
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reductive cyclotrimerization of CO and isonitriles with a highly reactive CaI synthon†
Stefan Thum ,
Jonathan Mai,
Marcel A. Schmidt,
Jens Langer and
Sjoerd Harder*
,
Jonathan Mai,
Marcel A. Schmidt,
Jens Langer and
Sjoerd Harder*
Inorganic and Organometallic Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstrasse 1, 91058 Erlangen, Germany. E-mail: sjoerd.harder@fau.de
First published on 27th May 2025
Abstract
Whereas the small molecule activation with β-diketiminate (BDI)MgI complexes of type (BDI)Mg–Mg(BDI) is extensively investigated, lack of similar CaI reagents prevents studies on CaI reactivity. Herein, we report on small molecule activation with dinitrogen complexes of type (BDI)Ca(N2)Ca(BDI) which acts as CaI synthon by release of N2 and two electrons. Reaction of [(BDI*)Ca(THP)]2(N2) with CO led to formation of a deltate product with the cyclic C3O32− dianion (1); BDI* = HC[C(Me)N(DIPeP)]2 (DIPeP = 2,6-(Et2CH)-phenyl) and THP = tetrahydropyran. Reaction with the isonitrile CyN![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C gave as the major product a complex with the triimino deltate C3(NCy)32− dianion (2) which is unstable in solution. Isolation of the side-product (BDI*)2Ca·(CN–Cy) (3) indicates dynamic ligand exchange and Schlenk equilibria. Variation of the isonitrile reagent led to isolation of (BDI*)2Ca·(CN–R) (4: R = xylyl, 5: R = tBu). Crystal structures and NMR studies in solution are discussed for complexes 1–5. We also report an extensive DFT study on the reductive trimerization of MeNC with this CaI synthon. The key intermediate (BDI)Ca(MeNC)Ca(BDI) contains dianionic MeNC2−. Contrary to expectation, C–C coupling does not proceed by nucleophilic attack at a second MeNC reagent. Electron transfer results in two bridging MeNC˙− radical anions. This rare singlet diradicaloid reacts further by radical coupling to [MeNC–CNMe]2−. Differences with MgI reactivity are discussed.
C gave as the major product a complex with the triimino deltate C3(NCy)32− dianion (2) which is unstable in solution. Isolation of the side-product (BDI*)2Ca·(CN–Cy) (3) indicates dynamic ligand exchange and Schlenk equilibria. Variation of the isonitrile reagent led to isolation of (BDI*)2Ca·(CN–R) (4: R = xylyl, 5: R = tBu). Crystal structures and NMR studies in solution are discussed for complexes 1–5. We also report an extensive DFT study on the reductive trimerization of MeNC with this CaI synthon. The key intermediate (BDI)Ca(MeNC)Ca(BDI) contains dianionic MeNC2−. Contrary to expectation, C–C coupling does not proceed by nucleophilic attack at a second MeNC reagent. Electron transfer results in two bridging MeNC˙− radical anions. This rare singlet diradicaloid reacts further by radical coupling to [MeNC–CNMe]2−. Differences with MgI reactivity are discussed.
Introduction
Carbon monoxide (CO) and isoelectronic isonitriles (RNC) are important C1 feedstocks for the preparation of fine chemicals.1–5 Especially the bulk reagent CO is a key building block in numerous industrial processes mediated by transition metal (TM) complexes such as the Cativa process for manufacturing acetic acid6 and the Fischer–Tropsch process for liquid hydrocarbons.7 Recent years have seen rapid growth of the field of main-group metal complexes mimicking TM complexes in their reactivity.8 Main group metal-mediated C–C bond formation is of particular interest due to the metal's high natural abundance, low toxicity and eco-friendly reputation compared to late TMs. Within this field, we are particularly interested in low oxidation state s-block chemistry.9–12 Reductive C–C homologation of CO by low oxidation state magnesium complexes [(ArBDI)Mg]2 (ArBDI = HC[C(Me)N(Ar)]2; Ar = aryl) is known to give deltates [C3O3]2− (I, Scheme 1),13,14 squarates [C4O4]2− (II),15 benzenehexolates [C6O6]2− (III),16 or more recently, reductive dimerization of CO to ethynediolates [O–CC–O]2− (IV).17–20 Product selectivity strongly depends on ligand bulk, the presence of catalytic amounts of Mo(CO)6 and activation of the Mg–Mg bond by asymmetric solvation and polarization (vide infra). There is also rapid development of low-valent p-block chemistry: heterobimetallic alkali metal aluminyl complexes have been investigated for CO homologation.21,22
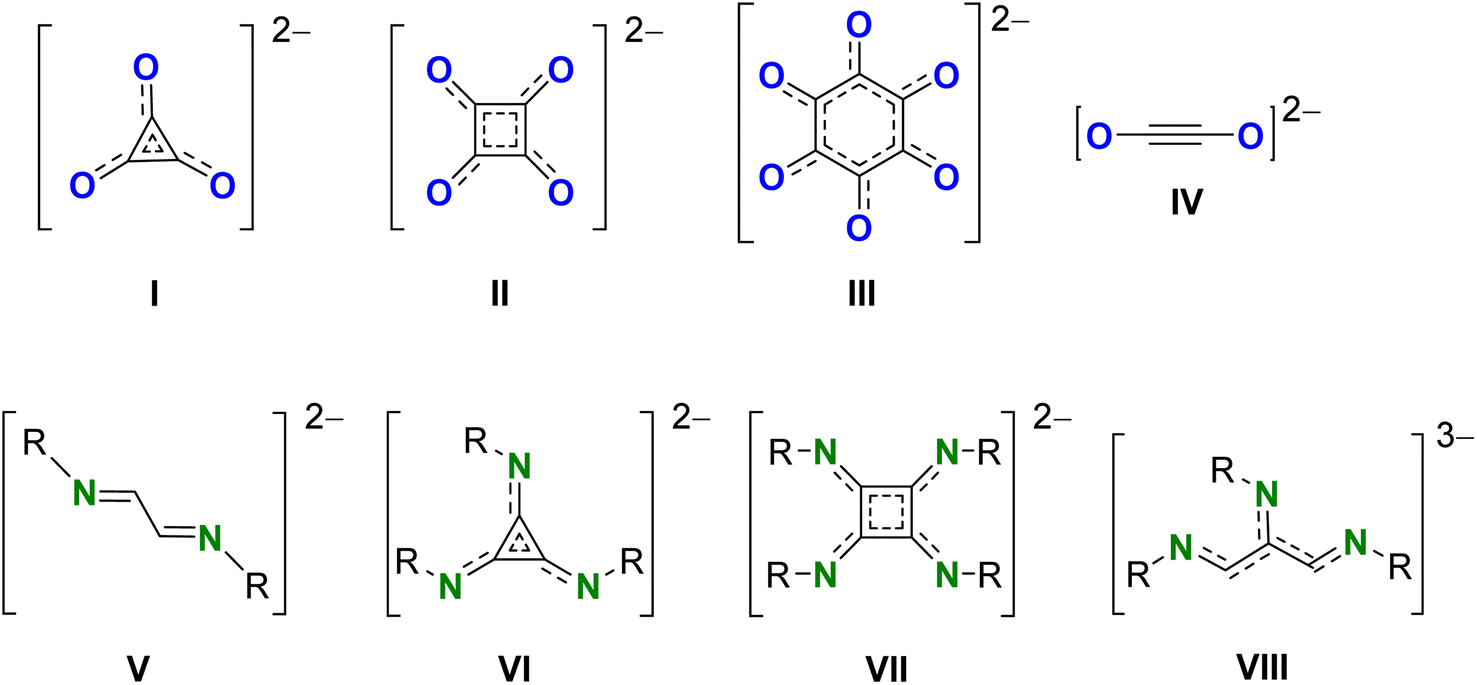 | ||
| Scheme 1 Reductive coupled CO and isonitrile R–NC products (I–VIII). | ||
As isonitriles are isoelectronic to CO, also the activation of this important feedstock with low oxidation state main group complexes is emerging.23 Reductive dimerization of isonitriles to corresponding 1,4-diazabutadiene-2,3-diyl [RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CNR]2− (V) has been reported for low-valent dialanes,24–26 digallanes,27 MgI dimers,28 and GeI dimers.29 However, examples for the reductive cyclomerization of isonitriles are limited. Low-valent transition metal complexes have shown reductive isonitrile coupling to triimino deltate C3(NR)32− (VI), squaramidinate [C4N4R4]2– (VII) or linear higher homologues (e.g. VIII).26,30–32 However, to the best of our knowledge, in main group metal chemistry formation of cyclic products could only be achieved with low-valent AlII reagents. Reaction of a dialumane system with isonitriles gave reductive trimerization to triimino deltate C3(NR)32− (VI).26
C–CNR]2− (V) has been reported for low-valent dialanes,24–26 digallanes,27 MgI dimers,28 and GeI dimers.29 However, examples for the reductive cyclomerization of isonitriles are limited. Low-valent transition metal complexes have shown reductive isonitrile coupling to triimino deltate C3(NR)32− (VI), squaramidinate [C4N4R4]2– (VII) or linear higher homologues (e.g. VIII).26,30–32 However, to the best of our knowledge, in main group metal chemistry formation of cyclic products could only be achieved with low-valent AlII reagents. Reaction of a dialumane system with isonitriles gave reductive trimerization to triimino deltate C3(NR)32− (VI).26
The reduction of CO or isonitriles with low-valent alkaline-earth (Ae) metal reagents is so far limited to reactivity studies on MgI complexes like [(ArBDI)Mg]2. This limitation is mainly due to the fact that the low oxidation chemistry of the early main group metals is hardly developed. Apart from complexes of BeI,33,34 Be0,35,36 and Mg0,37–39 there are currently no examples of heavier AeI or Ae0 reagents which are notoriously unstable and highly reactive. Attempts to prepare a [(ArBDI)Ca]2 reagent led either to reduction of the aromatic solvent or of the inert gas N2 to give [(ArBDI)Ca]2(C6H6) or [(ArBDI)Ca]2(N2), respectively.40 However, it was found that such complexes react like a strongly reducing synthon for [(ArBDI)Ca]2 by releasing C6H6 or N2 and two electrons.40–43 These complexes therefore enable reduction chemistry with well-defined heavier Ae metal reagents. We herein report the reduction of CO and isonitriles (R–NC) with different substituents R with the CaI synthon [(ArBDI)Ca]2(N2). Comparing the outcome with well-established MgI reduction chemistry will demonstrate the influence of the Ae metal centre in such reactions. Our findings will be supported by computational studies.
Results and discussion
Reaction with CO
In our reactivity studies we used the CaI synthon [(BDI*)Ca(THP)]2(N2); BDI* = HC[C(Me)N(DIPeP)]2 (DIPeP = 2,6-(Et2CH)-phenyl) and THP = tetrahydropyran (Scheme 2a). In contrast to the THF adduct, which decomposes slowly at room temperature, this THP adduct shows increased stability and can be easily obtained in crystalline purity.40 The THP ligand also affects the selectivity of the conversion. Reaction of the non-solvated complex [(BDI*)Ca]2(N2) with CO in methylcyclohexane-d14 led at −85 °C already to rapid conversion (Fig. S30†). However, this reaction is not very selective and many products formed. In contrast, the reaction of the THP adduct is much more selective. | ||
| Scheme 2 (a) Reaction of [(BDI*)Ca(THP)]2(N2) with CO. (b) Reaction of MgI complexes with CO after prior polarization of Mg–Mg bond.13,14 DEP = 2,6-Et-phenyl, DIPP = 2,6-iPr-phenyl, DMAP = 4-Me2N-pyridine, TMC = :C[N(Me)CMe]2. (c) Reaction of a heterobimetallic Mg/K dinitrogen complex with CO.18 (d) Reaction of a heterobimetallic Mg/Na complex with CO.17 | ||
Reacting a red-brown suspension of the CaI synthon [(BDI*)Ca(THP)]2(N2) in pentane at −85 °C with 1 bar of CO led upon warming to room temperature to an orange solution and precipitation of [(BDI*)Ca]4(THP)3(C3O3)2 (1) in form of microcrystalline white solid. After concentration and cooling to −25 °C, colourless crystals suitable for X-ray diffraction analysis were obtained. To monitor the formation of 1 and trap possible intermediates, the reaction was repeated in methylcyclohexane-d14. However, 1H NMR monitoring only showed quantitative formation of deltate complex 1, indicating that intermediates are likely to be short-lived, transient species.44 The 1H NMR spectrum of the reaction mixture shows two sets of signals for chemically inequivalent BDI* ligands assigned to isolated 1 (Fig. S29†). Despite the high selectivity of the reaction, the very good solubility induced by the flexible Et2CH-groups allowed isolation of the Ca deltate complex 1 in only 20% crystalline yield. Poor yields to high product solubility of complexes with the BDI* ligand is a known problem we already observed in various investigations.41,42,45
Product 1 crystallized in the centrosymmetric triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two independent, but similar [(BDI*)Ca]4(THP)3(C3O3)2 aggregates in the asymmetric unit (Fig. 1). The molecular structure consists of two deltate dianions C3O32− bound to two bridging [(BDI*)Ca]+ fragments and two terminal [(BDI*)Ca]+ units. The Ca atoms in the terminal [(BDI*)Ca]+ units are additionally saturated by either one or two THP ligands. Both deltate dianions are planar with C–C–C angles close to 60° (59.8(2)–60.5(2)°). The C–C bond lengths between 1.397(3) Å and 1.410(3) Å and C–O bond lengths ranging from 1.276(3) Å to 1.289(7) Å are in agreement with the expected delocalization of π-electrons over the C3O3 core. Reports about the reductive trimerization of CO to C3O32− are limited to UIII46 and MgI13,14 complexes. The aromatic character of the C3 cycle in 1 is similar to the deltate dianions in (BDI)Mg complexes with C–C bond lengths (1.391(2)–1.402(2) Å;13 1.396(3)–1.399(3) Å;13 1.385(5)–1.399(6) Å)14 and C–O bond lengths (1.269(2)–1.288(2) Å;13 1.276(3)–1.280(3) Å;13 1.273(5)–1.278(5) Å)14 in comparable ranges. However, the highly symmetric deltate structure in 1 differs from the irregular deltate structure in an uranium complex which is caused by different coordination modes.46
with two independent, but similar [(BDI*)Ca]4(THP)3(C3O3)2 aggregates in the asymmetric unit (Fig. 1). The molecular structure consists of two deltate dianions C3O32− bound to two bridging [(BDI*)Ca]+ fragments and two terminal [(BDI*)Ca]+ units. The Ca atoms in the terminal [(BDI*)Ca]+ units are additionally saturated by either one or two THP ligands. Both deltate dianions are planar with C–C–C angles close to 60° (59.8(2)–60.5(2)°). The C–C bond lengths between 1.397(3) Å and 1.410(3) Å and C–O bond lengths ranging from 1.276(3) Å to 1.289(7) Å are in agreement with the expected delocalization of π-electrons over the C3O3 core. Reports about the reductive trimerization of CO to C3O32− are limited to UIII46 and MgI13,14 complexes. The aromatic character of the C3 cycle in 1 is similar to the deltate dianions in (BDI)Mg complexes with C–C bond lengths (1.391(2)–1.402(2) Å;13 1.396(3)–1.399(3) Å;13 1.385(5)–1.399(6) Å)14 and C–O bond lengths (1.269(2)–1.288(2) Å;13 1.276(3)–1.280(3) Å;13 1.273(5)–1.278(5) Å)14 in comparable ranges. However, the highly symmetric deltate structure in 1 differs from the irregular deltate structure in an uranium complex which is caused by different coordination modes.46
 | ||
| Fig. 1 Molecular structure of [(BDI*)Ca]4(THP)3(C3O3)2 (1). The H atoms and Et2CH-substituents have been omitted for clarity. | ||
One remarkable difference between reduction of CO with MgI reagents or the CaI synthon is the considerable higher reactivity of the latter. Symmetric MgI complexes of type (BDI)MgMg(BDI) do not react with CO and need to be activated with a Lewis base (LB) (Scheme 2b).13,14 Whereas addition of two LB ligands does result in elongation of the Mg–Mg bond, it also gives steric congestion, preventing Mg–CO coordination and further reactivity. However, addition of one equivalent of LB was shown to give a polarized, activated Mg–Mg bond (BDI)Mgδ−–δ+Mg(LB)(BDI) but leaves room for CO coordination at the second Mg centre. In contrast, the non-solvated CaI synthon [(BDI*)Ca]2(N2) does not need any activation and reacts instantaneously with CO. The role of the THP ligands is to control the selectivity of the reaction to deltate formation (1). The observed deltate formation contrasts with the CO reactivity of heterobimetallic Mg/K dinitrogen complex which acts as a synthon for a MgI radical and readily reduced CO to ethynediolate [O–CC–O]2− (Scheme 2c).18 It also contrasts with the MgI reactivity of Hill's heterobimetallic Mg/Na complex which similarly led to [O–CC–O]2− formation (Scheme 2d).17 Noteworthy, Jones and coworkers recently introduced a highly reactive heterobimetallic Ca/K dinitrogen complex that instantly reacts at room temperature with CO but only gave intractable product mixtures.47
Reaction with isonitriles (R–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C)
C)
Dropwise addition of a hexanes solution of cyclohexyl isonitrile (Cy–NC, 3.2 equivalents) to a red-brown suspension of [(BDI*)Ca(THP)]2(N2) in hexanes at −85 °C resulted upon warming to 0 °C in a dark yellow-brown solution. The 1H NMR spectrum of the crude reaction mixture in benzene-d6 showed relatively selective formation of one major product with one major sharp singlet for the methine in the ligand backbone among smaller signals of minor side products (Fig. S31†). Changing the stoichiometry did not improve selectivity. The product could be isolated in the form of orange crystals suitable for X-ray diffraction analysis by storing a concentrated hexanes solution at −25 °C. This revealed reductive trimerization of Cy–NC to the triimino deltate dianion C3(NCy)32− and gave [(BDI*)Ca]2(C3(NCy)3)(THP) (2) in 27% isolated yield (Scheme 3a). A second crop of crystals gave a mixture of 2 and (BDI*)2Ca·(CN–Cy) (3). The latter side-product could be obtained in 10% isolated yield by isolation of a third crop of crystals. The formation of complex 3 shows that ligand exchange by Schlenk equilibria are operative. The reactant stoichiometry changes the product selectivity. Reaction of [(BDI*)Ca(THP)]2(N2) with only two equivalents gave less of the triimino deltate product 2 but more of 3 (Fig. S35†). Alternatively, complex 3 could be obtained in a high yield of 81% by reacting the homoleptic complex (BDI*)2Ca with one equivalent of Cy–NC in benzene (Scheme 3b).
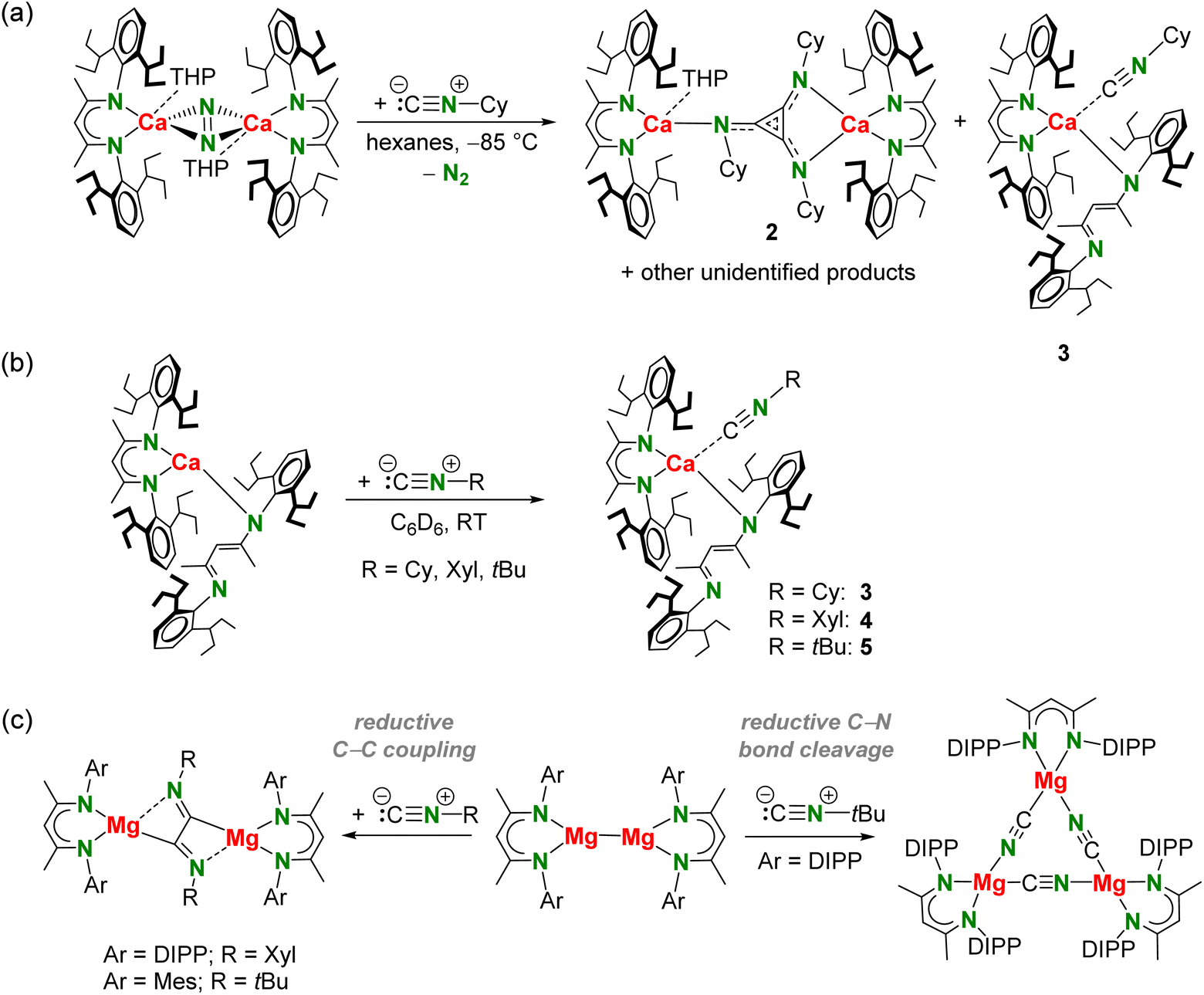 | ||
| Scheme 3 (a) Reaction of CaI synthon [(BDI*)Ca(THP)]2(N2) with Cy–NC. (b) Reaction of (BDI*)2Ca with R–NC. (c) Reactivity of [(ArBDI)Mg]2 with isonitrile R–NC depending on substituents Ar and R.28 Xyl = 2,6-dimethyl-phenyl, Mes = 2,4,6-trimethyl-phenyl. | ||
The activation of Cy–NC with the CaI synthon stands in contrast to reported isonitrile reactivity with MgI complexes. The course of the reaction with low oxidation state Mg complexes of type [(ArBDI)Mg]2 depends on the steric bulk of the Ar substituent and on the organic moiety R of the R–NC reagent (Scheme 3c).28 A bulky DIPP substituent and bulky tBu groups resulted in a trimeric Mg cyanide complex which was formed by reductive R–NC bond cleavage. Small Ar (Ar = Mes) and bulky R (R = tBu) as well as the combination of bulky Ar (Ar = DIPP) and smaller R (R = Xyl) groups led to reductive dimerization, i.e. [RNC–CNR]2− flanked by two [(ArBDI)Mg]+ units.
Since product formation by reduction of R–NC with MgI dimers strongly depends on the steric bulk of the Ar and R substituents,28 the influence of the substituent R in reduction of R–NC with the CaI synthon was investigated. Addition of a Xyl–NC solution in hexanes to a red-brown suspension of [(BDI*)Ca(THP)]2(N2) in hexanes at −85 °C resulted upon warming to 0 °C in a dark purple solution. 1H NMR spectroscopy indicated formation of several products (Fig. S37†) and only (BDI*)2Ca·(CN–Xyl) (4) could be isolated in 12% crystalline yield. Using bulky tBu–NC resulted in a more selective reaction outcome with one major backbone signal for the methine in the ligand backbone but prolonged crystallization times led to formation of several side-products (Fig. S38 and S39†) and only (BDI*)2Ca·(CN–tBu) (5) could be isolated. Complexes 4 and 5 could be isolated in high yields (85–91%) from a solution of homoleptic complex (BDI*)2Ca and the corresponding R–NC in benzene (Scheme 3b). All complexes 2–5 have been fully characterized by NMR methods, elemental analyses and single crystal X-ray diffraction.
The crystal structure of 2 shows a C3(NCy)32− dianion that is bridging two [(BDI*)Ca]+ fragments in an asymmetric η1:η2 fashion (Fig. 2a). A similar coordination mode was recently found in an aluminium triimino deltate complex.26 The C3(NCy)32− dianion in 2 is disordered over an inversion centre in the middle of the molecule (Fig. S45†). One of the Ca centres is additionally coordinated by one THP molecule. The C3(NR)3 unit, a [3]radialene derivative, in 2 is of great interest as building block for polymers, organic conductors and ferromagnets.48–51 The expected aromatic character of the dianion is confirmed by inspection of the bond distances in C3(NCy)32−. The C–C bond distances ranging from 1.397(8)–1.423(8) Å and C–N bond distances from 1.334(7)–1.353(8) Å are in between corresponding double and single bonds,52 thus supporting electronic delocalization over the entire C3N3 core. The bond distances for the triimino deltate dianion are in similar ranges to those observed in the dialumane system for C3(NtBu)32− (C–C: 1.381(4)–1.402(4) Å, C–N: 1.341(3)–1.366(3) Å)26 and in the vanadium complex of C3(NXyl)32− (C–C: 1.388(5)–1.427(5) Å, C–N: 1.324(5)–1.369(4) Å).31 The C3 ring in 2 exhibits an almost perfect triangular geometry with internal C–C–C angles ranging from 59.1(4)° to 61.0(4)°, similar to those reported for a dialumane system (59.3(2)–60.8(2)°).26 The Ca complex 2 represents the first reductive cyclo-trimerization of an isonitrile promoted by an s-block metal complex. Despite its flexible Et2CH-groups, complex 2 shows only moderate solubility in aromatic and aliphatic solvents. Moreover, 2 is not stable in solution and decomposes at room temperature to unidentified products and meaningful 13C NMR data could not be obtained (Fig. S32 and S33†). The 1H NMR spectrum of 2 shows one characteristic signal for the methine in the ligand backbone indicating rapid fluctuation of the THP molecule between the two Ca centres in solution. Cooling the sample to −20 °C led to appearance of several new methine backbone signals (Fig. S34†) indicating formation of various species. Warming to room temperature resulted in coalescence to one signal, showing that this process is reversible. However, further heating to +80 °C gave irreversible decomposition of 2 in various unidentified species. These combined observations show that the products are not very stable and in dynamic exchange.
 | ||
| Fig. 2 (a) Crystal structure of [(BDI*)Ca]2(C3(NCy)3)(THP) (2) and a detailed view of the triimino deltate dianion C3(NCy)32− with bond distances in Å. H atoms and Et2CH-groups have been omitted for clarity. (b) Comparison of the crystal structures of (BDI*)2Ca40 with the isonitrile adducts 3–5. H atoms and the Et groups of the Et2CH substituents have been omitted for clarity. | ||
The crystal structures of (BDI*)2Ca·(CN–R) (R = Cy, Xyl, tBu) (3–5, Fig. 2b) have in common that one BDI* ligand coordinates in η2-fashion while the other one is only η1-coordinated due to the steric demand of the bulky DIPeP-substituents. As the reported crystal structure of (BDI*)2Ca40 shows a similar combination of η1- and η2-coordination, this comes as no surprise. Comparing the four structures shows remarkable similarities (Fig. 2b, Table 1). Due to the needle-like form of the isonitrile ligands, coordination to Ca only needs minimal space. In all complexes 3–5, the isonitrile ligand hovers above the bidentate N,N-chelating BDI* ligand. Coordination of an additional isonitrile ligand hardly affects the Ca–N bond distances in (BDI*)2Ca. Elongations between 0.022 and 0.075 Å are observed (Table 1). The Ca–C bond distances range from 2.578(2) Å for the Cy–NC complex (3) to 2.664(2) Å in the Xyl–NC complex (4). Since the Ca–C bond in 5 with the bulky tBu–NC ligand falls in between (2.599(2) Å) the poorer donor ability of Xyl–NC may be due to differences in electronic effects, i.e. partial delocalization of π-electron density of the CN bond in the xylyl ring.
| Complex | (BDI*)2Ca | (BDI*)2Ca·(CN–Cy) (3) | (BDI*)2Ca·(CN–Xyl) (4) | (BDI*)2Ca·(CN–tBu) (5) |
|---|---|---|---|---|
| a Average value.b Large standard deviation due to disorder. | ||||
| Ca–N (η2–BDI*)a | 2.330 | 2.355 | 2.354 | 2.352 |
| Ca–N (η1–BDI*) | 2.290(1) | 2.365(2) | 2.362(1) | 2.340(1) |
| Ca–C (isonitrile) | — | 2.578(2) | 2.664(2) | 2.599(2) |
| CN |
— | 1.147(2) | 1.156(2) | 1.180(8)b |
Solutions of complexes 3–5 in C6D6 show 1H NMR spectra (Fig. S11, S17 and S23†) which show strong similarity to that of (BDI*)2Ca. However, all signals are shifted and partially broadened which indicates that there is in solution dynamic coordination of the isonitrile ligands.
Computational and mechanistic studies
The complete structures of the Ca deltate 1 and Ca triimino deltate 2 were optimized at the B3PW91/def2tzvp//def2svp level of theory. The calculated geometries of the full aggregates fit reasonably well with the crystal structures (Fig. S49 and S50†), indicating a sufficient level of theory. Natural Population Analysis (NPA) (Fig. S51 and S52†) confirms that both complexes are ionically bound (NPA charges in 1: Ca +1.82, deltate anion −1.86; NPA charges in 2: Ca +1.79, triimino deltate −1.78). The C–C bonds in the C3-ring in the deltate anion C3O32− show Wiberg Bond Indices (WBI's) between 1.18–1.22, supporting delocalized single/double bonds (Fig. S53†). The C–O bonds are part of the delocalized system and show WBI's in the range of 1.23–1.29. A similar bonding situation is found in the triimino deltate C3(NCy)32− (Fig. S54†) with C–C bonds WBI's in the range of 1.15–1.20, and WBI's for deltate C–N bonds in the range of 1.25–1.38. Atoms-In-Molecule (AIM) analysis for Ca deltate 1 shows that the C3O32− dianion is not only bound to the Ca2+ cations but is also integrated in a network of O⋯H–C bonding interaction with the BDI* and THP ligands (Fig. S56†). Although such non-classical hydrogen bonding was once ridiculed,53 these weak interactions have been shown important in determining structure and reactivity.54 Such non-classical hydrogen bonds are enforced by high electron density on the acceptor side.55 As the O atoms in C3O32− carry most of the negative charge (NPA charges range from −0.88 to −0.93), the O⋯H–C bonding network in 1 should be considered important.Although there are several computational studies on reductive CO homologation with low-valent MgI complexes,13,14,16 reductive trimerization of isonitrile with s-block metal reagents is so far unexplored. To gain further insights in the mechanism of isonitrile reduction with a CaI synthon, a DFT study at the B3PW91-GD3BJ/def2tzvp//B3PW91-GD3BJ/def2svp level of theory was conducted. Due to size limitations the DIPeP-substituents in the BDI* ligand have been replaced with smaller DIPP-substituents and Cy–NC was modelled with Me–NC.
The energy profile for reaction of (DIPPBDI)Ca(N2)Ca(DIPPBDI) with MeNC is shown in Scheme 4. The N2 complex reacts as a synthon for (DIPPBDI)Ca–Ca(DIPPBDI) by releasing N2 and transferring two electrons to the isonitrile (A–B). This formal N22−/MeNC to N2/[MeNC]2− exchange is exothermic by ΔH = −6.3 kcal mol−1 (ΔG = −4.4 kcal mol−1). Note that reaction of the CaI complex (DIPPBDI)Ca–Ca(DIPPBDI) with MeNC would be considerably more exothermic: ΔH = −40.6 kcal mol−1, ΔG = −25.9 kcal mol−1. The NPA charge on the bridging isonitrile in B is −1.60, reflecting its dianionic state (Fig. S64†). The [MeNC]2− anion is bent (C–N–C 121.0°) and bridges asymmetrically between the Ca2+ cations with two unequal Ca–C contacts and one Ca–N bond (Fig. 3a). This differs from the reaction of (MesBDI)Mg–Mg(MesBDI) with CO which is endothermic by +9.4 kcal mol−1 and forms a product with a symmetrically bridging CO2− anion between the Mg2+ centres (side-on bonding with two equal Mg–C and two equal Mg–O contacts).16 However, when the bulkier DIPPBDI is used and one of the Mg centres is solvated with a N-heterocyclic carbene (NHC),13 this reaction becomes slightly exothermic (ΔH = −7.0 kcal mol−1) and CO bridges asymmetrically like the [MeNC]2− anion in B (see inset in Scheme 4). For comparison, the reduction of ArNC with (DIPPBDI)AlI was calculated to be endothermic by more than 10 kcal mol−1.56 The exothermic reduction of MeNC by (DIPPBDI)Ca(N2)Ca(DIPPBDI) is therefore a demonstration of the considerable reducing power of this N2 complex.
 | ||
| Scheme 4 Reductive coupling of isonitrile. Energy profile for triimino deltate formation calculated at the B3PW91-GD3BJ/def2tzvp//B3PW91-GD3BJ/def2svp level of theory for a model system with L = DIPPBDI and MeNC. ΔH in kcal mol−1. Between brackets: ΔG (298 K) in kcal mol−1. Selected bond lengths are given in Å. Inset: For comparison, reductive CO dimerization with (DIPPBDI)Mg–Mg(DIPPBDI) activated with a N-heterocyclic carbene (NHC) follows a different pathway.13 | ||
 | ||
| Fig. 3 (a) Calculated intermediates on the pathway for reductive trimerization of MeNC with (DIPPBDI)Ca(N2)Ca(DIPPBDI). Selected bond distances in Å. (b) Atoms-In-Molecules (AIM) analysis for the singlet diradicaloid D showing bond paths and bond-critical-points (bcp in blue). NPA charges are shown in green boxes. The electron density ρ(r) and Laplacian ∇2ρ(r) are shown in a.u. in red and orange boxes, respectively. (c) HOMO and LUMO for singlet diradicaloid D. Ca is shown in green and [MeNC˙]– in yellow. | ||
The next step in the reaction (B–C) is coordination of a second MeNC reagent to one of the Ca centres which is exothermic by ΔH = −20.3 kcal mol−1. Starting from this coordination complex C, we searched for the transition state for C–C bond formation, assuming C-nucleophilic attack of [MeNC]2− at the C atom in the neutral MeNC ligand. To our surprise, we located an energy minimum which is highly symmetric, showing two nearly identical MeNC moieties bridging between [(BDI*)Ca]+ fragments (D); Fig. 3a. The negative charges on the fragments are similar (−0.80 and −0.83) and there is a rather short C⋯C distance of 2.934 Å, which is substantially shorter than the van der Waals equilibrium distance for two non-bound C atoms, i.e. the layer distance in graphite (3.35 Å). AIM analysis of D (Fig. 3b) shows indeed a clear bond path and bond-critical-point (bcp) between these C atoms. The electron density (ρ(r) = 0.021 e × B−3) and Laplacian (∇2ρ(r) = +0.027 e × B−5) are small but significant and confirm weak bonding. Optimization of this minimum in the triplet state resulted in a much longer C⋯C bond separation of 3.518 Å (Fig. S60†), i.e. an interatomic distance just above the van der Waals equilibrium distance of 3.35 Å. The unrestricted triplet state is only +3.1 kcal mol−1 higher in energy than the restricted singlet state. Optimization as an unrestricted singlet gave a similar minimum and energy as found in the restricted singlet optimization. The similar negative charges (−0.80 and −0.83) on both nearly identical MeNC moieties indicate bridging radical anions [MeNC˙]− with anti-ferromagnetically coupled radical centres. Such singlet diradicals are known to be highly reactive intermediates in bond-breaking and bond-formation processes.57–60 Starting with the Niecke diradical,61,62 many examples of such fascinating species based on group 13, 14 or 15 elements could be isolated as stable diradicaloids.63 The highly reactive intermediate D with its open-shell singlet biradical character represents the first example of such a s-block substituted transient diradicaloid. The HOMO of diradicaloid D shows a bonding interaction between the C atoms which involves overlap of C p-orbitals but also has contributions of Ca d-orbitals (Table S2†). The LUMO is antibonding in respect of the two C atoms.
Although D is a minimum on the energy surface, isolation of such a diradicaloid is not possible. Coupling of the [MeNC˙]− radical anions is nearly barrier-free (D–E*: +2.0 kcal mol−1) and generates a complex with a bridging [MeNC–CNMe]2− anion with a C–C bond length of 1.407 Å (F). This reactivity is fundamentally different from the reductive coupling of CO with (DIPPBDI)Mg–Mg(DIPPBDI) which starts with formation of the dianion [CO]2− that as a nucleophile attacks a neutral CO ligand (see inset in Scheme 4).13
After formation of F, coordination of the third MeNC reagent (G) is followed by immediate insertion, again a process with hardly any barrier (G–H*: +2.6 kcal mol−1). This gives a species with a linear triimino dianion (I). The ring closure in the final step requires only an activation enthalpy of ΔH = +8.9 kcal mol−1 (I–J*) to form the triimino deltate dianion stabilized by two [(BDI)Ca]+ fragments (K). Considering the highly strained nature of the cyclopropane framework, this is a very modest energy barrier. As often observed in cyclopropane synthesis, this ring closing step proceeds through a carbene intermediate (I). A recent computational study by Munz and Chu also provided evidence for carbene intermediates in isonitrile homologation by (DIPPBDI)AlI.56 However, similar as reported for reductive C–C coupling of isonitriles promoted by aluminyl anions,64 also in this case cyclopropane formation was not observed. Although there are examples of reductive isonitrile trimerizations to triimino deltates with transition metals30,31 or dialumane species,26 this is a first example for an s-block mediated reaction. The energy profile in Scheme 4 demonstrates that alternative C–C coupling products like the product from reductive isonitrile dimerization (intermediate F) or the open trimer (I) are higher in energy than the closed deltate trimer (K). Overall, the Ca-mediated reductive trimerization of MeNC to the deltate complex K is a highly exothermic process with ΔH = −107.7 kcal mol−1 (ΔG = −81.9 kcal mol−1).
Conclusion
In contrast to the comprehensively investigated reactivity of (BDI)Mg–Mg(BDI) complexes,9 the chemistry of similar but hypothetical (BDI)Ca–Ca(BDI) complexes is fully unknown. Reaction of a closely related CaI synthon, [(BDI*)Ca(THP)]2(N2), with CO led to reductive trimerization and gave a product with the deltate dianion C3O32− (1). A similar product was observed for reductive coupling of CO with (BDI)Mg–Mg(BDI) complexes. However, the important difference is that the CaI synthon is even at low temperature highly reactive.Reaction of CaI synthon [(BDI*)Ca(THP)]2(N2) with CyNC also led to a cyclic product (2) and a complex with the triimino deltate dianion C3(NCy)32− was isolated. This reaction is not fully selective and the isolation of the side-product (BDI*)2Ca·(CN–Cy) (3) shows that the reaction products are in dynamic exchange by Schlenk equilibria. Although reductive coupling of isonitriles to triimino deltates has been demonstrated for transition metal reagents or a dialumane, this reactivity represents a first example for such products formed by s-block metal mediated isonitrile homologation. For comparison, reaction of isonitriles with (BDI)Mg–Mg(BDI) complexes led to reductive dimerization to [RNC–CNR]2– or reductive R–NC bond cleavage.
A computational study on Ca mediated triimino deltate formation showed a mechanism in which the first C–C coupling proceeds through a singlet diradical minimum. This is fundamentally different from CO coupling by low oxidation state (BDI)Mg–Mg(BDI) complexes which has been calculated to go through CO2− → CO nucleophilic attack.
As low oxidation state (BDI)Ca–Ca(BDI) reagents are currently not accessible, direct comparison with (BDI)Mg–Mg(BDI) reactivity is not possible. However, the highly reducing CaI synthon [(BDI*)Ca(THP)]2(N2) provides a good alternative to study metal influences in s-block metal mediated reduction reactions.
Author contributions
S. Thum: conceptualization, investigation, validation, formal analysis, writing – original draft, visualization. J. Mai: investigation, validation, formal analysis. M. A. Schmidt: investigation, validation, formal analysis. J. Langer: formal analysis, validation. Sjoerd Harder: conceptualization, writing – original draft – review and editing, visualization, validation, supervision, project administration.Data availability
All primary data are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mrs A. Roth for CHN analyses, Dr C. Färber and J. Schmidt for assistance with NMR analyses and L. Klerner for assistance with DFT calculations. The Deutsche Forschungsgemeinschaft is acknowledged for funding (HA 3218/11-1).References
- A. V. Lygin and A. de Meijere, Angew. Chem., Int. Ed., 2010, 49, 9094–9124 CrossRef CAS PubMed.
- R. M. Wilson, J. L. Stockdill, X. Wu, X. Li, P. A. Vadola, P. K. Park, P. Wang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2012, 51, 2834–2848 CrossRef CAS PubMed.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC.
- M. Giustiniano, A. Basso, V. Mercalli, A. Massarotti, E. Novellino, G. C. Tron and J. Zhu, Chem. Soc. Rev., 2017, 46, 1295–1357 RSC.
- A. Massarotti, F. Brunelli, S. Aprile, M. Giustiniano and G. C. Tron, Chem. Rev., 2021, 121, 10742–10788 CrossRef CAS PubMed.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- B. Rösch and S. Harder, Chem. Commun., 2021, 57, 9354–9365 RSC.
- L. A. Freeman, J. E. Walley and R. J. Gilliard, Nat. Synth., 2022, 1, 439–448 CrossRef CAS.
- M. J. Evans and C. Jones, Chem. Soc. Rev., 2024, 53, 5054–5082 RSC.
- K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS PubMed.
- K. Yuvaraj, I. Douair, D. D. L. Jones, L. Maron and C. Jones, Chem. Sci., 2020, 11, 3516–3522 RSC.
- K. Yuvaraj, J. C. Mullins, T. Rajeshkumar, I. Douair, L. Maron and C. Jones, Chem. Sci., 2023, 14, 5188–5195 RSC.
- A. Paparo, K. Yuvaraj, A. J. R. Matthews, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 630–634 CrossRef CAS PubMed.
- H.-Y. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, T. Rajeshkumar, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2023, 62, e202308347 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, D. T. Nguyen, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2023, 60, 1016–1019 RSC.
- A. O'Reilly, M. D. Haynes, Z. R. Turner, C. L. McMullin, S. Harder, D. O'Hare, J. R. Fulton and M. P. Coles, Chem. Commun., 2024, 60, 7204–7207 RSC.
- M. J. Evans, M. G. Gardiner, M. D. Anker and M. P. Coles, Chem. Commun., 2022, 58, 5833–5836 RSC.
- A. Heilmann, M. M. D. Roy, A. E. Crumpton, L. P. Griffin, J. Hicks, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2022, 144, 12942–12953 CrossRef CAS.
- S. Mukhopadhyay, A. G. Patro, R. S. Vadavi and S. Nembenna, Eur. J. Inorg. Chem., 2022, e202200469 CrossRef CAS.
- W. Uhl, U. Schütz, W. Hiller and M. Heckel, Chem. Ber., 1994, 127, 1587–1592 CrossRef CAS.
- W. Haider, D. M. Andrada, I.-A. Bischoff, V. Huch and A. Schäfer, Dalton Trans., 2019, 48, 14953–14957 RSC.
- W. Chen, Y. Zhao, W. Xu, J. H. Su, L. Shen, L. Liu, B. Wu and X. J. Yang, Chem. Commun., 2019, 55, 9452–9455 RSC.
- W. Uhl, I. Hahn, U. Schütz, S. Pohl, W. Saak, J. Martens and J. Manikowski, Chem. Ber., 1996, 129, 897–901 CrossRef CAS.
- M. Ma, A. Stasch and C. Jones, Chem.–Eur. J., 2012, 18, 10669–10676 CrossRef CAS PubMed.
- J. Li, M. Hermann, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8611–8614 CrossRef CAS PubMed.
- J. Shen, G. P. A. Yap and K. H. Theopold, J. Am. Chem. Soc., 2014, 136, 3382–3384 CrossRef CAS PubMed.
- S. Hasegawa, Y. Ishida and H. Kawaguchi, Chem. Commun., 2021, 57, 8296–8299 RSC.
- K. Gour, D. Pramanik, S. R. Dash, D. D. Shinde, G. Venugopal, K. Vanka, A. K. Rath and S. S. Sen, Angew. Chem., Int. Ed., 2025, 64, e202417052 CrossRef CAS.
- J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Science, 2023, 380, 1147–1149 CrossRef CAS.
- J. T. Boronski, A. E. Crumpton, A. F. Roper and S. Aldridge, Nat. Chem., 2024, 16, 1295–1300 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS.
- C. Berthold, J. Maurer, L. Klerner, S. Harder and M. R. Buchner, Angew. Chem., Int. Ed., 2024, 63, e202408422 CrossRef CAS.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef.
- J. Mai, J. Maurer, J. Langer and S. Harder, Nat. Synth., 2023, 3, 368–377 CrossRef.
- J. Maurer, L. Klerner, J. Mai, H. Stecher, S. Thum, M. Morasch, J. Langer and S. Harder, Nat. Chem., 2025, 17, 703–709 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef.
- J. Mai, B. Rösch, J. Langer, S. Grams, M. Morasch and S. Harder, Eur. J. Inorg. Chem., 2023, 26, e202300421 CrossRef CAS.
- S. Thum, O. P. E. Townrow, J. Langer and S. Harder, Chem. Sci., 2025, 16, 4528–4536 RSC.
- S. Thum, J. Mai, N. Patel, J. Langer and S. Harder, ChemistryEurope, 2025, e202500080 Search PubMed.
- R. Y. Kong and M. R. Crimmin, Dalton Trans., 2020, 49, 16587–16597 RSC.
- J. Mai, B. Rösch, N. Patel, J. Langer and S. Harder, Chem. Sci., 2023, 14, 4724–4734 Search PubMed.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829–831 CrossRef CAS PubMed.
- R. Mondal, K. Yuvaraj, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2022, 58, 12665–12668 RSC.
- H. Hopf and G. Maas, Angew. Chem., Int. Ed., 1992, 31, 931–954 CrossRef.
- T. Höpfner, P. G. Jones, B. Ahrens, I. Dix, L. Ernst and H. Hopf, Eur. J. Org Chem., 2003, 2596–2611 CrossRef.
- M. Gholami and R. R. Tykwinski, Chem. Rev., 2006, 106, 4997–5027 CrossRef CAS.
- N. Liu, S. Yu and Y. Ding, J. Organomet. Chem., 2017, 828, 75–82 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc. Perkin Trans. 2, 1987, S1–S19 RSC.
- M. Huggins, in Structural Chemistry and Molecular Biology, ed. A. Rich and N. Davidson, Freeman W. H., San Francisco, 1968, p. 768 Search PubMed.
- G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290–296 CrossRef CAS.
- S. Harder, Chem.–Eur. J., 1999, 1852–1861 CrossRef CAS.
- C. Zhang, F. Dankert, Z. Jiang, B. Wang, D. Munz and J. Chu, Angew. Chem., Int. Ed., 2023, 62, e202307352 CrossRef CAS.
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS.
- M. Abe, J. Ye and M. Mishima, Chem. Soc. Rev., 2012, 41, 3808 RSC.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- J. Rosenboom, A. Villinger, A. Schulz and J. Bresien, Dalton Trans., 2022, 51, 13479–13487 RSC.
- W. W. Schoeller and E. Niecke, Phys. Chem. Chem. Phys., 2012, 14, 2015–2023 RSC.
- W. W. Schoeller, Eur. J. Inorg. Chem., 2019, 2019, 1495–1506 CrossRef CAS.
- A. Hinz, J. Bresien, F. Breher and A. Schulz, Chem. Rev., 2023, 123, 10468–10526 CrossRef CAS PubMed.
- M. J. Evans, M. D. Anker, C. L. McMullin and M. P. Coles, Chem. Sci., 2023, 14, 6278–6288 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H and 13C NMR spectra, crystallographic details including ORTEP presentations, details for the DFT calculations including XYZ-files. CCDC 2428665–2428669. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02829a |
| This journal is © The Royal Society of Chemistry 2025 |