamp, Two photochemical systems in photosynthesis, Nature, 1961, 190, 510–511 CrossRef CAS.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, 2004 Search PubMed.
- M. Calvin, The path of carbon in photosynthesis, Science, 1962, 135, 3507 CrossRef.
- S. H. Lee, J. H. Kim and C. B. Park, Coupling photocatalysis and redox biocatalysis toward biocatalyzed artificial photosynthesis, Chem.–Eur. J., 2013, 19, 4392–4406 CrossRef CAS PubMed.
- T. E. Miller, T. Beneyton, T. Schwander, C. Diehl, M. Girault, R. Mclean, T. Chotel, P. Claus, N. S. Cortina, J.-C. Baret and T. J. Erb, Light-powered CO2 fixation in a chloroplast mimic with natural and synthetic parts, Science, 2020, 368, 649–654 CrossRef CAS PubMed.
- L. S. Vidal, C. L. Kelly, P. M. Mordaka and J. T. Heap, Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 327–347 CrossRef.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Photobiocatalysis: Activating redox enzymes by direct or indirect transfer of photoinduced electrons, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS.
- H. K. Chenault and G. M. Whitesides, Regeneration of nicotinamide cofactors for use in organic synthesis, Appl. Biochem. Biotechnol., 1987, 14, 147–197 CrossRef CAS PubMed.
- F. Hollmann, D. J. Opperman and C. E. Paul, Biocatalytic reduction reactions from a chemist's perspective, Angew. Chem., Int. Ed., 2021, 60, 5644–5665 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Enzymatic reductions for the chemist, Green Chem., 2011, 13, 2285–2314 RSC.
- C.-H. Yun, J. Kim, F. Hollmann and C. B. Park, Light-driven biocatalytic oxidation, Chem. Sci., 2022, 13, 12260–12279 RSC.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Cofactor NAD(P)H regeneration inspired by heterogeneous pathways, Chem, 2017, 2, 621–654 CAS.
- Q. Shi, D. Yang, Z. Jiang and J. Li, Visible-light photocatalytic regeneration of NADH using P-doped TiO2 nanoparticles, J. Mol. Catal. B: Enzym., 2006, 43, 44–48 CrossRef CAS.
- J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Self-assembled light-harvesting peptide nanotubes for mimicking natural photosynthesis, Angew. Chem., Int. Ed., 2012, 51, 517–520 CrossRef CAS PubMed.
- D. H. Nam and C. B. Park, Visible light-driven NADH regeneration sensitized by proflavine for biocatalysis, ChemBioChem, 2012, 13, 1278–1282 CrossRef CAS.
- J. Liu and M. Antonietti, Bio-inspired NADH regeneration by carbon nitride photocatalysis using diatom templates, Energy Environ. Sci., 2013, 6, 1486–1493 RSC.
- D. H. Nam, S. H. Lee and C. B. Park, CdTe, CdSe, and CdS nanocrystals for highly efficient regeneration of nicotinamide cofactor under visible light, Small, 2010, 6, 922–926 CrossRef CAS PubMed.
- K. A. Brown, M. B. Wilker, M. Boehm, H. Hamby, G. Dukovic and P. W. King, Photocatalytic regeneration of nicotinamide cofactors by quantum dot–enzyme biohybrid complexes, ACS Catal., 2016, 6, 2201–2204 CrossRef CAS.
- A. Sánchez-Iglesias, J. Barroso, D. M. Solís, J. M. Taboada, F. Obelleiro, V. Pavlov, A. Chuvilin and M. Grzelczak, Plasmonic substrates comprising gold nanostars efficiently regenerate cofactor molecules, J. Mater. Chem. A, 2016, 4, 7045–7052 RSC.
- A. Rogolino, N. Claes, J. Cizaurre, A. Marauri, A. Jumbo-Nogales, Z. Lawera, J. Kruse, M. Sanromán-Iglesias, I. Zarketa, U. Calvo, E. Jimenez-Izal, Y. P. Rakovich, S. Bals, J. M. Matxain and M. Grzelczak, Metal–polymer heterojunction in colloidal-phase plasmonic catalysis, J. Phys. Chem. Lett., 2022, 13, 2264–2272 CrossRef CAS PubMed.
- S. Roy, V. Jain, R. K. Kashyap, A. Rao and P. P. Pillai, Electrostatically driven multielectron transfer for the photocatalytic regeneration of nicotinamide cofactor, ACS Catal., 2020, 10, 5522–5528 CrossRef CAS.
- S. Singh, S. Kumari, M. Ahlawat and V. G. Rao, Photocatalytic NADH regeneration employing Au–Pd core–shell nanoparticles: Plasmonic modulation of underlying reaction kinetics, J. Phys. Chem. C, 2022, 126, 15175–15183 CrossRef CAS.
- A. Dhankhar, V. Jain, I. N. Chakraborty and P. P. Pillai, Enhancing the photocatalytic regeneration of nicotinamide cofactors with surface engineered plasmonic antenna-reactor system, J. Photochem. Photobiol., A, 2023, 437, 114472 CrossRef CAS.
- A. Dhankhar and P. P. Pillai, Plasmonic antenna–reactor photocatalysts based on anisotropic gold–rhodium superstructures for biological cofactor regeneration, Chem. Mater., 2024, 36, 10227–10237 CrossRef CAS.
- N. Deepak, V. Jain and P. P. Pillai, Metal-semiconductor heterojunction accelerates the plasmonically powered photoregeneration of biological cofactors, Photochem. Photobiol., 2024, 100, 1000–1009 CrossRef CAS.
- M. R. Schreier, B. Pfund, D. M. Steffen and O. S. Wenger, Photocatalytic regeneration of a nicotinamide adenine nucleotide mimic with water-soluble iridium(III) complexes, Inorg. Chem., 2023, 62, 7636–7643 CrossRef CAS PubMed.
- C. J. Seel and T. Gulder, Biocatalysis fueled by light: On the versatile combination of photocatalysis and enzymes, ChemBioChem, 2019, 20, 1871–1897 CrossRef CAS PubMed.
- J. A. Maciá-Agulló, A. Corma and H. Garcia, Photobiocatalysis: The power of combining photocatalysis and enzymes, Chem.–Eur. J., 2015, 21, 10940–10959 CrossRef PubMed.
- Y. Zhang, Y. Zhao, R. Li and J. Liu, Bioinspired NADH regeneration based on conjugated photocatalytic systems, Sol. RRL, 2021, 5, 2000339 CrossRef CAS.
- G. Zhao, C. Yang, W. Meng and X. Huang, Recent advances in porous materials for photocatalytic NADH regeneration, J. Mater. Chem. A, 2024, 12, 3209–3229 RSC.
- N. Pollak, C. Dölle and M. Ziegler, The power to reduce: pyridine nucleotides – small molecules with a multitude of functions, Biochem. J., 2007, 402, 205–218 CrossRef CAS PubMed.
- W. Hummel, Large-scale applications of NAD(P)-dependent oxidoreductases: recent developments, Trends Biotechnol., 1999, 17, 487–492 CrossRef CAS.
- J. B. Jones, D. W. Sneddon, W. Higgins and A. J. Lewis, Preparative-scale reductions of cyclic ketones and aldehyde substrates of horse liver alcohol dehydrogenase with in situ sodium dithionite recycling of catalytic amounts of NAD, J. Chem. Soc., Chem. Commun., 1972, 15, 856–857 RSC.
- K. E. Taylor and J. B. Jones, Nicotinamide coenzyme regeneration by dihydropyridine and pyridinium compounds, J. Am. Chem. Soc., 1976, 98, 5689–5694 CrossRef CAS PubMed.
- O. Abril and G. M. Whitesides, Hybrid organometallic/enzymic catalyst systems: regeneration of NADH using dihydrogen, J. Am. Chem. Soc., 1982, 104, 1552–1554 CrossRef CAS.
- R. Raunio and E. M. Lilius, Effect of dithionite on enzyme activities in vivo, Enzymologia, 1971, 40, 360–368 CAS.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Efficient catalytic interconversion between NADH and NAD+ accompanied by generation and consumption of hydrogen with a water-soluble iridium complex at ambient pressure and temperature, J. Am. Chem. Soc., 2011, 134, 367–374 CrossRef PubMed.
- C. Kohlmann, W. Märkle and S. Lütz, Electroenzymatic synthesis, J. Mol. Catal. B:Enzym., 2008, 51, 57–72 CrossRef CAS.
- V. K. Sharma, J. M. Hutchison and A. M. Allgeier, Redox biocatalysis: quantitative comparisons of nicotinamide cofactor regeneration methods, ChemSusChem, 2022, 15, e202200888 CrossRef CAS PubMed.
- J. Cantet, A. Bergel and M. Comtat, Coupling of the electroenzymatic reduction of NAD+ with a synthesis reaction, Enzyme Microb. Technol., 1996, 18, 72–79 CrossRef CAS.
- Y. Kashiwagi, Y. Yanagisawa, N. Shibayama, K. Nakahara, F. Kurashima, J. Anzai and T. Osa, Preparative, electroenzymatic reduction of ketones on an all components-immobilized graphite felt electrode, Electrochim. Acta, 1997, 42, 2267–2270 CrossRef CAS.
- S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J.-K. Lee and C. B. Park, Photoelectrochemical reduction of carbon dioxide to methanol through a highly efficient enzyme cascade, Angew. Chem., Int. Ed., 2017, 56, 3827–3832 CrossRef CAS PubMed.
- D. H. Nam, S. K. Kuk, H. Choe, S. Lee, J. W. Ko, E. J. Son, E.-G. Choi, Y. H. Kim and C. B. Park, Enzymatic photosynthesis of formate from carbon dioxide coupled with highly efficient photoelectrochemical regeneration of nicotinamide cofactors, Green Chem., 2016, 18, 5989–5993 RSC.
- X. Wang and H. H. P. Yiu, Heterogeneous catalysis mediated cofactor NADH regeneration for enzymatic reduction, ACS Catal., 2016, 6, 1880–1886 CrossRef CAS.
- T. Saba, J. Li, J. W. H. Burnett, R. F. Howe, P. N. Kechagiopoulos and X. Wang, NADH regeneration: A case study of Pt-catalyzed NAD+ reduction with H2, ACS Catal., 2021, 11, 283–289 CrossRef CAS.
- J. W. H. Burnett, R. F. Howe and X. Wang, Cofactor NAD(P)H regeneration: How selective are the reactions, Trends Chem., 2020, 2, 488–492 CrossRef CAS.
- J. A. Farrington, E. J. Land and A. J. Swallow, The one-electron reduction potentials of NAD, Biochim. Biophys. Acta, Bioenerg., 1980, 590, 273–276 CrossRef CAS.
- R. F. Anderson, Energetics of the one-electron steps in the NAD+/NADH redox couple, Biochim. Biophys. Acta, Bioenerg., 1980, 590, 277–281 CrossRef CAS PubMed.
- F. Liu, C. Ding, S. Tian, S.-M. Lu, C. Feng, D. Tu, Y. Liu, W. Wang and C. Li, Electrocatalytic NAD+ reduction via hydrogen atom-coupled electron transfer, Chem. Sci., 2022, 13, 13361–13367 RSC.
- M. Medina, Structural and mechanistic aspects of flavoproteins: photosynthetic electron transfer from photosystem I to NADP+, FEBS J., 2009, 276, 3942–3958 CrossRef CAS.
- H. C. Lo, O. Buriez, J. B. Kerr and R. H. Fish, Regioselective reduction of NAD+ models with [Cp*Rh(bpy)H]+: Structure-activity relationships and mechanistic aspects in the formation of the 1,4-NADH derivatives, Angew. Chem., Int. Ed., 1999, 38, 1429–1432 CrossRef CAS.
- F. Hildebrand, C. Kohlmann, A. Franz and S. Lütz, Synthesis, characterization and application of new rhodium complexes for indirect electrochemical cofactor regeneration, Adv. Synth. Catal., 2008, 350, 909–918 CrossRef CAS.
- J. Canivet, G. Süss-Fink and P. Štěpnička, Water-soluble phenanthroline complexes of rhodium, iridium and ruthenium for the regeneration of NADH in the enzymatic reduction of ketones, Eur. J. Inorg. Chem., 2007, 2007, 4736–4742 CrossRef.
- S. L. Johnson and P. T. Tuazon, Acid-catalyzed hydration of reduced nicotinamide adenine dinucleotide and its analogs, Biochemistry, 1977, 16, 1031–1250 CrossRef PubMed.
- H. K. Chenault, E. S. Simon and G. M. Whitesides, Cofactor regeneration for enzyme-catalysed synthesis, catalysed synthesis, Biotechnol. Genet. Eng. Rev., 1988, 6, 221–270 CrossRef CAS.
- C. H. Wong and G. M. Whitesides, Enzyme-catalyzed organic synthesis: NAD(P)H cofactor regeneration by using glucose 6-phosphate and the glucose-6-phosphate dehydrogenase from leuconostoc mesenteroides, J. Am. Chem. Soc., 1981, 103, 4657–4988 CrossRef.
- W. Ge, H. Peng, J. Dong, G. Wang, L. Cui, W. Sun, X. Ma and J. Yang, Zn(002)-preferred and pH-buffering triethanolamine as electrolyte additive for dendrite-free Zn anodes, Chem. Commun., 2024, 60, 750–753 RSC.
- K. Kinastowska, J. Liu, J. M. Tobin, Y. Rakovich, F. Vilela, Z. Xu, W. Bartkowiak and M. Grzelczak, Photocatalytic cofactor regeneration involving triethanolamine revisited: The critical role of glycolaldehyde, Appl. Catal., B, 2019, 243, 686–692 CrossRef CAS.
- C. B. Park, S. H. Lee, E. Subramanian, B. B. Kale, S. M. Lee and J.-O. Baeg, Solar energy in production of L-glutamate through visible light active photocatalyst-redox enzyme coupled bioreactor, Chem. Commun., 2008, 42, 5423–5425 RSC.
- Y.-Z. Wang, Z.-P. Zhao, R.-L. Zhou and W.-F. Liu, Optimization of a photoregeneration system for NADH using pristine TiO2 as a catalyst, J. Mol. Catal. B: Enzym., 2016, 133, S188–S193 CrossRef.
- R. K. Yadav, A. Kumar, N.-J. Park, K.-J. Kong and J.-O. Baeg, A highly efficient covalent organic framework film photocatalyst for selective solar fuel production from CO2, J. Mater. Chem. A, 2016, 4, 9413–9418 RSC.
- Y. Pellegrin and F. Odobel, Sacrificial electron donor reagents for solar fuel production, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- X.-G. Zhu, S. P. Long and D. R. Ort, What is the maximum efficiency with which photosynthesis can convert solar energy into biomass?, Curr. Opin. Biotechnol., 2008, 19, 153–159 CrossRef CAS.
- X.-G. Zhu, S. P. Long and D. R. Ort, Improving photosynthetic efficiency for greater yield, Annu. Rev. Plant Biol., 2010, 61, 235–261 CrossRef CAS.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- J. Huang, M. Antonietti and J. Liu, Bio-inspired carbon nitride mesoporous spheres for artificial photosynthesis: photocatalytic cofactor regeneration for sustainable enzymatic synthesis, J. Mater. Chem. A, 2014, 2, 7686–7693 RSC.
- S. Wang, X. Wu, J. Fang, F. Zhang, Y. Liu, H. Liu, Y. He, M. Luo and R. Li, Direct Z-scheme polymer/polymer double-shell hollow nanostructures for efficient NADH regeneration and biocatalytic artificial photosynthesis under visible light, ACS Catal., 2023, 13, 4433–4443 CrossRef CAS.
- H. Zhao, L. Wang, G. Liu, Y. Liu, S. Zhang, L. Wang, X. Zheng, L. Zhou, J. Gao, J. Shi and Y. Jiang, Hollow Rh-COF@COF S-scheme heterojunction for photocatalytic nicotinamide cofactor regeneration, ACS Catal., 2023, 13, 6619–6629 CrossRef CAS.
- I. N. Chakraborty, V. Jain, P. Roy, P. Kumar, C. P. Vinod and P. P. Pillai, Photocatalytic regeneration of reactive cofactors with InP quantum dots for the continuous chemical synthesis, ACS Catal., 2024, 14, 6740–6748 CrossRef CAS.
- Y. Wu, J. Shi, D. Li, S. Zhang, B. Gu, Q. Qiu, Y. Sun, Y. Zhang, Z. Cai and Z. Jiang, Synergy of electron transfer and electron utilization via metal–organic frameworks as an electron buffer tank for nicotinamide regeneration, ACS Catal., 2020, 10, 2894–2905 CrossRef CAS.
- S. Zhang, S. Liu, Y. Sun, S. Li, J. Shi and Z. Jiang, Enzyme-photo-coupled catalytic systems, Chem. Soc. Rev., 2021, 50, 13449–13466 RSC.
- Y. Sun, J. Shi, Z. Wang, H. Wang, S. Zhang, Y. Wu, H. Wang, S. Li and Z. Jiang, Thylakoid membrane-inspired capsules with fortified cofactor shuttling for enzyme-photocoupled catalysis, J. Am. Chem. Soc., 2022, 144, 4168–4177 CrossRef CAS PubMed.
- R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-J. Kong and J.-O. Baeg, Highly selective solar-driven methanol from CO2 by a photocatalyst/biocatalyst integrated system, J. Am. Chem. Soc., 2014, 136, 16728–16731 CrossRef CAS.
- S. Zhang, J. Shi, Y. Sun, Y. Wu, Y. Zhang, Z. Cai, Y. Chen, C. You, P. Han and Z. Jiang, Artificial thylakoid for the coordinated photoenzymatic reduction of carbon dioxide, ACS Catal., 2019, 9, 3913–3925 CrossRef CAS.
- S. Morra and A. Pordea, Biocatalyst–artificial metalloenzyme cascade based on alcohol dehydrogenase, Chem. Sci., 2018, 9, 7447–7454 RSC.
- M. Poizat, I. W. C. E. Arends and F. Hollmann, On the nature of mutual inactivation between [Cp*Rh(bpy)(H2O)]2+ and enzymes – analysis and potential remedies, J. Mol. Catal. B: Enzym., 2010, 63, 149–156 CrossRef CAS.
- S. Zhang, Y. Zhang, Y. Chen, D. Yang, S. Li, Y. Wu, Y. Sun, Y. Cheng, J. Shi and Z. Jiang, Metal hydride-embedded titania coating to coordinate electron transfer and enzyme protection in photo-enzymatic catalysis, ACS Catal., 2021, 11, 476–483 CrossRef CAS.
- Y. Chen, P. Li, J. Zhou, C. T. Buru, L. Đorđević, P. Li, X. Zhang, M. M. Cetin, J. F. Stoddart, S. I. Stupp, M. R. Wasielewski and O. K. Farha, Integration of enzymes and photosensitizers in a hierarchical mesoporous metal-organic framework for light-driven CO2 reduction, J. Am. Chem. Soc., 2020, 142, 1768–1773 CrossRef CAS PubMed.
- V. Jain, S. Roy, P. Roy and P. P. Pillai, When design meets function: The prodigious role of surface ligands in regulating nanoparticle chemistry, Chem. Mater., 2022, 34, 7579–7597 CrossRef CAS.
- L. Jayasinghe, J. Wei, J. Kim, E. Lineberry and P. Yang, Particle on a Rod: Surface-tethered catalyst on CdS nanorods for enzymatically active nicotinamide cofactor generation, Nano Lett., 2024, 24, 13269–13276 CrossRef CAS PubMed.
- L. Tensi and A. Macchioni, Extremely fast NADH-regeneration using phosphonic acid as hydride source and iridium-pyridine-2-sulfonamidate catalysts, ACS Catal., 2020, 10, 7945–7949 CrossRef CAS.
- C. Trotta, G. M. Rodriguez, C. Zuccaccia and A. Macchioni, Electrochemical NADH regeneration mediated by pyridine amidate iridium complexes interconverting 1,4- and 1,6-NADH, ACS Catal., 2024, 14, 10334–10343 CrossRef CAS.
- L.-J. Zhao, Z. Yin, Y. Shi, W. Sun, L. Sun, H. Su, X. Sun, W. Zhang, L. Xia and C. Qi, A highly active Cp*Ir complex with an anionic N,N-donor chelate ligand catalyzes the robust regeneration of NADH under physiological conditions, Catal. Sci. Technol., 2021, 11, 7982–7991 RSC.
- L.-J. Zhao, C. Zhang, S. Zhang, X. Lv, J. Chen, X. Sun, H. Su, T. Murayama and C. Qi, High selectivity cofactor NADH regeneration organic iridium complexes used for high-efficiency chem-enzyme cascade catalytic hydrogen transfer, Inorg. Chem., 2023, 62, 17577–17582 CrossRef CAS PubMed.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Using nature's blueprint to expand catalysis with Earth-abundant metals, Science, 2020, 369, 6505 CrossRef.
- J. A. Kim, S. Kim, J. Lee, J. O. Baeg and J. Kim, Photochemical production of NADH using cobaloximes catalysts and visible-light energy, Inorg. Chem., 2012, 51, 8057–8063 CrossRef CAS.
- Y. H. Hong, M. Nilajakar, Y. M. Lee, W. Nam and S. Fukuzumi, Artificial photosynthesis for regioselective reduction of NAD(P)+ to NAD(P)H using water as an electron and proton source, J. Am. Chem. Soc., 2024, 146, 5152–5161 CrossRef CAS PubMed.
- C. L. Kwok, S. C. Cheng, P. Y. Ho, S. M. Yiu, W. L. Man, V. K. M. Au, P. K. Tsang, C. F. Leung, C. C. Ko and M. Robert, Precious-metal free photocatalytic production of an NADH analogue using cobalt diimine–dioxime catalysts under both aqueous and organic conditions, Chem. Commun., 2020, 56, 7491–7494 RSC.
- N. H. A. Besisa, K.-S. Yoon and M. Yamauchi, In situ electrochemical regeneration of active 1,4-NADH for enzymatic lactic acid formation via concerted functions on Pt-modified TiO2/Ti, Chem. Sci., 2024, 15, 3240–3248 RSC.
- M. Wang, X. Ren, M. Guo, J. Liu, H. Li and Q. Yang, Chemoselective NADH regeneration: the synergy effect of TiOx and Pt in NAD+ hydrogenation, ACS Sustainable Chem. Eng., 2021, 9, 6499–6506 CrossRef CAS.
- A. Bianco, M. Zaffagnini and G. Bergamini, Mediator-free NADH photochemical regeneration with the aid of the amino acid L-cysteine, Sustainable Energy Fuels, 2022, 6, 4393–4397 RSC.
- B. O. Burek, S. R. de Boer, F. Tieves, W. Zhang, M. van Schie, S. Bormann, M. Alcalde, D. Hotlmann, F. Hollmann, D. W. Bahnemann and J. Z. Bloh, Photoenzymatic hydroxylation of ethylbenzene catalyzed by unspecific peroxygenase: Origin of enzyme inactivation and the impact of light intensity and temperature, ChemCatChem, 2019, 11, 3093–3100 CrossRef CAS.
- M. M. Van Schie, W. Zhang, F. Tieves, D. S. Choi, C. B. Park, B. O. Burek, J. Z. Bloh, I. W. Arends, C. E. Paul, M. Alcalde and F. Hollmann, Cascading g-C3N4 and peroxygenases for selective oxyfunctionalization reactions, ACS Catal., 2019, 9, 7409–7417 CrossRef CAS.
- Y. Ma, Y. Wang, B. Wu, J. Zhou, S. Yang, F. Zhang, K. Luo, Y. Wang and F. Hollmann, Photobiocatalysis: More than just an interesting lab curiosity?, Chem Catal., 2024, 4, 101077 CrossRef CAS.
- J. Barber, Photosynthetic energy conversion: natural and artificial, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- M. F. Hohmann-Marriott and R. E. Blankenship, Evolution of photosynthesis, Annu. Rev. Plant Biol., 2011, 62, 515–548 CrossRef CAS PubMed.
- Y. Sun, W. Li, Z. Wang, J. Shi and Z. Jiang, General framework for enzyme-photo-coupled catalytic system toward carbon dioxide conversion, Curr. Opin. Biotechnol., 2022, 73, 67–73 CrossRef CAS PubMed.
- Y. Tian, Y. Zhou, Y. Zong, J. Li, N. Yang, M. Zhang, Z. Guo and H. Song, Construction of functionally compartmental inorganic photocatalyst–enzyme system via imitating chloroplast for efficient photoreduction of CO2 to formic acid, ACS Appl. Mater. Interfaces, 2020, 12, 34795–34805 CrossRef CAS.
- N. Zhang, S. Trépout, H. Chen and M.-H. Li, AIE polymer micelle/vesicle photocatalysts combined with native enzymes for aerobic photobiocatalysis, J. Am. Chem. Soc., 2023, 145, 288–299 CrossRef CAS.
- Y. Bai, P. Luan, Y. Bai, R. N. Zare and J. Ge, Enzyme-photo-coupled catalysis in gas-sprayed microdroplets, Chem. Sci., 2022, 13, 8341–8348 RSC.
- K. Li, H. Zhou, X. Tong and H. Yang, Enhanced photobiocatalytic cascades at pickering droplet interfaces, J. Am. Chem. Soc., 2024, 146, 17054–17065 CrossRef CAS.
- M. Pera-Titus, L. Leclercq, J. M. Clacens, F. De Campo and V. Nardello-Rataj, Pickering interfacial catalysis for biphasic systems: From emulsion design to green reactions, Angew. Chem., Int. Ed., 2015, 54, 2006–2021 CrossRef CAS.
- L. Ni, C. Yu, Q. Wei, D. Liu and J. Qiu, Pickering emulsion catalysis: Interfacial chemistry, catalyst design, challenges, and perspectives, Angew. Chem., Int. Ed., 2022, 61, e202115885 CrossRef CAS PubMed.
- M. Zhang, R. Ettelaie, T. Li, J. Yang, L. Dong, N. Xue, B. P. Binks, F. Cheng and H. Yang, Pickering emulsion droplets and solid microspheres acting synergistically for continuous-flow cascade reactions, Nat. Catal., 2024, 7, 295–306 CrossRef CAS.
- L. Velasco-Garcia and C. Casadevall, Bioinspired photocatalytic systems towards compartmentalized artificial photosynthesis, Commun. Chem., 2023, 6, 263 CrossRef CAS.
- Y. Okamoto, V. Kohler and T. R. Ward, An NAD(P)H-dependent artificial transfer hydrogenase for multienzymatic cascades, J. Am. Chem. Soc., 2016, 138, 5781–5784 CrossRef CAS.
- A. Kirschning, On the evolution of coenzyme biosynthesis, Nat. Prod. Rep., 2022, 39, 2175–2199 RSC.
- R. J. Mayer and J. Moran, Metal ions turn on a stereoselective nonenzymatic reduction of keto acids by the coenzyme NADH, Chem, 2024, 10, 2564–2576 CAS.
- S. Zhang, J. Shi, Y. Chen, Q. Huo, W. Li, Y. Wu, Y. Sun, Y. Zhang, X. Wang and Z. Jiang, Unraveling and manipulating of NADH oxidation by photogenerated holes, ACS Catal., 2020, 10, 4967–4972 CrossRef CAS.
- I. Zachos, M. Doring, G. Tafertshofer, R. C. Simon and V. Sieber, carba nicotinamide adenine dinucleotide phosphate: robust cofactor for redox biocatalysis, Angew. Chem., Int. Ed., 2021, 60, 14701–14706 CrossRef CAS PubMed.
- A. Alvarez-Martin, S. Trashin, M. Cuykx, A. Covaci, K. De Wael and K. Janssens, Photodegradation mechanisms and kinetics of Eosin-Y in oxic and anoxic conditions, Dyes Pigm., 2017, 145, 376–384 CrossRef CAS.
- J. R. Fisher and D. J. Cole-Hamilton, Photochemical hydrogen production from ascorbic acid catalysed by tris(2,2’-bipyridyl) ruthenium (II) and hydridotris(triethylphosphine)-palladium(II), J. Chem. Soc., Dalton Trans., 1984, 809–813 RSC.
- F. Camara, J. S. Aguirre-Araque, J. Fortage and M. N. Collomb, Enhancing the stability of photocatalytic systems for hydrogen evolution in water by using a tris-phenyl-phenanthroline sulfonate ruthenium photosensitizer, Sustainable Energy Fuels, 2024, 8, 1457–1472 RSC.
- X. Yang and D. Wang, Photocatalysis: From fundamental principles to materials and applications, ACS Appl. Energy Mater., 2018, 1, 6657–6693 CrossRef CAS.
- S. Chen and L. W. Wang, Thermodynamic oxidation and reduction potentials of photocatalytic semiconductors in aqueous solution, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- H. Baek, S. Kang, J. Heo, S. Choi, R. Kim, K. Kim, N. Ahn, Y. G. Yoon, T. Lee, J. B. Chang, K. S. Lee, Y. G. Park and J. Park, Insights into structural defect formation in individual InP/ZnSe/ZnS quantum dots under UV oxidation, Nat. Commun., 2024, 15, 1671 CrossRef CAS PubMed.
- K. Sun, Y. Kuang, E. Verlage, B. S. Brunschwig, C. W. Tu and N. S. Lewis, Sputtered NiOx films for stabilization of p+n-InP photoanodes for solar-driven water oxidation, Adv. Energy Mater., 2015, 5, 1402276 CrossRef.
- C. Li, T. Hisatomi, O. Watanabe, M. Nakabayashi, N. Shibata, K. Domen and J. J. Delaunay, Positive onset potential and stability of Cu2O-based photocathodes in water splitting by atomic layer deposition of a Ga2O3 buffer layer, Energy Environ. Sci., 2015, 8, 1493–1500 RSC.
- B. Koo, S. Byun, S. W. Nam, S. Y. Moon, S. Kim, J. Y. Park, B. T. Ahn and B. Shin, Reduced graphene oxide as a catalyst binder: Greatly enhanced photoelectrochemical stability of Cu(In,Ga)Se2 photocathode for solar water splitting, Adv. Funct. Mater., 2018, 28, 1705136 CrossRef.
- S. Huang, Y. Lin, J. Yang, X. Li, J. Zhang, J. Yu, H. Shi, W. Wang and Y. Yu, Enhanced photocatalytic activity and stability of semiconductor by Ag doping and simultaneous deposition: The case of CdS, RSC Adv., 2013, 3, 20782–20792 RSC.
- S. He, A. Ni, S. T. Gebre, R. Hang, J. R. McBride, A. L. Kaledin, W. Yang and T. Lian, Doping of colloidal nanocrystals for optimizing interfacial charge transfer: A double-edged sword, J. Am. Chem. Soc., 2024, 146, 24925–24934 CrossRef CAS PubMed.
- S. Roy, S. Roy, A. Rao, G. Devatha and P. P. Pillai, Precise nanoparticle-reactant interaction outplays ligand poisoning in visible-light photocatalysis, Chem. Mater., 2018, 30, 8415–8419 CrossRef CAS.
- M. Rahaman, C. Pulignani, M. Miller, S. Bhattacharjee, A. B. M. Annuar, R. R. Manuel, I. A. C. Pereira and E. Reisner, Solar-driven paired CO2 reduction–alcohol oxidation using semiartificial suspension, photocatalyst sheet, and photoelectrochemical devices, J. Am. Chem. Soc., 2025, 147, 8168–8177 CrossRef CAS PubMed.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Interfacing nature's catalytic machinery with synthetic materials for semi-artificial photosynthesis, Nat. Catal., 2018, 13, 890–899 CAS.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Bacteria photosensitized by intracellular gold nanoclusters for solar fuel production, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed.
- S. Cestellos-Blanco, S. Louisia, M. B. Ross, Y. Li, N. E. Soland, T. C. Detomasi, J. N. Cestellos Spradlin, D. K. Nomura and P. Yang, Toward abiotic sugar synthesis from CO2 electrolysis, Joule, 2022, 6, 2304–2323 CrossRef CAS.
- A. Sánchez-Iglesias, J. Kruse, A. Chuvilin and M. Grzelczak, Coupling plasmonic catalysis and nanocrystal growth through cyclic regeneration of NADH, Nanoscale, 2021, 13, 15188–15192 RSC.
- A. Bianco, A. H. McMillan, C. Giansante and G. Bergamini, NADH photoregeneration in a fully automated microfluidic setup, Energy Fuels, 2024, 38, 12078–12086 CrossRef CAS.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjose-Orduna, D. Pintossi, T. D. Hartog and T. Noël, Automated self-optimization, intensification, and scale-up of photocatalysis in flow, Science, 2024, 383, 6681 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |