
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntermediate knowledge enhanced the performance of the amide coupling yield prediction model†
Chonghuan
Zhang‡
ab,
Qianghua
Lin‡
b,
Chenxi
Yang
c,
Yaxian
Kong
c,
Zhunzhun
Yu
*b and
Kuangbiao
Liao
 *ab
*ab
aGuangzhou Municipal and Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, The NMPA and State Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences, Guangzhou Laboratory, Guangzhou Medical University, Guangzhou, Guangdong, PR China 511436. E-mail: yu_zhunzhun@gzlab.ac.cn; liao_kuangbiao@gzlab.ac.cn
bGuangzhou National Laboratory, No. 9 Xingdaohuanbei Road, Guangzhou International Bio Island, Guangzhou, Guangdong, PR China 510005
cAIChemEco Inc., Guangzhou, Guangdong, PR China 510005
First published on 5th June 2025
Abstract
Amide coupling is an important reaction widely applied in medicinal chemistry. However, condition recommendation remains a challenging issue due to the broad condition space. Recently, accurate condition recommendation via machine learning has emerged as a novel and efficient method to find suitable conditions to achieve the desired transformations. Nonetheless, accurately predicting yields is challenging due to the complex relationships involved. Herein, we present our strategy to address this problem. Two steps were taken to ensure the quality of the dataset. First, we selected a diverse and representative set of substrates to capture a broad spectrum of substrate structures and reaction conditions using an unbiased machine-based sampling approach. Second, experiments were conducted using our in-house high-throughput experimentation (HTE) platform to minimize the influence of human factors. Additionally, we proposed an intermediate knowledge-embedded strategy to enhance the model's robustness. The performance of the model was first evaluated at three different levels—random split, partial substrate novelty, and full substrate novelty. All model metrics in these cases improved dramatically, achieving an R2 of 0.89, MAE of 6.1%, and RMSE of 8.0% in the full substrate novelty test dataset. Moreover, the generalization of our strategy was assessed using external datasets from reported literature, delivering an R2 of 0.71, MAE of 7%, and RMSE of 10%. Meanwhile, the model could recommend suitable conditions for some reactions to elevate the reaction yields. Besides, the model was able to identify which reaction in a reaction pair with a reactivity cliff had a higher yield. In summary, our research demonstrated the feasibility of achieving accurate yield predictions through the combination of HTE and embedding intermediate knowledge into the model. This approach also has the potential to facilitate other related machine learning tasks.
Introduction
The amide coupling reaction is one of the most critical transformations in drug discovery,1,2 playing a pivotal role in the synthesis of numerous pharmaceutical compounds. Its significance in pharmaceutical chemistry is underscored by surveys indicating that amide coupling reactions frequently dominate among all reaction types.3 Despite the development of over 200 activators for amide coupling reactions,4 only a few are commonly used in large-scale amide synthesis due to their proven reliability, affordability, ease of handling, and other favorable attributes.5 However, when these coupling reagents are applied to challenging target substrates, it remains difficult to predict their effectiveness—particularly in couplings between acids and aromatic amines with weak nucleophilicity. The deactivation of aromatic amines by the aromatic system, combined with steric hindrance and electronic demands imposed by various substituents, complicates the selection of efficient coupling reagents. Furthermore, other aspects of reaction conditions, such as the choice of base and solvent, can also impact reactivity. Therefore, there is a pressing need for a strategy that can quickly explore chemical space and identify suitable conditions for amide coupling of challenging substrates, without resorting to time-consuming and labor-intensive experimental screening processes.To address these challenges, researchers have increasingly turned to machine learning (ML) models to enhance reaction yield predictions and streamline the selection of optimal reaction conditions. Recent advancements highlight the transformative impact of ML in this domain.6–9 Notably, Doyle et al. pioneered the use of Random Forest algorithms in predicting yields for C–N cross-coupling reactions, showcasing ML's capability to significantly reduce experimental workloads and expedite the discovery process.10 Building on this foundation, the Sigman and Denmark groups have extended these predictive models to encompass broader reaction scopes, thereby increasing their robustness and applicability across diverse chemical landscapes.11,12 These efforts represent a substantial leap forward in the practical deployment of ML, moving beyond proof-of-concept to tools that offer real-world utility. Schwaller et al. introduced an approach using Bidirectional Encoder Representations from Transformers (BERT) neural networks13 to predict yield based on textual descriptions of chemical reactions, which leverages natural language processing (NLP) algorithms to interpret reaction transformation outcomes.14
The first amide coupling reaction yield prediction model by Isayev et al. utilized literature-based reactions curated from Reaxys15 to build predictive models, but highlighted the inherent difficulties of using such data. Literature reactions often suffer from inconsistencies in reporting, variability in experimental conditions, and a lack of comprehensive datasets, making it challenging to build robust and generalizable models.16 Additionally, literature sources typically report only successful reactions with high yields, neglecting low-yield and negative data that are crucial for creating well-distributed and accurate predictive models.17 It is essential to curate relevant datasets for model development and to identify and control factors that complicate yield prediction. The variability in data sources, reaction scales, and structural diversity reported in the literature further complicates the development of reliable models.18 Sigman et al. explored a related challenge in predicting reaction rates for amide coupling using linear free-energy relationships, emphasizing the importance of understanding underlying reactivity trends. Although their work focused on rates rather than yields, the overarching goal of providing actionable insights for the synthetic community is similar to our presented work.19 Very recently, Doyle et al. further explored the optimization of reaction conditions through the bandit optimization technique to efficiently navigate the vast chemical space, balancing the exploration of new conditions with the exploitation of known successful ones.20 This approach is particularly powerful for the case study amide coupling reaction, where multiple variables—such as reagent, solvent, and temperature—must be optimized simultaneously. By using bandit optimization, the researchers were able to significantly reduce the number of experimental trials needed to identify optimal conditions, showcasing yet another example of how ML can accelerate and enhance the process of chemical discovery. However, for new substrate pairs, we were unable to directly obtain yields under different conditions and had to perform the reactions following the corresponding workflow, which differs from the role of a yield prediction model in recommending conditions.
High-throughput experimentation (HTE)21 has emerged as a powerful alternative to traditional literature-based approaches for building reaction yield prediction models.22–24 HTE techniques generate large datasets through automated, parallelized experiments, offering a more consistent, comprehensive, and controlled data source with a broader range of reaction conditions, including low-yield and negative outcomes. This systematic approach helps in developing more robust and generalizable models. With our in-house automated HTE platform, we have successfully optimized reaction conditions, explored the reaction space, and collected standardized experimental data for machine learning studies, resulting in a series of related publications.25–31
Despite these advantages, many HTE-based models achieve high accuracy but are limited to a narrow range of substrate and reaction condition spaces. This limitation has been well documented by several research groups, including those led by Doyle, Sigman, and Denmark.6,32 Additionally, a common issue with these models is the evaluation methodology. Often, models are tested using data splits that include test substrates seen by the model in the training set, resulting in overly optimistic performance metrics. However, when evaluated using a strict test set—where the model must predict yields for entirely new combinations of substrates—the performance typically drops. This strict testing better reflects real-world applications where chemists need to predict reaction yields for novel substrate pairs. Therefore, creating a reaction dataset with diversified substrates and conditions, implementing rigorous testing protocols and curating relevant datasets are crucial for developing reliable and accurate predictive models. This issue has been widely recognized in the literature, with prior studies highlighting the pitfalls of inadequate dataset partitioning.6,32 Recent discussions in the chemical engineering field further emphasize the necessity of rigorous testing protocols to avoid overfitting and ensure model generalizability.33 The flaws in evaluation methodologies have been acknowledged in various domains, including C–N coupling yield predictions, where flaws in dataset partitioning have been publicly debated and addressed.10,34,35
In this context, we aim to build a high-quality dataset on amide coupling and develop a high-performance yield prediction model that can accurately recommend optimal conditions for novel substrate pairs in the training dataset. In this work, we first demonstrate our efforts to prepare the dataset. We selected substrate pairs according to structures reported in the USPTO reaction dataset36 and a virtual commercial available space to ensure potential application and structural diversity. Our method employs a machine-based sampling approach to systematically explore the chemical space of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 virtual compounds, complemented by a small number of manually selected substrates to ensure diversity and practicality. Second, our in-house HTE platform was utilized to collect data, incorporating control strategies including duplicate conditions to detect variability, repeating selected plates for consistency checks, and employing internal standards for accurate yield measurement to improve reproducibility. With the dataset in hand, we then focused on developing a robust prediction model. Given the challenge to develop a robust model under 95 conditions, we transformed our goal into an iterative prediction task across the list of 95 conditions. Meanwhile, intermediate knowledge was embedded into the model to enhance its performance. The distinguishing feature of this strategy is that the model does not need to learn the relationships among different conditions, while still retaining condition information, thereby providing better performance with high probability. The results of a series of studies revealed that the generalization ability of the model could be significantly improved after applying this strategy (Fig. 1). The model had a good performance toward a fully unseen test set from the literature, achieving an R2 of 0.71, MAE of 7%, and RMSE of 10%. Meanwhile, the model could recommend more suitable conditions for some reactions with low yield, indicating the potential application of our work. Additionally, our strategy achieved satisfactory prediction results for reaction pairs with reactivity cliffs, delivering an accuracy of 0.73 in binary classification.
000 virtual compounds, complemented by a small number of manually selected substrates to ensure diversity and practicality. Second, our in-house HTE platform was utilized to collect data, incorporating control strategies including duplicate conditions to detect variability, repeating selected plates for consistency checks, and employing internal standards for accurate yield measurement to improve reproducibility. With the dataset in hand, we then focused on developing a robust prediction model. Given the challenge to develop a robust model under 95 conditions, we transformed our goal into an iterative prediction task across the list of 95 conditions. Meanwhile, intermediate knowledge was embedded into the model to enhance its performance. The distinguishing feature of this strategy is that the model does not need to learn the relationships among different conditions, while still retaining condition information, thereby providing better performance with high probability. The results of a series of studies revealed that the generalization ability of the model could be significantly improved after applying this strategy (Fig. 1). The model had a good performance toward a fully unseen test set from the literature, achieving an R2 of 0.71, MAE of 7%, and RMSE of 10%. Meanwhile, the model could recommend more suitable conditions for some reactions with low yield, indicating the potential application of our work. Additionally, our strategy achieved satisfactory prediction results for reaction pairs with reactivity cliffs, delivering an accuracy of 0.73 in binary classification.
 | ||
| Fig. 1 Studies on condition recommendation for amide coupling. | ||
Results and discussion
HTE substrate selection
The selection of substrates for HTE in amide coupling reactions was guided by a systematic approach that integrates virtual compound generation, dimensionality reduction, clustering, virtual compound filtering and stratified sampling, as described below.Virtual compound generation
The substrates include one amine and one acid, forming a product whose chemical structure allows for back-tracing the corresponding amine and acid. Therefore, we focused on selecting substrates within the chemical space of the virtual products, which inherently includes both amines and acids.We first used the USPTO 50k reaction dataset,36 encompassing 50000 synthetic reactions derived from published US patents, to compile a comprehensive dataset focusing on amide coupling reactions. To achieve this, as shown in Fig. 2a, we first composed a reaction template in SMiles ARbitrary Target Specification (SMARTS) syntax,37 following the general equation of the amide coupling reaction. We used RDKit38 to filter amide coupling reactions from the USPTO dataset, identifying 11663 entries of amide coupling reactions.
 | ||
| Fig. 2 (a) General equation and SMILES template of the amide coupling reaction. (b) The chemical space of USPTO amide coupling products, products from commercially available substrates and products of self-developed HTE reactions reduced from PCA. | ||
The product SMILES strings were then converted into extended connectivity fingerprints (1024 bit ECFP)39 with a radius of 2, serving as numerical representations of the molecular structures. To manage the high-dimensional nature of the Morgan fingerprints and facilitate analysis, we employed Principal Component Analysis (PCA),40 an unsupervised learning technique, to reduce the dimensionality of the data while preserving the variance inherent in the molecular descriptors. The data were reduced to a two-dimensional space primarily for visualization purposes and to observe the overall distribution of amide coupling reactions within the chemical space. It is important to note that this dimensionality reduction was not used for clustering but rather for visualization. While PCA was chosen for this purpose, other dimensionality reduction techniques, such as t-distributed stochastic neighbor embedding (t-SNE)41 or uniform manifold approximation and projection (UMAP),42 which were often better at preserving distances in high-dimensional spaces20,43,44 were also attempted as alternatives for PCA. However, t-SNE and UMAP visualizations both exhibit a globular structure, which indicates that neither t-SNE nor UMAP can provide a uniform distribution like PCA does, as shown in Fig. S19 and S20 of the ESI.†
The derived USPTO amide coupling dataset is notable for its open-source availability, enabling reproducibility by others. We focus on the biological activity and practical applications of the product space, as shown by the grey scatters in Fig. 2b, underscoring their potential significance in various fields. However, given that many USPTO substrates are derived from patented molecules, synthesizing them can be difficult. We could only use the USPTO space to calibrate the size of the virtual product space. To develop a virtual product space for the purpose of strategic HTE substrate selection, we focused on buyable substrates. We identified amines and carboxylic acids from our in-house commercial molecule database, curated from various chemical providers. Due to the reliance on DMF as the solvent in our HTE workflow, substrates with poor solubility in DMF were systematically excluded from the dataset. While this exclusion ensures experimental feasibility, it may limit the chemical diversity of certain subclasses of acids or amines. A virtual product space of 71000 products was developed. The yellow, red and green scatters in Fig. 2b cover most of the virtual product space, with products outside the edge of the USPTO space excluded, as described in the “Virtual compound filtering” section.
Clustering
The reduced-dimension data (including the USPTO products and the virtual products) were clustered using K-means clustering,45 resulting in 10 clusters. This partitioning of the dataset was essential for capturing the structural diversity of the virtual amide products. The choice of 10 clusters was based on the need to balance the structural differences within the dataset while maintaining an appropriate cluster size that avoids both oversimplification and overfitting.Virtual compound filtering
To further refine the virtual compound library, we filtered out virtual compounds whose distance from the cluster centers exceeded the maximum distance of 1.37, the maximum edge of the USPTO product space. This filtering step ensured that the remaining virtual compounds were structurally similar to the USPTO chemical space, thereby removing outliers and compounds far removed from the core chemical space of interest.Stratified sampling
We applied a stratified sampling approach within each cluster to capture a wide range of compounds with varying distances from the cluster centers. The compounds in each cluster were divided into 10 strata based on their distance from the cluster center. This stratification allowed us to account for internal variability within each cluster by sampling both near-center and far-center compounds. A random selection was made from each stratum, ensuring that the entire chemical space within each cluster was adequately represented.The use of 10 strata provided sufficient granularity to ensure a comprehensive representation of chemical diversity while avoiding selection bias. This stratified approach also helped ensure that compounds selected for the final HTE library were diverse in their chemical features. Some of the sampled products were discarded due to their high price and complex structures, which were not suitable for quantitative analysis via NMR. The distribution of these discarded virtual products was illustrated by yellow scatters in Fig. 2b.
Using this approach, we selected 447 HTE products from the virtual product space, as shown in the “sampling-selected HTE products” of Fig. 2b. Glorius et al.17 emphasized that robust models generally require a dataset comprising at least 500 substrate combinations and sufficient diversity. We also manually supplemented 186 products with additional compounds that were cost-effective and structurally appropriate, as shown in the “expert-selected HTE products” of Fig. 2b. The expert-selected HTE products cover some extent of the space that was not addressed by the machine-selected region. Eventually, this yields a final dataset of 632 products corresponding to 632 unique substrate pairs, which include a total of 70 amines and 66 acids. This final selection of a subset of the original virtual compound space was designed to capture the chemical diversity from the entire virtual library. Our approach aimed to enhance representativeness while ensuring practicality for high-throughput experimentation.
The comparison of this chemical space indicates that our self-developed HTE reactions encompass a breadth of chemical coverage comparable to that of the virtual space, which is recognized for its extensive coverage in reaction modeling. Although it does not capture the entire chemical space historically explored by chemists,46 it represents a robust and comprehensive starting point due to its open-access nature. This facilitates the replication of our study and underscores that our strategies to select substrates are effectively aligned with ensuring practical applications, which would benefit the development of a robust model.
HTE condition selection
According to the results of previous related studies,25,26,28,31 we preparerd 95 different conditions for HTE. The details of the 95 conditions are shown in Table S1 of the ESI.† It should be noted that all commercial coupling reagents were involved in the condition set except for acyl chloride, because it is not compatible with DMF solvent. With the condition–substrate pairs in hand, we performed the HTE to collect reaction yield data. Our HTE workflow comprises several key steps: experiment design, high-throughput experimental preparation, high-throughput reaction setup, high-throughput reaction work-up, and high-throughput detection and analysis. The experimental design was tailored for HTE, with careful consideration of well placement to minimize variability. Following this, automated systems were used for the precise preparation and setup of a large number of reactions in parallel. Post-reaction, automated work-up processes were employed to quench reactions and prepare samples for analysis. Finally, high-throughput detection methods, including UPLC, MS, and NMR, were utilized to analyze reaction outcomes efficiently. Detailed protocols and specific conditions for each step of the HTE process are provided in Section S2 of the ESI.† As a result, more than 47000 yield data were collected, excluding those discarded data, where overlapping of chromatography peaks and difficulty in NMR analysis usually result in the failure to obtain the corresponding yield data. This dataset was designed to be rich and diverse, providing a robust foundation for training machine learning models. The aim was to ensure that our HTE data would be sufficiently comprehensive to enable the models to understand and predict the yields of amide coupling reactions across the entire chemical space covered by our study.
Multi-condition model development and assessment
In this section, we describe the development and assessment of our multi-condition models for predicting amide coupling reaction yields. As shown in Fig. 3, we employed several machine learning algorithms, including XGBoost,47 Support Vector Machine (SVM),48 Random Forest,49 and AutoGluon,50 utilizing 1024-bit ECFP fingerprints as descriptors.39 Additionally, we explored the use of advanced deep learning textual methods such as Yield-BERT14 and T5-Chem,51 which leverage reaction SMILES strings for yield prediction. | ||
| Fig. 3 Schematics of the multi-conditional model workflow, using methylamine reacting with acetic acid as an example. | ||
First, we generated 1024-bit ECFP descriptors from the SMILES strings of reactants and products. These ECFP descriptors, with 1024 bits and a radius of 2, capture the structural features of the molecules involved in the reactions. These fingerprints of the substrates and product of a reaction were concatenated to create a vector of 3072 or point-wise added to keep a size of 1024, as the reaction fingerprint. Each of the different reaction conditions was encoded in a unique integer (1–95). We then used these descriptors to train the XGBoost,47 SVM,48 Random Forest,49 and AutoGluon50 models. Each model was fine-tuned to optimize hyperparameters, ensuring the best performance for yield prediction. AutoGluon, a robust ensemble model, combines the strengths of various machine learning algorithms to improve predictive performance and model robustness.52Fig. 3 uses the modelling workflow of AutoGluon to exemplify the machine learning approaches.
In parallel, we implemented deep learning Yield-BERT14 and T5-Chem51 models. Yield-BERT, based on the BERT architecture13 was trained on reaction SMILES to predict yields by understanding the sequence-to-sequence relationships within the reaction data. Similarly, T5-Chem, a variant of the T5 transformer model,53 was also trained to capture contextual information from reaction SMILES strings, enabling it to predict yields by considering the entire reaction context. To do this, we first tokenized the reaction SMILES strings, converting them into a sequence of tokens that represent the individual components of the molecules. These tokens were then fed through the transformer processes, which include multiple layers of attention mechanisms and feed-forward neural networks. Next, we incorporated the categorical features of reaction conditions into the model. These categorical features were combined with the text features output by the transformer through a combining module. This module integrated the encoded textual information from the SMILES strings with the reaction condition data. Finally, the combined features were passed through fully connected layers to predict the reaction yield. These layers consisted of several dense neural network layers that progressively refined the combined features into a single output value representing the predicted yield. In the above methods, we tried using fingerprints or reaction SMILES to represent the reaction conditions, but these did not represent the conditions well. Since we do not intend to predict outside these conditions, we opted for a categorical encoding approach to maintain clarity and consistency.
For the assessment of HTE-based reaction models, it is important to evaluate the model's performance in a way that reflects real-world applications. While the conventional approach involves using a random split to build a test dataset, recent studies have highlighted that this method can result in overly optimistic performance metrics and may not accurately reflect the challenges faced by chemists in practice. For instance, Doyle and co-workers have argued that random splits allow models to benefit from familiar substrate combinations that might appear in both training and test sets, thereby inflating performance metrics.6 Similarly, Denmark et al. emphasized that random splitting does not represent real-world scenarios where chemists often encounter novel substrate pairs, leading to a significant drop in model performance when faced with external validation.12,32 These findings underscore the importance of more stringent evaluation methods, such as using partially and fully external test sets.
In line with these recommendations, our study adopted a more rigorous assessment strategy by developing three levels of test sets, as shown in Fig. 4. The “random split” involves randomly dividing the dataset into training and test sets, and while it ensures exposure to a broad range of substrates and conditions, it may still present overly optimistic results. To address this, we also created a “partial substrate novelty” test set, which excludes any test cases where both substrates were seen during training. This approach ensures that at least one novel substrate is present, offering a more challenging and realistic evaluation of the model's predictive capabilities. Finally, the “full substrate novelty” test set consists entirely of new substrate combinations that the model has not encountered during training, providing the most rigorous assessment of its generalizability. These three levels of testing—random split, partial substrate novelty, and full substrate novelty—offer a comprehensive framework to evaluate the model's robustness and applicability in real-world chemical spaces, aligning with the best practices recommended in recent literature.
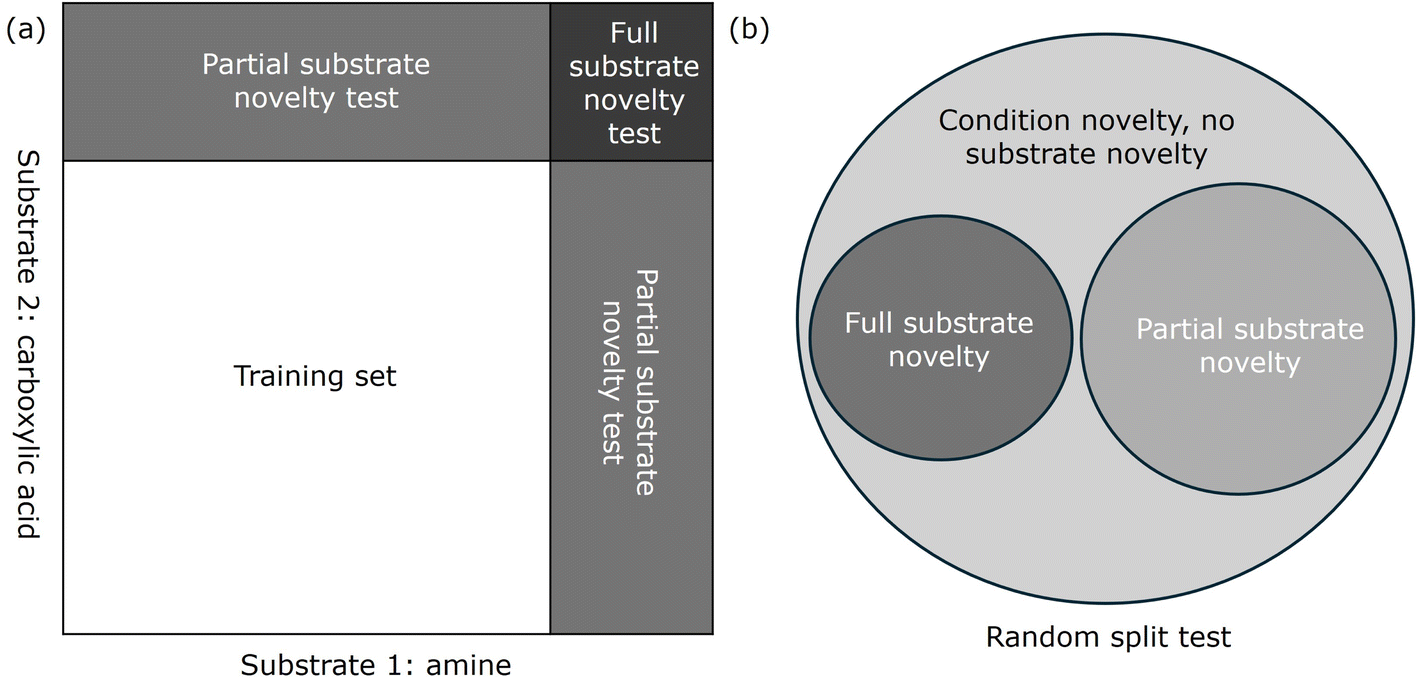 | ||
| Fig. 4 Schematics of three levels of test sets – random split, partial substrate novelty, and full novelty test: (a) split of training and test sets in the dimension of two substrates and (b) Venn diagram of the three levels of test sets. | ||
The results indicate that models performed better on the random split and partial substrate novelty test sets compared to the full substrate novelty test set. This could be inferred from the lower MAE and RMSE values and higher R2 values for the first two splits (shown in Table 1). These findings align with our expectations that models trained on datasets where they are exposed to a broad range of substrates and reaction conditions perform better on familiar substrates, but their performance drops when predicting yields for entirely new substrate pairs.
| Test data split | Metrics | XGBoost | SVM | RF | AutoGluon | Yield-BERT | T5-Chem |
|---|---|---|---|---|---|---|---|
| Random split | R 2 | 0.32 | 0.25 | 0.35 | 0.55 | 0.66 | 0.53 |
| MAE | 18% | 19% | 17% | 15% | 10% | 16% | |
| RMSE | 22% | 23% | 21% | 20% | 15% | 22% | |
| Partial substrate novelty | R 2 | 0.26 | 0.23 | 0.26 | 0.66 | 0.68 | 0.58 |
| MAE | 4% | 16% | 14% | 13% | 14% | 20% | |
| RMSE | 20% | 21% | 19% | 18% | 10% | 15% | |
| Full substrate novelty | R 2 | 0.25 | 0.22 | 0.26 | 0.42 | 0.63 | 0.58 |
| MAE | 20% | 22% | 19% | 17% | 15% | 22% | |
| RMSE | 24% | 27% | 23% | 22% | 11% | 17% |
In the random split test dataset, AutoGluon and Yield-BERT achieved the best results, with R2 values of 0.55 and 0.66, respectively. These models outperformed SVM, Random Forest, T5-Chem, and XGBoost in terms of predictive accuracy. Notably, Yield-BERT consistently demonstrated strong performance, retaining a relatively high R2 of 0.63 even on the full substrate novelty dataset, followed by T5-Chem with an R2 of 0.58 and AutoGluon at 0.42. This suggests that transformer-based models such as Yield-BERT and T5-Chem, along with ensemble methods such as AutoGluon, exhibit greater robustness and generalizability across varying substrate combinations. To further assess the novelty of the test sets, we quantified molecular similarity using the Tanimoto coefficient. A pairwise comparison of product molecules in the training and random split test datasets yielded an average similarity of 0.20, indicating considerable structural diversity between the two sets. Full details of this calculation are provided in Section S3.7 of the ESI.†
Selected condition model embedded with intermediate knowledge
The results from models trained under 95 different conditions demonstrated their reliability in accurately recommending conditions to facilitate reactions proceeding at an acceptable yield, especially as model performance improved further. Given the complex structure–yield relationship (SYR) and the cost of data collection via HTE, we decided not to generate more data to enhance model performance. Inspired by the concepts of knowledge embedding,54 we proposed achieving our goal through model development embedded with intermediate knowledge and selected six conditions to exemplify the method, as presented below.We could transform the yield prediction under multiple conditions into an iterative prediction within a condition list, a method we termed selected condition model prediction. In this approach, all reaction data within a single model were generated under the same set of conditions, thus eliminating reaction contexts such as condensation reagents, catalysts, bases, and solvents. This allowed the model to focus solely on the relationship between substrates and products, leading to improved learning and predictive accuracy. However, a significant challenge with this method is the potential loss of critical reaction condition information. Since reaction conditions play a crucial role in determining the outcome of chemical reactions, ignoring them can lead to incomplete models that do not accurately reflect real-world scenarios. To address this issue, we incorporated intermediate information based on reaction mechanisms into our model.
To evaluate our concept, we chose six different conditions with various coupling reagents that are frequently used in the literature and have well-defined intermediates. For the condition selection, we performed a statistical analysis of the literature-reported amide coupling reactions curated from Reaxys.15 We identified the 25 most frequently used conditions, as shown in Table S11 of the ESI,† and selected six for model development and assessment, as shown in Table 2. These conditions were chosen based on their prevalence in the dataset, ensuring that our HTE conditions were both representative and relevant to a wide range of amide coupling reactions. In our investigation of selected condition models, we followed the meticulous approach in preparing our dataset, ensuring it mirrored the model's rigor through three distinct datasets: random split, partial substrate novelty, and full substrate novelty.
| Condition # | Activation reagent | Additive | Base | Solvent |
|---|---|---|---|---|
| 1 | HATU | DIPEA | DMF | |
| 6 | TBTU | DIPEA | DMF | |
| 13 | EDC.HCl | HOBT | DIPEA | DMF |
| 21 | HBTU | DIPEA | DMF | |
| 34 | PyBOP | DIPEA | DMF | |
| 79 | DCC | HOBT | DMF |
To simplify the complexity of the reaction system in multi-condition amide coupling reaction modeling, we developed multiple single-condition models for the selected conditions by removing condition variables as mentioned above. Meanwhile, we incorporated intermediate information based on reaction mechanisms by using reaction SMARTS templates to represent the formation of activated acid intermediates. For example, in the presence of HATU as a condensation reagent, the transformation of an acid to its activated intermediate was represented using the following template shown in Fig. 5(a).
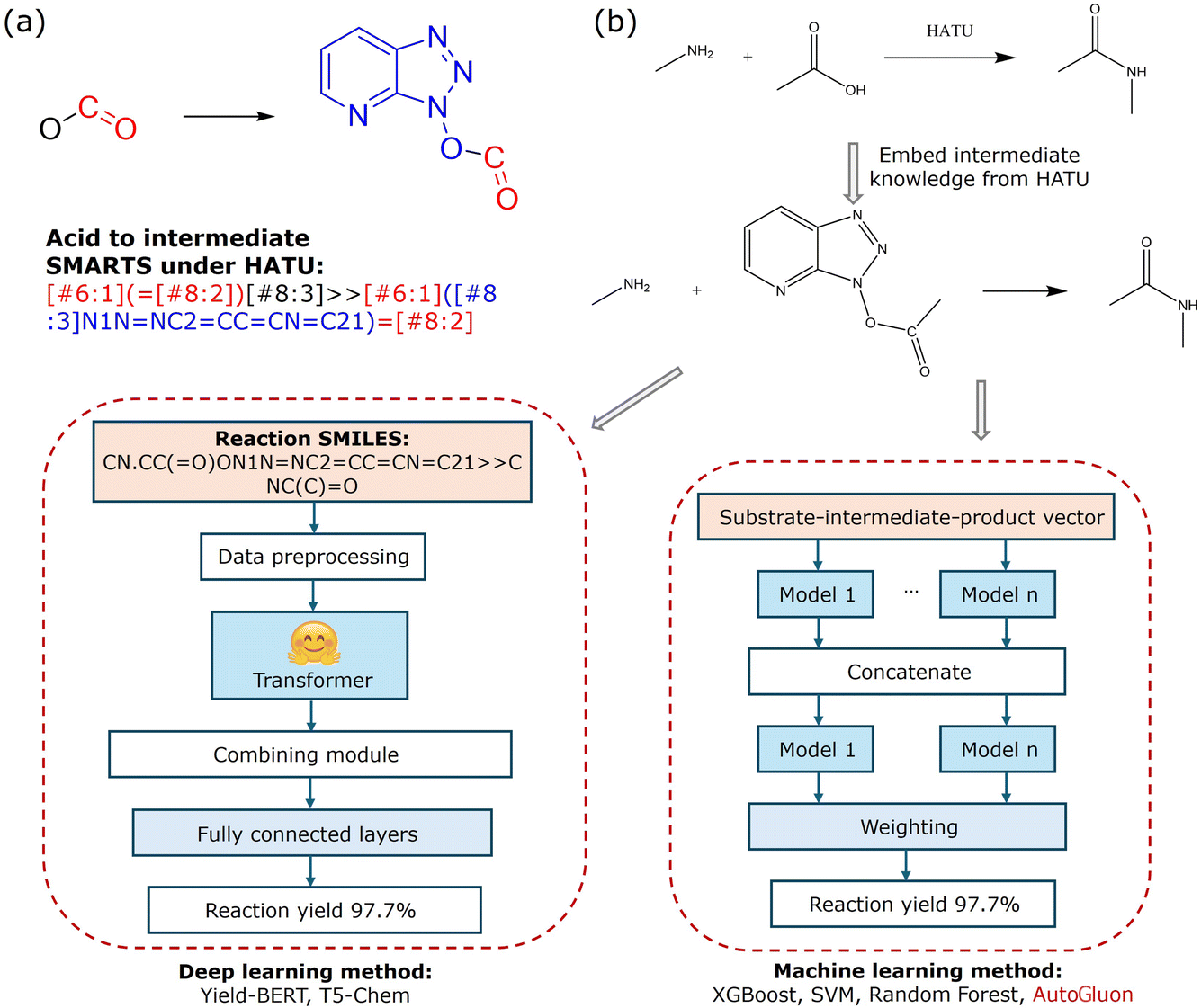 | ||
| Fig. 5 (a) Transformation of an acid into an intermediate SMARTS pattern using HATU as the activation reagent, and (b) schematics of the selected single-conditional model workflow. | ||
This template converts the acid into the activated acid. We applied specific SMARTS templates for all six conditions, which are detailed in the code repository. Next, we added the intermediate information into the reaction contexts, allowing the model to learn the effect of intermediates on the reaction outcome. To generate descriptors, we experimented with three approaches for generating the reaction context for the selected condition model, using the following patterns:
(1) No intermediate.
(2) Amine + acid + intermediate → amide.
(3) Amine + intermediate → amide.
As shown in Fig. 5(b), the reaction contexts were vectorized into ECFP fingerprints and also converted into reaction SMILES, effectively capturing the structural features of the reactants and products, along with crucial intermediate information. This approach ensured that the model considered the essential reaction conditions indirectly through the intermediate representation. The ECFP fingerprints and reaction SMILES were then used to train the selected condition models using similar machine learning and deep learning algorithms, respectively, as those employed for multi-condition models (Fig. 3). However, in this case, the reaction conditions were no longer concatenated with the reaction vector. We employed the same rigorous testing protocols as used for the multi-condition models, evaluating performance across random split, partial substrate novelty, and full substrate novelty datasets.
Our results reveal that the BERT model trained on the random split dataset usually delivered superior performance, characterized by lower Mean Absolute Error (MAE) and Mean Squared Error (MSE), alongside higher R2 values. This trend indicates that fewer variables enhance model accuracy. Moreover, descriptors incorporating intermediate information indeed enhanced performance. Specifically, under HATU and TBTU conditions, R2 values surged from 0.69 and 0.71 to 0.86 and 0.84, respectively, with corresponding decreases in errors, underscoring the efficacy of our intermediate strategy. This robust performance of intermediate-inclusive descriptors persisted even in the full substrate novelty dataset, where the BERT model retained an R2 value of approximately 0.8 across all reaction conditions, albeit with slight reductions (Table 3). Among the intermediate-inclusive descriptors, the amine + intermediate approach usually outperformed the amine + acid + intermediate strategy across all reaction conditions when using the BERT algorithm (more metric details can be found in Table S13 of the ESI†). This observation aligns with the reaction mechanism, where amines and acids form intermediates before converting to products. Since our intermediate is represented as an activated acid, it already encapsulates acid information, making the amine + acid + intermediate descriptors redundant. Consequently, the more precise amine + intermediate descriptors yield better results by avoiding redundant information and focusing on the critical reaction components. To ensure data consistency, we performed 5-fold cross-validation on the randomly split test datasets, as detailed in Section 3.9 of the ESI.† However, 5-fold cross-validation was not applicable to test sets with partial or full substrate novelty due to dataset constraints; these were evaluated using a single data split. The 5-fold cross-validation results, which are illustrated through scatter plots in Section 3.9 of the ESI,† comparing model predictions to actual yields, validate the reliability of our random split data and further demonstrate the performance of our model.
| Test data split | Metrics | TBTU | HATU | PyBOP | DCC | HBTU | EDC |
|---|---|---|---|---|---|---|---|
| Random split | R 2 | 0.84 (0.71) | 0.86 (0.69) | 0.90 (0.80) | 0.86 (0.80) | 0.89 (0.83) | 0.89 (0.82) |
| RMSE | 10% (13%) | 9% (14%) | 8% (11%) | 9% (11%) | 9% (11%) | 8% (11%) | |
| MAE | 7% (10%) | 6% (10%) | 5% (8%) | 7% (8%) | 6% (8%) | 6.1% (7%) | |
| Partial substrate novelty | R 2 | 0.77 (0.57) | 0.78 (0.53) | 0.82 (0.63) | 0.81 (0.74) | 0.86 (0.72) | 0.88 (0.79) |
| MAE | 12% (16%) | 12% (17%) | 10% (14%) | 11% (13%) | 9% (13%) | 9% (12%) | |
| RMSE | 8% (12%) | 8% (13%) | 7% (11%) | 8% (9%) | 7% (10%) | 6% (8%) | |
| Full substrate novelty | R 2 | 0.85 (0.66) | 0.84 (0.39) | 0.89 (0.40) | 0.67 (0.1) | 0.83 (0.68) | 0.75 (0.46) |
| MAE | 9% (13%) | 7% (14%) | 8% (18%) | 7% (12%) | 10% (14%) | 14% (18%) | |
| RMSE | 7% (11%) | 6% (11%) | 6% (12%) | 5% (10%) | 7% (8%) | 11% (13%) |
In contrast, the XGBoost algorithm's performance lagged behind the multi-condition model, and the inclusion of intermediate descriptors did not enhance results, resulting in marginal declines (details shown in Table S13 of the ESI†). This discrepancy between BERT and XGBoost is likely attributable to algorithmic differences. XGBoost, a machine learning algorithm, excels in learning simple reactions but struggles with the complexity added by intermediate descriptors. In contrast, the deep learning-based BERT model thrives on this additional complexity, leveraging it to improve predictive accuracy. Besides XGBoost, we also investigated other algorithms' performance after intermediate knowledge was embedded into the model. However, no better result was obtained in all cases (details shown in Table S13 of the ESI†).
By incorporating intermediate information, our selected condition models demonstrated significantly improved performance. The intermediate-powered model achieved an R2 of 0.86, compared to an R2 of 0.69 for the model without intermediate incorporation. This innovative strategy not only enhanced model accuracy but also provided a balanced approach that integrates condition-specific data with broader chemical knowledge, ultimately improving the robustness and generalizability of yield predictions for amide coupling reactions. This comprehensive approach ensures that our selected condition models are capable of accurately predicting reaction yields while considering the crucial role of reaction conditions through intermediate representations, thereby providing reliable and practical tools for chemists in optimizing amide coupling reactions. Additionally, when we encoded the reaction conditions using a one-hot approach, the model's performance significantly decreased, underscoring the conclusion that this method lacks meaningful chemical information (details as shown in Section S3.10 of the ESI†), and indicating the importance of intermediate knowledge (details as shown in Section S3.8 of the ESI†).
Having identified the power of intermediate knowledge embedded in a model, we next aim to determine whether our strategy would also work well in the case of combining data from all six different conditions into one dataset. Indeed, the performance of the BERT model enhanced by intermediate knowledge improved, but the growth rate in performance was less than that observed in selected condition predictions, especially in cases of complete novelty splitting, as shown in Table 4. This may be because the model needs to learn the relationships among the six different conditions, but the dataset is insufficient for the model to learn these relationships effectively.
| Intermediate information | Test data split | R 2 | RMSE | MAE |
|---|---|---|---|---|
| Without intermediate knowledge | Random split | 0.77 | 12% | 9% |
| Partial novelty | 0.71 | 14% | 10% | |
| Full novelty | 0.62 | 10% | 8% | |
| With embedded intermediate knowledge | Random split | 0.85 | 10% | 7% |
| Partial novelty | 0.8 | 11% | 8% | |
| Full novelty | 0.65 | 9% | 8% |
In summary, it is evident that intermediate-inclusive descriptors yield better results not only in selected condition predictions but also in multi-condition predictions. The absence of intermediate descriptors leads the model to erroneously assume that reactions depend solely on substrates, ignoring the significant impact of reaction conditions. This misassumption explains why models without intermediate descriptors perform well in the random split and known single-substrate datasets but experience sharp performance drops in the full novelty substrate dataset.
Prediction of external literature reactions
To demonstrate the generalization ability of our yield prediction model, enhanced by incorporating intermediate knowledge, we evaluated its performance on a novel external dataset sourced from the literature. We used SciFinder55 to identify relevant amide coupling reactions by drawing a general structural formula and applying the “Structure Match” filter to find reactions based on substructure similarity. To ensure relevance, we focused on journals prominent in drug discovery and biological studies, such as the Journal of Medicinal Chemistry, Bioorganic & Medicinal Chemistry and ACS Medicinal Chemistry Letters. Given the large number of identified reactions (over 19000), we employed a randomized selection process, choosing more than ten reactions from each journal for every reaction condition. Importantly, all selected reactions were entirely distinct from those in our HTE dataset, ensuring that the model's performance was tested on truly novel substrate combinations. Subsequently, we collected data from 257 reactions relevant to medicinal chemistry and biochemistry to showcase the potential application of our model in this field. We then used the corresponding models embedded with intermediate knowledge, which exhibited the best performance on the full novelty dataset, to predict the yields of these reactions. The results in Fig. 6 showed an R2 of 0.71, MAE of 07%, RMSE of 10%, and MSE of 1%. Considering the size of the training dataset (approximately 400), the performance of our model was quite strong, and its generalization ability would likely improve with additional data.
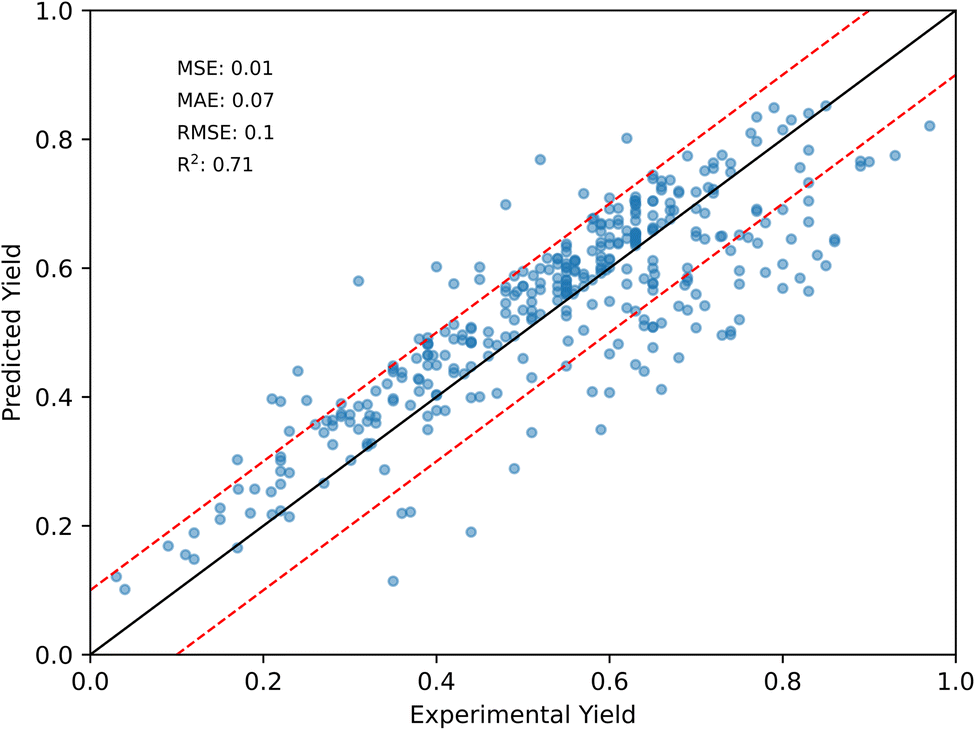 | ||
| Fig. 6 Predicted vs. experimental yields for 257 external literature reaction examples. | ||
A series of functional groups, such as alkyne (5, 11, 12), azide (2, 14), hydroxyl (11, 16), halide (4, 6, 8, 9, 18), carboxyl (7), phosphonate (16), aldehyde (6), and others, were tolerated by our prediction model, indicating its broad applicability. For some substrates, the amidation transformations were challenging, with complex relationships due to chemoselectivity arising from special functional groups, such as amine (1, 7), hydroxyl (11, 16), and carboxyl groups (7). Thus, achieving accurate yield predictions for these transformations was difficult. Nevertheless, the model provided rather accurate predictions, suggesting that it effectively learned the complex structure–yield relationships. Our model also performed well with heterocycles (4, 7, 9, 15, 18) and the sulfonamide group (12), which are commonly found in drugs. Moreover, the model appeared to have no bias toward yield distribution during prediction. Given the structural diversity and highly accurate predictions for the aforementioned reactions, the model appears to have achieved a considerable balance between sensitivity and robustness (Fig. 7).
 | ||
| Fig. 7 Prediction results of some external literature reaction examples. | ||
The above series of studies have shown that the generalization ability of the yield prediction model could indeed be greatly improved after embedding intermediate knowledge. Therefore, we were particularly interested in whether we can recommend suitable conditions for some reactions with low yields to improve their yields through the model. With this question in mind, we selected 5 reactions with yields below 40% from the above 257 reactions, and their corresponding substrates were all commercially available. Subsequently, we used the prediction model to predict the yields of these 5 reactions under the selected 6 conditions, and repeated the experiments under the condition with the highest predicted yield. As shown in Fig. 8, the yields of compounds 20, 21, and 22 were all improved dramatically. Although the yield of compound 23 did not increase under the top one condition, it was also significantly improved under the top two conditions. More results can be found in Tables S18 and S38 of the ESI.† These results indicated that our model can indeed recommend appropriate reaction conditions for some reactions, helping chemists to synthesize corresponding amide compounds with higher yields.
 | ||
| Fig. 8 The literature reported yield of five selected external literature reactions vs. experimental yield from model recommended conditions. | ||
Model performance evaluation on a benchmark dataset
After gaining a clear understanding of the generalization ability of the model trained on the HTE dataset, we aimed to evaluate the performance of the BERT model enhanced with intermediate knowledge on a benchmark dataset from Isayev's work. We prepared the dataset according to the list of reaction IDs provided in the report. The model's performance was evaluated via a random split. Initially, we studied the performance of the standard BERT framework, which delivered modest metrics with an R2 of 0.37, MAE of 13%, and RMSE of 18%. However, the BERT model enhanced by intermediate knowledge improved performance to some extent, achieving an R2 of 0.42, MAE of 12%, and RMSE of 16%. Isayev disclosed that reactivity cliffs were a reason for the poor performance of the model. Reactions were considered “cliffs” when their similarity surpassed 0.9, yet the yield difference was greater than 30. We were curious about whether our model's performance was affected by reactivity cliffs. Therefore, we predicted the yield of reactions identified as reactivity cliffs in Isayev's work (Fig. 9). The prediction error averaged 0.34, indicating that the reactivity cliff may also weaken the performance of our model. Although the performance of BERT in regression was not optimal, the model could extrapolate which reaction from a reaction pair with a reactivity cliff would achieve a greater yield, with a classification accuracy of 0.73 (details on prediction results, please see Table S11 in the ESI†). This analysis demonstrates the model's potential to handle challenging reaction scenarios such as reactivity cliffs while highlighting areas for further improvement. | ||
| Fig. 9 Prediction performance toward a reaction pair with a reactivity cliff. | ||
Conclusions
Accurate yield prediction is a crucial objective among many reaction-related prediction tasks, as several tasks can be viewed as yield prediction problems, including selectivity, condition recommendation, catalyst design, ligand design, and more. Despite its importance, it remains a challenging issue due to the impact of both data quality and the generalization ability of the model. During the process of substrate pair selection, our goal was to match the diversity found in literature-reported reactions. This targeted method ensured that our data collection was comprehensive and purposeful, rather than arbitrary. The data were then collected utilizing our in-house high-throughput experimentation (HTE) platform to ensure its quality. Our model's performance was validated through three levels of test sets — from random splits to strict tests—and further calibrated using recent unbiased external literature datasets. To address the challenges observed with strict test results, we proposed a strategy that enhances model performance by embedding domain-specific knowledge about reaction intermediates and dimension reduction. We evaluated our concept from different aspects, and the results revealed the importance of intermediate knowledge in elevating the model's performance. Excitingly, the model could even provide quite accurate predictions for some useful reactions reported in the literature and recommend better conditions for some reactions with low yields. In summary, we developed an amide coupling yield prediction model with high performance by embedding intermediate knowledge into the model and employing dimension reduction, using computationally economical SMILES as input. Our strategy can also be applied to other related machine learning tasks to enhance model performance.Data availability
https://www.github.com/aichemeco/amide_coupling/tree/main.Author contributions
Z. Yu and K. Liao conceived and supervised the project. C. Zhang, Q. Lin, C. Yang and Z. Yu built the model and performed the evaluation. Y. Kong and Z. Yu design the HTE. The manuscript was written through contributions of C. Zhang, Q. Lin, Z. Yu, and K. Liao. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China, Grant No. 22071249 and 22393892. This work was also supported by the Major Project of Guangzhou National Laboratory, Grant No. GZNL2025C01022, GZNL2024A01005 and GZNL2023A02012.References
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Expanding the medicinal chemistry synthetic toolbox, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef PubMed.
- V. R. Pattabiraman and J. W. Bode, Rethinking amide bond synthesis, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- D. G. Brown and J. Bostrom, Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone? Miniperspective, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- A. El-Faham and F. Albericio, Peptide coupling reagents, more than a letter soup, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- A. M. Żurański, J. I. Martinez Alvarado, B. J. Shields and A. G. Doyle, Predicting Reaction Yields via Supervised Learning, Acc. Chem. Res., 2021, 54, 1856–1865 CrossRef PubMed.
- M. Saebi, B. Nan, J. E. Herr, J. Wahlers, Z. Guo, A. M. Zurański, T. Kogej, P.-O. Norrby, A. G. Doyle and N. V. Chawla, On the use of real-world datasets for reaction yield prediction, Chem. Sci., 2023, 14, 4997–5005 RSC.
- V. Voinarovska, M. Kabeshov, D. Dudenko, S. Genheden and I. V. Tetko, When yield prediction does not yield prediction: an overview of the current challenges, J. Chem. Inf. Model., 2023, 64, 42–56 CrossRef PubMed.
- D. Fooshee, A. Mood, E. Gutman, M. Tavakoli, G. Urban, F. Liu, N. Huynh, D. Van Vranken and P. Baldi, Deep learning for chemical reaction prediction, Mol. Syst. Des. Eng., 2018, 3, 442–452 RSC.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Predicting reaction performance in C-N cross-coupling using machine learning, Science, 2018, 360, 186–190 CrossRef CAS PubMed.
- M. H. Samha, L. J. Karas, D. B. Vogt, E. C. Odogwu, J. Elward, J. M. Crawford, J. E. Steves and M. S. Sigman, Predicting success in Cu-catalyzed C–N coupling reactions using data science, Sci. Adv., 2024, 10, eadn3478 CrossRef CAS PubMed.
- N. I. Rinehart, R. K. Saunthwal, J. Wellauer, A. F. Zahrt, L. Schlemper, A. S. Shved, R. Bigler, S. Fantasia and S. E. Denmark, A machine-learning tool to predict substrate-adaptive conditions for Pd-catalyzed C–N couplings, Science, 2023, 381, 965–972 CrossRef CAS.
- J. Devlin, M. Chang, K. Lee and K. Toutanova, BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding, CoRR, 2018, 04805 Search PubMed.
- P. Schwaller, A. C. Vaucher, T. Laino and J.-L. Reymond, Prediction of chemical reaction yields using deep learning, Mach. Learn.: Sci. Technol., 2021, 2, 015016 Search PubMed.
- Elsevier Reaxys, https://www.reaxys.com, accessed 2024-06-28.
- Z. Liu, Y. S. Moroz and O. Isayev, The challenge of balancing model sensitivity and robustness in predicting yields: a benchmarking study of amide coupling reactions, Chem. Sci., 2023, 14, 10835–10846 RSC.
- F. Strieth-Kalthoff, F. Sandfort, M. Kühnemund, F. R. Schäfer, H. Kuchen and F. Glorius, Machine learning for chemical reactivity: the importance of failed experiments, Angew. Chem., Int. Ed., 2022, 61, e202204647 CrossRef CAS PubMed.
- P. Raghavan, A. J. Rago, P. Verma, M. M. Hassan, G. M. Goshu, A. W. Dombrowski, A. Pandey, C. W. Coley and Y. Wang, Incorporating Synthetic Accessibility in Drug Design: Predicting Reaction Yields of Suzuki Cross-Couplings by Leveraging AbbVie's 15-Year Parallel Library Data Set, J. Am. Chem. Soc., 2024, 146, 15070–15084 CrossRef CAS.
- B. C. Haas, A. E. Goetz, A. Bahamonde, J. C. McWilliams and M. S. Sigman, Predicting relative efficiency of amide bond formation using multivariate linear regression, Proc. Natl. Acad. Sci., 2022, 119, e2118451119 CrossRef CAS.
- J. Y. Wang, J. M. Stevens, S. K. Kariofillis, M.-J. Tom, D. L. Golden, J. Li, J. E. Tabora, M. Parasram, B. J. Shields and D. N. Primer, Identifying general reaction conditions by bandit optimization, Nature, 2024, 626, 1025–1033 CrossRef CAS.
- S. W. Krska, D. A. DiRocco, S. D. Dreher and M. Shevlin, The Evolution of Chemical High-Throughput Experimentation To Address Challenging Problems in Pharmaceutical Synthesis, Acc. Chem. Res., 2017, 50, 2976–2985 CrossRef CAS PubMed.
- Z. Fu, X. Li, Z. Wang, Z. Li, X. Liu, X. Wu, J. Zhao, X. Ding, X. Wan and F. Zhong, Optimizing chemical reaction conditions using deep learning: a case study for the Suzuki–Miyaura cross-coupling reaction, Org. Chem. Front., 2020, 7, 2269–2277 RSC.
- J. Götz, M. K. Jackl, C. Jindakun, A. N. Marziale, J. André, D. J. Gosling, C. Springer, M. Palmieri, M. Reck and A. Luneau, High-throughput synthesis provides data for predicting molecular properties and reaction success, Sci. Adv., 2023, 9, eadj2314 CrossRef PubMed.
- M. Fitzner, G. Wuitschik, R. Koller, J.-M. Adam and T. Schindler, Machine learning C–N couplings: Obstacles for a general-purpose reaction yield prediction, ACS Omega, 2023, 8, 3017–3025 CrossRef CAS PubMed.
- Y. Xu, Y. Gao, L. Su, H. Wu, H. Tian, M. Zeng, C. Xu, X. Zhu and K. Liao, High-Throughput Experimentation and Machine Learning-Assisted Optimization of Iridium-Catalyzed Cross-Dimerization of Sulfoxonium Ylides, Angew. Chem., Int. Ed., 2023, 62, e202313638 CrossRef CAS PubMed.
- J. Qiu, Y. Xu, S. Su, Y. Gao, P. Yu, Z. Ruan and K. Liao, Auto Machine Learning Assisted Preparation of Carboxylic Acid by TEMPO-Catalyzed Primary Alcohol Oxidation, Chin. J. Chem., 2023, 41, 143–150 CrossRef CAS.
- Y. Xu, F. Ren, L. Su, Z. Xiong, X. Zhu, X. Lin, N. Qiao, H. Tian, C. Tian and K. Liao, HTE and machine learning-assisted development of iridium (I)-catalyzed selective O–H bond insertion reactions toward carboxymethyl ketones, Org. Chem. Front., 2023, 10, 1153–1159 RSC.
- Z. Yu, Y. Kong, B. Li, S. Su, J. Rao, Y. Gao, T. Tu, H. Chen and K. Liao, HTE-and AI-assisted development of DHP-catalyzed decarboxylative selenation, Chem. Commun., 2023, 59, 2935–2938 RSC.
- J. Qiu, J. Xie, S. Su, Y. Gao, H. Meng, Y. Yang and K. Liao, Selective functionalization of hindered meta-C–H bond of o-alkylaryl ketones promoted by automation and deep learning, Chem, 2022, 8, 3275–3287 CAS.
- B. Li, S. Su, C. Zhu, J. Lin, X. Hu, L. Su, Z. Yu, K. Liao and H. Chen, A deep learning framework for accurate reaction prediction and its application on high-throughput experimentation data, J. Cheminform., 2023, 15, 72 CrossRef PubMed.
- A. Lin, J. Liu, Y. Xu, H. Wu, Y. Chen, Y. Zhang, L. Su, X. Zhao and K. Liao, High-throughput experimentation and machine learning-promoted synthesis of α-phosphoryloxy ketones via Ru-catalyzed P(O)-OH insertion reactions of sulfoxonium ylides, Sci. China: Chem., 2025, 68, 679–686 CrossRef CAS.
- A. F. Zahrt, J. J. Henle and S. E. Denmark, Cautionary Guidelines for Machine Learning Studies with Combinatorial Datasets, ACS Comb. Sci., 2020, 22, 586–591 CrossRef CAS PubMed.
- B. Meredig, E. Antono, C. Church, M. Hutchinson, J. Ling, S. Paradiso, B. Blaiszik, I. Foster, B. Gibbons, J. Hattrick-Simpers, A. Mehta and L. Ward, Can machine learning identify the next high-temperature superconductor? Examining extrapolation performance for materials discovery, Mol. Syst. Des. Eng., 2018, 3, 819–825 RSC.
- K. V. Chuang and M. J. Keiser, Comment on “Predicting reaction performance in C–N cross-coupling using machine learning”, Science, 2018, 362, eaat8603 CrossRef PubMed.
- J. G. Estrada, D. T. Ahneman, R. P. Sheridan, S. D. Dreher and A. G. Doyle, Response to Comment on “Predicting reaction performance in C–N cross-coupling using machine learning”, Science, 2018, 362, eaat8763 CrossRef PubMed.
- D. Lowe, Chemical reactions from US patents (1976-Sep2016), https://figshare.com/articles/dataset/Chemical_reactions_from_US_patents_1976-Sep2016_/5104873, 2017, accessed 12 June 2024 Search PubMed.
- Daylight Chemical Information Systems, I. SMiles ARbitrary Target Specification (SMARTS), http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html, 2023, accessed 2024-06-12 Search PubMed.
- RDKit RDKit: cheminformatics and machine learning software, 2023, https://www.rdkit.org/ Search PubMed.
- D. Rogers and M. Hahn, Extended-connectivity fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- I. T. Jolliffe, Mathematical and statistical properties of sample principal components, Principal component analysis, 2002, 29–61 Search PubMed.
- L. van der Maaten and G. Hinton, Visualizing Data using t-SNE, J. Mach. Learn. Res., 2008, 9, 2579–2605 Search PubMed.
- L. McInnes, J. Healy and J. Melville, U.M.A.P.: Uniform Manifold Approximation and Projection for Dimension Reduction, arXiv, preprint, 2018, arXiv:1802.03426, DOI:10.48550/arXiv.1802.03426.
- P. Raghavan, B. C. Haas, M. E. Ruos, J. Schleinitz, A. G. Doyle, S. E. Reisman, M. S. Sigman and C. W. Coley, Dataset design for building models of chemical reactivity, ACS Cent. Sci., 2023, 9, 2196–2204 CrossRef CAS PubMed.
- D. Rana, P. M. Pfluger, N. P. Holter, G. Tan and F. Glorius, Standardizing Substrate Selection: A Strategy toward Unbiased Evaluation of Reaction Generality, ACS Cent. Sci., 2024, 10, 899–906 CAS.
- S. P. Lloyd, Least Squares Quantization in PCM, IEEE Trans. Inf. Theor., 1982, 28, 129–137 CrossRef.
- A. Thakkar, T. Kogej and J.-L. Reymond, Datasets and their influence on the development of computer assisted synthesis planning tools in the pharmaceutical domain, Chem. Sci., 2019, 10, 10302–10313 Search PubMed.
- T. Chen and C. Guestrin, XGBoost: a scalable tree boosting system, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785–794 Search PubMed.
- C. Cortes and V. Vapnik, Support-vector networks, Mach. Learn., 1995, 20, 273–297 Search PubMed.
- L. Breiman, Random forests, Mach. Learn., 2001, 45, 5–32 CrossRef.
- N. Erickson, J. Mueller, S. R. Gupta, et al., AutoGluon-Tabular: Robust and accurate AutoML for structured data, arXiv, preprint, 2020, arXiv:2003.06505, DOI:10.48550/arXiv.2003.06505.
- J. Lu and Y. Zhang, Unified Deep Learning Model for Multitask Reaction Predictions with Explanation, J. Chem. Inf. Model., 2022, 62, 1376–1387 CrossRef CAS PubMed.
- AutoGluon AutoML for text, image, and tabular data, 2023, https://auto.gluon.ai/ Search PubMed.
- C. Raffel, N. Shazeer, A. Roberts, K. Lee, S. Narang, M. Matena, Y. Zhou, W. Li and P. J. Liu, Exploring the Limits of Transfer Learning with a Unified Text-to-Text Transformer, CoRR, 2019, 10683 Search PubMed.
- T. Stuyver and C. W. Coley, Quantum chemistry-augmented neural networks for reactivity prediction: performance, generalizability, and explainability, J. Chem. Phys., 2022, 156, 084104 CrossRef CAS PubMed.
- CAS SciFinder, https://scifinder.cas.org, 2024, accessed 2024-09-01 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03364k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |