
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-inspired total synthesis of daphnepapytone A†
Joan
Pereira
 a,
Nicolas
Casaretto
b,
Gilles
Frison
c and
Bastien
Nay
*a
a,
Nicolas
Casaretto
b,
Gilles
Frison
c and
Bastien
Nay
*a
aLaboratoire de Synthèse Organique, Ecole Polytechnique, ENSTA Paris, CNRS, Institut Polytechnique de Paris, Route de Saclay, F-91128 Palaiseau Cedex, France. E-mail: bastien.nay@polytechnique.edu
bLaboratoire de Chimie Moléculaire, Ecole Polytechnique, ENSTA Paris, CNRS, Institut Polytechnique de Paris, Route de Saclay, F-91128 Palaiseau Cedex, France
cLaboratoire de Chimie Théorique, Sorbonne Université, CNRS, F-75005 Paris, France
First published on 19th May 2025
Abstract
Daphnepapytone A (1) is an unprecedented guaiane-derived sesquiterpene characterized by a bridged and highly substituted cyclobutane. We describe its total synthesis through a bio-inspired sequence of skeleton construction and late-stage oxidation. After the Eschenmoser–Tanabe fragmentation of (R)-carvone epoxide, the allenylation of the resulting aldehyde was followed by an allenic Pauson–Khand reaction with distal regioselectivity in the presence of [Rh(CO)2Cl]2 to give the guaiane skeleton. Oleodaphnone (3) was identified as a key intermediate of this strategy and was engaged in a biomimetic [2 + 2]-photocycloaddition, leading to the bridged cyclobutane of the title compound. Finally, a late-stage C–H oxidation chemoselectively released a triketone intermediate (15), which was reduced in a remarkably chemo- and stereoselective manner to furnish target compound 1. During this work, complex rearrangements of the bridged skeleton were observed. Beside the total synthesis of daphnepapytone A, this paper also describes the total synthesis of three guaiane natural products (oleodaphnone, diarthroncha C, daphnenicillata W), one of them being structurally revised.
Introduction
Since Komppa's pioneering synthesis of camphor in 1903,1 bridged terpenes have always been considered as challenging targets in total synthesis (Fig. 1a). In particular, sesquiterpenes offer an infinite structural playground for this purpose, often with a strong medicinal importance. Longifolene, one of the flavouring molecules of the lapsang souchong tea, is a famous representative example of synthetic target that inspired various strategies, like those of Corey2 or Oppolzer.3 More recently, the enantioselective total synthesis of artatrovirenol A, a potentially anticancer guaiane-derived compound with a rare and complex caged structure, was independently reported by Zhu,4 and by Xie and She,5 the last one involving a biomimetic intramolecular Diels–Alder reaction to install the bridged system. Beside complex carbocyclic skeletons, the degree of functionalization of terpenes adds a significant level of synthetic complexity, especially in terms of oxidation levels. The development of late-stage oxidative C–H functionalization represents a breakthrough in total synthesis, allowing circumvention of long multistep sequences.6–15 This strategy was referred to as a bio-inspired “two-phase” approach by Baran,16–19 involving the “cyclase phase” aimed at skeleton construction and the “oxidase phase” aimed at skeleton functionalization (Fig. 1b). Although it is straightforward and applicable to synthetic targets as complex as taxol,19 this strategy is characterized by a significant challenge related to reactivity and selectivity issues during the oxidase phase. | ||
| Fig. 1 (a) Selected examples of bridged terpenoids (the authors of the first synthesis are italicized), and (b) general two-phase biosynthetic origin of oxygenated sesquiterpenes as an inspiration for total synthesis. | ||
Daphnepapytone A (1, Fig. 1a) is a guaiane-derived sesquiterpene isolated in 2022 by Zhao, Dai and co-workers from Daphne papyracea (“Xuehuagou”), an ornamental plant also used to treat diabetes and inflammatory diseases in Southern China.20 This oxygenated cage structure holds a rare bridged and highly substituted cyclobutane ring. It thus constitutes an unprecedented target for total synthesis, which is further justified by its α-glycosidase inhibitory properties and a limited extraction yield (5.3 mg out of 12.3 kg of air-dried plant stems). Indeed, considering this challenging synthetic target, a straightforward synthetic route is awaited if we are to use 1 as a lead compound to design new natural product-based antidiabetic treatments.
To synthesize daphnepapytone A, we envisaged a bio-inspired strategy21–25 combining a highly efficient skeleton construction from (R)-carvone and a challenging late-stage C–H oxidation performed on bridged intermediate 2 (Scheme 1). Compound 2 was expected to be formed in a biomimetic manner from the [2 + 2] cycloaddition of oleodaphnone 3. This HIV-targeting guaiane sesquiterpene26 could be the product of an allenic Pauson–Khand reaction (APKR) with a distal regioselectivity permitted by Rh(I)-catalysis.27,28 Finally, allene intermediate 4 would be available from the allenylation of aldehyde 5, a product of the Eschenmoser–Tanabe fragmentation of carvone (6) epoxide.29,30 Importantly, while we were submitting this work for publication, an elegant alternative to the APKR was disclosed by Stoltz and co-workers through the Pauson–Khand reaction of a methylidenecyclobutane intermediate.31,32 This approach was consecutive to a limited success in the biomimetic [2 + 2] cycloaddition.
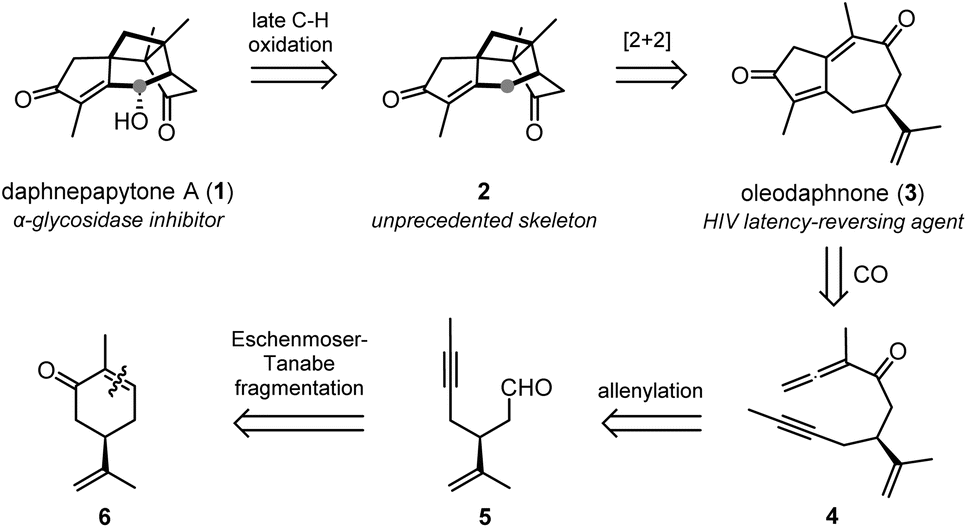 | ||
| Scheme 1 Retrosynthetic analysis of daphnepapytone A (1) through oleodaphnone (3). | ||
Results and discussion
To start this synthetic sequence, (R)-carvone (6) was readily epoxidized in presence of H2O2 under basic (NaOH) assistance (Scheme 2). The Eschenmoser–Tanabe fragmentation of the resulting epoxycarvone was conducted in presence of tosylhydrazide in a CH2Cl2/AcOH mixture, giving the branched 5-heptynal (5) in 43% yield on a decagram scale.33 The efficient In(0)-promoted allenylation of aldehyde 5 in presence of 2-butynyl bromide34 afforded a diastereomeric mixture of gem-disubstituted allenols 7a and 7b (dr4S/4R = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5, partially separable). Noteworthily, the diastereoselective synthesis of allenol 7a and 7b was also successfully achieved in presence of (2-butynyl)pinacolborane and chiral phosphoric acids (R)- and (S)-TRIP (5 mol%),35 respectively, paving the way to a stereoselective access to 10 and 11. Alternatively, the diastereomeric mixture of allenols could be oxidized in presence of Dess–Martin periodinane (DMP), furnishing ketone 4 (Scheme 1). This volatile ketone, however, turned out to be unreactive towards the next APKR step (not shown).
:1.5, partially separable). Noteworthily, the diastereoselective synthesis of allenol 7a and 7b was also successfully achieved in presence of (2-butynyl)pinacolborane and chiral phosphoric acids (R)- and (S)-TRIP (5 mol%),35 respectively, paving the way to a stereoselective access to 10 and 11. Alternatively, the diastereomeric mixture of allenols could be oxidized in presence of Dess–Martin periodinane (DMP), furnishing ketone 4 (Scheme 1). This volatile ketone, however, turned out to be unreactive towards the next APKR step (not shown).
 | ||
| Scheme 2 Total synthesis of oleodaphnone (3), diarthroncha C (10, structure as reported in the literature), and daphnenicillata W (11), through an APKR performed in COware®. Notes: (a) COgen (2.5 equiv.), PdCl2(cod) (10 mol%), [(t-Bu)3PH]BF4 (10 mol%), MeN(Cy)2 (4.0 equiv.), toluene (0.02 M);36 (b) KOH (30 equiv.), CHCl3 (10 equiv.), toluene (0.05 M).37 Note: ORTEP structure of 9b at 50% probability level (CCDC number: 2419409).‡ | ||
After TBS-protection of the secondary alcohols, the Pauson–Khand reaction was envisaged on intermediates 8a and 8b, and directly on the diastereomeric mixture (Scheme 2, step 6). It is known that the APKR faces a regioselectivity issue due to the two reactive, distal or proximal alkenes on allenes, depending on the catalytic system and the substitution pattern of the allene.27,28 Brummond38–43 and Mukai44–47 previously showed that a Rh(I)-based catalytic system exclusively results in distal selectivity. To perform this reaction, a Skrydstrup's two-chamber COware® system was used, with CO being safely generated from 9-methyl-9H-fluorene-9-carbonyl chloride (COgen) under Pd-catalysis in chamber A (Scheme 2).36 Thus, the APKR of substrates 8a and 8b (added slowly to chamber B)43 in presence of [Rh(CO)2Cl]2 (5 mol%) exclusively afforded the 5/7-fused TBS-protected products 9a and 9b possessing a guaiane skeleton. The relative stereochemistry of these compounds was deduced from the X-ray crystallographic structure of 9b (CCDC 2419409). During the optimisation (see Tables S1–S3†), while isomer 8a gave cleaner and faster reactions, isomer 8b provided more by-products on similar reaction times, supposedly formed from intermolecular processes.43 This problem was solved by increasing the addition time of substrate 8b onto the catalytic system. Furthermore, the APKR on the diastereomeric mixture 8a/8b could be achieved in a 75% yield after an addition time of 20 h on a 1.87 mmol scale. Finally, the catalyst [Rh(CO)(dppp)2]Cl tested on 8b gave a lower yield (54%) than [Rh(CO)2Cl]2, while Pauson–Khand reagents like Mo(CO)6 or Co2(CO)8 led to degradation or no reaction. To improve the atom-economy and reduce the cost of this two-chamber process, CO was also generated from the reaction of CHCl3 with KOH in chamber A,37,48 yet with a drop of the APKR yield (55% for mixture 9a/9b).
The deprotection of the diastereomeric mixture 9a/9b under acidic condition (aqueous HCl, CH3CN), followed by the oxidation of the secondary alcohol with Dess–Martin periodinane (DMP), in 50% yield over two steps, completed the first total synthesis of pale green compound oleodaphnone (3, see Table S4† for NMR data comparison with the literature). Alternatively, since this acidic condition was found to induce epimerization of the C-9 stereocenter, the deprotection of pure diastereoisomers 9a and 9b in presence of tetrabutylammonium fluoride (TBAF) afforded the two natural products diarthroncha C (10, structure shown as reported)49 and daphnenicillata W (11),50 in 73% and 61% yields, respectively. While the NMR data of 11 matched those of natural daphnenicillata W50 in CDCl3 (Table S6†), significant deviations were observed for 10 compared to those of natural diarthroncha C49 in DMSO-d6 (Table S5†). Fortunately, we also recorded the NMR spectra of 11 in DMSO-d6, and observed striking similarities with the NMR data of natural diarthroncha C (Table S7†). Consequently, we propose to revise the reported stereochemistry of diarthroncha C as structure 11. In other words, diarthroncha C and daphnenicillata W are the same compound 11.
Interestingly, the X-ray crystallographic structure of 3 (CCDC 2418579, Scheme 3) showed an equimolar distribution of two conformers in the crystal, either with a pseudo-equatorial or a pseudo-axial orientation of the isopropenyl group (see also Fig. S1†). The proximity of the isopropenyl olefin with that of the cycloheptenone ring (distance < 4 Å) was observed on the pseudo-axial conformer, supporting the feasibility of the photochemical enone–alkene [2 + 2] cycloaddition.51 To finally elaborate the carbocyclic skeleton of daphnepapytone A, a solution of oleodaphnone 3 in CH2Cl2 was irradiated at 370 nm (Kessil lamp), giving the bridged cyclobutane intermediate 2 in 73% yield (Scheme 3, and Fig. S2a† for key NMR correlations). This structure was confirmed by X-ray crystallography (CCDC 2419407). It seems reasonable to suggest that this [2 + 2] cycloaddition is “biomimetic” in the sense that it could occur in the Daphne plant under the sunlight. To test this hypothesis—since Paris weather in 2024 did not allow us to copy Ciamician's rooftop condition used in 1908 to photocyclize carvone52—a sample of 3 in CDCl3 was treated under white light (LED). NMR monitoring confirmed the formation of 2, yet in low yield (10%) and with marked degradation after 18 days (Fig. S3†).
 | ||
| Scheme 3 Biomimetic [2 + 2] cycloaddition of 3. Note: ORTEP structures at 50% probability level (CCDC numbers for 2: 2419407; 3: 2418579).‡ | ||
Finally, we turned our attention to the late-stage oxidative functionalization of 2, targeting the γ-position (C-6) of the remaining enone (Scheme 4). This transformation had to be regioselective and stereoselective to install the secondary alcohol of daphnepapytone A (1). The reactivity of caged intermediate 2 was thus investigated under various conditions. Compound 2 turned out to be neither reactive under Riley oxidation condition in presence of SeO2, nor toward the formation of a dienol silyl ether targeting a vinylogous Rubottom reaction toward 1 (Table S8†). While photooxygenation attempts53 in presence of singlet oxygen led to degradation (see Table S9† for other attempts), the application of autooxidation conditions in presence of tBuOK under air (route a) provided a new, rearranged but non-oxidized structure (13, see Fig. S2b† for key NMR correlations) in 40% yield (or 67% when performed under argon). The 1,2-migration of C-10, with concomitant ring expansion of the cyclobutane ring, was supposed to proceed through a base-promoted retro-Michael addition followed by a new Michael addition on C-5 of the cyclopentadienone intermediate 12. X-ray crystallographic analysis confirmed structure 13 (CCDC 2419408).
 | ||
| Scheme 4 Late-stage functionalization of 2. Note: ORTEP structures at 50% probability level (CCDC numbers for 13: 2419408; 14: 2428644; 15: 2418568).‡ | ||
Most interestingly, based on Newhouse's precedent, the application of the 2-hydroxy-2-methyl-butanoic acid chromium complex Cr(V) (route b) not only afforded 6-epi-daphnepapytone A (epi-1) as a major compound (28% yield, or 46% b.r.s.m.), but also the rearranged triketone 14 (12%), a substantial amount of remaining starting material (40%) and traces of ketone 15. Despite its appealing character confirming the possibility to directly hydroxylate position C-6, this result also highlighted the unfavourable stereoselectivity of the α-hydroxylation to reach 1, explained by the higher steric hindrance of the α face. The surprising structure of triketone 14, confirmed by X-ray crystallography (CCDC 2428644), was supposed to result from a complex rearrangement mechanism (Scheme 4) involving the 1,2-shift of C-10 (similar to that leading to 13), followed by the 1,2-carbonyl shift of C-6. This proposition is however highly speculative since radical intermediates could also be generated in presence of Cr(V) reagents.
Gratifyingly, oxidation attempts in presence of CrO3 under acidic condition (CH2Cl2/AcOH)8 furnished enedione 15 in 62% yield (see Table S10† for optimisation). The regioselectivity of this transformation, confirmed by X-ray crystallography (CCDC 2418568), was rationalized by the preferred hydrogen abstraction at C-6, leading to the formation of stabilized radical intermediate 16 (Scheme 4). Reaction of 16 with CrO3 could form the C–O bond and a Cr(V) species (17), while decomposition of 17 would release ketone 15 and Cr(III).17,54
Compound 15 harbours three ketones, two of them embedded in an enedione motif. This structure brought a new challenge for the chemo- and stereoselective reduction of the newly installed ketone on C-6, which was initially overestimated by the apparent accessibility of the cyclopentenone carbonyl on C-3. In fact, this reduction turned out to target exclusively the cyclohexanone ring at C-6, in presence of NaBH4 (0.75 equiv.) in MeOH, delivering daphnepapytone A (1) in a 73% yield (Scheme 5) and thus achieving the total synthesis of this natural product. All spectroscopic data of 1 were consistent with those of the literature (Table S11†),20 including crystallographic analysis (CCDC 2418571). The Luche conditions (CeCl3, NaBH4, MeOH)55 resulted in poor chemo- and stereoselectivity, giving a mixture of 1 (16%), diol 19 (19%) and other non-identified products. The relative stereochemistry of diol 19 (CCDC 2423044) was explained by the stereocontrolled reduction of the cyclopentanone ring at C-9, after the reduction of C-6, following a Saksena mechanism through boron hydride complex 18.56
 | ||
| Scheme 5 Final reduction to daphnepapytone A. Note: ORTEP structure at 50% probability level (CCDC numbers for 1: 2418571; 19: 2423044).‡ | ||
Despite an apparent steric hindrance of the ketone on C-6, the high chemoselectivity observed after the reduction could be explained by the faster reaction of cyclohexanones over cyclopentanones.57,58 The exclusive π-facial stereoselectivity was explained by the favoured axial approach of the hydride nucleophile onto the chair cyclohexanone (green cycle, Fig. 2). Furthermore, not only the disfavoured equatorial approach, but also steric hindrance of the other face imposed by the cage structure, are likely to preclude the formation of the C-6 epimer (epi-1 not observed experimentally). DFT calculations confirmed these tendencies. The energy of the transition state TSax resulting from the axial approach at C-6 (71.4 kJ mol−1) is the lowest among all possible transition states calculated for the reduction of each carbonyl of 1. In particular, it is lower than TSeq resulting from the equatorial approach (77.9 kJ mol−1). The reduction of the carbonyl groups at C-3 or C-9, within five-membered rings, proves to be more energetic, with activation barriers ranging from 92.8 to 101.3 kJ mol−1 (Fig. S5†).
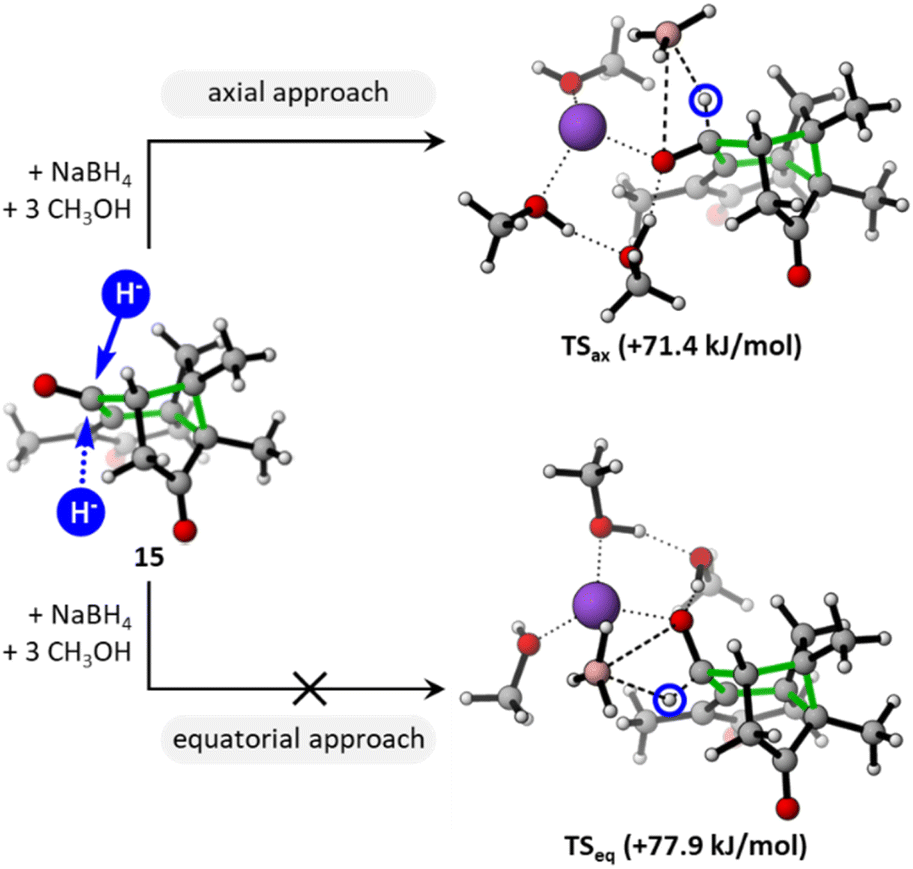 | ||
| Fig. 2 DFT-supported rationalization of the stereoselectivity observed during the reduction of 15, favouring an axial approach of the reducing agent (pink: B; purple: Na) onto the cyclohexanone ring (green). DFT approach based on Tomoda's work, considering three explicit solvent molecules (the free energies of transitions states TSax and TSeq are given relatively to the separated reactants).59 Calculations performed at the SMD(methanol)-M06-2X/aug-cc-pVTZ//SMD(methanol)-B3LYP-D3/6-311++G(d,p) level (see ESI† for details).‡ | ||
Conclusions
This work represents the first biomimetic total synthesis of daphnepapytone A (1). Noteworthily, it was concurrently disclosed at the same time as a non-biomimetic approach by Stoltz.31,32 We employed a straightforward and highly efficient two-phase strategy, involving a Pauson–Khand reaction to construct the guaiane skeleton, and a biomimetic [2 + 2] cycloaddition to achieve the bridged structure of the target natural product. Remarkably, the late-stage C–H oxidation step was performed in a highly chemoselective manner, resulting in triketone intermediate 15. The newly installed ketone, the only one localized in a cyclohexanone ring, was chemo- and stereoselectively reduced through an axial approach of the hydride, to release natural product 1. During this work, peculiar rearrangements of the bridged skeleton were observed, highlighting its reactivity and the possibility to discover new natural product skeletons with similar structures in the near future. Furthermore, three other guaiane products with medicinally relevant properties were synthesized: oleodaphnone (3), diarthroncha C (10, structure initially reported for the natural product), and daphnenicillata W (11). On this occasion, we revised the structure of diarthroncha C, showing that it is the same as daphnenicillata W (11). Overall, this work brings a rapid access to the complex caged structure of a structurally unique natural product (1) and its biomimetic precursor (3) with biological perspectives towards anti-diabetes or anti-HIV treatments.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
B. N. and J. P. designed the synthetic strategy. J. P. performed synthetic experiments. N. C. performed crystallographic analyses. G. F. performed DFT calculations. B. N. supervised the research project and wrote the manuscript. J. P. and G. F. contributed to the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Ecole Polytechnique, the Centre National de la Recherche Scientifique and the Agence Nationale de la Recherche (ANR-22-CE44-0006) for financial support. We acknowledge ResoMag facility (IP-Paris/CNRS) for the support with NMR facility. We thank Julian Quévarec for his technical support to the Eschenmoser–Tanabe fragmentation.Notes and references
- G. Komppa, Ber. Dtsch. Chem. Ges., 1903, 36, 4332–4335 Search PubMed.
- E. J. Corey, M. Ohno, R. B. Mitra and P. A. Vatakencherry, J. Am. Chem. Soc., 1964, 86, 478–485 CrossRef CAS.
- W. Oppolzer and T. Godel, J. Am. Chem. Soc., 1978, 100, 2583–2584 CrossRef CAS.
- R. Lavernhe, P. Domke, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2023, 145, 24408–24415 CrossRef CAS PubMed.
- Y. Yang, D. Xie, L. Huo, Y. Liu, J. Duan, H. Li, P.-P. Zhou, X. Xie and X. She, Nat. Commun., 2025, 16, 322 CrossRef CAS PubMed.
- K. Chen and P. S. Baran, Nature, 2009, 459, 824–828 CrossRef CAS PubMed.
- R. Marín-Barrios, A. L. García-Cabeza, F. J. Moreno-Dorado, F. M. Guerra and G. M. Massanet, J. Org. Chem., 2014, 79, 6501–6509 CrossRef PubMed.
- Z. Yuan, X. Hu, H. Zhang, L. Liu, P. Chen, M. He, X. Xie, X. Wang and X. She, Chem. Commun., 2018, 54, 912–915 RSC.
- Y. Kuroda, K. J. Nicacio, I. A. da Silva-Jr, P. R. Leger, S. Chang, J. R. Gubiani, V. M. Deflon, N. Nagashima, A. Rode, K. Blackford, A. G. Ferreira, L. D. Sette, D. E. Williams, R. J. Andersen, S. Jancar, R. G. S. Berlinck and R. Sarpong, Nat. Chem., 2018, 10, 938–945 CrossRef CAS.
- X. Hu, A. J. Musacchio, X. Shen, Y. Tao and T. J. Maimone, J. Am. Chem. Soc., 2019, 141, 14904–14915 CrossRef CAS PubMed.
- L. A. Wein, K. Wurst, P. Angyal, L. Weisheit and T. Magauer, J. Am. Chem. Soc., 2019, 141, 19589–19593 CrossRef CAS PubMed.
- M. Berger, C. Knittl-Frank, S. Bauer, G. Winter and N. Maulide, Chem, 2020, 6, 1183–1189 CAS.
- L. A. Wein, K. Wurst and T. Magauer, Angew. Chem., Int. Ed., 2022, 61, e202113829 CrossRef CAS PubMed.
- Y. Qiu and S. Gao, Nat. Prod. Rep., 2016, 33, 562–581 RSC.
- I. Bakanas, R. F. Lusi, S. Wiesler, J. Hayward Cooke and R. Sarpong, Nat. Rev. Chem., 2023, 7, 783–799 CrossRef CAS PubMed.
- Y. Ishihara and P. S. Baran, Synlett, 2010, 2010, 1733–1745 Search PubMed.
- N. C. Wilde, M. Isomura, A. Mendoza and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4909–4912 CrossRef CAS PubMed.
- H. Chu, J. M. Smith, J. Felding and P. S. Baran, ACS Cent. Sci., 2017, 3, 47–51 CrossRef CAS PubMed.
- Y. Kanda, H. Nakamura, S. Umemiya, R. K. Puthukanoori, V. R. Murthy Appala, G. K. Gaddamanugu, B. R. Paraselli and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 10526–10533 CrossRef CAS PubMed.
- S.-Z. Huang, Q. Wang, J.-Z. Yuan, C.-H. Cai, H. Wang, A. Mándi, T. Kurtán, H.-F. Dai and Y.-X. Zhao, J. Nat. Prod., 2022, 85, 3–14 CrossRef CAS.
- M. C. de la Torre and M. A. Sierra, Angew. Chem., Int. Ed., 2004, 43, 160–181 CrossRef CAS PubMed.
- Biomimetic Organic Synthesis (1 & 2), ed. E. Poupon and B. Nay, Wiley-VCH Verlag, 2011 Search PubMed.
- M. Razzak and J. K. De Brabander, Nat. Chem. Biol., 2011, 7, 865–875 CrossRef CAS.
- R. Bao, H. Zhang and Y. Tang, Acc. Chem. Res., 2021, 54, 3720–3733 CrossRef CAS.
- L. Chen, P. Chen and Y. Jia, Acc. Chem. Res., 2024, 57, 3524–3540 CrossRef CAS PubMed.
- S. Li, X. Wang, Y. Yang, X. Wu and L. Zhang, Int. J. Mol. Sci., 2023, 24, 7357 CrossRef CAS PubMed.
- S. Kitagaki, F. Inagaki and C. Mukai, Chem. Soc. Rev., 2014, 43, 2956–2978 RSC.
- E. D. Deihl, F. Haghighi and K. M. Brummond, Org. Synth., 2023, 100, 29–47 CrossRef CAS.
- A. Eschenmoser, D. Felix and G. Ohloff, Helv. Chim. Acta, 1967, 50, 708–713 CrossRef CAS.
- M. Tanabe, D. F. Crowe and R. L. Dehn, Tetrahedron Lett., 1967, 8, 3943–3946 Search PubMed.
- E. C. Gonzalez, I. d. l. T. Roehl and B. M. Stoltz, ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2025-z55dj.
- J. Pereira, N. Casaretto, G. Frison and B. Nay, ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2025-0gds5.
- Z. Meng and A. Fürstner, J. Am. Chem. Soc., 2019, 141, 805–809 CrossRef CAS PubMed.
- M. B. Isaac and T.-H. Chan, J. Chem. Soc. Chem. Commun., 1995, 1003–1004 RSC.
- M. Wang, S. Khan, E. Miliordos and M. Chen, Adv. Synth. Catal., 2018, 360, 4634–4639 CrossRef CAS.
- S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594–605 CrossRef CAS PubMed.
- P. Halder, K. Mondal, A. Jash and P. Das, J. Org. Chem., 2024, 89, 9275–9286 CrossRef CAS PubMed.
- K. M. Brummond, H. Chen, K. D. Fisher, A. D. Kerekes, B. Rickards, P. C. Sill and S. J. Geib, Org. Lett., 2002, 4, 1931–1934 CrossRef CAS PubMed.
- K. M. Brummond and D. Gao, Org. Lett., 2003, 5, 3491–3494 CrossRef CAS PubMed.
- A. S. Bayden, K. M. Brummond and K. D. Jordan, Organometallics, 2006, 25, 5204–5206 Search PubMed.
- F. Grillet, C. Huang and K. M. Brummond, Org. Lett., 2011, 13, 6304–6307 CrossRef CAS PubMed.
- B. Wen, J. K. Hexum, J. C. Widen, D. A. Harki and K. M. Brummond, Org. Lett., 2013, 15, 2644–2647 CrossRef CAS PubMed.
- S. M. Wells and K. M. Brummond, Tetrahedron Lett., 2015, 56, 3546–3549 CrossRef CAS PubMed.
- C. Mukai, I. Nomura, K. Yamanishi and M. Hanaoka, Org. Lett., 2002, 4, 1755–1758 CrossRef CAS PubMed.
- C. Mukai, I. Nomura and S. Kitagaki, J. Org. Chem., 2003, 68, 1376–1385 CrossRef CAS PubMed.
- T. Hirose, N. Miyakoshi and C. Mukai, J. Org. Chem., 2008, 73, 1061–1066 CrossRef CAS PubMed.
- Y. Hayashi, K. Ogawa, F. Inagaki and C. Mukai, Org. Biomol. Chem., 2012, 10, 4747–4751 Search PubMed.
- S. N. Gockel and K. L. Hull, Org. Lett., 2015, 17, 3236–3239 CrossRef CAS.
- D.-X. Sun, D. Zhao, H.-Y. Wei, X.-L. Ma, L.-L. Shi and J. Zhang, Molecules, 2018, 23, 1383 CrossRef PubMed.
- P. Zhao, B.-S. Xin, S.-Y. Qin, Z.-Y. Li, B. Lin, G.-D. Yao, S.-J. Song and X.-X. Huang, Org. Chem. Front., 2022, 9, 6213–6222 RSC.
- M. T. Crimmins and T. L. Reinhold, in Organic Reactions, John Wiley & Sons, Ltd, 2004, pp. 297–588 Search PubMed.
- G. Ciamician and P. Silber, Ber. Dtsch. Chem. Ges., 1908, 41, 1928–1935 Search PubMed.
- C.-Y. Zheng and J.-M. Yue, Nat. Commun., 2023, 14, 2399 Search PubMed.
- P. Mueller and J. Rocek, J. Am. Chem. Soc., 1974, 96, 2836–2840 CrossRef CAS.
- A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS.
- A. K. Saksena and P. Mangiaracina, Tetrahedron Lett., 1983, 24, 273–276 CrossRef CAS.
- H. C. Brown and K. Ichikawa, Tetrahedron, 1957, 1, 221–230 CrossRef CAS.
- D. E. Ward and C. K. Rhee, Can. J. Chem., 1989, 67, 1206–1211 CrossRef CAS.
- Y. Suzuki, D. Kaneno and S. Tomoda, J. Phys. Chem. A, 2009, 113, 2578–2583 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6, Tables S1–S23, detailed experimental procedures, copies of NMR and HRMS spectra, computational details and crystallographic data (.pdf). Cartesian coordinates for computational studies are provided in a separated file (.xyz). CCDC 2418568 (15), 2418571 (1), 2418579 (3), 2419407 (2), 2419408 (13), 2419409 (9b), 2423044 (19) and 2428644 (14). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02953h |
| ‡ X-ray and model structures were generated with CYLview20, C. Y. Legault, Université de Sherbrooke, 2020 (https://www.cylview.org). |
| This journal is © The Royal Society of Chemistry 2025 |