
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-metal synergistic catalysis for promoting electrocatalytic CO2 reduction
Peng-Yu
Shi†
 ,
Yan
Yan†
,
Si-Yuan
Yang
,
Jing-Jing
Hao
,
Mei
Wang
* and
Tong-Bu
Lu
*
,
Yan
Yan†
,
Si-Yuan
Yang
,
Jing-Jing
Hao
,
Mei
Wang
* and
Tong-Bu
Lu
*
School of Materials Science and Engineering, Institute for New Energy Materials & Low Carbon Technologies, Tianjin University of Technology, Tianjin 300384, China. E-mail: meiwang@email.tjut.edu.cn; lutongbu@tjut.edu.cn
First published on 11th June 2025
Abstract
The increasing emphasis on carbon neutrality has driven significant research into the electrochemical CO2 reduction reaction (CO2RR), aiming to convert CO2 into value-added chemicals and fuels. Dual-metal catalysts, known for their synergistic effects, have garnered considerable attention due to their enhanced electrocatalytic performance for the CO2RR by providing more active sites and optimizing intermediate interactions. Herein, this review will comprehensively explore how the synergistic effect between the two metal centers embedded within atomic and nanoparticle dual-metal catalysts facilitates the electrocatalytic CO2RR, elucidating the structure–activity correlations. Recent significant progress of dual-metal catalysts with synergistic effects for promoting electrocatalytic CO2 reduction will be summarized. Moreover, we will explore the design strategies of dual-metal catalysts and examine the influence of different types of metal active centers in the catalysts on the reaction pathway of the electrocatalytic CO2RR, aiming to uncover profound insights for catalyst optimization and deepen mechanistic understanding of the catalytic process. Finally, the review identifies current research gaps and outlines future directions, emphasizing the need for innovative techniques to enhance catalytic stability and achieve multi-carbon products from the CO2RR using dual-metal catalysts with synergistic effects. This topic could inspire extensive interest to further accelerate and explore the innovations of catalysts in energy conversion.
1 Introduction
The combustion of fossil fuels leads to substantial emissions of carbon dioxide and other greenhouse gases, exacerbating global warming and posing a grave threat to human survival and sustainable development.1–6 Consequently, achieving carbon neutrality and mitigating temperature rise has garnered global consensus.7–11 The conversion of CO2 into high-value-added chemicals and fuels offers a promising pathway to close the artificial carbon cycle and address the environmental challenges caused by excessive CO2 emissions.12–16In CO2, the carbon atom forms two strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds, each comprising a sigma (σ) and a pi (π) bond, ensuring its high chemical stability. This results in significant thermodynamic and kinetic barriers for the activation of CO2, making its conversion pathways both critical and challenging.17–20 Hence, developing effective methodologies and efficient catalysts holds great significance in advancing the carbon dioxide reduction reaction (CO2RR). Various chemical methods have been utilized for CO2 reduction, including electrocatalysis, thermal catalysis, photocatalysis, and bioconversion.21–26 Electrocatalytic (CO2RR) technology offers advantages such as simple equipment requirements, high environmental compatibility, and potential integration with intermittent or renewable electricity sources, making it a promising strategy for efficient CO2 conversion.27–29
O double bonds, each comprising a sigma (σ) and a pi (π) bond, ensuring its high chemical stability. This results in significant thermodynamic and kinetic barriers for the activation of CO2, making its conversion pathways both critical and challenging.17–20 Hence, developing effective methodologies and efficient catalysts holds great significance in advancing the carbon dioxide reduction reaction (CO2RR). Various chemical methods have been utilized for CO2 reduction, including electrocatalysis, thermal catalysis, photocatalysis, and bioconversion.21–26 Electrocatalytic (CO2RR) technology offers advantages such as simple equipment requirements, high environmental compatibility, and potential integration with intermittent or renewable electricity sources, making it a promising strategy for efficient CO2 conversion.27–29
To date, significant advancements have been made in the field of the electrocatalytic CO2RR. Various catalyst systems, ranging from nanomaterial catalysts to atomic catalysts, have been meticulously developed to tackle this complex process.30–34 However, the intricacies of the multi-electron and multi-proton transfer reactions involved in the CO2RR have posed numerous challenges, such as low efficiency, sluggish reaction rates and insufficient stability.35–37 Among all types of catalysts, dual-metal catalysts have garnered particular attention. The modulation of CO2RR catalysts with different active sites plays a vital role in solving the existing critical issues.38 By introducing dual metal active sites into the catalysts, the electronic and geometric properties of the catalysts can be manipulated.1,9,37,39 Crucially, this approach enables the realization of synergistic catalytic effects. For instance, such synergistic effects can be achieved wherein one metal site facilitates CO2 activation while another one provides H+ ions for CO2 protonation.40 Thereby, the synergistic effects between different active centres can lead to the optimization of the adsorption and activation of reactants, transition state stabilization, and the desorption of products, as well as the refinement of the catalytic pathway for optimal performance for CO2 reduction. The dual-metal catalysts can be broadly classified into two types: dual-atom metal catalysts and dual-metal nanomaterial catalysts.1,9,41,42 Dual-atom catalysts can further be subdivided into homonuclear and heteronuclear dual-atom catalysts. Compared to single-atom catalysts, dual-atom catalysts offer much more advantages such as higher metal loading, more active sites, and more flexible adjustability for the CO2RR.43,44 By tuning the composition and arrangement of the two metal atoms in dual-atom catalysts, enhanced catalytic efficiency can be achieved compared to single-atom catalysts for the CO2RR.45,46 The synergistic effect between the two metal atoms can decrease the activation barriers for CO2, directing the reaction pathway towards high selectivity. For example, Lu et al. reported an asymmetric TeN2–CuN3 catalyst with a synergistic mechanism: Te activates CO2, while Cu aids H2O dissociation, which significantly reduces the energy barrier and improves proton transfer kinetics for CO2-to-CO reduction.40 Dual-metal nanomaterial catalysts constitute another crucial class of catalysts for the electrochemical CO2RR, experiencing notable advancements.31 The copper metal exhibits weak hydrogen adsorption but moderate CO adsorption, effectively suppressing the hydrogen evolution (HER) and uniquely facilitating efficient production of multi-carbon products in CO2 electroreduction.47 Therefore, copper-based dual-metal nanomaterial catalysts stand out due to their distinct advantages in the electrocatalytic CO2RR, demonstrating remarkable potential in facilitating the production of high-value C2+ products. For instance, Wang et al. constructed Ag–Cu dual sites in Ag-doped Cu3N NC (Cu3N–Ag NC), where CO is produced on Ag sites, and asymmetric C–C coupling to COCHO is promoted on Cu sites, leading to a marked increase in Faraday efficiency (FE) for C2H4 production compared to that of pure Cu3N NC, which highlights the crucial role of the synergistic effects between the dual-metal Ag and Cu in the formation of multicarbon products.48 There have been many reviews on electrocatalytic CO2 reduction. Nevertheless, to date, there has been no review devoted to the impact of the synergistic effect of dual-active sites in the catalysts on the reaction pathway for the CO2RR, which is of great significance in this area.
In this review, our primary goal is to offer a thorough examination of how dual-metal systems leverage their synergistic effects to optimize both electronic and geometric structures. Concurrently, these optimizations serve to enhance the functionality of dual-active sites, thereby achieving superior electrocatalytic performance towards the CO2RR. First, we will delve into uncovering the design principles underlying dual-metal systems for the CO2RR, with a primary focus on two key aspects: the precise modulation of the electronic structure and the control of the geometric configuration. In terms of electronic structure regulation, we will scrutinize three primary approaches such as chemical state, vacancy, and coordination environment modifications. Regarding geometric structure control, we will focus on several key parameters such as crystal facet, nanoparticle size, and morphology adjustments. Furthermore, we will systematically probe the way varying dual-metal active sites embedded within atomic (notably dual-atom catalysts, DACs) and nanoparticle catalysts affect CO2RR catalytic performance, elucidating the pivotal role of synergistic effects in the fundamental reaction mechanisms. Specifically, we will investigate the synergistic dynamics within homonuclear and heteronuclear DACs systems. Additionally, an in-depth analysis will be conducted on nanomaterial catalysts that pair copper with main-group elements, non-precious metals, precious metals, and lanthanides, to elucidate their effects on catalytic mechanisms and overall efficiency in the CO2RR. Finally, we identify the prevalent challenges and promising directions for the ongoing advancement of dual-metal catalysts, with the goal of enhancing electrocatalytic efficiency and stability for the production of multi-carbon compounds through the CO2RR. This review will provide profound and insightful perspectives, thereby facilitating the rapid acceleration and in-depth exploration of innovations in dual-metal catalysts within the domain of energy conversion (Scheme 1).
 | ||
| Scheme 1 Different types of dual-metal catalysts with synergistic effects for promoting electrocatalytic CO2 reduction. Reproduced with permission.124 Copyright 2023, Elsevier. Reproduced with permission.96 Copyright 2023, Wiley-VCH Verlag. Reproduced with permission.89 Copyright 2024, John Wiley and Sons Ltd. | ||
2 Design principles of dual-metal catalysts with synergistic effects for the electrocatalytic CO2RR
A vast array of design approaches has been meticulously investigated to enhance the electrocatalytic performance for the CO2RR. The key design principles for dual-metal catalysts in the electrocatalytic CO2RR involve electronic and geometric aspects, as these factors critically govern the formation of key intermediates, the pathway of C–C coupling, and the ability to suppress hydrogen evolution.49 The electronic structure dictates the intrinsic catalytic properties by modulating the binding energy of reaction intermediates and steering the reaction pathway, while the geometric structure controls the physical arrangement of active sites, influencing reactant accessibility, mass transport, and the exposure of specific catalytic facets. By optimizing both of these aspects, dual-metal catalysts can achieve enhanced efficiency and selectivity for converting CO2 into valuable products. Therefore, prioritizing electronic and geometric structure regulation is essential for designing advanced catalysts that maximize performance and contribute to sustainable CO2 utilization.502.1 Electronic structure regulation
Electronic structure regulation is a critical aspect of dual-metal catalyst design. By adjusting the chemical state of the metals, the valence state of the active sites can be stabilized or altered, which in turn influences their catalytic activity for the electrocatalytic CO2RR.51 Additionally, the incorporation of dual-metal catalysts can lead to the formation of vacancies or defects within the catalyst structure, which can highly tune the electronic structure of the catalyst.52,53 Furthermore, coordination environment regulation can alter the local electronic sphere of metal active sites with synergistic interactions between the two metals, enhancing catalytic performance by tuning the energy levels of key intermediates during the CO2RR process.54Dual-metal catalysts can leverage the tight interactions between two distinct metals to stabilize and/or modulate the chemical state of active sites. The electron density around the active sites can be modulated by the synergistic interaction between the two metals. Fine-tuning the valence states in dual-metal catalysts can lead to optimal adsorption and activation of CO2 molecules, as well as the stabilization of key intermediates, thereby influencing its catalytic activity and product efficiency for the electrocatalytic CO2RR.58
Many non-noble transition metals, such as Fe, Co, Ni, and Zn are used to as the second metal to stabilize the oxidation state of active metal sites, leveraging the synergistic catalytic effect of the two metals to enhance the performance of the electrocatalytic CO2RR.59–61 For example, Ma and Zhang's group reported that incorporating Ni into CuO nanosheets can stabilize the chemical state of Cu, meanwhile reinforcing the connections between Cu atoms located on the surface and those beneath the surface, resulting in a sustained FEC2+ and improved stability for the electrocatalytic CO2RR compared to pure CuO nanosheets (Fig. 1a).62 Choi et al. demonstrated that incorporating Zn into Cu2O achieves two significant effects: it can stabilize Cu in a lower valence state (Cu0), which is more active for CO2 reduction; and it can lower the energy barrier for the OHC–CHO C–C coupling pathway, thereby promoting the formation of C2H4. These beneficial effects can be attributed to the charge transfer from Zn to Cu, which reduces Cu+ to a lower and stable valence state and facilitates a more favorable reduction reaction at the Cu site (Fig. 1b).56 Therefore, the combination of a second metal with the copper based catalyst can stabilize the valence state of the active site, leading to improve catalytic performance for the CO2RR due to the synergistic effect.
 | ||
| Fig. 1 (a) Different Ni-incorporated Cu catalysts. (b) Activation barrier diagram of C–C coupling on Zn-doped Cu2O. (c) Potential-dependent faradaic efficiencies (FEs) for CO2RR products and total current densities obtained by using Cu0.48Ir0.52. (d) Scheme of the reconstruction process to obtain Cu/CaCO3. (a) Reproduced with permission.62 Copyright 2025, Elsevier. (b) Reproduced with permission.56 Copyright 2023, Wiley. (c) Reproduced with permission.63 Copyright, 2023, Wiley. (d) Reproduced with permission.64 Copyright 2025, John Wiley and Sons Ltd. | ||
In addition to maintaining the constant chemical state of the active sites, the second metal can also effectively regulate the chemical state of the active metal centers. Many noble metals or main group metals are used to modulate the oxidation state of the metal active centers. Kim et al. underscored the pivotal role of modulating the oxidation state by decorating copper (Cu) nanoparticles with iridium (Ir), leading to the charge transfer from Cu to Ir, which indicates that the oxidation state of Cu in the CuIr alloy is higher than that in Cu nanoparticles. By exploiting the robust affinity of Ir for oxygen, the electronic configuration of Cu is effectively modulated, thereby stabilizing oxygen-bound intermediates like CH3CHO* and CH3CH2O* during the CO2RR that are otherwise unstable on bare Cu catalysts. This valence state adjustment strategy markedly boosted the FE to 14.8% for reducing CO2 to tert-butanol (t-BuOH), which represents a substantial improvement compared to that of pure Cu catalysts (Fig. 1c).63 Loh's group constructed a Cu/CaCO3 catalyst for the CO2RR by integrating Cu nanoclusters with CaCO3, in which the metal Ca can regulate the formation of dynamically stable Cu0/Cuδ+ pair sites and maintain the Cuδ+ valence state. Consequently, the synergistic effect of the dual metal Cu and Ca can enhance adsorption capacity of CO2 intermediate *CO and lower the C–C coupling energy barriers, achieving a remarkable FE of 83.7% for C2+ at a partial current density of 393 mA cm−2 (Fig. 1d).64 The strategy of introducing a second metal to stabilize or modulate the valence state of catalytic centers is of great significance in designing and synthesizing highly efficient dual-metal catalysts. This approach can optimize intermediate binding, lower energy barriers and significantly enhance the electrocatalytic performance for the CO2RR.
Of particular note are cationic vacancies, which have unique electronic structures and orbital distributions, and can greatly affect the properties of the metal catalysts for CO2 reduction.66–70 For instance, Sun et al. reported that introducing Sn dopants into a copper catalyst could modify the lattice structure of CuO, giving rise to the formation of oxygen vacancies, which have a significant impact on the surface electron distribution of the CuO nanosheets. DFT calculations confirmed that the synergistic effect of oxygen vacancies and the metals can strengthen CO2 adsorption and reduce the energy barrier for the formation of intermediate species such as *COOH and *CO during the electroreduction process, leading to a substantial enhancement in the efficiency for electrocatalytic CO2 reduction to CO (Fig. 2a).71 Similarly, He, Xu, et al. constructed a series of dual-metal Cu/Ce catalysts with different concentrations of oxygen vacancies, in which the oxygen vacancies could alter the electronic microenvironment of the catalyst, leading to the introduction of uncoordinated metal site Cu+ active centers. This revealed that the high concentration of oxygen vacancies in the dual-metal catalyst is crucial for facilitating the adsorption of *COOH and *CO intermediates, resulting in the efficient production of CH4 with a FE of 70.03% and a TOF of 9946.7 h−1 (Fig. 2b).70
 | ||
| Fig. 2 (a) Schematic illustration of the synthesis of Vo-CuO (Sn). (b) Schematic illustration for the synthesis of the Cu/ceria-H2 catalyst. (c) Corresponding energy diagrams of C–C coupling for pristine Li2CuO2, Li2−xCuO2-2%, Li2−xCuO2-4%, and Li2−xCuO2-8%. (d) Faradaic efficiencies for CO2RR products and TOF values for C2+ products on the Li2−xCuO2 catalyst. (e) Scheme of Li+ vacancy-tuned [CuO4] sites that promote C–C coupling. (a) Reproduced with permission.71 Copyright 2022, American Chemical Society. (b) Reproduced with permission.70 Copyright 2025, John Wiley and Sons Ltd. (c–e) Reproduced with permission.73 Copyright 2022, Wiley-VCH Verlag. | ||
In addition to tuning the electronic structure of the catalyst, these vacancies in dual-metal catalysts can also act as active sites for the adsorption and conversion of CO2. Xiao et al. doped Cu into ZnO, inducing a synergistic effect that generated abundant oxygen vacancies. These vacancies can not only modulate the local electronic structure of the active sites but also serve as electron-trapping centers, facilitating CO2 adsorption and promoting electron transfer for its reduction. Additionally, they could also suppress the HER, thereby lowering the energy barrier for electrocatalytic CO2 conversion to CO, leading to a high FECO > 80% within a wide potential range.72 In dual-metal catalysts, the cooperation between the two metals may also lead to the formation of vacancies within certain metal cations. These vacancies, akin to those in anion sites, possess the ability to regulate the electronic structure and also serve as catalytic centers, thereby influencing the adsorption and activation of CO2. Zheng, Li, et al. incorporated the second metal lithium (Li) into the copper oxides to construct the dual-metal catalyst Li2CuO2, constructing Li+ vacancies, which can tailor the electronic structure and regulate the adsorption energies of intermediate species. It can lower the coordination number of Li+ around each Cu atom, which highly facilitated the CO–CO coupling, while impeding the competitive HER through suppressing the adsorption of *H intermediates, resulting in the highest FE of 90.6% for C2+ products (Fig. 2c–e).73
The design and synthesis of dual-metal catalysts containing vacancies with synergistic catalytic effects offer distinct advantages in optimizing the electronic structure of the catalyst and enhancing catalytic efficiency for the CO2RR. These vacancies not only improve the interaction with reactants but also serve as active sites for catalyzing the CO2RR. Precise control over the vacancy position, quantity, and type can thus optimize the dual-metal catalyst's electronic structure and performance, offering a novel strategy for efficient CO2 conversion.
Ligands with varying electronegativities such as N, O, S, and P can alter the electron density of metal active centers. For example, electron-withdrawing ligands can lower the energy of metal d-orbitals, weakening intermediate adsorption, while electron-donating ligands can enhance adsorption and promote CO2 activation.38,77–79 Voiry et al. introduced electron-withdrawing ligand groups such as N2SN–, N3N– and C2N– to tailor the surface coordination environment of a dual-metal Cu–Ag electrocatalyst for CO2 reduction. Notably, the ligands N2SN– and N3N– were found to stabilize the Cu center in a low oxidation state (0 < δ < +1), which critically modulated the electronic structure of the dual-metal Ag–Cu catalyst. This ligand-induced electronic perturbation enhanced the synergistic cooperation between Ag and Cu, promoting CO2 adsorption and regulating the reduction pathways. Consequently, the optimized catalyst achieved efficient electrocatalytic conversion of CO2 into high-value multicarbon products (Fig. 3a and b).80 Zhang et al. constructed a redox-active cobalt (Co) protoporphyrin-modified MIL-101(Cr)–NH2 material for efficient CO2 electroreduction. The electron-donating ligand –NH2 group on the coordinated sphere can not only link the Co and Cr sites, increasing the active sites and tuning their electronic structures, but also improve the CO2 absorption ability, highly optimizing the intermediate *CO binding energy, leading to high CO2-to-CO conversion (Fig. 3c).81
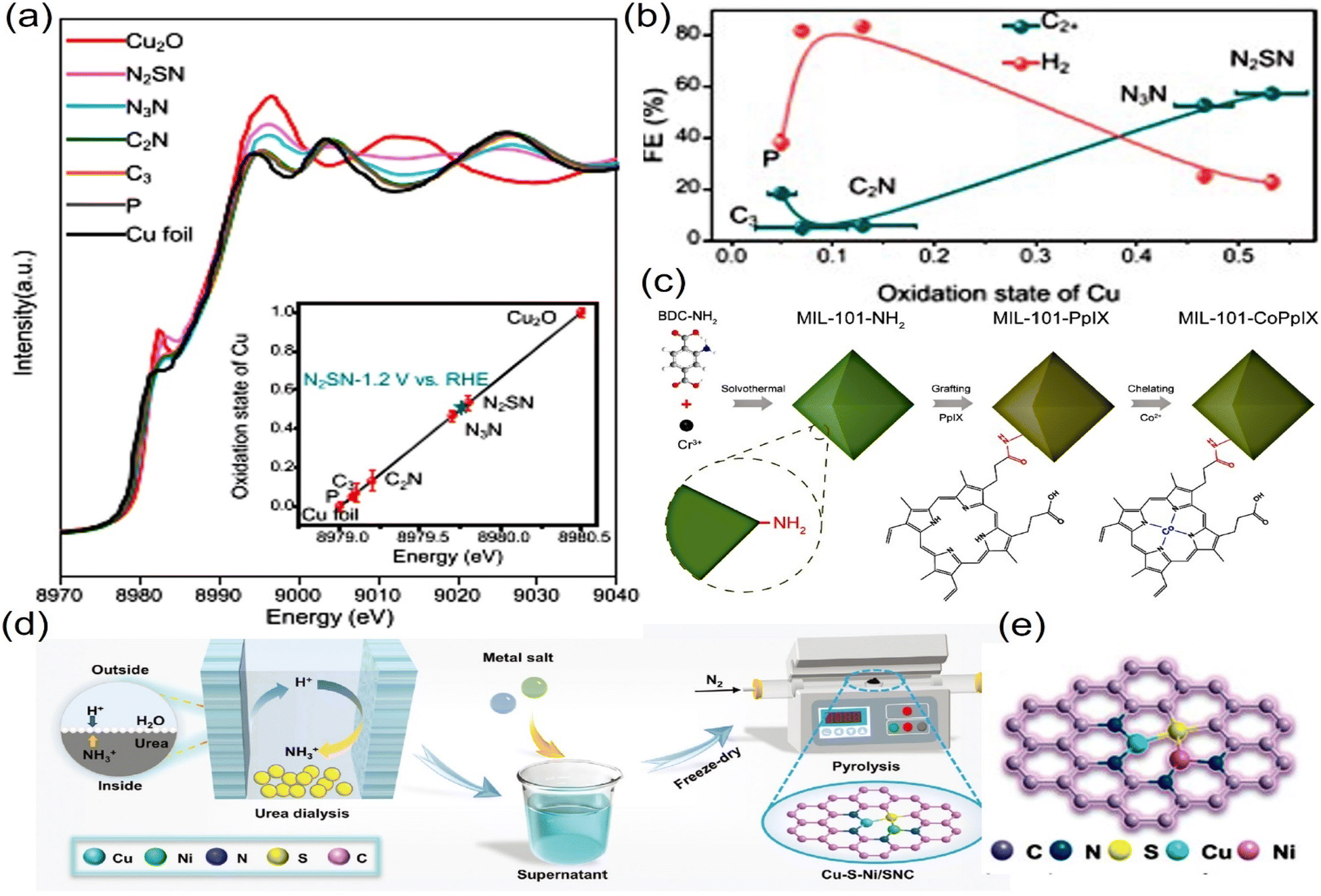 | ||
| Fig. 3 (a) XANES spectra at the Cu K-edge of the Ag–Cu catalyst. (b) Evolution of the faradaic efficiency for C2+ and H2 measured at −1.2 V vs. RHE with the oxidation state of Cu. (c) Synthesis of MIL-101-CoPpIX. (d) Schematic illustration of the synthesis of the Cu–S–Ni/SNC catalyst. (e) Schematic illustration of the synthesis of the Cu–S–Ni/SNC catalyst. (a and b) Reproduced with permission.80 Copyright 2021, Springer Nature. (c) Reproduced with permission.81 Copyright, 2024, Elsevier. (d and e) Reproduced with permission.84 Copyright 2024, Wiley. | ||
Bridging coordination ligands like N, O, S or halogens can enable electron transfer between the dual metals, creating electron transmission channels, leading to a modified electronic structure and superior catalytic performance for CO2 reduction.38 For instance, Zn–N4-coordinated Cu single atoms in tandem catalysts can facilitate electron flow from Zn to Cu, enhancing CO2 activation.82 In another study, Sakamoto, Sekizawa et al. developed a robust Br-bridged dinuclear Cu(I) complex, which can efficiently catalyze CO2 reduction to produce C3H7OH with high selectivity. The Br bridge can not only modulate the electronic configurations of the dual Cu active sites by stabilizing their low-spin states, but also strengthen the synergistic collaboration between the bridged Cu centers. This dual modulation mechanism effectively promoted C–C coupling reactions, leading to significantly enhanced C2+ hydrocarbon productivity.83 Chen, Zhang, and colleagues developed a dual-atom catalyst featuring sulfur-bridged Cu–S–Ni motifs. The S-bridge architecture could modulate the electronic charge distribution across the Cu and Ni dual-atom centers, while enhancing synergistic effects between the dual-metal sites. This atomic-level cooperation facilitates the critical COOH* intermediate formation during CO2 electroreduction, resulting in a remarkable FE of 98.1% for CO production at −0.65 V in a H-cell (Fig. 3d and e).84
The synergistic interaction between dual metals, mediated by tailored coordination environments, is critical for optimizing electrocatalytic performance for the CO2RR. This interaction not only enhances CO2 adsorption and activation but also promotes favorable reduction pathways, leading to high-value product formation. The regulation of coordination environments has provided a theoretical and experimental basis for designing high-performance dual-metal catalysts, elucidating the mechanisms of metal interactions and improving overall catalytic efficiency. Therefore, the strategic design of coordination environments in dual-metal catalysts, through the careful selection of central metals, ligands, bridging atoms and other coordination parameters, is essential for achieving efficient and selective electrocatalytic CO2 reduction.
2.2 Geometric structure regulation
Geometric structure regulation is another important aspect of dual-metal catalyst design. By controlling the crystal facet of the metal active sites, the catalyst can be optimized to facilitate the formation of desired products. The incorporation of a second metal can help stabilize these favorable crystal facets, thereby enhancing the catalyst's selectivity for specific products in CO2 conversion. Additionally, the size of the nanoparticles used in CO2 reduction can be controlled by the addition of a second metal. Smaller nanoparticles tend to increase the number of active sites and promote the catalytic performance. Morphology regulation is also crucial, as different morphologies such as polyhedra, nanosheets, hierarchical nanoflowers, and porous structures can exhibit unique physicochemical properties that affect the catalyst's performance. By carefully designing the morphology of the dual-metal catalysts through the addition of a second metal, the catalyst can be optimized for high selectivity towards the electrocatalytic CO2RR.85,86In the design and preparation of dual-metal catalysts, controlling the crystal planes of metal active sites is a crucial step. This strategy focuses on selectively exposing crystal planes that are most favorable for specific chemical reactions, thereby enhancing catalytic performance and significantly promoting the formation of desired products. Specifically, introducing a second metal can stabilize the crystal planes conducive to target product formation, enabling the catalyst to exhibit higher selectivity—preferentially generating the desired product over by-products. The integration of crystal plane regulation with secondary metal incorporation offers an innovative framework for the development of efficient and highly selective catalysts. Fan, Huang, Xi et al. achieved well-defined Ag–Cu Janus nanostructures with {100} facets (JNS-100) through the confined growth of Cu on Ag nanocubes (NCs). The introduction of Ag into the Cu catalyst system created a synergistic catalytic effect. DFT calculations revealed that the electronic structure of Ag–Cu JNS-100 was optimized due to the electron transfer from Ag to Cu domains, which enhanced the catalytic activity. The Ag domains efficiently converted CO2 to CO at lower overpotentials, while the generated CO then spilled over to the adjacent Cu domains, where it was further reduced and C–C coupling was realized toward C2H4 production with high efficiency. This tandem mechanism, combined with the exposed {100} facets on the Cu domains, significantly improved the selectivity and efficiency of the CO2RR toward C2+ products. Specifically, the Ag65–Cu35 JNS-100 catalyst demonstrated a FE of 54% for C2H4 and 72% for C2+ products at −1.2 V (vs. RHE), outperforming pure Cu nanocubes and physical mixtures of Ag and Cu (Fig. 4a and b).90 Zheng, Kuang et al. successfully incorporated the metal Mg into the Cu catalyst to promote the formation and stabilization of Cu (111) facets, which were particularly effective in promoting C–C coupling, and enhancing the production of ethanol with a high FEethanol of 76.2 ± 4.8% at 600 mA cm−2. It was found that the Cu (111) facets provided a high density of active sites and optimal adsorption energies for key intermediates such as *CO. These facets also lowered the energy required for *CO–CO coupling, and improved the stabilization of the *CHCHOH intermediate, which were critical for ethanol production. Moreover, the synergistic effect between Cu and Mg in the dual-metal material facilitated the electron transfer from Mg to Cu, leading to a negative shift in the d-band center compared to that of commercial Cu, indicating a more favorable electronic environment for C–C coupling (Fig. 4c).91
 | ||
| Fig. 4 (a) Schematic illustration for the synthesis of three kinds of Ag–Cu JNS-100. (b) Faradaic efficiencies (FEs) for CO2RR products obtained on Ag65–Cu35 JNS-100. (c) Electronic localization function of Cu2Mg (111). (d) Optimized structure of cluster Cu (111), cluster In (101), and cluster CuIn alloy (200). (e) Schematic diagram of the fabrication process of the CuIn electrode. (a and b) Reproduced with permission.90 Copyright 2021, Wiley. (c) Reproduced with permission.91 Copyright 2023, John Wiley and Sons Ltd. (d and e) Reproduced with permission.92 Copyright 2023, Elsevier. | ||
In addition to the conventional approach of modulating copper crystal facets, such as the (100) plane, the regulation of other types of metal facets through the synergistic effect of dual-metal systems is also considered as a promising approach for enhancing the electrocatalytic CO2RR. Qiao, Zhang and their team developed CuIn dual-metal catalysts featuring co-exposed CuIn (200) and In (101) facets, which exhibited a high FE up to 80% for formate production at relatively low overpotentials, along with high stability. They discovered that the synergistic effect of the two metals played a crucial role, where the CuIn (200) facet can specifically reduce the overpotential for formate production, while the In (101) facet can enhance the overall CO2 adsorption capacity. Density functional theory (DFT) calculations further revealed that the CuIn (200) and In (101) lattice facets collaborate to stabilize the key OCHO* intermediate (Fig. 4d and e).92 Xiong et al. achieved highly efficient electrocatalytic CO2RR to produce formate by fabricating dual-metal Bi5In5 nanofibers (NFs) with precise control over the growth of InBi (200) crystal facets, through a synthetic approach combining electrospinning and electrochemical reduction techniques. It was found that attributed to the dual-metal sites formed by In and Bi atom, the (200) crystalline plane of Bi5In5 could promote the electrocatalytic CO2RR process by strongly adsorbing the key *OCHO intermediate and reducing the reaction energy barrier, thereby enhancing catalytic selectivity for HCOOH production.93
In summary, the precise regulation of crystal facets in dual-metal catalysts has emerged as a highly effective approach to enhance the selectivity and efficiency of electrocatalytic CO2 reduction. By tailoring specific facets and leveraging synergistic effects in dual-metal systems, remarkable control over reaction pathways has been achieved, enabling high FE for target products. These advancements highlight the critical role of facet engineering in optimizing intermediate adsorption and lowering energy barriers for key steps.
As for dual-atom catalysts (DACs) consisting of two heterometallic active centers, they represent an emerging class of electrocatalysts that combine the advantages of single-atom catalysts (SACs) and subnanometer clusters for the CO2RR. By precisely engineering dual-metal sites, DACs can achieve maximized metal atom utilization while enabling synergistic electronic and geometric effects that are unattainable with isolated SACs or larger clusters.94 Lu, Liu, et al. employed a molten-salt-assisted strategy to synthesize Fe–Zn dual-atom catalysts (FeZnNC) on N-doped carbon supports. The study demonstrated that due to the electronegativity difference between Fe and Zn (Fe: 1.83; Zn: 1.65), electrons preferentially transferred from Zn to Fe, which resulted in charge redistribution and modified electronic environments of the two active sites, creating optimal conditions for the CO2-to-CO conversion pathway. The synergistic effect of dual-atom catalysts entailed the Zn site functioning as the primary active center for CO2 activation and *COOH intermediate formation, with the Fe site stabilizing intermediates and weakening *CO binding to avert site poisoning, thereby achieving FECO > 94% in acidic media at industrial current densities ranging from 100 to 400 mA cm−2 (Fig. 5a and b).95 Wang et al. developed a heterodimetallic Co–Ni–N–C dual-atom catalyst with neighboring Co–Ni diatomic sites, which attained a remarkable CO yield rate of 53.36 mA mg cat.−1, along with an outstanding FECO of 94.1%. DFT calculations have revealed that the Co–Ni synergistic catalysis mechanism for the CO2RR entails the effective activation of CO2 at dual Co–Ni sites and also the formation of CO via optimized pathways where another CO2 molecule undergoes reduction while a CO* intermediate remains adsorbed at the site (Fig. 5c and d).96
 | ||
| Fig. 5 (a) Scheme diagram for the synthesis of a dual-atom FeZnNC catalyst. (b) Faradaic efficiencies of CO at different potentials by using FeZnNC. (c) Schematic diagram of the fabrication process of Co–N–C, Ni–N–C, Co–Ni–N–C, and Co–N–C (NP) (NP: nanoparticle). (d) Catalytic pathway on the dual Co–Ni site based on the optimized structures of adsorbed intermediates COOH*and CO*. (e) Faradaic efficiencies for CO2RR products and H2 on the Cu1Ni–BDP MOF at different cathode potentials. (f) Total structure of Au8Ag55, Au8Ag57, and Au12Ag60 NC. (a and b) Reproduced with permission.95 Copyright 2024, John Wiley and Sons Inc. (c and d) Reproduced with permission.96 Copyright 2024, the Royal Society of Chemistry. (e) Reproduced with permission.99 Copyright 2023, Wiley-VCH Verlag, Wiley. (f) Reproduced with permission.98 Copyright 2023, American Chemical Society. | ||
The metal clusters bridge atomic and nanoscale catalysts, as their well-defined yet tunable structures – ranging from a few to dozens of metal centers – combine atomic-level precision with nanoparticle-like ensemble effects. They can offer superior metal utilization like atomic catalysts, while avoiding the low metal loading limitations of atomic catalysts. Moreover, their multinuclear active sites can mimic the selectivity of nanoparticles, enabling tailored control over the C–C coupling reaction pathway for the CO2RR. The dual-metal clusters can offer superior catalytic performance compared to their monometallic systems by synergistically integrating the properties of different metal components, which can modify charge distribution and orbital hybridization, enabling precise optimization of intermediate binding energies for enhanced electrocatalytic CO2RR activity.97 Zhu, Jin, Du, et al. precisely engineered the structures of dual-metal clusters (Au8Ag55, Au8Ag57, and Au12Ag60) with different sizes, which enabled tailored catalytic performance for the CO2RR by manipulating geometric properties at the atomic level. They discovered that the smaller Au8Ag55 NC exhibits a distinctive charge segregation phenomenon, where electrons preferentially gather around the core Au atoms while inducing an electron-deficient condition on the outer Ag atoms. This reduction in electron density on the Ag atoms effectively diminishes the competition from the HER, thereby enhancing the CO2RR process (Fig. 5f).98 Metal–organic frameworks (MOFs) are unique cluster-based materials, in which atomically precise metal clusters are interconnected by well-ordered organic ligands, enabling outstanding electrocatalytic CO2RR performance. Lu, Francisco, Han, Zhu, et al. successfully constructed a Cu–Ni dual-metal MOF cluster with an asymmetric structure modulated by a pyrazolate ligand, namely Cu1Ni–BDP, which achieved a high CO2-to-C2H4 conversion with a FE of 52.7% along with great stability. DFT calculations revealed that electron transfer from Cu to Ni generates electron-deficient Cu and electron-rich Ni sites in the asymmetric Cu1Ni cluster. The unique asymmetric Cu–Ni electronic configuration creates synergistic effects that precisely modulate *CO adsorption energy while reducing the activation barrier for *COH–COH formation, thereby enabling exceptional CO2 reduction performance (Fig. 5e).99
In addition to dual-metal atomic catalysts and dual-metal clusters, the size regulation of dual-metal nanoparticle catalysts also has a significant impact on electrocatalytic CO2 reduction. Although bulkier nanoparticles suffer from decreased atomic utilization efficiency, the complex surface architecture containing multiple active site types can directly offer high catalytic performance towards the CO2RR. Chen, Feng, et al. innovatively designed dual-metal ZnO/Ag twinned-phase ultrasmall nanoparticles (≤3 nm) uniformly embedded in a nanoporous carbon matrix, achieving 60.9% energy conversion efficiency for CO production with exceptional stability over six days. They discovered that nanopore confinement ensures the small particle size of the catalysts, leading to high active site density and agglomeration resistance. DFT calculations revealed that the charge redistribution from Zn/Ag to O atoms preferentially stabilizes the *COOH intermediate while energetically disfavoring competing HCOO* and *H pathways, resulting in highly selective (98.1%) CO production.100 Byon, Yang, et al. developed a series of dual-metal CuxIr1–x alloy nanoparticles with a small size of about 1.6 nm, which could achieve unprecedented performance in CO2-to-t-BuOH conversion, delivering a FE of 14.8%—very high C4 production. Studies revealed that the high C4 selectivity arises from the synergistic Cu–Ir interaction, combined with the oxophilic nature of the Ir-rich surface, which stabilizes critical oxygen-bound intermediates (e.g., CH2CHO*, CH3CHO, and CH3CH2O) and thereby boosts CO2 reduction efficiency.63
The size regulation of dual-metal catalysts is a powerful lever to tailor the synergistic effect on catalytic properties for the electrocatalytic CO2RR. The interplay of electronic and geometric effects across scales will drive advancements in high-performance CO2RR catalysts.
By utilizing the modulation of two unique metals, dual-metal catalysts can be morphologically optimized into diverse nanostructures, ranging from distinct shapes like polyhedra, nanosheets, hierarchical nanoflowers or dendrites, to porous structures like core–shell or hollow structures. This approach enhances mass transport efficiency and promotes electrolyte infiltration during the CO2RR by establishing directional charge/mass transfer pathways and increasing the surface area available for reactant interaction.102,103
Polyhedral structures in nanomaterials offer numerous advantages that make them highly attractive for various applications. The unique shape and arrangement of atoms in polyhedral nanomaterials often lead to tailored electronic properties and enhanced catalytic activity for the electrocatalytic CO2RR.104 For instance, Zhang, Wang, and colleagues reported that the incorporation of Ag into Cu3N nanocubes (NCs) leads to a well-defined nanocube (NC) structure, where Ag atoms are successfully integrated into some Cu sites within the Cu3N unit cell, resulting in Cu3N–Ag NCs. The dual Ag–Cu sites exhibit a cascade catalysis strategy that markedly boosts the electrochemical CO2 reduction process, resulting in significantly enhanced FE and partial current density for C2H4 production, which are much higher compared to those of pure Cu3N nanocubes (NCs). They discovered the synergistic effect between Cu and Ag, wherein the relatively weak binding strength of *CO on Ag sites aids in the generation and subsequent transfer of CO species to Cu sites. At these Cu sites, the elevated local CO coverage promotes asymmetric C–C coupling, leading to the formation of key intermediate species *COCHO (Fig. 6a).48 In another example, Gong's group reported that incorporating dual-metal doping into catalytic materials presents a potent strategy for modulating their structural attributes and substantially impacting the efficiency of electrocatalytic CO2 reduction. By adjusting the ratios of Cu and Pd precursors, diverse morphological transformations of Pd–Cu nanocrystals (NCs) can be realized, spanning from concave rhombic dodecahedra to flower-like structures. The flower-like Pd3Cu (FL-Pd3Cu), composed of {111} and high-index step facets, contributed to their high selectivity for CO production. Conversely, the concave rhombic dodecahedral Cu3Pd (CRD-Cu3Pd) exposed high-index facets, which have demonstrated a reduced onset potential and increased current density for CH4 production compared to Cu foil. They discovered that attributed to the synergistic effects of Cu and Pd as well as the exposure of high-index facets, CRD-Cu3Pd offered more low coordinated active sites and modified the binding energies of reaction intermediates such as *CHO, thereby enhancing its catalytic performance for CH4 production (Fig. 6b).105
 | ||
| Fig. 6 (a) Structural characterization of Cu3N–Ag NCs. (b) Morphological evolution of Pd–Cu catalysts with different compositions. (c) Faradaic efficiencies for CO2RR products by using CuMnO2. (d) Structural characterization of Co0.05–BOON. (e) Synthesis schematic of CuMnO2-Vo. (f) SEM image of Cu1/SnS2. (g) Synthesis and characterization of dual-active site catalyst Bi2O3−x/Mg(OH)2. (h) Schematic illustration for the synthesis of Ag@Cu2O cascade nanoreactors. (i) Representative scheme of the microenvironment of Ag/Cu2O during the CO2 reduction process. (j) Schematic illustration for synthesizing Cu–Sn-based catalysts in the CO2RR. (a) Reproduced with permission.48 Copyright 2024, American Chemical Society. (b) Reproduced with permission.105 Copyright 2017, Wiley-VCH Verlag. (c and e) Reproduced with permission.109 Copyright,2025, Elsevier. (d) Reproduced with permission.108 Copyright 2024, John Wiley and Sons Ltd. (f) Reproduced with permission.113 Copyright 2024, American Chemical Society. (g) Reproduced with permission.114 Copyright 2025, Springer. (h) Reproduced with permission.103 Copyright 2023, American Chemical Society. (i) Reproduced with permission.115 Copyright 2024, American Chemical Society. (j) Reproduced with permission.118 Copyright 2024, Wiley. | ||
Due to their two-dimensional (2D) confinement and ultra-thin nature, nanosheets exhibit an enormously high surface area-to-volume ratio, which often leads to unique properties such as high carrier mobility and tunable band gaps, which are particularly advantageous for the electrocatalytic CO2RR.106,107 Wang and Che's group reported that metals like Co, In and Ru were successfully introduced into Bi materials via a facile hydrolysis method, resulting in the formation of metal-doped Bi nanosheets with unique structural and chemical properties compared to pristine Bi nanosheets. Notably, the Co0.05–BOON nanosheets significantly enhanced the electrocatalytic reduction of CO2 to HCOOH, thanks to the synergistic effect between cobalt and bismuth. The incorporation of cobalt atoms into the bismuth nanosheets modulated the electronic structure via electron transfer, leading to a high selectivity (∼90%) and stability for HCOOH production over 100 hours at 100 mA cm−2. Moreover, the Co-doped Bi (012) surface exhibited stronger binding energy towards the key OCHO* intermediates, facilitating the efficient conversion of CO2 to formic acid at a lower applied potential compared to the pure Bi surface (Fig. 6d).108 In another study, Zhang and colleagues reported on the CuMnO2 nanosheet structure, wherein the controlled synthesis method introduced Cu and Mn, yielding a material abundant in oxygen vacancy defects. The abundant oxygen vacancies in the CuMnO2-Vo nanosheets effectively shift the d-band center of the active Mn sites upwards. The synergistic effect of Cu and Mn in the CuMnO2 nanosheets, coupled with the presence of oxygen vacancies, significantly enhances the adsorption strength of the active sites for the intermediates *COOH and *CO, resulting in a high FE for ethylene production (93.4% at −1.0 V vs. RHE) and excellent stability over 192 hours (Fig. 6c and e).109
The branching structure of dendrites and the layered petals of nanoflowers offer excellent mass transport pathways, which can facilitate the efficient diffusion of CO2 to the active sites and the removal of reduction products from the catalyst surface, reducing mass transfer limitations and enhancing the overall reaction rate for the CO2RR.110–112 Liu et al. reported that the integration of Cu and Sn into the Cu1SnS2 catalyst leads to the formation of a unique microflower-like nanostructure, creating a hierarchical architecture with multiple layers and pores. The doping of Cu into SnS2 not only modifies the electronic structure of the material but also plays a pivotal role in shaping its morphological features. The operando detections confirmed that the incorporation of a single Cu atom into the Cu1/SnS2 catalyst could promote the reduction of SnS, resulting in the formation of Cu1/Sn during the CO2RR. This dynamic transformation effectively boosts the generation of CO2−/OCHO intermediates. DFT calculation found that Cu1 and Sn work together as active sites to reduce the Gibbs free energy for the formation of the *OCHO intermediate, facilitating the conversion of CO2 to formate (Fig. 6f).113 In another example, Lu, Jiao, et al. constructed a Bi2O3−x/Mg(OH)2 catalyst with a nanoflower-like structure, which can increase the surface area of the catalyst, providing more active sites for CO2 adsorption and reduction even for low-concentration CO2. They discovered that the dual-active-site catalyst leverages a synergistic effect between the bismuth oxide active sites and the magnesium hydroxide basic sites, in which a Lewis-base active site can facilitate the enrichment and activation of CO2, along with a highly selective catalytic site dedicated to CO2 conversion, leading to a high formic acid selectivity exceeding 97% across a wide potential range (Fig. 6g).114 There are also many dual-metal catalysts with dendrite structures for the electrocatalytic CO2RR, which exhibit excellent catalytic performance. For instance, Schultz et al. reported a dual-metal catalyst Ag/Cu2O with dendritic morphology, in which the combination of Ag and Cu2O leads to selective photoelectrochemical CO2 reduction to acetate, with a high FE of 54%. It was found that the Ag/Cu2O nanodendrite structure exhibits a unique combination of plasmonic and semiconductor properties that synergistically enhance the CO2 reduction reaction. The in situ tests revealed that the reaction rate entails the transfer of a single electron from the electrode to CO2, leading to its reduction into the *CO2− radical anion and subsequently generating adsorbed CO, which acts as a vital intermediate in the process of C–C coupling for acetate production (Fig. 6i).115
The porous structures like core–shell or hollow structures can not only offer a larger surface area and more active sites compared to their bulk counterparts, but also enhance CO2/product diffusion, significantly reducing mass transfer limitations.116,117 For instance, Zeng, Tuo, et al. developed a group of dual-metal Ag@Cu2O core–shell nanoreactors featuring adjustable Cu2O shell thicknesses, displaying a volcano-like pattern in the generation of C2+ products, in which Ag@Cu2O-40 with medium thickness exhibited the highest FEC2+ of 78.5%. The combination of Ag and Cu2O in a core–shell structure leads to synergistic effects, in which the CO generated at the Ag core spills over to the Cu2O shell, where it promotes the C–C coupling for C2+ production. They discovered that the porous Cu2O shell creates a confined space around the Ag core, promoting the local accumulation of CO intermediates. Meanwhile, the short diffusion pathways between the Ag core and Cu2O shell minimize the loss of reactive intermediates, ensuring efficient utilization of CO for subsequent C–C coupling (Fig. 6h).101 As for the hollow structure, Sun, Zhang, et al. developed two dual-metal Cu–Sn oxide based catalysts Cu–SnO2 and Cu2O–SnO2 with hollow spherical structures, of which the former was obtained under strong reduction conditions, while the latter was obtained under weaker reduction conditions. The findings reveal that the Cu–SnO2 catalyst exhibits remarkable selectivity towards ethanol, achieving a selectivity of 74.6%, while the Cu2O–SnO2 catalyst shows high selectivity towards ethylene, with a value of 71.4%. This underscores the crucial role played by the distinct interphases formed between Cu/Cu2O and SnO2 in determining the product distribution, demonstrating a significant impact on the catalytic outcomes. DFT calculations uncovered that the Cu–SnO2 interphase features robust electron interactions, facilitating the generation of essential intermediates such as *COH, which favors asymmetric C–C coupling to yield ethanol. Conversely, the Cu2O–SnO2 interphase is characterized by the presence of oxygen vacancies at both sites, enhancing the abundance of *CO intermediates, thereby promoting symmetric C–C coupling to produce ethylene (Fig. 6j).118
Precise morphological control in dual-metal catalysts—ranging from polyhedra and nanosheets to hierarchical and porous architectures—enables tailored electronic and geometric effects that govern intermediate binding, C–C coupling, and mass transport. This structural diversification, coupled with synergistic metal interactions, paves the way for industrial-scale CO2 conversion with target-product selectivity.
3 Influence of different types of dual-metal active sites with synergistic effects on the mechanism
The incorporation of dual-metal active sites in catalysts represents a highly effective strategy for enhancing catalytic performance, particularly in the field of electrocatalytic CO2 reduction (Tables 1 and 2). The synergistic interactions between dual-metal sites can optimize reaction pathways, significantly improving catalytic activity, selectivity, and stability. Based on the size scale and structural characteristics of the active sites, dual-metal active sites can be categorized into dual-atom metal catalysts and dual-metal nanoparticle catalysts.119| Catalyst | Dual metals | FE of C1 products (%) | Current density jC1 (mA cm−2) | Electrolyte | Ref. |
|---|---|---|---|---|---|
| Vo-CuO(Sn) | Cu and Sn | 99 (CO) | 38.65 | 0.1 M KHCO3 | 71 |
| Cu/CeO2 | Cu and Ce | 70.03 (CH4) | 150 | 1 M KOH | 63 |
| Cu–ZnO | Cu and Zn | 80 (CO) | 45 | 0.1 M KHCO3 | 65 |
| MIL-101-CoPpIX | Co and Cr | 97.1 (CO) | 5 | 0.1 M KHCO3 | 81 |
| Cu–S–Ni/SNC | Cu and Ni | 98.1 (CO) | 550 | 0.5 M KHCO3 | 84 |
| CuIn | Cu and In | 80 (HCOOH) | 13.84 | 0.5 M KHCO3 | 92 |
| Bi5In5 | Bi and In | 96.8 (HCOOH) | 250.9 | 0.1 M KHCO3 | 93 |
| FeZnNC | Fe and Zn | 94 (CO) | 100 | 1 M KOH | 95 |
| Co–Ni–N–C | Co and Ni | 94.1 (CO) | 13.34 | 0.1 M KHCO3 | 88 |
| Au8Ag55 | Au and Ag | 66 (CO) | 23 | 0.5 M KHCO3 | 98 |
| ZnO–Ag@UC | Zn and Ag | 98.1 (CO) | 22.3 | 0.5 M KHCO3 | 100 |
| Pd–Cu | Cu and Pd | 40.6 (CH4) | 16.7 | 0.1 M KHCO3 | 105 |
| Co0.05-BOOM | Co and Bi | ∼90 (HCOOH) | 200 | 1 M KOH | 108 |
| Cu1/SnS2 | Cu and Sn | ∼90.9 (HCOOH) | 158 | 0.5 M KHCO3 | 113 |
| Bi2O3−x/Mg(OH)2 | Bi and Mg | 97 (HCOOH) | 140 | 0.5 M KHCO3 | 114 |
| Ni2-NCNT | Ni andNi | 97 (CO) | 76 | 0.5 M KHCO3 | 126 |
| Ni2/N-CNTs | Ni and Ni | 90.7 (CO) | 64.5 | 0.5 M KHCO3 | 118 |
| Fe2–N–C | Fe and Fe | 80 (CO) | 32.04 | 0.5 M KHCO3 | 128 |
| Ni–Cu | Cu and Ni | ∼99 (CO) | 235 | Phosphate buffer, 1 M KOH and 0.5 M KHCO3 | 122 |
| Co–Cu DASC | Cu and Co | 99.1 (CO) | 483 | 0.5 M KHCO3 | 132 |
| Catalyst | Dual metals | FE of C2+ products (%) | Current density jC2+ (mA cm−2) | Electrolyte | Ref. |
|---|---|---|---|---|---|
| CuxIr1−x | Cu and Ir | 14.8 (t-BuOH) | 0.207 | 0.1 M KHCO3 | 63 |
| Cu/CaCO3 | Cu and Ca | 83.7 | 393 | 1 M KOH | 64 |
| Li2CuO2 | Cu and Li | 90.6 | 706 | 1 M KOH | 73 |
| Ag–Cu | Cu and Ag | 80 | 261.1 | 0.5 M KHCO3 | 81 |
| Ag65–Cu35 JNS-100 | Cu and Ag | 72 | 15.14 | 0.1 M KHCO3 | 90 |
| Cu2Mg(111) | Cu and Mg | 76.2 | 720 | 1 M KOH | 91 |
| Cu1Ni–BDP | Cu and Ni | 52.7 | 500 | 1 M KOH | 99 |
| Cu3N–Ag | Cu and Ag | 36.6 | 26.7 | 1 M KOH | 48 |
| CuMnO2 | Cu and Mn | 93.4 | 14.57 | 0.1 M KHCO3 | 109 |
| Ag@Cu2O-40 | Cu and Ag | 78.5 | 20 | 1 M KOH | 103 |
| Cu/Cu2O–SnO2 | Cu and Sn | 74.6 | 8.7 | 0.1 M KHCO3 | 118 |
| Cu2 | Cu and Cu | 51 | 496.4 | 1 M KOH | 122 |
| BiCu-SAA | Cu and Bi | 73.4 | 400 | 1 M KOH | 140 |
| CuxGax | Cu and Ga | 83 | 1000 | 1 M KOH and 0.5 M KHCO3 | 141 |
| CoCu composite | Cu and Co | 71.1 | 26.7 | 0.1 M KHCO3 | 52 |
| CuO + Ni-surface | Cu and Ni | 55.2 | 199 | 0.1 M KHCO3 | 62 |
| B–Cu–Zn | Cu and Zn | 79 | 200 | 1 M KOH | 60 |
| Ag/Cu2O–Cu | Cu and Ag | 76.5 | 1000 | 1 M KOH | 145 |
| Cu/Ag | Cu and Ag | ∼80 | 356.7 | 1 MKOH | 136 |
| Au–Cu Janus NSs | Cu and Au | 67 | 290 | 3 M KOH | 149 |
Dual-atom metal catalysts are composed of two isolated metal atoms, typically anchored on supports such as nitrogen-doped carbon or metal–organic frameworks. Their catalytic performance is finely tuned through electronic coupling and spatial proximity. The catalytic mechanism relies on atomic-level precision and the synergistic effects of dual-metal sites, particularly electronic synergy, which optimizes the adsorption of intermediates by modulating the d-band center. Specifically, electron transfer between the two metal atoms shifts their respective d-band centers, fine-tuning the adsorption strength: for instance, an upward shift in the d-band center of one metal atom enhances adsorption, while the other balances the adsorption strength through electron transfer or orbital hybridization, preventing efficiency loss caused by excessively strong or weak adsorption. In contrast, dual-metal nanoparticle catalysts feature active sites at the nanoscale (1–100 nm), with their catalytic performance governed by a combination of electronic effects, geometric effects, and interface effects. These include the optimization of intermediate adsorption through surface modification and electron transfer. Dual-atom metal catalysts demonstrate high selectivity in reduction reactions, predominantly yielding single products such as CO, while dual-metal nanoparticle catalysts are better suited for complex reactions, enabling the reduction of CO2 to a variety of hydrocarbons.120
3.1 Dual-atom metal synergistic catalysis for the electrocatalytic CO2RR
Dual-atom catalysts (DACs), characterized by their exceptional atomic efficiency, precisely defined electronic structures, and unique dual-atom synergistic effects, have risen as prominent candidates for electrocatalytic CO2 reduction. The precise arrangement of two metal atoms in DACs can create specific catalytically active sites with synergistic effects, which can lower the activation energy of the reaction, thereby accelerating the rate of CO2 reduction. DACs can be further distinguished into homonuclear and heteronuclear types, each offering unique advantages for their applications for the electrocatalytic CO2RR. Hence, in this section, we will explore the impact of synergistic catalysis between dual metal atoms on the efficiency and catalytic mechanism of electrocatalytic CO2 reduction, highlighting the distinct roles of homonuclear and heteronuclear dual-metal configurations.43Nickel, with its moderate d-band center position and rich valence state variability, is an ideal candidate for constructing highly efficient CO2 reduction catalysts. Particularly in homonuclear dual-atom configurations, the synergistic interaction between two nickel atoms creates a uniform and consistent electronic effect, significantly enhancing catalytic performance. In this research direction, Lu's group has made systematic contributions. Among the pioneering studies in this field, Lu and colleagues achieved a significant breakthrough for the electrocatalytic CO2RR in 2018 with the development of a dinuclear nickel complex, Ni2L1 (L1 = 1,2-bis((5,7-dimethyl-1,4,8,11-tetra-azacyclotetradecan-6-yl)methyl)benzene). This catalyst demonstrated remarkable performance in CO2 reduction, achieving a FECO of 95%, a turnover number (TON) of 4.1 × 106, and an impressive turnover frequency (TOF) of 190.0 s−1. The outstanding performance of complex Ni2L1 compared to mononuclear Ni molecular catalysts for the CO2-to-CO conversion stems from the synergistic catalytic effect between its two Ni centers. It was found that one Ni center can serve as a Lewis base, donating an electron to the CO2 molecule, while the other Ni center acts as a Lewis acid, stabilizing the partial negative charges on the O atom of CO2. This synergistic interaction significantly enhances the efficient conversion of CO2 into CO. These findings underscore the promising potential of dual-metal synergistic catalysis in the field of electrochemical CO2 reduction, establishing a new benchmark for deepening our understanding of the intricate structure-effect relationship (Fig. 7a).124 Building upon this foundation, Lu's group made substantial progress in understanding the synergetic effects in dual-metal catalysts. In 2022, they systematically investigated three Ni2 DACs, Ni2–N7, Ni2–N5C2 and Ni2–N3C4 with distinct coordination environments, identifying Ni2–N3C4 as the most effective configuration for CO2 reduction. The synergistic effect between the dual Ni centers in Ni2–N3C4, enabled by the fine-tuning of its electronic structure, plays a pivotal role in optimizing the binding energies to the COOH* and CO* intermediates, leading to the superior electrocatalytic CO2-to-CO reduction performance. This finding emphasizes the paramount importance of electronic structure modulation in the design of dual-atom catalysts (Fig. 7b).125 Further advancements were made in 2023 with the synthesis of a uniform Ni2-NCNT catalyst loaded on N-doped carbon nanotubes (NCNT), demonstrating an outstandingly high FE exceeding 90% for CO production across a wide range of potentials, accompanied by a significant partial current density for CO, which is much higher than that of single-atom catalyst Ni1-NCNT. DFT calculations revealed that the synergistic interaction between the two Ni atoms in the Ni2-NCNT configuration effectively stabilizes the *COOH intermediate and significantly reduces the free energy change in the rate-determining step when compared to the Ni1- NCNT SAC, thereby enhancing CO2 reduction efficiency (Fig. 7c and d).126 The most recent achievement of Lu's group in 2024 involved strategic coordination environment optimization of Ni2 dual-atoms through pyrrolic-N modification. They discovered that the pyrrolic-N-coordinated Ni2 catalysts showcase markedly enhanced electrocatalytic performance in the conversion of CO2-to-CO, outperforming pyridinic-N-coordinated low-valent Ni catalysts. This significant advancement primarily stems from the fact that the intrinsically weak electron-donating characteristic of pyrrolic-N facilitates the formation of a reduced active site, which is extremely advantageous for the CO2 electroreduction reaction. It was found that the reaction free energy pertaining to *COOH formation is notably lower on pyrrolic-N-coordinated Ni2 catalysts in contrast to their pyridinic-N-coordinated Ni counterparts. Furthermore, the water molecules bound to the catalyst during the catalytic process are conducive to the formation of the key intermediate *COOH and the desorption of CO, which greatly enhances the catalytic efficiency (Fig. 7e).127
 | ||
| Fig. 7 (a) Mechanism for the conversion of CO2 to CO, facilitated by a dinuclear nickel complex. (b) DFT calculations of Ni2N3C4. (c) Faradaic efficiencies of CO for NCNT, Ni1-NCNT and Ni2-NCNT catalysts. (d) Pathways of Ni2–N3 (Ni2-NCNT) for CO2 electroreduction to CO. (e) The optimized structures of Ni2/N-CNTs, Ni/N-CNTs, and Ni/N-CNTs-1000 (Ni2/N-CNTs-1000). (f) The free energy diagram of the CO2RR for Fe1–N4–C, Fe2–N6–C-o, Fe2–N6–C-p, Fe1–N3–C, and Fe2–N6–C-p-CO. (g) The energy barrier of Cu and Cu2. (a) Reproduced with permission.124 Copyright 2017, the Royal Society of Chemistry. (b) Reproduced with permission.125 Copyright 2022, John Wiley and Sons Ltd. (c and d) Reproduced with permission.126 Copyright, 2023, Elsevier. (e) Reproduced with permission.127 Copyright 2025, Springer. (f) Reproduced with permission.128 Copyright 2022, American Chemical Society. (g) Reproduced with permission.129 Copyright 2024, John Wiley and Sons Ltd. | ||
Advancements in dual-atom catalysts were notably achieved by Han, Lee, and their collaborators in the realm of iron-based catalysis. They engineered dual-atom Fe2 sites within the Fe2–N6–C catalyst, achieving a FE for CO production exceeding 80% over an extended range of applied potentials, in contrast to their single-atom Fe1–N4–C counterparts. The orbital coupling between the two adjacent Fe atoms in the Fe2 sites facilitates electron delocalization and reduces the energy levels of the Fe-3d orbitals. This phenomenon enables charge transfer between the Fe atoms and the CO intermediate during *CO adsorption, thereby modulating the *CO binding energy and promoting CO desorption (Fig. 7f).128 Another achievement was made by He, Wang et al., who developed a novel copper-based metal–organic polyhedral dual-atom catalyst Cu2, featuring a Cu–Cu bond distance of 2.95 Å, which demonstrated unprecedented C2H4 production performance in 1.0 M KOH solution. The catalyst achieved a FE of 51% and a current density of 450 mA cm−2, significantly outperforming both Cu single-atom catalysts and Cu nanoparticles. Mechanistic studies revealed that the unique Cu atom pairs in the catalyst can modulate *CO adsorption energy, promote C–C dimerization, and stabilize the *OCCO intermediate, thereby dramatically enhancing C2H4 production efficiency (Fig. 7g).129
These comprehensive studies have systematically elucidated the critical role of homonuclear dual-atom synergistic effects and coordination environment modulation in CO2 electrocatalysis. The cooperative interaction between identical metal atoms optimizes reaction pathways, stabilizes key intermediates, and enhances catalytic performance in CO2 reduction.
 | ||
| Fig. 8 (a) DFT calculations of NiN3–CuN3 and NiN3. (b) faradaic efficiencies of CO measured in a H-type cell. (c) Free energy profiles of the CO2RR on CoCu-DASC Co-SAC and Cu-SAC. (d) Calculated configurations for the conversions of CO2 → COOH* and COOH* → CO (IS, initial state; TS, transition state; FS, final state) over TeN2–CuN3 DAC. (e) A comparative study on the utilization degree of various materials. (f) Free energy diagrams of the CO2RR for production of formate. (a) Reproduced with permission.75 Copyright 2023, Wiley. (b and c) Reproduced with permission.132 Copyright 2022, John Wiley and Sons Ltd. (d) Reproduced with permission.40 Copyright, 2023, Springer Nature. (e and f) Reproduced with permission.134 Copyright 2021, John Wiley and Sons Ltd. | ||
Recent advancements in hetero-DACs without M–M bonds have also revealed remarkable synergistic catalytic effects in CO2 reduction, with multiple research teams developing high-performance catalysts through innovative design and synthesis strategies. Lu, Jiao, et al. reported a TeN2–CuN3 heteronuclear DAC with asymmetric coordination environments, offering a wide potential window for a high FE for CO production accompanied by enhanced reaction kinetics. It was confirmed that the Te center facilitates the activation of CO2, while the Cu center aids in the dissociation of H2O. Both experimental and theoretical findings indicate that the TeN2–CuN3 configuration can collaboratively reduce the energy barriers associated with the rate-limiting step, thereby enhancing proton transfer kinetics (Fig. 8d).40 In another example, Wei, Shao, et al. constructed a NiSn DAC featuring adjacent Ni and Sn atoms, each bonded to four nitrogen atoms, forming a N4–Ni–Sn–N4 configuration. The fabricated NiSn DAC showcased exceptional efficiency in the conversion of CO2 to formate via the CO2RR, attaining a remarkable TOF of 4752 h−1. It was found that the neighboring Ni–N4 configuration could facilitate electron redistribution within the Sn atom, with the synergistic catalytic effect leading to the lowered energy barrier for *OCHO key intermediate formation (Fig. 8e and f).134
In summary, recent advancements in heteronuclear DACs for CO2 reduction have demonstrated significant potential in achieving high efficiency and selectivity. Both DACs with M–M bonds and those without exhibit remarkable synergistic effects, which are primarily attributed to their unique electronic configurations. The presence of M–M bonds facilitates orbital overlap, electron delocalization, and modified d-band centers, optimizing the adsorption strength of key intermediates and lowering activation barriers. In contrast, DACs without M–M bonds often rely on asymmetric coordination environments or specific atomic pairings to enhance catalytic performance.
3.2 Dual-metal nanoparticle synergistic catalysis for the electrocatalytic CO2RR
Copper is widely regarded as one of the most promising candidates for the electrocatalytic CO2RR due to its suitable adsorption capacity for reaction intermediates. However, the performance of Cu-based catalysts is often limited by issues such as poor selectivity, low activity, and rapid deactivation. These challenges necessitate the development of advanced strategies to enhance their catalytic performance. One effective strategy to overcome these limitations involves modifying copper-based nanoparticle catalysts by incorporating a secondary metal into the system. The electronic coupling between Cu and the secondary metal can modulate the electronic density of states, facilitating the charge transfer between the catalyst and reactants.135 Most importantly, this approach can generate dual-active sites, creating a synergistic catalytic effect that enhances the overall catalytic performance.In this section, the main focus is on leveraging the synergistic effects between the copper host and guest metals to tune the electronic and geometric structures of the resulting nanoparticle catalysts. By modifying the dual-active sites, the catalytic performance can be significantly improved. A wide range of guest metals, encompassing main-group metals, non-precious transition metals, noble metals, and lanthanides, will be extensively studied as secondary metals incorporated into Cu-based catalysts to explore their potential for enhancing the product efficiency of the CO2RR. A particular focus will be placed on elucidating the mechanisms underlying synergetic catalysis in the context of the CO2RR. Understanding these cooperative mechanisms holds immense significance to elucidate the structure–activity relationships and identify the key factors that govern their catalytic performance.
The introduction of main-group elements can enhance the control of copper-based catalysts over the electrochemical CO2RR for selective generation of C1 products. For example, Pan et al. electrochemically deposited the main group metal indium (In) onto CuCl-decorated copper foil, successfully constructing a Cu–CuInO2 dual-metal material with a stable Cu0/Cu+ state. It was found that the high HER overpotential of In could minimize parasitic H2 production, directing more electrons to CO2 reduction, which enabled CuInO2 catalysts to achieve FEHER < 15% and FECO of 89% at −0.9 V vs. RHE. The CuInO2 catalyst demonstrated unique synergistic effects through electronic structure modulation, wherein p-orbitals of In hybridize with d-orbitals of Cu to redistribute charge density, optimize *CO binding energy, weaken *CO adsorption, and strengthen *COOH stabilization, thereby favoring CO production over C2 pathways (Fig. 9a).137 Numerous investigations have demonstrated that incorporating main group elements into copper-based catalysts can substantially enhance the catalytic pathway by lowering the energy barrier for C–C coupling, thereby promoting the formation of C2 products. For example, Wang, Jiang et al. reported the construction of a CuO/SnO2 dual-metal heterostructure through in situ electrochemical evolution, successfully regulating the C1/C2 product selectivity through the introduction of Sn. Most importantly, Sn2+/Sn0 in Cu/SnO2−x (oxygen-deficient SnO2) can stabilize *COOH and *CHOCO intermediates, thereby facilitating C–C coupling for CH3CH2OH production. Through strong d-p orbital hybridization between Cu (3d) and SnO2−x (O2p), key intermediates are synergistically stabilized, leading to a lowered energy barrier for C2 product formation. Besides, the introduction of Sn promoted the synergistic adsorption of *COOH and *CHOCO intermediates at the Cu/SnO2−x interface, significantly enhancing ethanol production efficiency (Fig. 9b).138 Meanwhile, Zhang's group introduced the main group metal Bi to prepare a single-atom Bi-modified Cu alloy (BiCu-SAA) for the first time. The high HER overpotential of Bi effectively suppresses competing reactions, while single-atom Bi doping modulates the electronic structure of Cu to stabilize *CO intermediates and facilitate efficient C–C coupling. Through this synergistic Bi–Cu interaction, the BiCu-SAA catalyst achieves exceptional performance at industrial current densities (400 mA cm−2), delivering a remarkable 73.4% FE for C2+ products (Fig. 9c and d).139 In another study, Wang et al. incorporated the main-group metal gallium (Ga) into a catalyst system, creating a dual-metal catalyst by combining it with copper (Cu). The resulting CuGa catalyst demonstrated a cathodic energy efficiency (EE) exceeding 50% for C2+ products at a current density of 1 A cm−2, significantly surpassing the performance of pure Cu, which exhibited an EE of approximately 30%. They discovered that Ga, with its lower electronegativity than Cu, can donate electrons to Cu, increasing the occupancy of Cu's sp-band, which strengthened σ-repulsion between Cu and adsorbed CO (*CO), weakening *CO binding energy. Besides, Ga can stabilize Cu+ states under ambient conditions, which may facilitate *CO adsorption and C–C coupling, leading to the high performance for C2+ production (Fig. 9e).140
 | ||
| Fig. 9 (a) XPS spectra of Cu–In-120 s before and after the e-CO2RR. (b) The comparison of free energy for the ethylene and ethanol pathways on Cu/SnO2−x. (c) FE values of various products on the BiCu-SAA at different applied potentials in an H-type cell. (d) The free energy diagram for the CO2RR to C2H4 on the BiCu(111)-SAA and Cu(111)-Nano. (e) The free energy surface for CO2 reduction to CO and CO–CO coupling with the Cu (100) surface with 0, 1/6, and 1/4 ML of Ga. (a) Reproduced with permission.137 Copyright 2023, American Chemical Society. (b) Reproduced with permission.138 Copyright 2023, John Wiley and Sons Ltd. (c and d) Reproduced with permission.139 Copyright, 2023, John Wiley and Sons Ltd. (e) Reproduced with permission.140 Copyright 2024, Springer Nature. | ||
In summary, the incorporation of main-group metals into copper-based catalysts offers a powerful strategy to optimize CO2 electroreduction by modulating electronic and geometric properties. These elements enhance selectivity and efficiency—either by suppressing the HER, stabilizing key intermediates, or lowering C–C coupling barriers—enabling high-performance C1 or C2+ production.
Many previous studies have shown that non-precious transition metals, owing to their distinctive d-electron configurations, can modulate the electronic structure and surface properties of Cu catalysts. Streb et al. developed a CuCo dual-metal electrocatalyst, formed under reaction conditions that drive Cu sites from single-atom states into small Cu clusters (Cu0) while maintaining Co sites in a monodispersed state, which enabled highly selective CO2-to-EtOH conversion with a FE > 70%. It was discovered that a synergistic CO spillover mechanism occurred where Co sites efficiently generated CO and created a high local CO concentration, while Cu sites received this CO and facilitate C–C coupling to produce EtOH, thereby significantly enhancing C2 product selectivity by reducing the CO transport distance. The close proximity of Co and Cu sites enables efficient CO2-to-ethanol conversion through the CO spillover mechanism, while inhibiting side reactions, demonstrating the importance of dual-metal catalysts in designing efficient CO2 reduction systems (Fig. 10a).59 In another study, Ma, Zhang, et al. added a trace amount of Ni to the surface of CuO nanosheets which markedly improved both the stability and FE for the electrochemical reduction of CO2 into C2 products. Ni doping into CuO nanosheets could significantly inhibit the dissolution of Cu during the CO2RR by strengthening the bonds between surface and subsurface Cu atoms. Hence, unlike pure CuO, which is prone to transforming into dendritic structures during prolonged reactions, the CuO + Ni-surface dual-metal system maintains its nanosheet architecture, ensuring consistent and reliable catalytic performance over time. DFT calculations and operando ATR-SEIRAS revealed that Ni-doped CuO surfaces exhibited stronger *CO adsorption and lower energy barriers for *CO dimerization, promoting C–C coupling, offering a cost-effective synergistic strategy to enhance CO2RR efficiency and durability for C2 production (Fig. 10b and c).62 The non-precious transition metal Zn is rich with d electrons. Schuhmann et al. doped the Cu-based catalyst with Zn to obtain the dual-metal gas diffusion electrodes (GDEs) B–Cu–Zn, which can enhance the electrocatalytic CO2RR activity toward multi-carbon C2+ products with high stability. Operando Raman spectroscopy confirmed that Zn incorporation helps retain Cu+ species under reduction conditions, which can suppress the HER and facilitate C–C coupling, while the Cu+ sites could lower the energy barrier for *OCCO intermediate formation, the key step in C2+ production. The synergistic interplay between Cu and Zn in B–Cu–Zn GDEs significantly improved the CO2RR performance mainly through the stabilization of Cu+ species by Zn and dynamic protection of active site Cu through Zn sacrificial oxidation (Fig. 10d and e).60
 | ||
Fig. 10 (a) The CO spillover mechanism for efficient ethanol formation at the CoCu electrocatalyst. (b) FT-IR characterization of CuO and the CuO + Ni-surface. (c) Relative Gibbs free energy diagrams for the CO2RR. (d) Operando electrochemical Raman spectra for the 0.5B–Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.025Zn GDE. (e) Faradaic efficiencies for CO2RR products at a catalyst loading of 0.5 mg cm−2 B–Cu at different potentials. (a) Reproduced with permission.59 Copyright 2024, American Chemical Society. (b and c) Reproduced with permission.62 Copyright 2025, Elsevier. (d and e) Reproduced with permission.60 Copyright 2021, John Wiley and Sons Ltd. :0.025Zn GDE. (e) Faradaic efficiencies for CO2RR products at a catalyst loading of 0.5 mg cm−2 B–Cu at different potentials. (a) Reproduced with permission.59 Copyright 2024, American Chemical Society. (b and c) Reproduced with permission.62 Copyright 2025, Elsevier. (d and e) Reproduced with permission.60 Copyright 2021, John Wiley and Sons Ltd. | ||
In summary, the incorporation of non-precious transition metals into Cu-based catalysts introduces synergistic effects that significantly enhance CO2 electroreduction toward C2+ products. These dopants modify the electronic structure of Cu through their distinct d-electron configurations and electronegativity differences, optimizing CO2 adsorption, stabilizing key intermediates like *CO and *OCCO, and lowering energy barriers for C–C coupling. Studies on CuCo, CuNi, and CuZn nanoparticle systems demonstrate that dual-metal interactions—such as CO spillover, strengthened Cu surface bonding, and sacrificial stabilization of Cu+—play pivotal roles in improving selectivity and durability at high current densities.61 These findings highlight the potential of cost-effective dual-metal catalysts to advance the CO2RR by precisely tuning electronic and geometric properties, offering a scalable pathway for sustainable C2+ production.
Compared to other noble metals like Au, Pd, and Pt for doping Cu-based CO2RR catalysts, Ag presents distinct and superior advantages, particularly in terms of cost-effectiveness. Moreover, Ag demonstrates weaker *CO adsorption compared to Cu but stronger adsorption than Au, thereby establishing an optimal intermediate state conducive to C–C coupling. For example, Wang, Ye, et al. developed a Ag-doped Cu2O–Cu catalyst, Ag/Cu2O–Cu, which can significantly enhance CO2 electroreduction CO2 to multi-carbon C2+ products at an industrial current density of 1.0 A cm−2 with a FE of 76.5%. Operando Raman spectroscopy and XPS analyses confirmed that Ag incorporation effectively stabilizes metastable Cu+ species (Cu2O) under reducing conditions, maintaining robust Ag/Cu+/Cu0 interfaces during the CO2RR, which is a critical feature for facilitating C–C coupling. The intermediate *CO binding strength of Ag (between Cu and Au) creates a synergistic system, where efficient *CO production couples with facile spillover to Cu+/Cu0 sites, lowering the *OCCO formation barrier by 56% versus Cu2O and dramatically boosting C2+ selectivity (Fig. 11a and b).144 Li's research group also developed a Ag-doped Cu-based material featuring (111)-facet-exposed Cu nanosheets decorated with Ag nanoparticles. This composite demonstrated remarkable efficiency in electrochemically converting CO2 to ethanol, achieving a FE of 56.5 ± 2.6% and a partial current density of 356.7 ± 9.5 mA cm−2. They discovered that the synergistic effect between Ag and Cu in the dual-metal catalyst lies in Ag's ability to supply CO to Cu(111) facets—which efficiently generate *CH2 intermediates—promoting asymmetric *CH2–CO coupling for ethanol production, while suppressing excessive *CO hydrogenation to methane (FE drops from 27.3% on bare Cu to 3.7%) and shifting selectivity away from symmetric CO–CO coupling that favors ethanol production. This dynamic interplay, enhanced by the layered Cu/Ag structure, optimizes local CO coverage and intermediate utilization, steering selectivity toward ethanol over hydrocarbons (Fig. 11c and d).146
 | ||
| Fig. 11 (a) Cu LMM spectra of Cu2O, Cu2O–Cu, and 5.6 atom% Ag/Cu2O–Cu catalysts. (b) Raman shift of the 5.6 atom% Ag/Cu2O–Cu catalyst. DFT calculations: barriers for *CHx (x = 1, 2) coupling with *CO/CO via (c) L–H and (d) E–R mechanisms. (e) The reaction free energies and activation barriers of C–C coupling on Cu (100) and Pd1Cu (100). (f) In situ ATR-IR spectra of Au–Cu Janus NSs. (a and b) Reproduced with permission.144 Copyright 2025, American Chemical Society. (c and d) Reproduced with permission.146 Copyright 2024, American Chemical Society. (e) Reproduced with permission.147 Copyright 2023, Springer Nature. (f) Reproduced with permission.148 Copyright 2022, Wiley-VCH Verlag. | ||
Many significant studies have highlighted the promotional role of doping other precious metals, such as Pd and Au, into Cu-based nanomaterials in facilitating the electrocatalytic reduction of CO2. For example, Yang, Che, et al. reported two platinum-group-metal doped Cu-based dual-metal catalysts Pd1Cu and Pt1Cu for the electrocatalytic CO2RR. It was found that both Pt and Pd single-atom dopants boost CO* adsorption on Cu, serving as CO* reservoirs that balance coverage and prevent poisoning, thereby enhancing C–C coupling for hydrocarbon production. In addition, Pt and Pd single-atom doping in Cu synergistically enhances the CO2RR by enriching CO* coverage while suppressing the HER and preserving facet-dependent selectivity (CH4 on Cu (111) and C2H4 on Cu (100)). Meanwhile, Pd1Cu demonstrates superior performance (CH4: 25%; C2H4: 33%) by optimally balancing CO* stabilization and hydrogenation kinetics compared to Pt1Cu (Fig. 11e).147 Huang et al. reported gold–copper (Au–Cu) Janus nanostructures as a dual-metal catalyst capable of efficiently electroreducing CO2 to C2+ products, achieving a C2+ selectivity of 67%. In situ ATR-IR spectroscopy revealed that the Au–Cu synergy enables selective CO2-to-CO* conversion at Au sites (the critical intermediate for C–C coupling) via weak CO* binding, while adjacent Cu sites facilitate CO* hydrogenation to hydrocarbons and C–C coupling. The enhanced CO* coverage on Au–Cu Janus nanostructures compared to pure Cu can be attributed to CO* spillover from Au to Cu domains. They discovered that fine-tuning Au–Cu interfaces of Janus structures could enhance selectivity toward C2+ products, which outperformed the physical mixtures or core–shell structures in the CO2RR, offering a general strategy for multi-step reaction catalysts (Fig. 11f).148
In summary, doping noble metals like Ag, Pd, and Au into Cu-based nanomaterials significantly enhances their performance in CO2 electroreduction.149 Ag stands out for its cost-effectiveness and optimal CO adsorption strength, facilitating C–C coupling and boosting C2+ selectivity. Pd and Au doping also promote CO adsorption and balance coverage, preventing poisoning and enhancing C–C coupling for hydrocarbon production.150 These findings underscore the critical role of precious transition metal doped Cu-based dual-metal synergies in modulating intermediate adsorption energies, stabilizing reactive species, and steering selectivity toward high-value C2+ products.
The incorporation of lanthanides into copper-based catalysts introduces lattice strain due to the substantial difference in ionic radii between lanthanides and copper.149,154,155 This strain can modulate the electronic structure and binding energies of reaction intermediates, thereby lowering the activation energy barriers and optimizing the reaction pathway for CO2 reduction.156 Han, Sun, et al. reported that the atomic doping of Gd into CuOx catalysts (Gd1/CuOx) induces tensile strain and stabilizes Cu+ species, enabling an exceptional FE of 81.4% for C2+ products with a partial current density of 444.3 mA cm−2 at −0.8 V vs. RHE, demonstrating its high efficiency in the CO2RR. They discovered that the unique 4f electron configuration and unfilled 5d orbitals of Gd can enhance spin–orbit coupling, modifying the local electron density around Cu atoms and inhibiting their reduction to Cu0. This Gd–Cu synergy facilitated efficient electron transfer during the CO2RR, significantly improving reaction kinetics. Additionally, the tensile strain induced through the large ionic radius of Gd introduced into the CuOx lattice could optimize the d-band center of Cu and fine-tune the binding energy of key intermediates *CO. This strain effect further reduces the Gibbs free energy barrier for the C–C coupling step (2CO → O*CCO), thereby promoting the selective formation of C2+ products (Fig. 12a and b).157 Liang et al. constructed a Cu/CeO2 dual-metal catalyst, in which the larger lattice spacing of the CeO2 (111) plane compared to Cu2O stretches Cu–O bonds, introducing tensile strain in the Cu domains. It was discovered that the tensile strain in the Cu/CeO2 heterostructure upshifted the d-band center of Cu, which can enhance *CO adsorption and promote dimerization, facilitating C–C coupling for multicarbon formation. DFT calculations revealed that the synergistic Cu and CeO2 interaction drives charge transfer from Cu to CeO2, creating electron-deficient interfacial Cu sites. By combining tensile strain and electron deficiency, Cu and CeO2 synergistically optimize CO coverage and stabilize key intermediates, collectively boosting selectivity toward C2+ products (Fig. 12c and d).158
 | ||
| Fig. 12 (a) Faradaic efficiencies for CO2RR products on 6.5% Gd1/CuOx and CuOx. (b) The free energy diagram of the CO2RR for Cu, CuOx, Gd1/CuOx, and Gd1/CuOx (2.5%). (c) Calculated *CO adsorption energies on standard Cu, stretched Cu, Cu/CeO2, the stretched Cu/CeO2 interface, and CeO2. (d) The transition barrier investigation for H2O dissociation on Cu (111) and CeO2 (111). (e) Adsorption configurations of different Pr6O11-Ovac–Cu models. (a and b) Reproduced with permission.157 Copyright 2023, American Chemical Society. (c and d) Reproduced with permission.158 Copyright 2023, American Chemical Society. (e) Reproduced with permission.159 Copyright, 2025, Springer Nature. | ||
Additionally, the unique electronic configurations of lanthanides can also regulate the electronic structure of active sites or modulate the d-band center of copper, enhancing *CO adsorption and stabilizing Cu+ sites critical for multicarbon product formation. Han, Zhu, et al. developed a Pr–Cu dual-metal oxide heterointerface, specifically Pr6O11–Cu-SS, which efficiently catalyzes the CO2RR to C2+ products, achieving a high FE of 71.3% for C2+ alcohols (ethanol and n-propanol) at a current density of 700mA cm−2. It was found that Pr doping in Cu oxides could generate an oxygen vacancy (Ovac)-rich interface, which modulated the electronic structure of Cu (Cuδ+/Cu0) to enhance CO adsorption and asymmetric C–C coupling. The Pr–O–Cu linkages further stabilized key intermediates, enabling sustained high activity and stability even at industrial current densities. DFT calculations and in situ Raman spectroscopy revealed that the Pr–Cu interface induced mixed *CO adsorption configurations, in which this asymmetry promoted C2+ alcohol formation over ethylene by stabilizing *OCHCH3 intermediates (Fig. 12e).159 In another study, Han, Sun, et al. developed a La-doped Cu electrocatalyst that achieved a remarkable FE of 86.2% for C2+ products at a partial current density of −775.8 mA cm−2 in acidic electrolyte during CO2 reduction. As validated by in situ SERS detection, owing to the synergistic effects of La and Cu, the mesoporous channels in La–Cu hollow spheres (La–Cu HS) could concentrate K+ and OH− near the catalyst surface, creating a localized alkaline microenvironment that can suppress the HER and promote C–C coupling. Electronic structure analysis reveals that due to the difference in electronegativity, La can donate electrons to Cu, which enhances *CO stabilization. DFT calculations further confirm that the resulting La–O–Cu linkages could stabilize metallic Cu during the CO2RR, while enabling synergistic catalysis, where La promoted CO generation and lowered C–C coupling barriers, while Cu maintained conductive pathways and active sites for CO2 reduction.160
In summary, the integration of lanthanide metals into copper-based nanomaterials presents a promising strategy for enhancing CO2 reduction performance. By leveraging the unique electronic properties of lanthanides, such as their f-orbital electrons and specific electron configurations, researchers have been able to modulate the reaction pathways of copper-based catalysts.158 The synergistic interactions between copper and lanthanides, driven by factors such as lattice strain, electron transfer, and interface engineering, have opened up new avenues for the development of highly efficient electrocatalysts for the CO2RR.161
4 Conclusions
The development of dual-metal synergistic catalysts has emerged as a powerful strategy to enhance the efficiency, selectivity, and stability of the electrocatalytic CO2RR. This review systematically explores the design principles, mechanistic insights, and catalytic performance of dual-metal catalysts, emphasizing the critical role of synergistic effects of different metal centers in optimizing CO2RR pathways, and elucidating the structure–activity correlations. Leveraging the unique synergistic effects of two metal centers has led to remarkable progress in tailoring catalyst properties to favor desired reaction intermediates and suppress competing HER.This review first focuses the design principles of dual-metal catalysts with synergistic effects for the CO2RR, focusing on electronic and geometric structure regulation. Electronic structure regulation involves chemical state, vacancy, and coordination environment modifications, while geometric structure regulation encompasses crystal facet, nanoparticle size, and morphology adjustments. These strategies are crucial for optimizing the adsorption and activation of reactants, stabilizing intermediate states, and refining the catalytic pathway. Next, the review delves into the influence of different types of dual-metal active sites, including dual-atom metal catalysts (DACs) and dual-metal nanomaterial catalysts. DACs are characterized by their atomic efficiency and precise electronic structures, while dual-metal nanomaterial catalysts offer more complex surface architectures and synergistic effects at the nanoscale. For dual-metal nanomaterial catalysts, various secondary metals, such as main-group metals, non-precious transition metals, noble metals and lanthanides, are studied for their potential to enhance copper-based catalysts for CO2RR performance. The review mainly explores various dual-metal catalysts with synergistic catalytic effects, highlighting their distinct advantages and mechanisms for promoting the CO2RR. The review also underscores the importance of rational design and innovative synthesis strategies in developing advanced CO2RR electrocatalysts.
Despite the significant progress made in dual-metal catalysis for the CO2RR, several challenges remain. Future research should focus on the following areas: (1) enhanced catalytic stability: improving the long-term stability of dual-metal catalysts is crucial for their practical application. Strategies such as surface passivation, defect engineering, and support optimization could be explored to mitigate deactivation pathways. (2) Developing scalable and cost-effective production methods for dual-metal catalysts is essential for their commercialization. Research on continuous flow reactors, high-throughput screening, and catalyst recycling could accelerate this process. (3) Deeper understanding of the synergistic mechanisms in dual-metal catalysts is needed to guide the rational design of more efficient catalysts. Advanced characterization techniques, such as operando spectroscopy and in situ electron microscopy, could provide valuable insights. (4) Achieving high selectivity towards multi-carbon products remains a challenge. The optimization of catalyst composition, structure and reaction conditions could pave the way for the efficient production of C2+ compounds from the CO2RR.
In summary, dual-metal catalysis with synergistic effects holds great promise for advancing the field of the CO2RR. With continued research efforts in catalyst design, synthesis, and mechanistic understanding, the efficient conversion of CO2 into value-added products could become a reality, contributing significantly to global carbon neutrality goals.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
M. W. and T. L. conceived the topic and revised the review. M. W., P. S. and Y. Y wrote and edited the original draft of the manuscript. All authors helped in literature search.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22375148) and the National Key R&D Program of China (2022YFA1502902).Notes and references
- A. Husile, Z. Wang and J. Guan, Chem. Sci., 2025, 16, 5413–5446 RSC.
- D. Suri, S. Das, S. Choudhary, G. Venkanna, B. Sharma, M. A. Afroz, N. K. Tailor, R. Joshi, S. Satapathi and K. Tripathi, Small, 2025, 21, 2408981 CrossRef CAS PubMed.
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43–50 CrossRef CAS PubMed.
- H. M. A. Sharif, M. Rashad, I. Hussain, A. Abbas, O. F. Aldosari and C. Li, Appl. Catal. B Environ. Energy, 2024, 344, 123585 CrossRef CAS.
- W. Lai, Z. Ma, J. Zhang, Y. Yuan, Y. Qiao and H. Huang, Adv. Funct. Mater., 2022, 32, 2111193 CrossRef CAS.
- M. J. Gidden, T. Gasser, G. Grassi, N. Forsell, I. Janssens, W. F. Lamb, J. Minx, Z. Nicholls, J. Steinhauser and K. Riahi, Nature, 2023, 624, 102–108 CrossRef CAS PubMed.
- B. Buchner, Science, 2024, 386, 601 CrossRef CAS PubMed.
- L. Wang, D. Wang and Y. Li, Carbon Energy, 2022, 4, 1021–1079 CrossRef CAS.
- W. Peng, F. Li, S. Kong, C. Guo, H. Wu, J. Wang, Y. Shen, X. Meng and M. Zhang, Carbon Energy, 2024, 6, e498 CrossRef.
- J. Han, X. Bai, X. Xu, X. Bai, A. Husile, S. Zhang, L. Qi and J. Guan, Chem. Sci., 2024, 15, 7870–7907 RSC.
- Z. Wang, Z. Sun, H. Yin, H. Wei, Z. Peng, Y. X. Pang, G. Jia, H. Zhao, C. H. Pang and Z. Yin, eScience, 2023, 3, 100136 CrossRef.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P.-C. Chen, H. Wang, C. J. Pollock, X. Huang, Y.-T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- N. Dutta and S. C. Peter, J. Am. Chem. Soc., 2025, 147, 9019–9036 CrossRef CAS PubMed.
- G. Liang, S. Yang, C. Wu, Y. Liu, Y. Zhao, L. Huang, S. Zhang, S. Dou, H. Du, D. Cui and L. Lin, J. Mater. Chem. A, 2025, 13, 11210–11235 RSC.
- J. Jia, Z. Zhang, A. Wang, Y. Liu, Z. Liu, J. Wang, Y. Guo, L. Peng, J. Li and J. Robertson, Adv. Funct. Mater., 2025, 35, 2412056 CrossRef CAS.
- W. Lai, Y. Qiao, Y. Wang and H. Huang, Adv. Mater., 2023, 35, 2306288 CrossRef CAS PubMed.
- F. Xie, Z. Wang, C.-W. Kao, J. Lan, Y.-R. Lu and Y. Tan, Angew. Chem., Int. Ed., 2024, 63, e202407661 CrossRef CAS PubMed.
- B. Lei, W. Cui, P. Chen, L. Chen, J. Li and F. Dong, ACS Catal., 2022, 12, 9670–9678 CrossRef CAS.
- J. Xiong, J. Ji, Q. Lei, X. Yang, Y. Bai, X. Zhang and H.-M. Cheng, eScience, 2024, 100306 Search PubMed.
- K. Wang, K. Huang, Z. Wang, G. An, M. Zhang, W. Liu, S. Fu, H. Guo, B. Zhang, C. Lian, J. Wu and L. Wang, Small, 2025, 2502733 CrossRef CAS.
- Q. Yang, H. Liu, Y. Lin, D. Su, Y. Tang and L. Chen, Adv. Mater., 2024, 36, 2310912 CrossRef CAS PubMed.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nat. Catal., 2022, 5, 818–831 CrossRef CAS.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS PubMed.
- Z. Wu, N. Meng, R. Yang, M. Chen, J. Pan, S. Chi, C. Wu, S. Xi, Y. Liu, Y. Ou, W. Wu, S. Han, B. Zhang, Q.-H. Yang and K. Ping Loh, Angew. Chem., Int. Ed., 2025, 64, e202420283 CrossRef CAS.
- C.-f. Li, R.-t. Guo, Z.-r. Zhang, T. Wu and W.-g. Pan, Small, 2023, 19, 2207875 CrossRef CAS PubMed.
- M. Kuang, B. Li, L. Zhou, Z. Huang, J. Wu, S. Wang and J. Yang, Angew. Chem., Int. Ed., 2025, 64, e202422357 CrossRef CAS PubMed.
- Y.-F. Tang, L.-B. Liu, M. Yu, S. Liu, P.-F. Sui, W. Sun, X.-Z. Fu, J.-L. Luo and S. Liu, Chem. Soc. Rev., 2024, 53, 9344–9377 RSC.
- T. Lu, T. Xu, S. Zhu, J. Li, J. Wang, H. Jin, X. Wang, J.-J. Lv, Z.-J. Wang and S. Wang, Adv. Mater., 2023, 35, 2310433 CrossRef CAS.
- H. Shang and D. Liu, Nano Res., 2023, 16, 6477–6506 CrossRef CAS.
- J. Liu, Y. Cai, R. Song, S. Ding, Z. Lyu, Y.-C. Chang, H. Tian, X. Zhang, D. Du, W. Zhu, Y. Zhou and Y. Lin, Mater. Today, 2021, 48, 95–114 CrossRef CAS.
- Y. Zhai, P. Han, Q. Yun, Y. Ge, X. Zhang, Y. Chen and H. Zhang, eScience, 2022, 2, 467–485 CrossRef.
- J. Wang, D. Deng, Q. Wu, M. Liu, Y. Wang, J. Jiang, X. Zheng, H. Zheng, Y. Bai, Y. Chen, X. Xiong and Y. Lei, ACS Nano, 2023, 17, 18688–18705 CrossRef CAS PubMed.
- C. Wang, Z. Lv, X. Feng, W. Yang and B. Wang, Adv. Energy Mater., 2024, 14, 2400160 CrossRef CAS.
- J. Huang, T. Yang, K. Zhao, S. Chen, Q. Huang and Y. Han, J. Energy Chem., 2021, 62, 71–102 CrossRef CAS.
- P. Sun, S. Liu, X. Zheng, G. Hu, Q. Zhang, X. Liu, G. Zheng and Y. Chen, Nano Today, 2024, 55, 102152 CrossRef CAS.
- P. Li, J. Liu, Y. Wang, X.-D. Zhang, Y. Hou, Y. Zhang, X. Sun, X. Kang, Q. Zhu and B. Han, J. Am. Chem. Soc., 2024, 146, 26525–26533 CrossRef CAS PubMed.
- Q. Zhang, D. Liu, Y. Zhang, Z. Guo, M. Chen, Y. Chen, B. Jin, Y. Song and H. Pan, J. Energy Chem., 2023, 87, 509–517 CrossRef CAS.
- Y. Li, H. Wang, X. Yang, T. O'Carroll and G. Wu, Angew. Chem., Int. Ed., 2024, 63, e202317884 CrossRef CAS PubMed.
- L. Shi, J. Song, Y. Yang, L. Yang, Z. Dai, L. Yao and W. Jiang, J. Mater. Chem. A, 2025, 13, 2478–2504 RSC.
- J. Jiao, Q. Yuan, M. Tan, X. Han, M. Gao, C. Zhang, X. Yang, Z. Shi, Y. Ma, H. Xiao, J. Zhang and T. Lu, Nat. Commun., 2023, 14, 6164 CrossRef CAS.
- B. Ren, G. Wen, R. Gao, D. Luo, Z. Zhang, W. Qiu, Q. Ma, X. Wang, Y. Cui, L. Ricardez-Sandoval, A. Yu and Z. Chen, Nat. Commun., 2022, 13, 2486 CrossRef CAS PubMed.
- W. Wu, J. Zhu, Y. Tong, S. Xiang and P. Chen, Nano Res., 2024, 17, 3684–3692 CrossRef CAS.
- A. R. Woldu, A. G. Yohannes, Z. Huang, P. Kennepohl, D. Astruc, L. Hu and X.-C. Huang, Adv. Mater., 2024, 36, 2414169 CrossRef CAS PubMed.
- Y. Gao, B. Liu and D. Wang, Adv. Mater., 2023, 35, 2209654 CrossRef CAS PubMed.
- Y. Yang, W. Zhang, G. Wu, Q. Huang, J. Wen, D. Wang and M. Liu, Angew. Chem., Int. Ed., 2025, e202504423 CAS.
- Y. Chen, J. Zhao, X. Pan, L. Li, Z. Yu, X. Wang, T. Ma, S. Lin and J. Lin, Angew. Chem., Int. Ed., 2024, 63, e202411543 CrossRef CAS PubMed.
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Nat. Commun., 2023, 14, 4615 CrossRef CAS PubMed.
- J. Li, Y. Chen, B. Yao, W. Yang, X. Cui, H. Liu, S. Dai, S. Xi, Z. Sun, W. Chen, Y. Qin, J. Wang, Q. He, C. Ling, D. Wang and Z. Zhang, J. Am. Chem. Soc., 2024, 146, 5693–5701 CrossRef CAS PubMed.
- J. Liu, P. Li, J. Bi, S. Jia, Y. Wang, X. Kang, X. Sun, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 23037–23047 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed.
- W. Xu, Y. Wang, C. Zhang, X. Ma, J. Wu, Y. Liu, B. Lu, H. Zhang, C. Ming and J. Xiang, Chem. Eng. J., 2023, 461, 141911 CrossRef CAS.
- R. Cao, S. Yin, Y. Han, J. Zhang, W. Jiang and G. Liu, Coord. Chem. Rev., 2025, 538, 216722 CrossRef CAS.
- J. Ji, L. Wu, S. Zhou, T. Qiu, Z. Li, L. Wang, L. Zhang, L. Ma, M. Ling, S. Zhou and C. Liang, Small Methods, 2022, 6, 2101511 CrossRef CAS PubMed.
- H. Shang, S. K. Wallentine, D. M. Hofmann, Q. Zhu, C. J. Murphy and L. R. Baker, Chem. Sci., 2020, 11, 12298–12306 RSC.
- B. Zhao, F. Chen, C. Cheng, L. Li, C. Liu and B. Zhang, Adv. Energy Mater., 2023, 13, 2204346 CrossRef CAS.
- M. Jun, J. Kundu, D. H. Kim, M. Kim, D. Kim, K. Lee and S.-I. Choi, Adv. Mater., 2024, 36, 2313028 CrossRef CAS.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu and S. Zhao, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
- M. Qi, M. J. Zachman, Y. Li, Y. Zeng, S. Hwang, J. Liang, M. Lyons, Q. Zhao, Y. Mao, Y. Shao, Z. Feng, Z. Wang, Y. Zhao and G. Wu, Energy Environ. Sci., 2025, 18, 5643–5656 RSC.
- S. A. Chala, R. Liu, E. O. Oseghe, S. T. Clausing, C. Kampf, J. Bansmann, A. H. Clark, Y. Zhou, I. Lieberwirth, J. Biskupek, U. Kaiser and C. Streb, ACS Catal., 2024, 14, 15553–15564 CrossRef CAS.
- Y. Song, J. R. C. Junqueira, N. Sikdar, D. Öhl, S. Dieckhöfer, T. Quast, S. Seisel, J. Masa, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 9135–9141 CrossRef CAS.
- Z. Zhang, S. Li, Y. Rao, L. Yang, W. Yan and H. Xu, Chem. Eng. J., 2024, 479, 147376 CrossRef CAS.
- M. Li, S. Kuang, Y. Jin, H. Chi, S. Zhang and X. Ma, Chem. Eng. J., 2025, 506, 160048 CrossRef CAS.
- M.-G. Kim, J. Park, Y. Choi, H. C. Song, S.-H. Kim, K.-M. Bang, H. C. Ham, N.-K. Kim, D. H. Won, B. K. Min, S. J. Yoo and W. Kim, Adv. Energy Mater., 2023, 13, 2300749 CrossRef CAS.
- R. Zhang, H. Ma, S. Han, Z. Wu, X. Zhou, Z. Chen, J. Liu, Y. Xiao, W. Chen and K. P. Loh, Angew. Chem., Int. Ed., 2025, 64, e202421860 CrossRef CAS PubMed.
- Z. Ren, B. Li, Y. Wu, H. Liu, F. Guo, S. Tian and J. Yang, Chem. Eng. J., 2025, 507, 160394 CrossRef CAS.
- L. Yan, W. Su, X. Cao, P. Zhang and Y. Fan, Chem. Eng. J., 2021, 412, 128718 CrossRef CAS.
- L. Xue, Q.-Y. Fan, Y. Zhao, Y. Liu, H. Zhang, M. Sun, Y. Wang and S. Zeng, J. Energy Chem., 2023, 82, 414–422 CrossRef CAS.
- S. You, J. Xiao, S. Liang, W. Xie, T. Zhang, M. Li, Z. Zhong, Q. Wang and H. He, Energy Environ. Sci., 2024, 17, 5795–5818 RSC.
- X. Shi, L. Shi, J. Wang, Y. Zhou and S. Zhao, Matter, 2024, 7, 4233–4259 CrossRef CAS.
- F. Huang, X. Chen, H. Sun, Q. Zeng, J. Ma, D. Wei, J. Zhu, Z. Chen, T. Liang, X. Yin, X. Liu, J. Xu and H. He, Angew. Chem., Int. Ed., 2025, 64, e202415642 CrossRef CAS.
- W. Guo, S. Liu, X. Tan, R. Wu, X. Yan, C. Chen, Q. Zhu, L. Zheng, J. Ma, J. Zhang, Y. Huang, X. Sun and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21979–21987 CrossRef CAS PubMed.
- K. Wang, D. Liu, L. Liu, J. Liu, X. Hu, P. Li, M. Li, A. S. Vasenko, C. Xiao and S. Ding, eScience, 2022, 2, 518–528 CrossRef.
- C. Peng, X. Zhu, Z. Xu, S. Yan, L. Y. Chang, Z. Wang, J. Zhang, M. Chen, T.-K. Sham, Y. Li and G. Zheng, Small, 2022, 18, 2106433 CrossRef CAS.
- C. Li, H. Yan, H. Yang, W. Zhou, C. Xie, B. Pan and Q. Zhang, Sci. China Mater., 2025, 68, 21–38 CrossRef CAS.
- L. Zhang, J. Feng, S. Liu, X. Tan, L. Wu, S. Jia, L. Xu, X. Ma, X. Song, J. Ma, X. Sun and B. Han, Adv. Mater., 2023, 35, 2209590 CrossRef CAS PubMed.
- X. Zhang, C. Liu, Y. Zhao, L. Li, Y. Chen, F. Raziq, L. Qiao, S.-X. Guo, C. Wang, G. G. Wallace, A. M. Bond and J. Zhang, Appl. Catal. B Environ. Energy, 2021, 291, 120030 CrossRef CAS.
- X. K. Lu, B. Lu, H. Li, K. Lim and L. C. Seitz, ACS Catal., 2022, 12, 6663–6671 CrossRef CAS.
- H. Zhang, Y. Cao, M. Sun, Y. Liu, Y. Wang, H. Li, R. Zhang, X. Gu and S. Zeng, Chem. Eng. J., 2024, 490, 151706 CrossRef CAS.
- K. Huang, R. Li, H. Qi, S. Yang, S. An, C. Lian, Q. Xu, H. Liu and J. Hu, ACS Catal., 2024, 14, 8889–8898 CrossRef CAS.
- H. Wu, J. Li, K. Qi, Y. Zhang, E. Petit, W. Wang, V. Flaud, N. Onofrio, B. Rebiere, L. Huang, C. Salameh, L. Lajaunie, P. Miele and D. Voiry, Nat. Commun., 2021, 12, 7210 CrossRef CAS PubMed.
- A. Bohan, X. Jin, M. Wang, X. Ma, Y. Wang and L. Zhang, J. Colloid Interface Sci., 2024, 654, 830–839 CrossRef CAS PubMed.
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Adv. Energy Mater., 2020, 10, 1902844 CrossRef CAS.
- N. Sakamoto, K. Sekizawa, S. Shirai, T. Nonaka, T. Arai, S. Sato and T. Morikawa, Nat. Catal., 2024, 7, 574–584 CrossRef CAS.
- Z. Sun, C. Li, Z. Wei, F. Zhang, Z. Deng, K. Zhou, Y. Wang, J. Guo, J. Yang, Z. Xiang, P. Ma, H. Zhai, S. Li and W. Chen, Adv. Mater., 2024, 36, 2404665 CrossRef CAS.
- X.-Q. Wang, Q. Chen, Y.-J. Zhou, H.-M. Li, J.-W. Fu and M. Liu, Adv. Sens. Energy Mater., 2022, 1, 100023 CrossRef.
- Y. Jia, F. Li, K. Fan and L. Sun, Adv. Powder Mater., 2022, 1, 100012 CrossRef.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS.
- H. Zhang, C. He, S. Han, Z. Du, L. Wang, Q. Yun, W. Cao, B. Zhang, Y.-H. Tian and Q. Lu, Chin. Chem. Lett., 2022, 33, 3641–3649 CrossRef CAS.
- D. Ma, C. Zhi, Y. Zhang, J. Chen, Y. Zhang and J.-W. Shi, ACS Nano, 2024, 18, 21714–21746 CrossRef CAS PubMed.
- Y. Ma, J. Yu, M. Sun, B. Chen, X. Zhou, C. Ye, Z. Guan, W. Guo, G. Wang, S. Lu, D. Xia, Y. Wang, Z. He, L. Zheng, Q. Yun, L. Wang, J. Zhou, P. Lu, J. Yin, Y. Zhao, Z. Luo, L. Zhai, L. Liao, Z. Zhu, R. Ye, Y. Chen, Y. Lu, S. Xi, B. Huang, C.-S. Lee and Z. Fan, Adv. Mater., 2022, 34, 2110607 CrossRef CAS.
- C. Peng, J. Ma, G. Luo, S. Yan, J. Zhang, Y. Chen, N. Chen, Z. Wang, W. Wei, T.-K. Sham, Y. Zheng, M. Kuang and G. Zheng, Angew. Chem., Int. Ed., 2024, 63, e202316907 CrossRef CAS PubMed.
- L. Li, Y. Zhang, X. Luo, I. Masood ul Hasan, K. Wu, B. Nan, Y. Zhang, N. Xu and J. Qiao, J. Energy Chem., 2023, 86, 569–578 CrossRef CAS.
- Y. Li, Y. Jin, X. Zong, X. Zhang, G. Li and Y. Xiong, J. Mater. Chem. A, 2023, 11, 11445–11453 RSC.
- K. H. Dinh, L. T. Menisa, H. Warkentin, T. N. Nguyen and C.-T. Dinh, ACS Catal., 2025, 15, 5731–5759 CrossRef.
- Q. Tang, Q. Hao, Q. Zhu, J. Wu, K. Huang, K. Liu and J. Lu, Adv. Energy Mater., 2025, 15, 2403778 CrossRef CAS.
- J. Chen, M. R. Ahasan, J.-S. Oh, J. A. Tan, S. Hennessey, M. M. Kaid, H. M. El-Kaderi, L. Zhou, K. U. Lao, R. Wang and W.-N. Wang, J. Mater. Chem. A, 2024, 12, 4601–4609 RSC.
- M. Wu, F. Dong, Y. Yang, X. Cui, X. Liu, Y. Zhu, D. Li, S. Omanovic, S. Sun and G. Zhang, Electrochem. Energy Rev., 2024, 7, 10 CrossRef CAS.
- J. Hu, M. Zhou, K. Li, A. Yao, Y. Wang, Q. Zhu, Y. Zhou, L. Huang, Y. Pei, Y. Du, S. Jin and M. Zhu, Small, 2023, 19, 2301357 CrossRef CAS PubMed.
- L. Huang, Z. Liu, G. Gao, C. Chen, Y. Xue, J. Zhao, Q. Lei, M. Jin, C. Zhu, Y. Han, J. S. Francisco and X. Lu, J. Am. Chem. Soc., 2023, 145, 26444–26451 CrossRef CAS PubMed.
- Z. Zhang, G. Wen, D. Luo, B. Ren, Y. Zhu, R. Gao, H. Dou, G. Sun, M. Feng, Z. Bai, A. Yu and Z. Chen, J. Am. Chem. Soc., 2021, 143, 6855–6864 CrossRef CAS PubMed.
- Y. Lu, H. Li, H. Sun, J. Zhao, Y. Zhang, Y. Wang, C. Zhu, D. Gao, Y. Tuo, J. Zeng, D. Chen and Z. Yan, ACS Catal., 2024, 14, 14744–14753 CrossRef CAS.
- H. Yu, W. Zhao, X. Dong, J. Wang, W. Wang, L.-L. Shen, G.-R. Zhang and D. Mei, Appl. Catal. B Environ. Energy, 2025, 363, 124805 CrossRef CAS.
- Y. Hu, D. Lu, W. Zhou, X. Wang and Y. Li, J. Mater. Chem. A, 2023, 11, 1937–1943 RSC.
- S. Yan, Z. Chen, Y. Chen, C. Peng, X. Ma, X. Lv, Z. Qiu, Y. Yang, Y. Yang, M. Kuang, X. Xu and G. Zheng, J. Am. Chem. Soc., 2023, 145, 26374–26382 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, P. Yang, X. Chang, H. Dong, A. Li, C. Hu, Z. Huang, Z.-J. Zhao and J. Gong, Small, 2018, 14, 1703314 CrossRef PubMed.
- X. Yao, D. Cui, C. Zhu, J. He, F. Meng, S. Yang, M. Dong, G. Shan, M. Zhang, C. Sun, X. Wang and Z. Su, ACS Mater. Lett., 2024, 6, 5112–5119 CrossRef CAS.
- H. Wang, G. Zhan, C. Tang, D. Yang, W. Liu, D. Wang, Y. Wu, H. Wang, K. Liu, J. Li, M. Huang and K. Chen, ACS Nano, 2023, 17, 4790–4799 CrossRef CAS PubMed.
- R. Nankya, Y. Xu, A. Elgazzar, P. Zhu, T.-U. Wi, C. Qiu, Y. Feng, F. Che and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202403671 CrossRef CAS PubMed.
- Q. Zhang, J. Peng, S. Jiang, H. Xiong, X. Fu, S. Shang, J. Xu, G. He and P.-C. Chen, Appl. Catal. B Environ. Energy, 2025, 371, 125255 CrossRef CAS.
- Z. Zhang, Q. Fang, X. Yang, S. Zuo, T. Cheng, Y. Yamauchi and J. Tang, Adv. Mater., 2025, 37, 2411498 CrossRef CAS PubMed.
- Z. Niu, X. Gao, S. Lou, N. Wen, J. Zhao, Z. Zhang, Z. Ding, R. Yuan, W. Dai and J. Long, ACS Catal., 2023, 13, 2998–3006 CrossRef CAS.
- R. Belgamwar, R. Verma, T. Das, S. Chakraborty, P. Sarawade and V. Polshettiwar, J. Am. Chem. Soc., 2023, 145, 8634–8646 CrossRef CAS PubMed.
- R. Chen, J. Zhao, X. Zhang, Q. Zhao, Y. Li, Y. Cui, M. Zhong, J. Wang, X. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2024, 146, 24368–24376 CrossRef CAS PubMed.
- M. Qi, Y. Ma, C. Zhang, B. Li, X. Yang, Z. Shi, S. Liu, C. An, J. Jiao and T. Lu, Sci. China Chem., 2025, 68, 1620–1626 CrossRef CAS.
- E. Landaeta, N. I. Kadosh and Z. D. Schultz, ACS Catal., 2023, 13, 1638–1648 CrossRef CAS.
- Y. Hu, D. Lu, W. Zhou, X. Wang and Y. Li, J. Mater. Chem. A, 2023, 11, 1937–1943 RSC.
- X. Dong, S. Li, C. Zhu, J. Mao, G. Wu, G. Li, G. Feng, A. Chen, Y. Wei, X. Liu, J. Wang, Y. Song, W. Chen and W. Wei, Appl. Catal. B Environ. Energy, 2023, 336, 122929 CrossRef CAS.
- H. Liu, C. Yang, T. Bian, H. Yu, Y. Zhou, Y. Zhang and L. Sun, Adv. Energy Mater., 2025, 2405658 CrossRef CAS.
- Y. Shao, Q. Yuan and J. Zhou, Small, 2023, 19, 2303446 CrossRef CAS PubMed.
- J. Zhao and S. Lin, J. Colloid Interface Sci., 2025, 680, 257–264 CrossRef CAS PubMed.
- H. Liu, C. Yang, T. Bian, H. Yu, Y. Zhou and Y. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202404123 CrossRef CAS PubMed.
- Q. Hao, H.-x. Zhong, J.-z. Wang, K.-h. Liu, J.-m. Yan, Z.-h. Ren, N. Zhou, X. Zhao, H. Zhang, D.-x. Liu, X. Liu, L.-w. Chen, J. Luo and X.-b. Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef CAS.
- X. Zhao, K. Zhao, Y. Liu, Y. Su, S. Chen, H. Yu and X. Quan, ACS Catal., 2022, 12, 11412 CrossRef CAS.
- L.-M. Cao, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Green Chem., 2018, 20, 798–803 RSC.
- Y.-N. Gong, C.-Y. Cao, W.-J. Shi, J.-H. Zhang, J.-H. Deng, T.-B. Lu and D.-C. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202215187 CrossRef CAS PubMed.
- X.-M. Liang, H.-J. Wang, C. Zhang, D.-C. Zhong and T.-B. Lu, Appl. Catal. B Environ. Energy, 2023, 322, 122073 CrossRef CAS.
- Z.-W. Chen, H.-J. Wang, C. Liu, X.-L. Lu and T.-B. Lu, Sci. China Chem., 2025, 68, 570–579 CrossRef CAS.
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K.-J. Noh, K.-S. Lee and J. W. Han, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS.
- S. Chen, X. Zheng, P. Zhu, Y. Li, Z. Zhuang, H. Wu, J. Zhu, C. Xiao, M. Chen, P. Wang, D. Wang and Y.-L. He, Angew. Chem., Int. Ed., 2024, 63, e202411591 CrossRef CAS PubMed.
- G. Yasin, A. Kumar, S. Ajmal, M. Asim Mushtaq, M. Tabish, A. Saad, M. A. Assiri, M. Tariq Nazir and Q. Zhuo, Coord. Chem. Rev., 2024, 501, 215589 CrossRef CAS.
- L. Zhang, J. Feng, L. Wu, X. Ma, X. Song, S. Jia, X. Tan, X. Jin, Q. Zhu, X. Kang, J. Ma, Q. Qian, L. Zheng, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 21945–21954 CrossRef CAS.
- J.-d. Yi, X. Gao, H. Zhou, W. Chen and Y. Wu, Angew. Chem., Int. Ed., 2022, 61, e202212329 CrossRef CAS PubMed.
- Z. Guo, H. Zhu, G. Yang, A. Wu, Q. Chen, Z. Yan, K. Loon Fow, H. Do, J. D. Hirst, T. Wu and M. Xu, Chem. Eng. J., 2023, 476, 146556 CrossRef CAS.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed.
- H. Zhang, X. Wang, Y. Sun, X. Wang, Z. Tang, S. Li, X. Gao, J. Wang, Z. Hou, K. Nie, J. Xie, Z. Yang and Y.-M. Yan, Appl. Catal. B Environ. Energy, 2024, 351, 123992 CrossRef CAS.
- T. Zhang, B. Yuan, W. Wang, J. He and X. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202302096 CrossRef CAS.
- L. Yin, Z. Li, J. Feng, P. Zhou, L. Qiao, D. Liu, Z. Yi, W. F. Ip, G. Luo and H. Pan, ACS Appl. Mater. Interfaces, 2023, 15, 47135–47144 CrossRef CAS PubMed.
- M. Wang, H. Chen, M. Wang, J. Wang, Y. Tuo, W. Li, S. Zhou, L. Kong, G. Liu, L. Jiang and G. Wang, Angew. Chem., Int. Ed., 2023, 62, e202306456 CrossRef CAS.
- Y. Cao, S. Chen, S. Bo, W. Fan, J. Li, C. Jia, Z. Zhou, Q. Liu, L. Zheng and F. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303048 CrossRef CAS PubMed.
- L. Chen, J. Chen, W. Fu, J. Chen, D. Wang, Y. Xiao, S. Xi, Y. Ji and L. Wang, Nat. Commun., 2024, 15, 7053 CrossRef CAS PubMed.
- A. Saxena, S. Kapila, J. E. Medvedeva and M. Nath, ACS Appl. Mater. Interfaces, 2023, 15, 14433–14446 CAS.
- F. Chang, K. Zhu, C. Liu, J. Wei, S. Yang, Q. Zhang, L. Yang, X. Wang and Z. Bai, Adv. Funct. Mater., 2024, 34, 2400893 CrossRef CAS.
- R. Yun, F. Zhan, X. Wang, B. Zhang, T. Sheng, Z. Xin, J. Mao, S. Liu and B. Zheng, Small, 2021, 17, 2006951 CrossRef CAS.
- Y. Jiang, C. Lv, B. Lu, Y. Song, T. Liu, X. Zhang, D. Gao, K. Ye and G. Wang, ACS Nano, 2025, 19, 11263–11272 CrossRef CAS.
- S. Shen, X. Peng, L. Song, Y. Qiu, C. Li, L. Zhuo, J. He, J. Ren, X. Liu and J. Luo, Small, 2019, 15, 1902229 CrossRef.
- P. Luan, X. Dong, L. Liu, J. Xiao, P. Zhang, J. Zhang, H. Chi, Q. Wang, C. Ding, R. Li and C. Li, ACS Catal., 2024, 14, 8776–8785 CrossRef CAS.
- M. Chhetri, M. Wan, Z. Jin, J. Yeager, C. Sandor, C. Rapp, H. Wang, S. Lee, C. J. Bodenschatz, M. J. Zachman, F. Che and M. Yang, Nat. Commun., 2023, 14, 3075 CrossRef CAS PubMed.
- Y. Zheng, J. Zhang, Z. Ma, G. Zhang, H. Zhang, X. Fu, Y. Ma, F. Liu, M. Liu and H. Huang, Small, 2022, 18, 2201695 CrossRef CAS PubMed.
- X. Li, M. Qin, X. Wu, X. Lv, J. Wang, Y. Wang and H. B. Wu, Small, 2023, 19, 2302530 CrossRef CAS PubMed.
- Z. Wei, S. Yue, S. Gao, M. Cao and R. Cao, Nano Res., 2023, 16, 7777–7783 CrossRef CAS.
- T. Wan, C. Lv, K. Ye, M. Ma, D. Hu, J. Xiao and W. Xiao, Small Methods, 2025, 2500005 CrossRef PubMed.
- X. Liu, T. Liu, T. Ouyang, J. Deng and Z.-Q. Liu, Angew. Chem., Int. Ed., 2025, 64, e202419796 CrossRef CAS PubMed.
- H. Zhang, X. Wang, Y. Sun, X. Wang, Z. Tang, S. Li, X. Gao, J. Wang, Z. Hou, K. Nie, J. Xie, Z. Yang and Y.-M. Yan, Appl. Catal. B Environ. Energy, 2024, 351, 123992 CrossRef CAS.
- Y. Xiao, F. Yu, C. Xia, D. Zhu, J. Chen, N. Liu, Y. Zhao, R. Qi, W. Guo, B. You, T. Yao, Y. Pang, Z. Wang, H. Wang, F. Song and B. Y. Xia, J. Am. Chem. Soc., 2025, 147, 15654–15665 CrossRef CAS PubMed.
- Z. Guo, H. Zhu, Z. Yan, L. Lei, D. Wang, Z. Xi, Y. Lian, J. Yu, K. L. Fow, H. Do, J. D. Hirst, T. Wu and M. Xu, Appl. Catal. B Environ. Energy, 2025, 364, 124839 CrossRef CAS.
- Z. Yang, D. Ji, Z. Li, Z. He, Y. Hu, J. Yin, Y. Hou, P. Xi and C.-H. Yan, Small, 2023, 19, 2303099 CrossRef CAS.
- J. Feng, L. Wu, S. Liu, L. Xu, X. Song, L. Zhang, Q. Zhu, X. Kang, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS.
- H. Wang, H. Zhang, Y. Huang, H. Wang, A. Ozden, K. Yao, H. Li, Q. Guo, Y. Liu, A. Vomiero, Y. Wang, Z. Qian, J. Li, Z. Wang, X. Sun and H. Liang, ACS Nano, 2023, 17, 346–354 CrossRef CAS.
- J. Liu, P. Li, S. Jia, Y. Wang, L. Jing, Z. Liu, J. Zhang, Q. Qian, X. Kang, X. Sun, Q. Zhu and B. Han, Nat. Synth., 2025 DOI:10.1038/s44160-025-00752-4.
- J. Feng, L. Wu, X. Song, L. Zhang, S. Jia, X. Ma, X. Tan, X. Kang, Q. Zhu, X. Sun and B. Han, Nat. Commun., 2024, 15, 4821 CrossRef CAS PubMed.
- X. Zhou, j. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |