
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcceleration and regioselectivity switching in 1,3-dipolar cycloaddition reactions confined in a bis-calix[4]pyrrole cage†
Yifan Li ab,
Gemma Aragaya and
Pablo Ballester*ac
ab,
Gemma Aragaya and
Pablo Ballester*ac
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), 43007 Tarragona, Spain. E-mail: pballester@iciq.es
bDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, 43007 Tarragona, Spain
cICREA, 08010 Barcelona, Spain
First published on 7th July 2025
Abstract
We report the results of our investigations on the role of the octa-imine bis-calix[4]pyrrole cage 1 in mediating confined 1,3-dipolar cycloaddition reactions of a series of 4-azido(alkyl)-pyridine-N-oxides (alkyl = null, Me, Et; 2a–c) with 1-(2-propynyl)-4(1H)-pyridinone (4). We performed 1H NMR binding studies of the different substrates with the octa-imine cage, evidencing the formation of thermodynamically and kinetically highly stable inclusion homo-complexes featuring 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 stoichiometry. We used simulated speciation profiles and performed ITC experiments to thermodynamically characterize the formed complexes. Adding mixtures of an azido-derivative with the propynyl–pyridinone in a solution of cage 1 resulted in the formation of the expected 1:1 and 2:1 homo-complexes, and allowed the detection and characterization of the corresponding ternary hetero-complexes (Michaelis) in solution, 2·4⊂1. The azide–alkyne cycloaddition reactions are significantly accelerated by confinement of the reactants in cage 1. We assessed the reactions' acceleration factor by determining their effective molarities (EM = kintra/kbulk). We derived the kintra values from the best-fit computer simulation of an elaborated theoretical kinetic model to the experimental kinetic data. The determined EM values range from 2000 to 70 M depending on the length of the azido(alkyl) spacer. Notably, we observed a complete switching in the regioselectivity of the confined cycloaddition reactions. That is, the confined reaction is stereoselective for the 1,4-isomer of the triazole adduct of the 4-azido pyridine-N-oxide derivative 2a, but turns stereoselective for the 1,5-counterpart for the azido(methyl) and azido(ethyl) derivatives, 2b and 2c. We used the computed DFT structures of the inclusion complexes to rationalize our findings.
:1 and 2:1 stoichiometry. We used simulated speciation profiles and performed ITC experiments to thermodynamically characterize the formed complexes. Adding mixtures of an azido-derivative with the propynyl–pyridinone in a solution of cage 1 resulted in the formation of the expected 1:1 and 2:1 homo-complexes, and allowed the detection and characterization of the corresponding ternary hetero-complexes (Michaelis) in solution, 2·4⊂1. The azide–alkyne cycloaddition reactions are significantly accelerated by confinement of the reactants in cage 1. We assessed the reactions' acceleration factor by determining their effective molarities (EM = kintra/kbulk). We derived the kintra values from the best-fit computer simulation of an elaborated theoretical kinetic model to the experimental kinetic data. The determined EM values range from 2000 to 70 M depending on the length of the azido(alkyl) spacer. Notably, we observed a complete switching in the regioselectivity of the confined cycloaddition reactions. That is, the confined reaction is stereoselective for the 1,4-isomer of the triazole adduct of the 4-azido pyridine-N-oxide derivative 2a, but turns stereoselective for the 1,5-counterpart for the azido(methyl) and azido(ethyl) derivatives, 2b and 2c. We used the computed DFT structures of the inclusion complexes to rationalize our findings.
Introduction
Chemists have designed and synthesized molecular containers inspired by enzymes,1,2 and used their cavities as nanometric flasks to mediate intra- and inter-molecular reactions.3,4 Similarly to enzymes' active sites, the simple inclusion of two reacting substrates within the confined space of a synthetic molecular flask increases their local concentration. Considering identical bulk concentrations, this simple confinement effect can significantly increase the bimolecular reaction rate compared to that in solution. However, confined reactions might experience other effects.5,6 The confinement process can arrange and orient the reacting groups of the substrates in a geometry close to that of the reaction's transition state (TS). In doing so, the binding of the substrates in the container's cavity can compensate for additional entropic costs of the reaction, thereby further reducing the energy barrier of the TS (ΔS‡).2Enthalpic stabilization of the TS, desolvation, and others have also been postulated to explain the reaction's acceleration induced by confinement.7,8 Controlling the relative orientation of the reacting groups within the confined reaction space can significantly influence the reaction's selectivity.9,10 This control is mainly exerted by size, shape, and functional groups complementarity between the substrates and cavities, as well as the minimization of steric clashes. Examples were reported in which the selectivity of the confined reaction was significantly altered11,12 or even completely changed13 compared to the bulk.
In short, the simultaneous binding of two reacting substrates in a molecular container leads to the formation of a ternary hetero-complex (a.k.a. Michaelis complex). This is a key feature of the confinement effect in bimolecular reactions, which, due to multiple factors, influences the reaction's rate and selectivity.9,14
Designing tailored molecular containers to facilitate specific chemical reactions is a complex task.15 Typically, these containers are synthetically challenging to access, and their customization involves intricate procedures.16 Additionally, predicting the influence of a molecular container on the confined chemical reaction (i.e., acceleration and selectivity) is not straightforward.
In the last few decades, the azide–alkyne Huisgen cycloaddition reaction has been widely studied. This 1,3-dipolar cycloaddition reaction grants access to triazole derivatives, which are highly relevant to medicinal chemistry and pharmacology.17 In the bulk solution, the inter-molecular version of the reaction is typically very slow, even at high temperatures, producing a mixture of two regioisomeric products: 1,4- and 1,5-disubstituted 1,2,3-triazoles.18 The Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction is a well-established and highly efficient procedure for the regioselective synthesis of 1,4-substituted 1,2,3-triazoles.19–21 On the other hand, an analogous method for obtaining 1,5-disubstituted triazoles is less developed and generally less efficient. A common approach is the Ru(II)-catalyzed azide–alkyne cycloaddition (RuAAC).22,23
The literature provides few examples of azide–alkyne cycloaddition (AAC) reactions mediated by molecular containers.24–26 These reactions were regioselective, producing the 1,4-disubstituted triazole isomer, and experienced significant accelerations. The acceleration factors, quantified as effective molarity (EM = kintra/kbulk), were in the range of 10–104 M.27
We recently described the acceleration of AAC reactions, Huisgen reactions, included in the cavity of the self-assembled [4 + 2] octa-imine bis-calix[4]pyrrole cage 1.28 The AAC reactions involved 4-azido(alkyl) pyridine-N-oxide derivatives, 2b–c, and the 4-ethynyl pyridine-N-oxide, 3 (Fig. 1). The functionalization of the cavity of cage 1 with eight inwardly directed NH polar groups constitutes the most significant difference with respect to other molecular flasks used in mediating the AAC reaction. This particularity allowed using highly directional interactions, i.e., hydrogen bonds, to drive the inclusion of the substrates and suitably orient their reacting groups in a productive geometry. In turn, it produced acceleration factors, quantified as EM, larger than 103 M. These represent the largest accelerations reported to date for bimolecular reactions included in a molecular container, in which the direct detection of the ternary Michaelis complex simplifies the analysis of the kinetic data. As in other examples, the mediated AAC reaction was regioselective, producing exclusively the 1,4-disubstituted isomer (i.e. 5b and 5c).
 | ||
| Fig. 1 Molecular structure of octa-imine cage 1 and scheme of the 1,3-dipolar cycloaddition reaction between azido-(2a–2c) and ethynyl (3 or 4) producing the two regioisomeric 1,4- (5 or 7) and 1,5- (6 or 8) disubstituted 1,2,3-triazoles. The corresponding proton assignments for octa-imine 1, substrates 2, and 4, and products 7 and 8 are also shown. | ||
We undertook this work because the AAC reaction between 2a and 3 was not noticeably accelerated when it was included in 1 (Fig. 1).28 In contrast, and as mentioned above, that of 2b with 3 was indeed significantly accelerated. The only difference between 2a and 2b was the incorporation of a methylene unit spacing the reacting azide group and the pyridine-N-oxide knob.
We became interested in evaluating the reactivity and acceleration effect caused by adding the methylene spacer unit in the alkyne substrate instead of the azide. Herein, we report our findings for analogous AAC reactions confined in cage 1 using 1-(2-propynyl)-4-pyridinone, 4, as the alkyne substrate. Pyridinone 4 is the surrogate of the desired homologous of pyridine-N-oxide 3 that was synthetically elusive for us.
We use 1H NMR titrations to characterize the formation of 1:1 and 2:1 homo-complexes by including 4 in cage 1. The results of related experiments with the series of azido(alkyl) pyridine-N-oxides, 2a–c, were already reported.28 Mixing equimolar amounts of an azido-derivative, 2a–c, with 1-(2-propynyl)-4-pyridinone 4 and cage 1 allowed the detection and characterization of the corresponding ternary hetero-complexes (Michaelis) in solution: (2·4)⊂1. We investigate the kinetics and regioselectivities of the AAC reactions emanating from the (2·4)⊂1 complexes. The obtained results demonstrate acceleration of the reactions and change in regioselectivity. The cycloaddition reactions of 4 with 2b–c mediated by cage 1 are highly regioselective for the 1,5-disubstituted triazole isomers, 8b–c (Fig. 1). This isomer is challenging to synthesize in a good yield and regioselective manner using metal-catalyzed approaches (e.g. RuAAC). Our findings represent the first reported case of 1,5-regioselectivity in AAC reactions mediated by molecular containers, marking a significant and novel contribution to the field.
Results and discussion
Synthesis
Octa-imine cage 1 and azido pyridine-N-oxide derivatives 2a–c were prepared following reported procedures.28,29 Notably, in this work, we isolated the octa-imine cage 1. Following the quantitative self-assembly of the octa-imine cage 1 in solution, the solvent was removed under vacuum. The reaction crude was dissolved in dichloromethane, and MeOH was added to precipitate pure 1 as a yellowish solid (DCM:MeOH 4:1).
The 1-(2-propynyl)-4-pyridinone 4 was prepared by reacting 4-hydroxypyridine with propargyl bromide in acetonitrile using potassium carbonate as a base.30 Compound 4 was isolated as a white solid after column chromatography purification. We used neutral alumina as the stationary phase and a DCM: isopropyl alcohol (IPA), 97:3, solvent mixture as eluent (see ESI for details and complete spectroscopic characterization of the compounds).
1,4-disubstituted 1,2,3-triazole derivatives, 7a–c, were synthesized via CuAAC reactions of the pyridine-N-oxides containing para-substituted azido(alkyl) residues, 2a–2c, and 1-(2-propynyl)-4-pyridinone 4. Analytical quantities of the 1,5-disubstituted counterparts, 8b–c, were obtained after HPLC purification of the reaction crudes of the uncatalyzed thermal cycloaddition reactions performed at 343 K in DMF solution. The reaction crudes contained the two regioisomeric 1,2,3-triazoles, 7 and 8, in a ∼3:1 molar ratio, respectively.‡ We used an analytical BEH HILIC column and a linear solvent gradient elution of CH3CN:H2O (98:2 to 60:40 in 15 minutes) for the isolation and purification of the two triazole isomers (see ESI for details and complete spectroscopic characterization of all compounds).
Binding studies with octa-imine cage 1
:1, (2·CD3CN)⊂1, and 2:1 (2)2⊂1, inclusion complexes of octa-imine cage 1 with azido pyridine-N-oxide derivatives, 2a–c.28 In the 1:1 complexes, one pyridine-N-oxide molecule 2 was bound in one hemisphere of the octa-imine cage 1 by establishing four convergent hydrogen bonding interactions between its oxygen atom and the pyrrole NHs. A molecule of solvent (i.e., CD3CN), was included in the opposite hemisphere through hydrogen bonding interactions. In the 2:1 complexes, both hemispheres of cage 1 included one molecule of pyridine-N-oxide 2. Their para-azido substituents converged in the center of the cage.:CD3CN, 9:1, solvent mixture. When approximately 1 equiv. of pyridinone 4 was added to a 2 mM solution of the octa-imine cage 1, two new sets of proton signals with different intensities appeared (Fig. S25†). We assigned these signals to the inclusion complexes: 1:1, (4·CD3CN)⊂1, and 2:1, (4)2⊂1. We also detected weak signals corresponding to the free guest 4 and cage 1.Upon adding nearly to 2 equiv. of 4, the signals of the 1:1 complex, (4·CD3CN)⊂1, disappeared, while those of the 2:1 complex, (4)2⊂1, increased significantly. At the same time, the signals of free 4 intensified, whereas those of free cage 1 became undetectable.
When more than 2 equiv. of 4 were added, the 1H NMR spectrum of the mixture remained largely unchanged, except for the increase in the intensity of the signals corresponding to the free 4. The presence of free 4, even after adding 1 equiv. to the 2 mM solution of cage 1, suggested that the overall binding constants for the 1:1 and the 2:1 complexes were lower than 104 M−1 and 108 M−2, respectively.
To obtain more accurate binding constants, we performed ITC experiments. The computer-controlled injections of aliquots (10 μL) of a 20 mM solution of guest 4 in a CHCl3:CH3CN 9:1 solvent mixture into a 1 mM solution of the octa-imine cage 1, in the same solvent mixture, placed in the calorimeter cell produced a gradual release of heat. The normalized areas of the integrated heat peaks produced a single sigmoidal binding isotherm, with inflection point centered close to a [4]/[1] molar ratio of 2 and not starting with a flat line (Fig. S52†). We fit the calorimetric data to the “two sets of sites” theoretical binding model implemented in the Microcal analysis software.31 The fit returned the macroscopic stepwise binding constants as K[(CH3CN)2⊂1 + 4 ⇌ (CH3CN·4)⊂1] = (4 ± 1) × 103 M−1 and K[(CH3CN·4)⊂1 + 4 ⇌ (4)2⊂1] = (1 ± 0.6) × 104 M−1. The calculated values agreed with our estimates derived from the 1H NMR titration experiment.
Study of the co-inclusion of 4-azido(alkyl) pyridine-N-oxides 2a–c with 1-(2-propynyl)-4-pyridinone 4
Next, we investigated the co-inclusion of para-azido-substituted pyridine-N-oxide derivatives 2 with 1-(2-propynyl)-4-pyridinone 4 in the octa-imine cage 1. An equimolar solution of the octa-imine cage 1 with guests 2a and 4 in CDCl3:CD3CN, 9:1 solvent mixture, produced immediately after its preparation a 1H NMR spectrum displaying four sets of proton signals for cage 1 (Fig. 2d). We assigned these signals as follows: three sets corresponded to the 1:1 complex (2a·CD3CN)⊂1 and the 2:1 homo-complexes (2a)2⊂1 and (4)2⊂1. We attributed the fourth set of signals to the ternary-hetero complex (2a·4)⊂1. Additionally, we detected residual signals from the aromatic protons, He and Hf, of free 4 (Fig. 2d).
 | ||
| Fig. 2 (Top) disproportionation equilibrium of homo-complexes producing the ternary hetero-complex with the assignment of proton signals. (Bottom) selected regions of the 1H NMR spectra (400 MHz, 298 K, CDCl3:CD3CN 9:1 solvent mixture) of (a) octa-imine cage 1; (b) equimolar mixture of cage 1 with 1-(2-propynyl)-4-pyridone 4; (c) equimolar mixture of cage 1 with 4-azido pyridine-N-oxide 2a; (d) equimolar mixture of 1, 4 and 2a immediately after its preparation. Proton assignments in blue and red correspond to the 1:1 and 2:1 homomeric complexes of 1-(2-propynyl)-4-pyridinone 4 and 4-azido pyridine-N-oxide 2a, respectively. Primed black protons correspond to the 2:1 heteromeric complex (2a·4)⊂1. | ||
From the integral values of selected proton signals, we estimated the equilibrium concentration of the (2a·4)⊂1 complex to be approximately 0.4 mM (Fig. S25†). Similar experiments with the azido(alkyl) pyridine-N-oxides 2b and 2c and 1-(2-propynyl)-4-pyridinone 4 yielded comparable concentrations (ca. 0.4 mM) for the corresponding ternary hetero-complexes (2b·4)⊂1 and (2c·4)⊂1 at equilibrium (Fig. S33 and S44†).
To further analyze the disproportionation equilibria ((2)2⊂1 + (4)2⊂1 ⇌ 2 (2·4)⊂1), we simulated the theoretical speciation using HySS2009 software.32 The stability constants for the 1:1 and 2:1 homo-complexes of cage 1 with 2b and 4 were fixed based on the values determined in separate titration experiments.28 We then manually fit the overall stability constant, β, for the formation of the hetero-ternary complex (CD3CN)2⊂1 + 2 + 4 ⇌ (2·4)⊂1, until the experimental and theoretical simulated concentrations of all the species aligned. From this analysis, we concluded that the stability constants of the three hetero-ternary complexes (2a·4)⊂1, (2b·4)⊂1, and (2c·4)⊂1, were similar, falling within the range of 8.0 × 107 – 1.3 × 108 M−2 (Table 1). Further details on the comparisons between experimental and theoretical speciation at equilibrium are provided in the ESI (Fig. S27, S30, S34, S37, S44, and S47).†
| Homo-complex | β2:1 (M−2) |
Hetero-complex | β2:1 (M−2) |
|---|---|---|---|
| (2a)2⊂1 | 6.2(±1.2) × 108 | (2a·4)⊂1 | 8.0(±1.6) × 107 |
| (2b)2⊂1 | 1.6(±0.3) × 109 | (2b·4)⊂1 | 1.3(±0.3) × 108 |
| (2c)2⊂1 | 4.0(±0.8) × 109 | (2c·4)⊂1 | 1.3(±0.3) × 108 |
| (4)2⊂1 | 4.0(±0.8) × 107 |
Using the determined β constants, the formula of the disproportionation equilibrium, Kdispro = [(2·4)⊂1]2/([(2)2⊂1] × [(4)2⊂1]) = [β(2·4)⊂1]2/([β(2)2⊂1] × [β(4)2⊂1]), and neglecting the presence of other equilibria in solution, we calculated an average value of Kdispro to be ca. 0.3. This value is significantly lower than the statistical estimate (Kdispro = 4) assuming isoenergetic 2:1 complexes. This difference derives mainly from a reduction in the stability of hetero-complexes (2·4)⊂1 compared to those of their 2:1 homo-analogs of the azido-derivatives 2. The stability reduction is likely due to steric clashes or/and repulsive dipole–dipole interactions between the two reacting groups in the hetero-complexes. This explanation also applies to the 2:1 complex of the 1-(2-propynyl)-4-pyridinone, (4)2⊂1, suggesting that the termolecular complexes involving 4 are generally less stable.
Monitoring the 1,3-dipolar cycloaddition reaction of 2a with 4 included in 1
We previously reported that the AAC reaction between 2b and 3 confined in cage 1 exhibited an exceptional acceleration factor (EM ∼103 M) and resulted in the exclusive formation of the 1,4-triazole isomer, 5b (Fig. 3).28 Given the strong structural similarity between the energy-minimized conformations of the 1,4-triazole isomers 5b and 7a included in 1 (Fig. 3b), we hypothesized that the analogous AAC reaction of 4-azido pyridine-N-oxide 2a and 1-(2-propynyl)-4-pyridone 4, when included in cage 1 (Fig. 3c), would be selective for the 1,4-triazole isomer, 7a, with a comparable acceleration factor.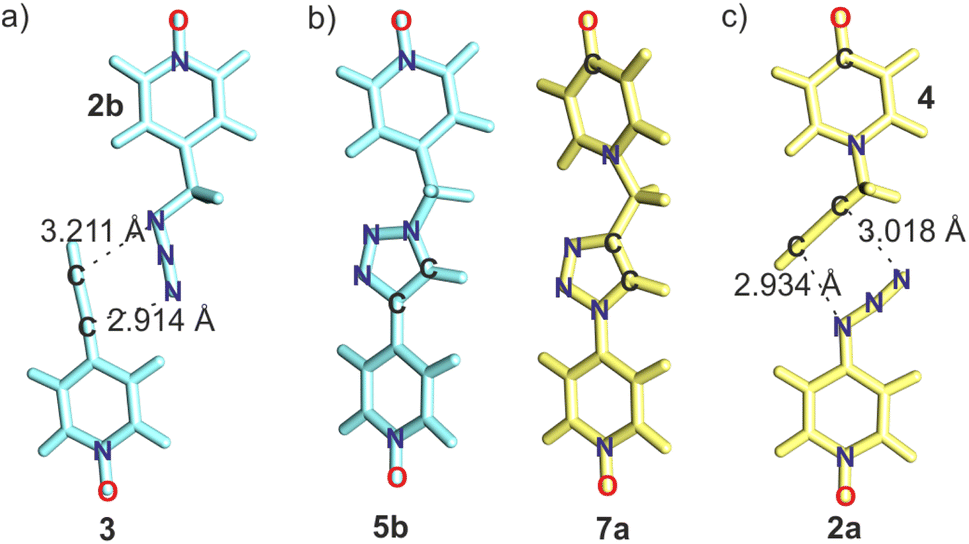 | ||
| Fig. 3 (a) Arrangement of the reacting substrates in the energy-minimized structures of: (a) the (2b·3)⊂1 ternary hetero-complex and (c) the (2a·4)⊂1 ternary hetero-complex. The Nazide–Cacetylide distances are shown. (b) Energy-minimized structures of the 1,4-triazole isomers 5b (light-blue) and 7a (light-yellow) in the complexes 5b⊂1 and 7a⊂1. For clarity the structure of cage 1 is not shown in the figures. | ||
We used 1H NMR spectroscopy to monitor the evolution of a nearly equimolar (2 mM) solution mixture of 2a, 4, and octa-imine cage 1. The 1H NMR spectrum acquired approximately 5 min after preparing the solution, showed the expected proton signals and distribution of homo-complexes and hetero-ternary complex: (2a·CD3CN)⊂1, (2a)2⊂1, (4)2⊂1 and (2a·4)⊂1, respectively (vide supra and Fig. 2d). After 6 h, the 1H NMR spectrum of the mixture revealed the emergence of a new set of signals for the protons of cage 1 (Fig. S27b†). Specifically, we detected two new broad signals resonating at δ = 8.8 and 8.9 ppm. We attributed these signals to the hydrogen-bonded pyrrole NHs of 1 in a new inclusion complex. The intensity of these NH signals increased over time, while the signals corresponding to the homo- and hetero-ternary complexes diminished. We concluded that the structure of the new inclusion complex should correspond to the 7a⊂1 complex, that is, the inclusion complex of the 1,4-disubstituted triazole isomer resulting from the AAC of 2a with 4 inside cage 1.
To further support our hypothesis, we performed a solid–liquid extraction of 7a using a mM solution of 1. The 1H NMR spectrum of the filtered solution was diagnostic of the quantitative formation of the 7a⊂1 complex. The spectrum totally matched the proton signals of the major species detected after monitoring the reaction of 2a with 4 in the presence of cage 1 for 16 days (Fig. S27e†). Notably, after 16 days and in the absence of 1, a control reaction of 2a with 4 at 2 mM concentration produced a 1H NMR spectrum that only displayed the proton signals of the reactants, with no indication of the presence of signals for the two triazole isomeric products.
Taken together, these results demonstrated that the AAC reaction of 2a with 4 was significantly accelerated in the presence of octa-imine cage 1. Moreover, the reaction was regioselective, forming the 1,4-triazole isomer 7a. The regioselective outcome aligned perfectly with our hypothesis, which was based on a related study's exclusive formation of the 5b⊂1 complex.28 The 1,2,3-triazole spacers of 7a and 5b only differ in the position of the methylene unit: 4 –C for 7a and 1 –N for 5b (Fig. 3b).
To assess the acceleration factor of the reaction of 2a with 4 exerted by confinement in cage 1, we monitored the changes in the concentration of the 7a⊂1 complex over time by integrating selected proton signals. We fitted the kinetic data to a theoretical kinetic model accounting for six binding equilibria involving three reactants (1, 2a, and 4) alongside the irreversible cycloaddition reaction occurring within cage 1 (Fig. S29 and S58†). The equilibria produced five species: the 1:1 and 2:1 homo-complexes, as well as the hetero-complex with 1 (Fig. 4). We considered the contribution of the uncatalyzed reaction (i.e., bulk reaction) to be negligible (vide infra), and therefore the model does not include the irreversible background thermal cycloaddition reaction. The kinetic model does not account for the dissociation of the cycloaddition product complex 7a⊂1 owing to its large thermodynamic stability (K7a⊂1 > 106 M−1, see ESI†).
 | ||
| Fig. 4 Theoretical model used for the non-linear mathematical analysis of the kinetic experimental data. The logK value is depicted on the top of each equilibrium. | ||
Using this kinetic analysis, the best fit of the data to the theoretical kinetic model yielded an optimized rate constant for the AAC reaction of 2a with 4 inside cage 1 k(7a–intra) = (4.4 ± 0.8) × 10−6 s−1. Separately, we determined the rate constant of the reaction producing 7a in the bulk solution to be k(7a–bulk) = 6.1 × 10−8 M−1 s−1. From these values, we quantified the acceleration factor induced by including the AAC reaction of 2a with 4 in cage 1 as EM = k(7a–intra)/k(7a–bulk) = 7 × 101 M The measured EM is two orders of magnitude lower than the EM > 103 M reported for the analogous reaction of the 4-azido(methyl) pyridine-N-oxide 2b with 4-ethynyl pyridine-N-oxide 3 included in cage 1 and exclusively producing 5b.28
In summary, replacing the azide–alkyne Huisgen-pair 2b–3 with its structural analog 2a–4 did not alter the 1,4-regioselectivity of the reactions within the octa-imine cage 1. However, it significantly impacted the reaction's acceleration factor. The TS occurring in the (2b·3)⊂1 is favored by 1.6 kcal mol−1 over the (2a·4)⊂1. However, we lack evidence to determine whether this additional stabilization originates from entropic/enthalpic factors or from a contribution of both.
Monitoring the 1,3-dipolar AAC reaction of 2b with 4 included in 1
We reported acceleration factors of EM ≥ 103 M for the AAC reaction of the 4-ethynyl pyridine-N-oxide 3 with 2b included in cage 1. These results contrasted with the lack of acceleration for the analogous AAC reaction of 3 with 2a within 1. We were intrigued by the acceleration factor caused by increasing one methylene unit in the azide-reacting substrate, that is, using 2b instead of 2a, in the AAC reaction with 4 within 1.To investigate this, we prepared an equimolar mixture of 2b, 4, and cage 1. The 1H NMR spectrum acquired immediately after the solution's preparation displayed four sets of separate signals for the protons of cage 1. We assigned these signals to the following cage complexes: (CD3CN·2b)⊂1, (2b)2⊂1, (4)2⊂1, and (2b·4)⊂1 (Fig. 5b). Additionally, we observed signals of reduced intensity corresponding to the protons of free 4.
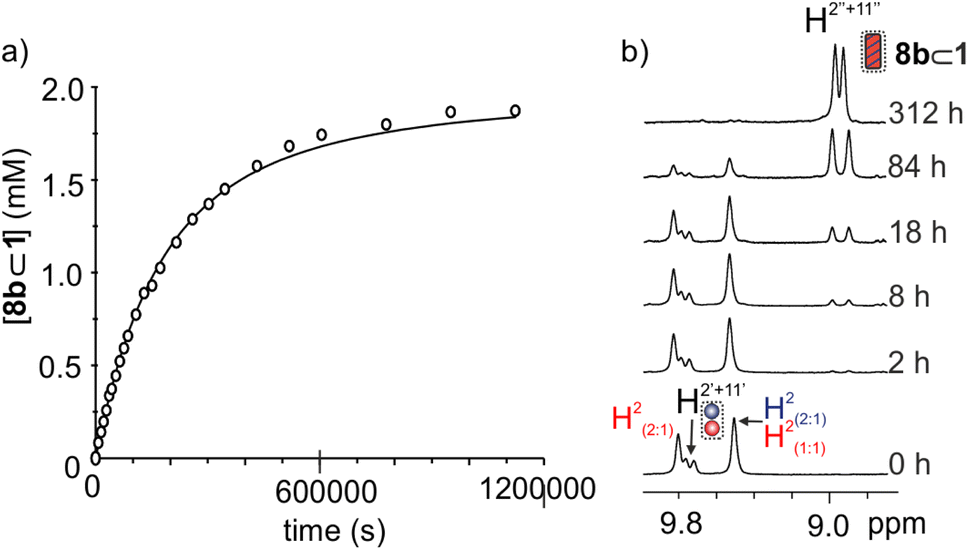 | ||
| Fig. 5 (a) Changes in the concentration of 8b⊂1 over time determined using the integral areas of protons H2′ and H11′. The solid line represents the fit of the experimental kinetic data to the theoretical model using COPASI V4.25. (b) Selected region of the 1H NMR spectra of the kinetic study of the formation of 8b⊂1 starting from a 1:1:1 mixture of 2b, 4, and octa-imine cage 1. The selected region corresponds to the area in the 1H NMR spectrum where the pyrrole NH protons resonate. | ||
Using 1H NMR spectroscopy, we monitored the solution mixture's evolution over a period of two weeks (Fig. 5). After 2 hours, we detected the emergence of two new signals for the NH protons of cage 1. We attributed these signals to the complex of the cycloaddition product. After 12 days, this complex became the predominant species in the solution (Fig. 5b). We no longer detected any of the proton signals of the initially formed cage complexes. We concluded that the confined AAC cycloaddition reaction between 4 and 2b had progressed nearly to completion within 12 days. This result represented a significant acceleration compared to the reaction of 4 with 2a in the cavity of 1 (vide supra, less than 50% of conversion after 12 days, Fig. S27d†). Nevertheless, both reactions were highly regioselective. We demonstrated that the AAC reaction of 4 with 2a confined in cage 1 selectively produced the 1,4-triazole isomer, 7a. Similarly, we hypothesized that the confinement of 4 with 2b in the cavity of 1 would lead to the selective formation of the 1,4-triazole isomer 7b as the primary cycloaddition product.
We added solid 7b to a mM solution of cage 1. After filtration, the 1H NMR spectrum of the filtered solution corresponding to the 7b⊂1 complex, did not match the proton signals of the major species detected after the quantitative reaction of 4 with 2b mediated by 1. In contrast, the proton signals of the major species of the reaction perfectly matched those of the 8b⊂1 complex (Fig. S34†).§ This observation indicated that, compared to previous examples of AAC reactions confined in cage 1, the reaction of 4 with 2b experienced a switch in regioselectivity. Notably, by enhancing the signal intensity of the 1H NMR spectrum registered at the end of the AAC reaction of 4 with 2b mediated by 1, we could detect the presence of traces of the 7b⊂1 complex in the solution (<3%) (Fig. S35a†). The significant change in triazole regioisomers' ratio, 7b:8b, from 75:25 in bulk§ to 3:97 in cage 1, demonstrated the strong influence of cage 1 in controlling the regioselectivity of the included AAC reaction.
To further investigate the change in the reaction's regioselectivity, we performed density functional theory (DFT) calculations at the RI33–35-BP8633-D3BJ36,37 def2-TZVP38,39 level, as implemented in Turbomole v.7.8,40 on the energies and structures of the inclusion complexes 7b⊂1 and 8b⊂1, and two conformers of the ternary hetero-complexes leading to their formation (2b·4)1,4⊂1 and (2b·4)1,5⊂1, respectively (Fig. 6 and 7). We also localized and optimized the transition state geometries leading to the two isomeric complexes 7b⊂1 and 8b⊂1 using DFT calculations at the RI33–35-BP8633-def-SV(P)38,39 level, as implemented in Turbomole v.7.0.40 The optimized structures are in agreement with those expected for late transition states.
 | ||
| Fig. 6 Equilibrium between two different conformers of the ternary hetero-complex (2b·4)⊂1 displaying the included reactive groups in different orientations favoring the 1,5- (left (2b·4)1,5⊂1) and the 1,4- (right (2b·4)1,4⊂1) disubstituted triazole product, respectively. Guests are shown in ball and stick representation, dark-blue for 1-(2-propynyl)-4-pyridinone 4 and light-blue for azido(methyl) pyridine-N-oxide 2a. In the equilibrium, cage 1 is shown in stick representation with one of the spacer linkers deleted to assist in visualizing the geometrical arrangement of the included reactive groups. The structure of the complexes with the cage rendered as CPK model is shown aside from each ball and stick representation. This representation evidences the opening of the cage portals allowing the protrusion of some sections of the included substrates through them. | ||
 | ||
| Fig. 7 (Top) energy-minimized structures (DFT) of inclusion complexes: (a) 7b⊂1 and (b) 8b⊂1. The average hydrogen bond distance (d(H–N⋯O)) for each hemisphere is depicted next to the complex's structure. (Bottom) 1H NMR spectra of the solutions obtained in the solid–liquid extraction experiments of the triazole isomers with a 2 mM solution of octa-imine cage 1 in a CDCl3:CD3CN 9:1 mixture: (c) 8b⊂1 and (d) 7b⊂1. | ||
The results of our calculations showed that the two triazole isomers, 7b and 8b, are size- and shape-compatible, as well as functionally complementary to the polar cavity of the octa-imine cage 1 (Fig. 7). Additionally, the energy-minimized structures of the two conformers of the ternary complex (2b·4)1,4⊂1 and (2b·4)1,5⊂1 (Fig. 6) showed that, in both of them, the reacting groups were well positioned to achieve the TS geometry. This was particularly evident for the (2b·4)1,5⊂1 conformer, where the distances between the carbon and nitrogen atoms of the dipolarophile (alkyne) and the 1,3-dipole (azide) were adequate for forming the two single bonds of the triazole in the TS (Fig. S72†). Moreover, the small computed energy difference (ΔE = 1.7 kcal mol−1) favoring the (2b·4)1,5⊂1 conformer suggested that it is likely to be more prevalent in solution.
Computational studies on 1,3-dipolar cycloaddition reactions supported the idea that the TS resembled products more than starting materials.41,42 In short, the TS of AAC reactions is product-like, mainly with respect to the bending of the 1,3-dipole.
Based on this premise, the energy difference between the products' complexes, 7b⊂1 and 8b⊂1, should reflect more accurately the energy difference between their corresponding TSs. The calculated energy difference between these two complexes was only ∼1 kcal mol−1, favoring the 1,5-triazole isomer complex, 8b⊂1.¶ This result contrasted with the 22.4 kcal mol−1 energy difference calculated for the product complexes, 7a⊂1 and 8a⊂1, which strongly favored the 1,4-triazole isomer complex, 7a⊂1. For context, 7a and 8a correspond to the 1,4- and 1,5-triazole isomers, respectively, formed in the AAC reaction of 4 with 2a (Fig. 1). While the computational results for the reaction of 4 with 2b within 1, did not fully account for the experimentally observed 3:97 ratio of 7b⊂1 to 8b⊂1 complexes (corresponding to a 2.0 kcal mol−1 difference), they explained the observed regioselectivity switch.||
Filtrated solutions of separate solid–liquid extraction experiments of 7b and 8b with mM CDCl3:CD3CN (9:1) solutions of octa-imine cage 1 yielded 1H NMR spectra displaying sharp and well-defined proton signals. This confirmed the good fit between the two triazole isomers, 7b, and 8b, and the octa-imine's 1 cavity. However, the 1H NMR spectra of the two inclusion complexes, 7b⊂1 and 8b⊂1, revealed differences in hydrogen-bonding distances. In the case of 7b⊂1, the pyrrole NHs of the cage exhibited a complexation-induced shift (CIS = ΔδNH(free–bound)) of 2.1 ppm. In contrast, the 8b⊂1 complex showed a smaller CIS of 1.4 ppm for the analogous protons (Fig. 7c and d, respectively). These chemical shift differences suggest that the distances of the hydrogen bonding interactions established between the NpyrroleH of the cage and the two oxygen atoms of the different polar ends O–N/O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the triazole isomers are shorter in the 7b⊂1 complex than in the 8b⊂1 counterpart.
C of the triazole isomers are shorter in the 7b⊂1 complex than in the 8b⊂1 counterpart.
This interpretation is supported by DFT-optimized structures of the two complexes, which reveal an average hydrogen bond distance of 2.863 Å for 7b⊂1 (Fig. 7a) and 3.001 Å for 8b⊂1 (Fig. 7b). These values underscore the limited structural flexibility of cage 1, which is unable to adapt efficiently to the different lengths of the guest isomers. Notably, in our previous studies on the binding of pyridine-N-oxide guests with calix[4]pyrroles, we found that optimal O⋯H–N hydrogen bond distances typically fall within the 2.9–3.0 Å range.43,44 Deviations from this range – particularly the shorter distance observed in the 7b⊂1 complex – suggests a geometric mismatch between host and guest, with the guest being slightly too long to fit ideally within the rigid cavity of the host.
To determine the acceleration factor of the reaction of 4 with 2b confined in 1, we fit the changes in concentration of the 8b⊂1 complex over the overall reaction to the elaborated kinetic theoretical model with kintra(8b) as single variable (Fig. S59†). The computer fit returned the optimized value of kintra(8b) as (5.1 ± 0.4) × 10−5 s−1 (Fig. 5a and Table 2). Separately, we determined the kbulk(8b) = 1.1 × 10−8 M−1s−1 for the thermal AAC reaction of 4 with 2b. We used the same kinetic methodology described in the previous section for the reaction between 2a and 4.
| Guest pair | Product | kbulkc (M−1s−1) | kintrae (s−1) | EMf (M) |
|---|---|---|---|---|
| a See ref. 28.b Traces of 1,4-7b (<3%) were detected in the 1H NMR at the end of the mediated reaction.c Determined by best-computer fit of the kinetic data using COPASI and a theoretical kinetic model for two competitive irreversible first-order bimolecular reactions.d n.d. = not determined.e Determined by best-computer fit of the kinetic data using COPASI and a theoretical kinetic model considering six binding equilibria, producing five different complexes and the irreversible pseudo-unimolecular reaction to produce the bound cycloaddition product.f EM = kintra/kbulkg We assumed that k(bulk–8c) can be approximated to k(bulk–8b). | ||||
| (2b·3) a | 1,4-5a | 5.6 × 10−8 | 5.0 × 10−5 | ∼1 × 103 |
| (2a·4) | 1,4-7a | 6.1 × 10−8 | (4.4 ± 0.8) × 10−6 | ∼7 × 101 |
| (2b·4) | 1,5-8b b | 2.3 × 10−8 | (5.1 ± 0.4) × 10−5 | ∼2 × 103 |
| (2c·4) | 1,5-8c | n.dd | (4.7 ± 0.4) × 10−6 | g∼4 × 102 |
Using these values, we determined an EM ∼2 × 103 M (Table 2). This represents an acceleration two figures larger than that observed for the analogous reaction involving 4-azido pyridine-N-oxide 2a instead of 4-azido(methyl) pyridine-N-oxide 2b. Moreover, this EM is in the same order of magnitude as the one determined in our previous work for accelerating the reaction between 2b and 4-ethynyl pyridine-N-oxide 3 (EM = 103 M).28
Taken in concert, the results of this and the previous sections highlight the strict requirements of size, shape, and function complementarity between the reacting pair of substrates and products with the container's cavity. In confined reactions, minor structural changes of the reacting substrates and products strongly influence the reaction's acceleration and regioselectivity.
To separate and compare the enthalpic and entropic components of the Gibbs energy barriers of the cycloaddition reaction between 2b and 4, both in the cage and in the bulk solution, we determined the reactions' rate constants (kbulk and kintra) at three different temperatures (298, 303, and 308 K). The activation reaction parameters, ΔH‡ and ΔS‡ derived from the corresponding Eyring plots (Fig. S71 and Table S5†) indicated that the observed acceleration is mainly caused by a reduction in the entropic cost associated with bringing the reactants together. Therefore, the binding of substrates in the cage primarily acts as an entropic reaction trap.
:CD3CN, 9:1, solvent mixture. The reaction also underwent a regioselectivity switch, yielding exclusively the inclusion complex of the 1,5-disubstituted 1,2,3-triazole isomer, 8c⊂1 (Fig. S45†).Using the previously described kinetic analysis, we determined the acceleration factor for the AAC reaction of 4 with 2c within 1, yielding an EM ∼4 × 102 M (Table 2). This EM value is one order of magnitude lower than that determined for the reaction of 4 with the shorter azido-derivative analog 2b confined within the polar cavity of cage 1. We attributed this difference to the expected entropic penalty associated with restricting an additional single-bond rotation in the TS of the reaction involving the ethyl-substituted substrate 2c. Moreover, the additional methylene in 2c compared to 2b allows a greater number of low-energy conformational isomers in the ternary complex. The increased flexibility reduces the degree of preorganization. It is likely that both factors, entropic effects at the TS and decreased preorganization of the ternary complex, jointly contribute to the lower reaction rate observed for 2c compared to 2b in its reaction with 4 within cage 1.
We performed DFT calculations of the two complexes, 7c⊂1 and 8c⊂1, that are produced by including the 1,2,3-triazole isomers of the cycloaddition reaction of 4 with 2c in cage 1. Likewise, we computed the two conformers of the ternary complex (4·2c)1,4⊂1 and (4·2c)1,5⊂1, featuring a relative arrangement of reacting groups suitable to produce the 7c⊂1 and 8c⊂1 complexes, respectively, upon cycloaddition reaction. The results of our calculations assigned an energetic preference of approximately 8.1 kcal mol−1 in favor of the complex of the 1,5-disubstituted isomeric product, 8c⊂1 (see dataset collection of computational results available at ioChem-BD repository).45,**,†† In contrast, the energy difference between the two conformers of the ternary complex was just 1.8 kcal mol−1, favoring the (4·2c)1,5⊂1 conformer having a suitable arrangement of reacting groups to produce the 1,5-triazole isomer upon cycloaddition reaction. As mentioned earlier, the regioselectivity level of the reaction can be better explained by correlating the energy difference between the cage complexes of the cycloaddition isomeric products, 7c⊂1 and 8c⊂1, to the energy difference of their corresponding TSs.
The reaction of 4 with 2c produced exclusively the 8c⊂1 complex. The proton signals of the 7c⊂1 complex were not detected in the 1H NMR spectrum acquired at the end of the reaction (8c⊂1:7c⊂1 < 98:2 molar ratio). This result allowed us to estimate a minimum value of 2.3 kcal mol−1 for the energy difference between the TSs yielding the 7c⊂1 and 8c⊂1 complexes, in favor of the latter.
Solid–liquid extraction experiments with guest 7c (Fig. S48†) and 8c (Fig. S49†) using a 2 mM solution of octa-imine cage 1 revealed marked differences in binding behavior. The 1H NMR spectrum of the filtered solution obtained after extraction with 8c showed a single set of signals corresponding to the cage protons, which we attributed to the formation of the 8c⊂1 complex. The absence of signals from the free cage suggests that the binding is essentially quantitative, allowing us to estimate a binding constant greater than 104 M−1 for the 8c⊂1 complex. In contrast, the 1H NMR spectrum of the filtered solution from the extraction with 7c showed two sets of protons for the cage, corresponding to both the 7c⊂1 complex and free cage 1. We also observed the proton signals of free 7c. These observations indicated a significantly weaker interaction, with a binding constant for the 7c⊂1 complex below 104 M−1.
Both experimental and theoretical studies thus confirm the preferential binding of cage 1 for the 1,5-triazole isomer 8c over the 1,4-isomeric counterpart 7c.
Conclusions
The reported results highlight the critical role of the container design and the complementary size, shape, and function between its cavity, and the substrates and products in accelerating and controlling the regioselectivity of the confined 1,3-dipolar cycloaddition reactions. The Huisgen cycloaddition reactions of azido pyridine-N-oxide 2a with 1-(2-propynyl)-4-pyridinone 4 within the octa-imine bis-calix[4]pyrrole cage 1 experienced an acceleration quantified with EM = 70 M. This acceleration factor is two orders of magnitude lower than the one previously determined using a related substrate pair (4-azido(methyl) pyridine-N-oxide 2b and 4-ethynyl pyridine-N-oxide 3). The two reactions are highly regioselective for the 1,4-triazole isomer, 7a and 5b, respectively, which are structurally very similar. However, the confinement of the two reactions within cage 1 produced significantly different acceleration factors, 70 vs. > 103 M.The cycloaddition reactions of 1-(2-propynyl)-4-pyridinone 4 with azido derivatives having one (2b), and two (2c), methylene units between the azido group and the pyridine-N-oxide ring increased their acceleration EM factors to 2000 or 400 M, respectively, when included in cage 1. Remarkably, the two reactions experienced a regioselectivity switch toward the 1,5-disubstituted 1,2,3-triazole isomer.
Binding studies confirmed that both, the 1,4-(8c–b) and 1,5-triazole (7c–b) isomers of the reactions are included in the cavity of the octa-imine cage 1. The results of DFT calculations assigned greater thermodynamic stability to the cage complexes with the 1,5-disubstituted isomers, 8c–b, over the 1,4-counterparts, 7c–b. Correlating the energy difference of the cage complexes of the cycloaddition isomeric triazole products, 7⊂1 and 8⊂1, with those of their corresponding TSs provided a straightforward method for explaining the observed regioselectivity.
We discovered the unprecedented switching of regioselectivity, favoring the 1,5-disubstituted triazole isomer, in AAC reactions mediated by a molecular container. Our findings emphasize the importance of molecular containers in controlling both the acceleration and product selectivity of confined reactions. We expect that our work will contribute to developing molecular reactor vessels to access challenging 1,5-disubstituted 1,2,3-triazole isomers, which are difficult to obtain in thermal or even metal-catalyzed reactions in bulk solution.
Data availability
All dataset collection of computational results of this manuscript is available in the ioChem-BD repository and can be accessed through this link: https://doi.org/10.19061/iochem-bd-1-388. The dataset collection includes two .mp4 files that animate the vibrational mode associated with the transition state (i.e., imaginary frequency) leading to the formation of the two isomeric complexes 7b⊂1 and 8b⊂1.Author contributions
Conceptualization, P.·B.; methodology, Y. L.; computational studies, P. B. and G.A.; formal analysis, P.·B., G. A. and Y. L.; writing – original draft, G. A.; writing – review and editing, P.·B., G. A., and Y. L.; supervision, P. B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by MICIU/AEI/10.13039/501100011033, FEDER/UE (PID2023-149233NB-I00) and MICIU/AEI/10.13039/501100011033 (Severo Ochoa Excellence Accreditations CEX2009-000925 S and CEX2024-001469 S), CERCA Programme/Generalitat de Catalunya, AGAUR (2021 SGR 00851), and the ICIQ Foundation.Notes and references
- J. P. Richard, Enzymatic Rate Enhancements: A Review and Perspective, Biochemistry, 2013, 52, 2009–2011 CrossRef CAS PubMed.
- M. I. Page and W. P. Jencks, Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1678–1683 CrossRef CAS PubMed.
- L.-D. Syntrivanis and K. Tiefenbacher, Reactivity Inside Molecular Flasks: Acceleration Modes and Types of Selectivity Obtainable, Angew. Chem., Int. Ed., 2024, 63, e202412622 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- Z. Ashbridge and J. N. H. Reek, The multifaceted roles of MnL2n cages in catalysis, Nat. Synth., 2024, 3, 1197–1207 CrossRef CAS.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Advances in supramolecular host-mediated reactivity, Nat. Cat., 2020, 3, 969–984 CAS.
- K. Wang, J. H. Jordan, X.-Y. Hu and L. Wang, Supramolecular Strategies for Controlling Reactivity within Confined Nanospaces, Angew. Chem., Int. Ed., 2020, 59, 13712–13721 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Chemical reactivity under nanoconfinement, Nat. Nanotech., 2020, 15, 256–271 CrossRef CAS PubMed.
- R. Wang and Y. Yu, Site-selective reactions mediated by molecular containers, Beilstein J. Org. Chem., 2022, 18, 309–324 CrossRef CAS PubMed.
- H. Takezawa, K. Iizuka and M. Fujita, Selective Synthesis and Functionalization of an Acyclic Methylene-Bridged-Arene Trimer in a Cage, Angew. Chem., Int. Ed., 2024, 63, e202319140 CrossRef CAS PubMed.
- X.-R. Mao, Q. Wang, S.-P. Zhuo and L.-P. Xu, Reactivity and Selectivity of the Diels–Alder Reaction of Anthracene in [Pd6L4]12+ Supramolecular Cages: A Computational Study, Inorg. Chem., 2023, 62, 4330–4340 CrossRef CAS PubMed.
- J. Huang and P. Ballester, A Bimolecular Diels–Alder Reaction Mediated by Inclusion in a Polar Bis-calix[4]pyrrole Octa-Imine Cage, J. Am. Chem. Soc., 2025, 147, 13962–13972 CrossRef CAS PubMed.
- M. Yoshizawa, M. Tamura and M. Fujita, Diels-Alder in Aqueous Molecular Hosts: Unusual Regioselectivity and Efficient Catalysis, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- C. Gaeta, P. La Manna, M. De Rosa, A. Soriente, C. Talotta and P. Neri, Supramolecular Catalysis with Self-Assembled Capsules and Cages: What Happens in Confined Spaces, ChemCatChem, 2021, 13, 1638–1658 CrossRef CAS.
- M. Otte, Reactions in Endohedral Functionalized Cages, Eur. J. Org Chem., 2023, 26, e202300012 CrossRef CAS.
- W. Liu and J. F. Stoddart, Emergent behavior in nanoconfined molecular containers, Chem, 2021, 7, 919–947 CAS.
- N. M. Grob, R. Schibli, M. Béhé, I. E. Valverde and T. L. Mindt, 1,5-Disubstituted 1,2,3-Triazoles as Amide Bond Isosteres Yield Novel Tumor-Targeting Minigastrin Analogs, ACS Med. Chem. Lett., 2021, 12, 585–592 CrossRef CAS PubMed.
- R. Huisgen, Kinetics and Mechanism of 1,3-Dipolar Cycloadditions, Angew. Chem., Int. Ed., 1963, 2, 633–645 CrossRef.
- J. E. Hein and V. V. Fokin, Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(i) acetylides, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- M. Meldal and C. W. Tornøe, Cu-Catalyzed Azide−Alkyne Cycloaddition, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides, J. Am. Chem. Soc., 2005, 127, 15998–15999 CrossRef CAS PubMed.
- M. De Tullio, A. M. Borys, A. Hernán-Gómez, A. R. Kennedy and E. Hevia, Regioselective synthesis of 1,5-disubstituted 1,2,3-triazoles catalyzed by cooperative s-block bimetallics, Chem Catal., 2021, 1, 1308–1321 CAS.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, Cycloaddition induced by cucurbituril. A case of Pauling principle catalysis, J. Org. Chem., 1983, 48, 3619–3620 CrossRef CAS.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, Catalysis by cucurbituril. The significance of bound-substrate destabilization for induced triazole formation, J. Org. Chem., 1989, 54, 5302–5308 CrossRef CAS.
- J. Chen and J. Rebek, Selectivity in an Encapsulated Cycloaddition Reaction, Org. Lett., 2002, 4, 327–329 CrossRef CAS PubMed.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, Effective Molarities in Supramolecular Catalysis of Two-Substrate Reactions, Acc. Chem. Res., 2004, 37, 113–122 CrossRef CAS PubMed.
- Y. Li, C. F. M. Mirabella, G. Aragay and P. Ballester, Acceleration and Selectivity of 1,3-Dipolar Cycloaddition Reactions Included in a Polar [4 + 2] Octa-imine Bis-calix[4]pyrrole Cage, JACS Au, 2025, 5, 902–912 CrossRef CAS PubMed.
- L. Adriaenssens, J. L. Acero Sánchez, X. Barril, C. K. O'Sullivan and P. Ballester, Binding of calix[4]pyrroles to pyridine N-oxides probed with surface plasmon resonance, Chem. Sci., 2014, 5, 4210–4215 RSC.
- C. Aubert, P. Betschmann, M. J. Eichberg, V. Gandon, T. J. Heckrodt, J. Lehmann, M. Malacria, B. Masjost, E. Paredes, K. P. C. Vollhardt and G. D. Whitener, Cobalt-Mediated [2+2+2] Cycloaddition versus C=H and N=H Activation of Pyridones and Pyrazinones with Alkynes: An Experimental Study, Chem. - Eur. J., 2007, 13, 7443–7465 CrossRef CAS PubMed.
- Microcal Origin Data Analysis v7.21, Malvern Instruments Limited Search PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Hyperquad simulation and speciation (HySS): a utility program for the investigation of equilibria involving soluble and partially soluble species, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef PubMed.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- M. Sierka, A. Hogekamp and R. Ahlrichs, Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- D. Rappoport and F. Furche, Property-optimized Gaussian basis sets for molecular response calculations, J. Chem. Phys., 2010, 133, 134105–134111 CrossRef PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- TURBOMOLE Program Package For Electronic Structure 817 Calculations. https://www.turbomole.com.
- H. Daver, J. N. Harvey, J. Rebek Jr and F. Himo, Quantum Chemical Modeling of Cycloaddition Reaction in a Self-Assembled Capsule, J. Am. Chem. Soc., 2017, 139, 15494–15503 CrossRef CAS PubMed.
- D. H. Ess and K. N. Houk, Theory of 1,3-Dipolar Cycloadditions: Distortion/Interaction and Frontier Molecular Orbital Models, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef CAS PubMed.
- A. Galan, G. Aragay and P. Ballester, A chiral “Siamese-Twin” calix[4]pyrrole tetramer, Chem. Sci., 2016, 7, 5976–5982 RSC.
- L. Escobar, E. C. Escudero-Adán and P. Ballester, Guest Exchange Mechanisms in Mono-Metallic PdII/PtII-Cages Based on a Tetra-Pyridyl Calix[4]pyrrole Ligand, Angew. Chem., Int. Ed., 2019, 58, 16105–16109 CrossRef CAS PubMed.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, Managing the computational chemistry big data problem: the ioChem-BD platform, J. Chem. Inf. Model., 2015, 55, 95–103 CrossRef PubMed.
- D. Clarke, R. W. Mares and H. McNab, Preparation and pyrolysis of 1-(pyrazol-5-yl)-1,2,3-triazoles and related compounds, J. Chem. Soc., Perkin Trans. 1, 1997, 1799–1804 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General information and instruments, synthetic procedures, characterization of compounds 4, 7a–c, and 8b–c, binding studies, theoretical kinetic models, kinetic and thermodynamic characterization of cycloaddition reactions. See DOI: https://doi.org/10.1039/d5sc03033a |
| ‡ The 3:1 ratio of 1,2,3-triazole regioisomers, 7:8, estimated for all bulk reactions, is greater than the 1.1–2.0 range commonly mentioned for Huisgen AAC reactions, unless the acetylene component is attached to an electron-withdrawing group.46 |
| § We acquired the 1H NMR spectrum of the 8b⊂1 complex by performing a solid–liquid extraction of the 1,5-triazole isomer 8b with a mM solution of cage 1. In turn, we isolated the 1,5-triazole 8b by purification of the crude of the thermally promoted AAC reaction of 4 with 2b. |
| ¶ The calculated energy between the two transition states, 7bTS⊂1 and 8bTS⊂1, leading to the corresponding inclusion complexes, 7b⊂1 and 8b⊂1, was 2.4 kcal mol−1 in favor of the latter. This result is in line with the experimentally observed ratio of products 7b⊂1:8b⊂1 (3:97; 2.0 kcal mol−1) and consistent with the energy difference of their DFT-optimized structures (∼1.0 kcal mol−1). |
| || We estimated K7b⊂1 ∼ 1 × 104 M−1 and K8b⊂1 ∼ 1 × 105 M−1 using ITC experiments. The estimated tenfold ratio of K as corresponds to an energy difference of 1.4 kcal mol−1, which agrees with the results derived from the kinetic experiments (3:97, ∼2 kcal mol−1). Additionally, the result of a competitive equimolar binding experiment between 7b and 8b with cage 1 extrapolated, using COPASI, to six months to reach equilibrium, yielded a 7b⊂1:8b⊂1 ratio of 25:75, or K8b⊂1/K7b⊂1 ∼ 10, supporting the greater thermodynamic stability of the 8b⊂1 complex (1,5-triazole). See ESI Fig. S56 and S57 for details. |
| ** Dataset collection can be accessed through this link: https://doi.org/10.19061/iochem-bd-1-388. |
| †† The calculated average hydrogen bond distances in the DFT-optimized 7c⊂1 (2.742 Å) and 8b⊂1 (2.932 Å) complexes suggest that guest 8c is a better geometric fit for the polar cavity of cage 1. |
| This journal is © The Royal Society of Chemistry 2025 |