
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic probes for NO-activatable biomedical imaging: NIR fluorescence, self-luminescence, and photoacoustic imaging
Weihao
An
b,
Zhongkun
Wang
ab,
Qingqing
Miao
 *b and
Qing
Li
*a
*b and
Qing
Li
*a
aSchool of Life Sciences, Anhui Agricultural University, Hefei 230036, People's Republic of China. E-mail: qli87@ahau.edu.cn
bSchool of Nuclear Science and Technology, University of Science and Technology of China, Hefei 230026, People's Republic of China. E-mail: qqmiao@ustc.edu.cn
First published on 14th July 2025
Abstract
Nitric oxide (NO) is a crucial signaling molecule involved in diverse physiological and pathological processes, making its precise detection essential for exploring its biological roles. Optical imaging is particularly attractive for NO detection due to its non-invasive nature, high sensitivity, and excellent spatial resolution. However, it suffers from limited tissue penetration and low signal-to-background ratios resulting from strong light scattering and autofluorescence. To overcome these challenges, several advanced imaging strategies have been developed, including near-infrared (NIR) fluorescence imaging that leverages optical regions with less light–tissue interactions, self-luminescence imaging that avoids the need for real-time light excitation, and photoacoustic imaging that detects acoustic signals with minimal attenuation. This review systematically summarizes recent advances in organic molecular probes for NO detection using these imaging modalities, focusing on their design strategies, recognition mechanisms, and biological applications. Finally, current challenges and future directions are discussed to guide the development of next-generation NO probes for both fundamental research and clinical translation.
1. Introduction
Nitric oxide (NO) is an indispensable gaseous signaling molecule that is critical for maintaining biological homeostasis.1–3 Generally, it is enzymatically synthesized from L-arginine by three isoforms of NO synthase (NOS): neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS).4,5 nNOS and eNOS produce low levels of NO (nanomolar range) to regulate normal physiological function, whereas iNOS is induced in response to proinflammatory stimuli and generates high concentrations of NO (micromolar range), contributing to host defense and inflammatory processes.6 Although NO is a metastable free radical, it is highly reactive and readily reacts with other free radicals in biological systems, thereby influencing multiple pathophysiological processes.7 Numerous studies have demonstrated that excessive NO production is implicated in various pathological conditions, such as liver damage, inflammation, tumours, and neurodegenerative diseases.8–12 Consequently, the precise detection and quantification of NO are essential for elucidating its potential functions, regulatory mechanisms, and synergistic interactions in biological systems. However, due to the complex physiological characteristics of NO, such as its low concentration,13 short lifetime,14 and wide distribution,15 visualizing and monitoring NO with high sensitivity and specificity in physiological and pathological processes remains a significant challenge.Various techniques have been developed for detecting NO in cells and biological systems, including colorimetry, magnetic resonance imaging (MRI), electron paramagnetic resonance (EPR) spectroscopy, electrochemistry, and optical imaging.16–19 Among them, optical imaging is preferred for its noninvasiveness, high sensitivity, and superior spatiotemporal resolution. Nevertheless, conventional optical imaging suffers from limitations in tissue penetration depth and low signal-to-background ratio owing to strong light scattering and endogenous autofluorescence. To address these issues, near-infrared (NIR) fluorescence imaging that leverages optical regions with reduced light–tissue interactions, self-luminescence imaging that eliminates the need for real-time light excitation, and photoacoustic imaging that detects acoustic signals with minimal attenuation, have emerged with improved optical imaging depth.20–26 In addition, compared with other types of probes, organic molecular probes exhibit exceptional structural versatility, tunable optical properties, and favorable biocompatibility, rendering them highly suitable for designing stimuli-responsive systems for biological applications. In this context, significant endeavors have shifted towards the development of activatable organic probes for NO detection based on NIR fluorescence, self-luminescence, and photoacoustic imaging techniques.
This review comprehensively summarizes the recent advancements in NO-activated organic molecular probes for first/second near-infrared (NIR-I/II) fluorescence, self-luminescence (including bioluminescence, chemiluminescence, and afterglow), and photoacoustic (PA) imaging. In the following, the design strategies of NO-activated molecules are first discussed. Then, molecular construction and applications of NIR-I/II fluorescence, self-luminescence, and PA imaging probes are introduced. Key examples of state-of-the-art probes are elaborated, showcasing their potential for highly sensitive and specific NO detection. Finally, the current challenges and future perspectives in this rapidly evolving field are highlighted, aiming to motivate researchers to further develop more advanced probes for NO detection.
2. Design strategies for NO-activatable organic probes
The rational design of responsive organic molecular probes is crucial to meet specific detection requirements. For NO-activated probes, the design strategies primarily focus on leveraging the chemical properties of NO and optimizing the molecular structure of the probe to achieve high selectivity and high sensitivity for NO detection. Typically, NO-activated organic probes consist of two fundamental components: a NO-specific recognition site and a dye.27–29 The selection of recognition groups capable of specific and efficient interaction with NO is the cornerstone of probe construction. Numerous reviews have extensively discussed NO recognition groups,30,31 such as aromatic amines, thiosemicarbazides, and selenium (Se)-based groups. These recognition groups can undergo specific chemical reactions with NO to induce measurable changes in the probe's optical properties, enabling NO detection.A series of NO-activatable organic molecular probes have been successively exploited.32–34 The most common approach is to utilize o-phenylenediamine that reacts with NO to form triazole derivatives in the presence of O2. This transformation activates fluorescence or photoacoustic signals by inhibiting photoinduced electron transfer (PET) or enhancing intramolecular charge transfer (ICT) (Scheme 1a and b). In addition to this classical mechanism, several alternative activation mechanisms for NO detection have also been reported, including the direct N-nitrosation of aromatic amines (Scheme 1c), the deamination of aromatic primary amines (Scheme 1d), the formation of the Se–NO bond (Scheme 1e), and the conversion of thiosemicarbazide into oxadiazole (Scheme 1f). Furthermore, some innovative strategies have been employed to construct advanced probes. For instance, ratiometric probes can be obtained by integrating o-phenylenediamine with two fluorophores capable of fluorescence resonance energy transfer (FRET) (Scheme 1g). To improve imaging specificity towards diseases, tandem activatable probes can be created by caging dyes with multi-recognition units of NO and other biomarkers (such as pH, enzymes). These probes undergo cleavage by NO and subsequent activation by the secondary biomarker to ultimately restore their optical signal (Scheme 1h and i).
 | ||
| Scheme 1 Design strategies for NO-activatable organic probes. Cyclization of o-phenylenediamine based on PET (a) and ICT (b) mechanisms. (c) N-Nitrosation of aromatic secondary amine. (d) Deamination of aromatic primary amine. (e) Formation of the Se–NO bond. (f) Conversion of thiosemicarbazide into oxadiazole. (g) Ratiometric fluorescence probe. (h) Tandem activatable probe. (i) Tandem bioluminescence probe. | ||
3. Organic probes for NO-activatable NIR fluorescence imaging
3.1 NIR-I fluorescence imaging
NIR-I light (650–900 nm), recognized as a biological transparency window, offers significant advantages over visible light, including deep tissue penetration, minimal photodamage, reduced scattering, and low background autofluorescence.35–37 These distinctive characteristics make NIR-I fluorescence probe a powerful tool for high-resolution biomedical imaging.38,39 To date, significant progress has been made in the development of activatable NIR-I fluorescent probes for NO imaging (Table 1). The design of these probes is based on NO-specific chemical reactions, with the most widely utilized strategies being the cyclization of o-phenylenediamine33,40–43 and the N-nitrosation of aromatic secondary amines.44–46 This investigation aims to introduce the design principles and response mechanisms of these probes, as well as their performance in cellular and in vivo imaging applications. Through a comprehensive analysis of these aspects, we seek to provide valuable insights for the development of next-generation probes with enhanced sensitivity, selectivity, and biocompatibility.| Probe name | Excitation wavelengths (nm) | Emission wavelengths (nm) | LOD | Application | References |
|---|---|---|---|---|---|
| DAC-P | 755 | 790 | — | Living cells and living mice | 48 |
| DAC-S | 755 | 790 | — | Living cells | 48 |
| Probe 1a | 600 | 681 | — | Living cells | 49 |
| Probe 1b | 666 | 750 | — | Living cells | 49 |
| SiRD | 680 | 710 | — | Living cells | 50 |
| deOxy-DALSiR | 645 | 680 | 0.12 nM | Living cells and living mice | 51 |
| Lyso-SiRB-NO | 667 | 680 | — | Living cells | 52 |
| LS-NO | 750 | 800 | 242 nM | Living cells and living mice | 53 |
| NRNO | 650 | 820 | 46 nM | Living cells and living mice | 41 |
| TAMPM1 | 680 | 810 | — | Living cells and living mice | 54 |
| BPTQ | 580 | 720 | 15 nM | In saliva and living cells | 55 |
| Cy7-MA | 760 | 800 | 11.3 nM | Living cells and living mice | 56 |
| SiRNO | 633 | 700 | 14 nM | Living cells and living mice | 46 |
| NIR-MA | 750 | 794 | 95 nM | Living cells and living mice | 57 |
| CS–Se | 680![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 740 740 |
780 | 28 nM | In saliva and living cells | 58 |
| DCM–NO | 436 | 661 | 17 nM | Living cells and living mice | 59 |
| TJ730-Golgi-NO | 622 | 780 | 0.54 μM | Living cells and living mice | 60 |
| AOSNP | 680 | 920 | 0.35 μM | Living mice | 71 |
| NRP@M-PHCQ | 780 | 950 | 5 μM | Living cells and living mice | 72 |
| BNDA | 808 | 938 | — | Living cells and living mice | 73 |
| P-SiRhd-7 | 800 | 1050 | 0.3 μM | Living mice | 74 |
| HC-N | 808 | 923 | 0.18 mM | Living mice | 75 |
Building upon this fundamental principle, Nagano and colleagues made a significant advancement in 2005 with the development of NIR fluorescent diaminocyanine (DAC) probes integrating tricarbocyanine with o-phenylenediamine (Scheme 2).48 The DAC probes were strategically designed with two distinct derivatives: DAC-P, containing two propyl groups for enhanced cell membrane permeability, and DAC-S, incorporating sulfonic acid groups to improve water solubility. In the initial state, these probes remained non-fluorescent due to the electron transfer from o-phenylenediamine to the excited state of the tricarbocyanine fluorophore. Upon reaction with NO under aerobic conditions, o-phenylenediamine was converted into triazole, which lacked sufficient electron-donating capacity to sustain PET, thereby restoring the tricarbocyanine fluorescence. To evaluate the effectiveness in vivo, the DAC-P probe was introduced into the kidneys by injection into the right renal artery along with the perfusion fluid, followed by administration of the NO donor NOC13. Near-infrared fluorescence imaging revealed a marked increase in fluorescence intensity, demonstrating the utility of DAC-P for NO imaging in isolated rat kidneys.
 | ||
| Scheme 2 Chemical structures of NO-activated NIR-I fluorescent probes. | ||
While cyanine-based NO probes function effectively under neutral to alkaline conditions (pH 6–11), their application in acidic environments remains limited. To overcome this constraint and develop pH-independent probes, Ge et al. incorporated 3,4-diaminophenyl groups into phenoxazine scaffolds to synthesize two dibenzo[c,h]phenoxazinium-based probes (probe 1a and probe 1b) for nitric oxide detection (Scheme 2).49 Upon NO reaction, probe 1a, featuring primary amino groups at 3- and 11-positions of the fluorophore, exhibited a 3-fold increase in the fluorescence intensity at 681 nm. In contrast, probe 1b, featuring tertiary amino groups at the same positions, displayed a remarkable 32-fold fluorescence enhancement at 750 nm. Notably, probe 1b showed good sensitivity and selectivity towards NO while maintaining its detection capability across a broad pH range (4.0–7.5). These advantageous properties enabled its successful application in NO imaging within HeLa cells, highlighting its potential for studying NO in diverse physiological and pathological contexts, particularly in acidic microenvironments.
Most existing NO-activated probes often suffer from relatively long response times, limiting their effectiveness in real-time imaging. To achieve rapid-response probes towards NO, Zhu et al. developed a silicon-substituted xanthene (SiR) NIR platform, which directly connected to an amino group of o-phenylenediamine to obtain a NO-activated probe SiRD (Scheme 2).50 The o-phenylenediamine moiety had a sufficiently high highest occupied molecular orbital (HOMO) energy level to quench fluorescence of silicon-substituted oxazine fluorophore through a PET process. The replacement of an oxygen atom with a silicon atom lowered the lowest unoccupied molecular orbital (LUMO) energy level, accelerating the reaction rate of SiRD with NO. Due to these significant features, the SiRD probe showed a rapid fluorescence turn-on at 710 nm within 1 minute upon NO addition, and was not interfered by other biologically relevant species. Furthermore, the positively charged SiRD probe could specifically localize in mitochondria, thereby enabling targeted imaging of mitochondrial NO in living cells.
Si-rhodamine has emerged as a superior alternative to traditional rhodamine dyes for in vivo bioimaging applications due to its advantageous photophysical properties, including longer emission wavelengths, high fluorescence brightness, and good photostability. Notably, Si-rhodamine contains a pH-sensitive spirolactam ring, which provides a valuable design feature for developing activatable NIR fluorescence probes for NO imaging in acidic microenvironments. In 2017, Guo et al. reported a Si-rhodamine-based NIR fluorescent probe deOxy-DALSiR for specific NO detection (Scheme 2).51 The probe maintained structural stability (ring-closed form) under alkaline conditions and opened the spirolactam ring under acidic conditions to generate a strong absorption peak at 650 nm, but it remained non-fluorescent. Upon exposure to NO, deOxy-DALSiR exhibited an extraordinary fluorescence enhancement (up to 6300-fold) at 680 nm, which was independent of pH (4.0–10.0). Its outstanding NO detection capability was successfully demonstrated in both living cells and an inflamed mouse model, highlighting its potential for precise NO imaging in biological systems.
To achieve more accurate detection of NO, exploiting organelle-targeted probes is an effective strategy. Lysosomes play a vital role in regulating diverse physiological and pathological processes, making them an important target for NO imaging. To enable real-time visualization of lysosomal NO, Wang et al. conjugated a morpholine group to Si-rhodamine to afford a lysosome-targeting NIR fluorescent probe, Lyso-SiRB-NO (Scheme 2).52 Under acidic conditions, Lyso-SiRB-NO could react with NO to generate strong red fluorescence emission at 680 nm. Due to minimal cytotoxicity and exceptional selectivity, the probe was further applied for real-time monitoring of lysosomal NO in RAW 264.7 macrophages. Recently, Xiong et al. reported a lysosome-targeting NIR fluorescent probe LS-NO (Scheme 2), designed with a donor–acceptor–donor (D–A–D) architecture to detect NO by harnessing the ICT mechanism.53 Initially, LS-NO was non-fluorescent because of weak ICT. Upon reaction with NO, the weak electron-accepting diaminobenzo-(1,2,5-thiadiazol) moiety in LS-NO was converted into the strong electron-accepting triazolo-benzo-(1,2,5-thiadiazol) moiety, significantly enhancing ICT and thereby triggering strong fluorescence in the NIR region. LS-NO had excellent water solubility, photostability, and specificity towards NO, with acid-promoted fluorescence well-suitable for lysosomal NO imaging. By virtue of the lysosome-targeting ability of the morpholine group, LS-NO successfully visualized NO within the lysosomes of RAW264.7 and HCC1299 cells. Remarkably, it also enabled noninvasive monitoring of endogenous NO levels in a lipopolysaccharide-induced inflammatory bowel disease (IBD) mouse model.
Two-photon fluorescence probes that simultaneously absorb two NIR photons offer deeper tissue penetration, reduced background fluorescence, and higher three-dimensional spatiotemporal resolution compared to conventional single-photon fluorescence probes. While various two-photon fluorescent probes have been developed for NO imaging, their relatively short emission wavelengths (<550 nm) significantly limit their effectiveness in deep-tissue applications. To increase the emission wavelength, Liu's group synthesized a two-photon NIR fluorescent probe (NRNO) by conjugating a Nile Red fluorophore with an o-phenylenediamine NO-responsive unit through an ethylenedioxy linker (Scheme 2).41 Upon excitation at 820 nm, the NRNO probe showed strong NIR fluorescence emission centered at 650 nm upon NO detection with a low detection limit (46 nM), rapid response rate, and excellent selectivity. Owing to its superior properties, NRNO successfully detected exogenous NO in HeLa cells and endogenously generated NO in RAW 264.7 cells under drug stimulation. Notably, NRNO enabled high-resolution imaging of NO at tissue depths up to 170 μm and was effectively applied to monitor NO generation in lipopolysaccharide (LPS)-mediated inflammation in mice.
Tandem-activated fluorescent probes represent advanced molecular imaging agents that are typically designed with multiple responsive sites. Unlike conventional single-activated probes, these sophisticated probes require two or more disease-related biomarkers to turn on fluorescence signals, thereby improving the detection accuracy of disease in complex biological environments and reducing false positives. For instance, Pu et al. reported a tandem-activated near-infrared fluorescence probe, TAMPM1 (Scheme 2), for imaging of tumour-infiltrating leukocytes.54 The TAMPM1 probe consisted of three key components: a polyvinylpyrrolidone tumour-targeting group, a hemicyanine NIR fluorophore (CyOH), and a triple-responsive module featuring an o-phenylenediamine group, a caspase-1 (Cas-1)-cleavable peptide N-acetyl-Tyr-Val-Ala-Asp-OH (YVAD) and an aminopeptidase N (APN)-cleavable alanine (Ala) substrate (Fig. 1a). TAMPM1 was non-fluorescent in its native state and opened its NIR fluorescence signal only in the presence of both M1 macrophage-associated biomarkers (NO and Cas-1) and tumour-related APN (Fig. 1b and c). Such a tandem design ensured that the probe was triggered only by tumour-infiltrating leukocytes in the tumour microenvironment, while remaining silent in normal tissues and inflammatory tissues (Fig. 1d). Such unprecedented specificity, achieved through rational molecular engineering of the activation cascade, establishes tandem-activated probes as powerful tools for precise disease imaging and diagnostic applications.
 | ||
| Fig. 1 (a) Chemical structure of TAMPM1 and its activation process in the presence of tumour and lymphocyte biomarkers. (b) NIR Fluorescence (NIRF) spectra of TAMPM1 in the presence of individual biomarkers or combined biomarkers in corresponding buffers at 37 °C for 2 h. (c) In vitro selectivity of TAMPM1. (d) NIRF image of the LPS-inflamed and CT26 tumour tissues after local injection of probe (0.1 mM, 10 μL) for 30 min. Reproduced from ref. 54 with permission from Springer Nature, copyright 2023. | ||
Based on this mechanism, Zhang et al. developed a NIR fluorescent probe (BPTQ) for highly sensitive NO detection (Fig. 2a).55 The probe capitalized on the electron-rich nitrogen of aromatic secondary amine to quench fluorescence through efficient photo-induced electron transfer to the fluorophore. Upon exposure to NO, the amine underwent specific nitrosation to form an electron-withdrawing N-nitroso group, which reduced nitrogen electron density, thereby effectively suppressing the PET process and restoring the fluorescence of the fluorophore (Fig. 2b). The probe exhibited exceptional selectivity towards NO, low detection limit (15 nM), and rapid response (30 s) (Fig. 2c). Cell experiments demonstrated that the probe had low toxicity and was able to detect endogenous NO in oral cancer Cal-27 cells. Li et al. synthesized a methylamine-functionalized cyanine dye (Cy7-MA, Scheme 2) as a NIR fluorescent probe to detect NO.56 Owing to the ICT effect, the Cy7-MA probe displayed extremely low fluorescence emission at 800 nm. The methylamine group could rapidly combine with NO to form a strong electron-withdrawing N-nitroso group, which inhibited the ICT process and induced a substantial red-shift of the fluorescence emission from the visible to the NIR region. This probe could rapidly and selectively detect NO with an impressive detection limit of 11.3 nM. Moreover, the Cy7-MA probe was successfully applied for in vivo imaging of endogenous NO in a LPS-induced inflammation mouse model.
 | ||
| Fig. 2 (a) Schematic diagram of the reaction mechanism between BPTQ and NO. (b) Fluorescence (FL) spectra of BPTQ (10 μM) incubated with varying NO concentrations (0–100 μM). (c) Linear correlation between BPTQ fluorescence intensity and NO concentration. Reproduced from ref. 55 with permission from Elsevier, copyright 2024. | ||
To enable deep-tissue imaging of NO, Liu's group rationally designed a two-photon NIR fluorescent probe (SiRNO, Scheme 2) by incorporating a Si-rhodamine derivative as the two-photon excitable fluorophore and a 4-methoxy-N-methylaniline group as the NO recognition site via a carbon–carbon single bond linker.46 This strategic design facilitated efficient PET to maintain the probe in a fluorescence-quenched state. Upon specific N-nitrosation of the aniline group by NO, SiRNO showed a strong NIR fluorescence emission at 672 nm with 440-fold enhancement in a maximal active cross section under excitation at 820 nm. Due to its high specificity, excellent sensitivity, rapid response, and NIR excitation/emission properties, SiRNO enabled effective visualization of NO in both cells and a xenograft tumour mouse model.
It is well known that NO can regulate various cellular functions through its interaction with the selenol (SeH) groups of selenoproteins, which results in the formation of Se–NO bonds. Inspired by this mechanism, Jiao et al. incorporated a selenium moiety into a rhodamine derivative to successfully develop a NIR probe (CS–Se).58 In the absence of NO, the CS–Se probe remained in its closed spirolactone form and was non-fluorescent. Upon the addition of NO, the Se-bond of the probe reacted with NO to form an unstable intermediate (CS–Se–NO), which subsequently underwent hydrolysis, triggering the open of spirolactone ring and resulting in a strong NIR fluorescence signal (Fig. 3a). Spectroscopic analysis confirmed this activation process: the probe in its closed form exhibited minimal background absorption, whereas NO exposure led to the emergence of distinct absorption peaks at 680 nm and 740 nm (Fig. 3b), along with strong fluorescence emission at 780 nm (Fig. 3c). Furthermore, CS–Se probe was employed to detect NO concentrations in the saliva samples from patients with oral diseases and oral cancer cells (Cal-27), demonstrating its potential as a promising tool for early diagnosis research in NO-related diseases.
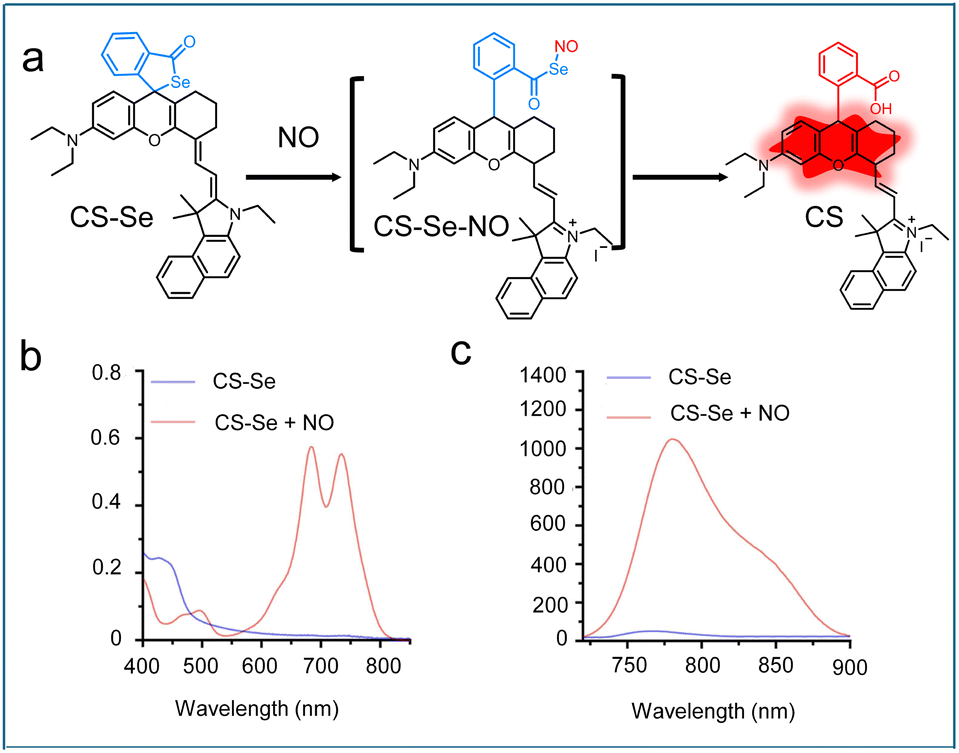 | ||
| Fig. 3 (a) Reaction mechanism of CS–Se with NO. (b) UV-vis and (c) fluorescence spectra of CS–Se before and after the reaction with NO. Reproduced from ref. 58 with permission from Elsevier, copyright 2023. | ||
Recently, the thiosemicarbazide moiety has emerged as a highly effective NO-responsive site for activatable fluorescent probe design. In 2022, Song's group synthesized a single-molecule fluorescent probe named DCM-NO (Scheme 2) by conjugating 4-(4-nitrophenyl) thiosemicarbazide to dicyanomethylene-4H-pyran (DCM) fluorophore.59 Owing to the rotational motion of the 4-(4-nitrophenyl) thiosemicarbazide moiety, the probe DCM-NO exhibited negligible fluorescence. In the presence of NO, the sensing unit 4-(4-nitrophenyl) thiosemicarbazide was cleaved, resulting in the formation of a NIR fluorophore. The DCM-NO probe displayed a large Stokes shift (198 nm), outstanding selectivity, a low detection limit (17 nM), and a fast response rate with complete activation within 60 s. These superior properties enabled DCM-NO to be successfully applied for visualizing elevated NO levels in pulmonary fibrosis cells and bleomycin-induced pulmonary fibrosis mouse models, providing a practical detection tool for the clinical diagnosis of idiopathic pulmonary fibrosis (IPF). To develop probes for accurate detection of NO in the Golgi apparatus, He et al. designed a Golgi-targeted NIR fluorescent probe, TJ730-Golgi-NO (Scheme 2).60 This probe consisted of a phenylsulfonamide group for Golgi targeting, rhodamine TJ730 as the fluorophore, and 4-(4-nitrophenyl) thiosemicarbazide as the NO-reactive site. TJ730-Golgi-NO is initially nonfluorescent because of its spirolactam ring structure. Upon reaction with NO, the sensing unit was transformed into an oxadiazole structure, which induced the opening of the spirolactam ring and activated the NIR fluorescence signal. Moreover, TJ730-Golgi-NO was successfully employed to monitor NO fluctuations in cellular models, including PC12 and RAW 264.7 cells, as well as in depression-like mouse models.
3.2 NIR-II fluorescence imaging
The second near-infrared window (NIR-II, 1000–1700 nm) fluorescence imaging technology has garnered considerable attention in the biomedical field due to its superior optical properties. Compared to traditional NIR-I and visible fluorescence, the extended excitation and emission wavelengths of NIR-II fluorescence significantly mitigate photon scattering and tissue autofluorescence,61–63 leading to unparalleled imaging performance, including improved imaging penetration depth (up to the centimeter level), improved spatial resolution, higher signal-to-background ratio (SBR), and increased imaging sensitivity.64–66 Moreover, NIR-II fluorescent probes exhibit exceptional photostability in vivo, effectively avoiding photobleaching and phototoxicity issues commonly associated with short-wavelength light exposure.67–70 These characteristics make NIR-II fluorescence a powerful tool for real-time monitoring and high-precision imaging of deep-seated diseases, providing a transformative approach for high-resolution in vivo molecular imaging in living systems.To develop NIR-II fluorescent probes for NO detection, a typical strategy is to employ benzo[c][1,2,5]thiadiazole-5,6-diamine as the NO-responsive moiety. Owing to the strong hydrophobicity of such structures, the probes are often encapsulated into nanoparticles. This nanoparticle encapsulation not only improves biocompatibility, stability, and water solubility, but also enhances pharmacokinetics and targeted delivery, thereby facilitating more precise NO detection in complex biological environments. For instance, Fan et al. designed an organic semiconducting compound (FTBD, Scheme 3) with a benzo[c][1,2,5]thiadiazole-5,6-diamine moiety, which was further conjugated with a poly(styrene-co-maleic anhydride) (PSMA) polymer and self-assembled into an NO-activated nanoprobe, designated as AOSNP.71 In the presence of NO, the benzo[c][1,2,5]thiadiazole-5,6-diamine unit was converted into a benzotriazole derivative, enhancing its electron-accepting ability and thereby resulting in a fluorescence shift from the NIR-I to the NIR-II region. In cellular experiments, AOSNP displayed significantly enhanced NIR-II fluorescence in acetaminophen (APAP)-treated cells compared to untreated controls, consistent with NO overproduction during APAP-induced hepatotoxicity. Upon treatment with the detoxifying agent N-acetylcysteine (NAC), the NIR-II fluorescence intensity notably decreased. Similar to the cellular experimental results, the APAP-treated mice exhibited a strong NIR-II fluorescence signal in the liver region compared to untreated mice and APAP/NAC-treated mice. These results indicate that the probe AOSNP can be used for noninvasive, real-time monitoring of drug-induced hepatotoxicity and for evaluating therapeutic efficacy in NO-related liver injury. In 2023, Tang et al. adopted a similar design strategy to synthesize a NO-activated NIR-II organic semiconducting compound (NRP, Scheme 3). To specifically track M2-like macrophages and monitor their polarization, NRP was self-assembled with an amphiphilic block copolymer bearing mannose and hydroxychloroquine groups (M-PHCQ), yielding a NO-activated nanoprobe NRP@M-PHCQ (Fig. 4a).72 The mannose group conferred the selective targeting towards M2-like macrophages, while the hydroxychloroquine could induce their polarization to M1-like macrophages with improved NO production. Upon exposure to NO, the benzo[c][1,2,5]thiadiazole-5,6-diamine unit in NRP was specifically oxidized into a stronger electron-accepting moiety 5H-[1,2,3]triazolo[4,5-f]-2,1,3-benzothiadiazole, thereby strengthening the ICT effect and resulting in red-shifted absorption and emission in the NIR-II region (900–1200 nm) (Fig. 4b and c). These features allowed NRP@M-PHCQ to visualize M2-like macrophage migration and polarization in real time and sensitively track early tumour metastasis in lymph nodes and lungs.
 | ||
| Scheme 3 Chemical structures of NO-activated NIR-II fluorescent probes. | ||
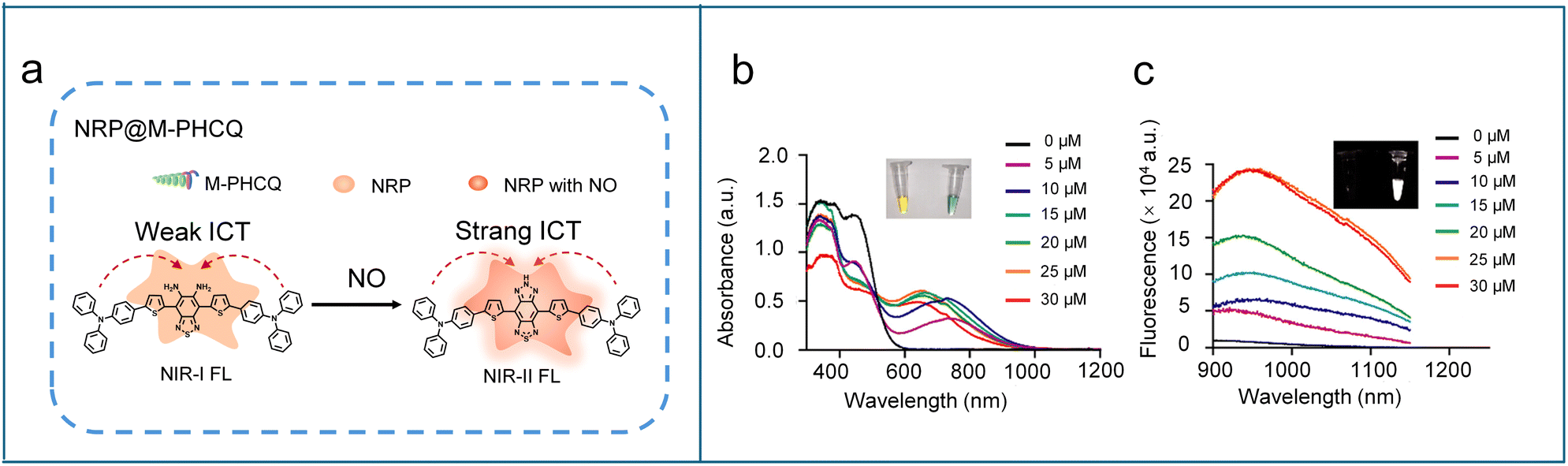 | ||
| Fig. 4 (a) Reaction mechanism of NRP@M-PHCQ with NO. Absorbance (b) and fluorescence (c) of NRP@M-PHCQ after the addition of NO concentrations (5–30 μM). The insets show the photos of NRP@M-PHCQ before (left) and after (right) the addition of NO (30 μM). Reproduced from ref. 72 with permission from American Chemical Society, copyright 2023. | ||
However, benzothiadiazole-based NIR-II dyes often suffer from poor water solubility, leading to fluorescence quenching in aqueous solution, reduced reactivity with NO, and consequently diminished detection performance. To overcome these limitations, Wu's group developed a supramolecular strategy by complexing a benzo[c][1,2,5]thiadiazole-5,6-diamine-core fluorophore BNDA (Scheme 3) with 2-hydroxypropyl-b-cyclodextrin (HbCD) to yield a host–guest complex BNDA-HbCD, which could self-assemble into nanoscale aggregates in aqueous solution.73 The incorporation of phenylnaphthalenamine groups as molecular rotors endowed the fluorophore with a typical aggregation-induced emission (AIE) feature, ensuring strong fluorescence in the aggregated state. As expected, the addition of NO triggered the BNDA-HbCD to emit strong fluorescence in 900–1100 nm. The resulting nanoprobe BNDA@HbCD was successfully employed for imaging and locating liver injury sites and monitoring therapeutic outcomes in living mice by detecting endogenous NO. Additionally, the probe demonstrated its versatility by enabling in situ detection and visualization of endogenous NO in soybean sprouts.
To explore high-performance NIR-II fluorescent scaffolds for in vivo NO imaging, Guo et al. introduced a bent-to-planar rehybridization design strategy to design an asymmetric Si-rhodamine probe B-SiRhd-7 (Scheme 3) with a NO-responsive o-phenylenediamine group.74 Upon reaction with NO, B-SiRhd-7 exhibited a significantly enhanced fluorescence emission at around 1050 nm, with a detection limit as low as 0.3 μM. To improve in vivo NO tracking, B-SiRhd-7 was encapsulated into liposomal nanoparticles, yielding B-SiRhd-7@liposome. The B-SiRhd-7@liposome could not only assess NO levels in RAW 264.7 cells but also effectively monitor in real-time the multi-organ inflammation caused by intraperitoneal injection of LPS. Importantly, B-SiRhd-7@liposome enabled direct in situ visualization of NO fluctuations in the brains of Alzheimer's disease model mice.
Building upon the design principles of the NIR-I fluorescent probe Cy7-MA, Wu et al. engineered a NO-activatable NIR-II fluorescent probe HC-N (Scheme 3) by modifying the heptamethine cyanine chromophore with methylamine.75 Two triethylene glycol chains were introduced to the probe to enhance its water solubility and biocompatibility. After reaction with NO, the secondary amine in the probe was converted into an electron-withdrawing N-nitroso group, generating a new absorption peak at 865 nm with obvious fluorescence enhancement in NIR-II range (900–1100 nm). As a result, the probe HC-N was able to detect in vivo NO and evaluate LPS-induced acute dermatitis and monosodium iodoacetate (MIA)-induced acute joint inflammation in living mice.
3.3 Ratiometric fluorescence imaging
Ratiometric fluorescence imaging, which simultaneously measures fluorescence signals at two distinct wavelengths, constitutes a major technological breakthrough compared to the traditional single-channel imaging method. The ratiometric fluorescent probes typically contain two distinct fluorophores or a single fluorophore with dual emission characteristics. When interacting with a specific analyte, the probe undergoes a chemical transformation to alter the fluorescence ratio between the two wavelengths, providing a direct correlation with the concentration of the analyte. This innovative methodology effectively minimizes interference from probe concentration variations, environmental factors, and instrument drift,76,77 while simultaneously eliminating background noise and significantly improving measurement accuracy.78 These superior performance characteristics render ratiometric fluorescent probes particularly valuable for conducting precise quantitative analyses in complex biological environments. Currently, two primary strategies are employed in the design of NO-activated ratiometric fluorescent probes, as shown in Fig. 5a. The commonly used strategy involves a single fluorophore that exhibits two reversible fluorescence signal changes upon reaction with NO. Another strategy utilizes a dual-fluorophore system, incorporating an NO-responsive fluorophore with an inert reference dye to enable ratiometric measurement. | ||
| Fig. 5 (a) The general strategies for constructing NO-responsive ratiometric fluorescent probes. (b) Ratiometric probe NFL-NH2. (c) Fluorescence intensity of NFL-NH2 after adding the NO donor (0–60 μM) in ethanol solution. (d) Linear relationship between fluorescence intensity ratio (F705/F780) and NO concentration (0–10 μM). Reproduced from ref. 32 with permission from American Chemical Society, copyright 2024. (e) The response mechanism of IRNO toward NO and the preparation of the NO-responsive nanoprobe DCNP@MPS@IRNO. (f) Illustration of the NO-responsive nanoprobe DCNP@MPS@IRNO for in vivo monitoring of liver injury induced by drug overdose. Reproduced from ref. 82 with permission from American Chemical Society, copyright 2021. | ||
Rheumatoid arthritis (RA) is a ruinous autoimmune disease in which NO plays a critical role in the progression of arthritic damage. To assess the severity of joint damage in RA, Liu's group developed a mitochondria-specific ratiometric NIR fluorescent probe (NFL-NH2) for monitoring the changes of NO levels.32 The fluorescent probe consisted of a hemicyanine derivative as the fluorescent moiety and an o-phenylenediamine as the recognition moiety (Fig. 5b). In the presence of NO, the o-phenylenediamine moiety was converted to a benzotriazole unit, leading to a fluorescence blue shift from 780 to 705 nm due to the ICT effect (Fig. 5c). The probe exhibited a 44-fold fluorescent intensity ratio (F705/F780) in response to NO, with high selectivity and a low detection limit of 0.536 nM (Fig. 5d). Thus, the probe enabled ratiometric fluorescence imaging of NO in macrophages and RA mouse models. NO levels within tumor-associated macrophages (TAMs) serve as a crucial biomarker for assessing the therapeutic efficacy of antineoplastic drugs. To precisely measure TAM-specific NO, Li et al. designed and synthesized a ratiometric fluorescence nanoprobe TAM-Cy-NO,79 which was composed of NO-responsive heptamethine cyanine-based probes (Cy-NO, Scheme 4) and TAM-targeting peptide-modified liposomes. Such a design allowed the nanoprobe TAM-Cy-NO to selectively enter TAMs, where Cy-NO reacted with NO to cause a dose-dependent ratiometric fluorescence response (F610/F815). Since antineoplastic drugs modulate NO production in TAMs, this probe was successfully applied to evaluate the efficacy of antitumor drugs by ratiometric fluorescence imaging, providing a reliable method for rapid screening of anticancer drugs.
 | ||
| Scheme 4 Chemical structures of NO-responsive ratiometric fluorescent probes. | ||
Since multi-biomarker responsive probes can significantly improve the specificity and accuracy of detection, efforts have been made to develop dual-activatable ratiometric fluorescence probes. For instance, Song et al. developed a boron dipyrromethene (BODIPY)-based ratiometric fluorescence probe (BDP, Scheme 4) capable of detecting overexpressed NO and high viscosity.80 The electron-rich aromatic secondary amine in the probe acted as a NO-reactive group that underwent N-nitrosation to regulate a push–pull system, generating a characteristic ratiometric fluorescence (F625/F715). Concurrently, the molecular rotor feature of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond between N-methylaniline and the BODIPY core enabled viscosity sensing, where free rotation in low-viscosity environments maintained relatively weak fluorescence, while restricted rotation in high-viscosity conditions significantly enhanced emission intensity. Due to overexpressed NO and increased viscosity in fibrous liver tissue, the probe effectively detected hepatic fibrosis in a mouse model using ratiometric fluorescence imaging. In 2024, Song's group combined acidity and NO, the two common tumour-related biomarkers, to construct a dual-activatable fluorescent probe (POZ-NO, Scheme 4).81 In the presence of NO under acidic conditions, the benzo[c][1,2,5]thiadiazole-5,6-diamine unit in the probe was nitrated to generate a stronger acceptor, 5H-[1,2,3]triazolo[4,5-f]-2,1,3-benzothiadiazole, enhancing the intramolecular charge transfer (ICT) effect and resulting in a significant red-shift in fluorescence emission from 630 nm to 810 nm. The fluorescence intensity ratio (F810/F630) was linearly correlated with the concentration of NO, and the detection limit for NO was calculated to be 12.6 nM. In vivo experiments further demonstrated that POZ-NO had the ability to accurately monitor tumours by ratiometric fluorescence imaging.
C bond between N-methylaniline and the BODIPY core enabled viscosity sensing, where free rotation in low-viscosity environments maintained relatively weak fluorescence, while restricted rotation in high-viscosity conditions significantly enhanced emission intensity. Due to overexpressed NO and increased viscosity in fibrous liver tissue, the probe effectively detected hepatic fibrosis in a mouse model using ratiometric fluorescence imaging. In 2024, Song's group combined acidity and NO, the two common tumour-related biomarkers, to construct a dual-activatable fluorescent probe (POZ-NO, Scheme 4).81 In the presence of NO under acidic conditions, the benzo[c][1,2,5]thiadiazole-5,6-diamine unit in the probe was nitrated to generate a stronger acceptor, 5H-[1,2,3]triazolo[4,5-f]-2,1,3-benzothiadiazole, enhancing the intramolecular charge transfer (ICT) effect and resulting in a significant red-shift in fluorescence emission from 630 nm to 810 nm. The fluorescence intensity ratio (F810/F630) was linearly correlated with the concentration of NO, and the detection limit for NO was calculated to be 12.6 nM. In vivo experiments further demonstrated that POZ-NO had the ability to accurately monitor tumours by ratiometric fluorescence imaging.
To enable deep-seated imaging of NO, Song et al. engineered an advanced ratiometric NIR-II fluorescent nanoprobe DCNP@MPS@IRNO, by integrating a NO-responsive organic dye (IRNO, Scheme 4) with a downconversion nanoparticle (DCNP, with 1550 nm emission when excited at 980 nm) as internal standard substance, into a mesoporous SiO2 carrier.82 The IRNO probe initially exhibited quenched fluorescence due to an intramolecular PET process, but upon NO exposure, its o-phenylenediamine group underwent cyclization to form a weaker electron-donating benzotriazole, suppressing the PET process and restoring NIR-II emission at 1050 nm when excited at 808 nm (Fig. 5e). As a result, the DCNP@MPS@IRNO nanoprobe could displayed a NO-activated fluorescence signal at 1050 nm (F1050 Em, 808 Ex) and an “always-on” fluorescence signal at 1550 nm (F1550 Em, 980 Ex), generating a ratiometric NIR-II fluorescence (F1050 Em, 808 Ex/F1550 Em, 980 Ex). In addition, DCNP@MPS@IRNO showed outstanding selectivity towards NO, with a low detection limit of 0.61 μM. In vivo experiments indicated that DCNP@MPS@IRNO was able to quantify NO levels in APAP-induced liver injury and screen potential APAP antidotes via NIR-II ratiometric fluorescence imaging (Fig. 5f).
4. Organic probes for NO-activatable self-luminescence imaging
Self-luminescence imaging represents a highly advanced optical technique that detects photons without requiring real-time light excitation, thereby eliminating tissue autofluorescence interference and enabling deep-tissue imaging with a significantly improved signal-to-background ratio (SBR) compared to conventional fluorescence imaging techniques. This imaging approach encompasses three distinct modalities: bioluminescence, chemiluminescence, and afterglow imaging.83,84 Bioluminescence is generated through enzyme-catalyzed reactions between luciferase and its substrate. Chemiluminescence capitalizes on direct chemical reactions between substrates and reactive species to emit light.85 Afterglow imaging fundamentally differs from other modalities in that it requires initial light excitation to create metastable energy states or chemical defects, followed by delayed photon emission after the cessation of light excitation.84,86–88 Owing to these unique features, bioluminescence, chemiluminescence, and afterglow imaging have emerged as powerful tools in biomedical research, offering distinct advantages for deep-tissue visualization, real-time molecular tracking, and high-sensitivity detection with minimal background interference.The rational design of activatable bioluminescence probes remains underdeveloped compared to fluorescence probes. Inspired by PET mechanisms widely used in the design of activatable fluorescence probes, Urano's group pioneered a bioluminescent enzyme-induced electron transfer (BioLeT) strategy for developing functional bioluminescent probes.89 Unlike conventional “caged luciferin” approaches, BioLeT leverages electron transfer from attached benzene substituents to the excited luminophore to control the luminescence of luciferin (Fig. 6a). To validate this strategy, a series of aminoluciferin derivatives bearing benzene moieties with different HOMO energy levels were synthesized. It was found that the luminescence intensity was highly dependent on the HOMO energy level of the benzene substituent, with efficient quenching occurring when the HOMO level exceeded −5 eV, supporting the BioLeT mechanism. Based on this principle, they designed a diaminophenylpropyl-aminoluciferin (DAL, Scheme 5) for NO detection. The o-phenylenediamine moiety in DAL probe could react selectively with NO to form benzotriazole, which significantly lowers the HOMO energy and thereby restored bioluminescence. This probe showed a 41-fold luminescence enhancement upon reaction with NO, with a detection limit of 1.51 μM (Fig. 6b). However, due to its emission in the visible range, this bioluminescent probe is not ideal for deep-tissue imaging. To address this limit, Chan et al. exploited the first NO-activated NIR-I bioluminescence probe (BL660-NO).90 This probe utilized o-phenylenediamine as the NO-responsive unit to mask the carboxylate group, thereby preventing enzymatic recognition by luciferase. Upon reaction with NO, the probe BL660-NO generated an acyl triazole intermediate, which spontaneously hydrolyzed to release the bioluminescence substrate BL660. The freed BL600 then reacted with luciferase to produce NIR-I bioluminescence (λem = 660 nm). This probe demonstrated excellent selectivity, biocompatibility, and sensitivity, enabling visualization of endogenous NO across systems ranging from cell cultures to various animal models. Importantly, the probe was successfully employed to investigate the effects of a high-fat diet on NO production within the tumour microenvironment.
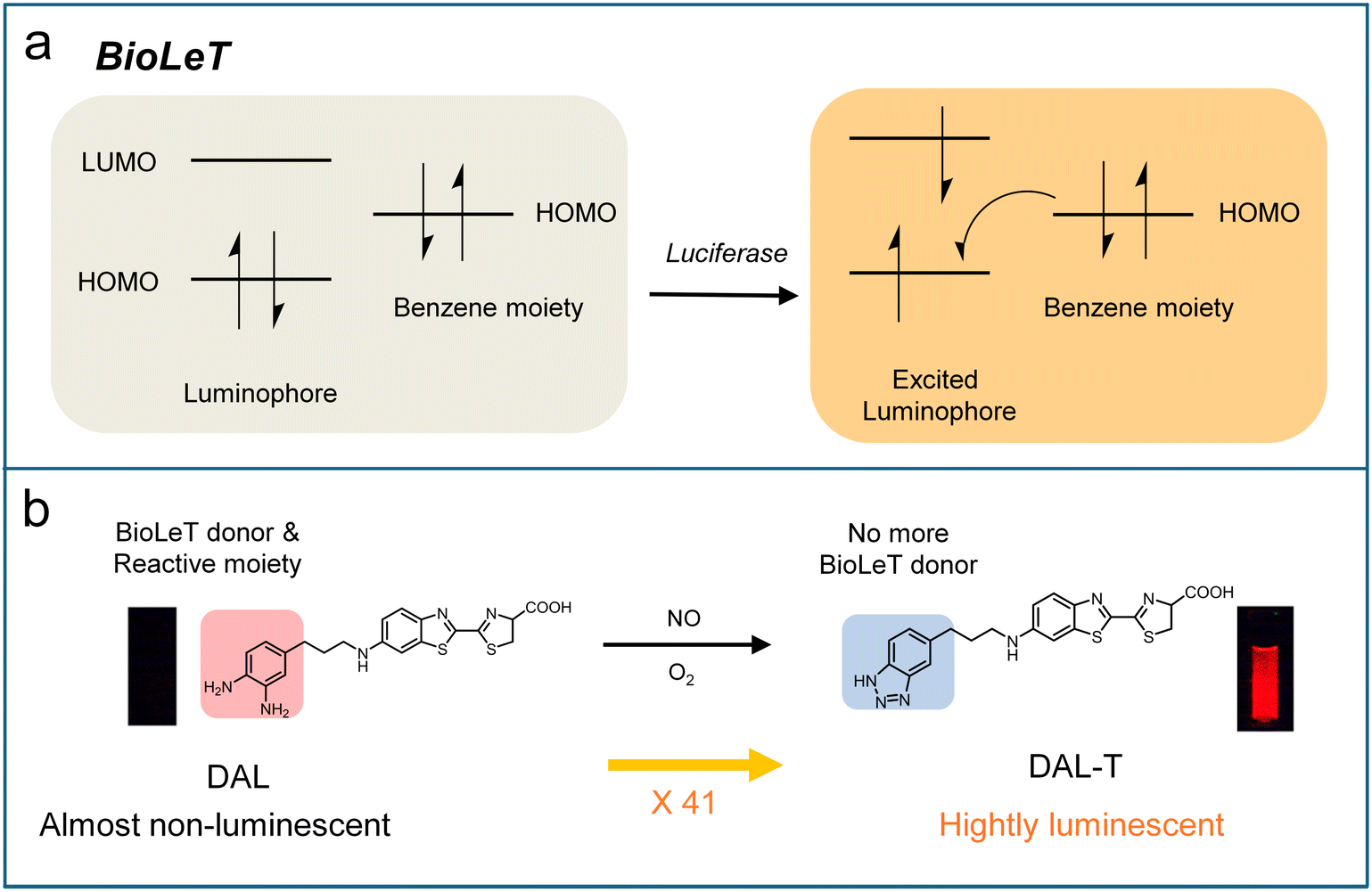 | ||
| Fig. 6 (a) Schematic diagram showing the BioLeT mechanism. (b) Molecular design of DAL based on the BioLeT mechanism. Reproduced from ref. 89 and with permission from American Chemical Society, copyright 2015. | ||
 | ||
| Scheme 5 Chemical structures of NO-responsive self-luminescence probes. | ||
Chemiluminescence offers greater flexibility than bioluminescence in probe design, as it does not rely on enzyme–substrate interactions. However, chemiluminescence probes generally suffer from short emission wavelengths, brief chemiluminescence half-lives, and low chemiluminescence quantum yields, severely restricting their utility in in vivo imaging applications. Recently, Pu et al. adopted a dual molecular engineering strategy on adamantylidene-1,2-dioxetane scaffolds by combining atomic substitution with the incorporation of electron-withdrawing groups to develop a series of highly efficient NIR-I chemiluminophores (Table 2).91 The optimized chemiluminophore DPDO (Fig. 7) showed a long emission (750 nm), an extended half-life (7.7 h), and a record-high quantum yield (2.7% einstein per mol). To enable specific monitoring of M1 TAMPs in the tumour microenvironment, DPDO was further modified with a 2 kDa polyethylene glycol (mPEG2k) and locked with gamma-glutamyl transferase (GGT)-cleavable γ-glutamate and NO-cleavable o-phenylenediamine, affording a hydrophilic tandem-locked chemiluminescence probe (DPDGN, Scheme 5). Notably, GGT and NO served as biomarkers of tumour and M1 macrophage, respectively. DPDGN intrinsically showed negligible chemiluminescence signals, but exhibited a 26.6-fold enhancement in chemiluminescence emission at 725 nm only upon exposure to both NO and GGT (Fig. 7a and b). Due to its high specificity, the DPDGN probe successfully realized the detection of M1 TAMs in the tumour-bearing mice undergoing R837 immunotherapy (Fig. 7c and d).
| Chemiluminophores | Adsorption λabs (nm) | Emission wavelength (nm) | Half-life | Quantum yield Φcl (×10−2 einstein per mol) |
|---|---|---|---|---|

|
502 | 750 | 7.7 h (PBS/DMSO = 6/4) | 2.7 |

|
507 | 800 | 5.7 h (PBS/DMSO = 6/4) | 1.0 |

|
484658 |
760 | 3.2 h (PBS/DMSO = 6/4) | 0.6 |

|
504664 |
820 | 0.8 h (PBS/DMSO = 6/4) | 0.1 |
 | ||
| Fig. 7 (a) Schematic illustration of the sequential chemiluminescence activation mechanism. (b) Chemiluminescence spectra of DPDGN (30 μM) in the presence of individual biomarkers or combined biomarkers in PBS (10 mM, pH 7.4) at 37 °C. (c) Real-time chemiluminescence imaging in orthotopic lung tumour after intratracheal injection of DPDGN. The inhibitor GGsTop was intraperitoneally injected 12 h before chemiluminescence imaging. (d) Quantified chemiluminescence signal from the lung area of 4T1-bearing mice in (b) (n = 3 independent mice, mean ± s.d.). Reproduced from ref. 91 and with permission from Wiley, copyright 2024. | ||
Both bioluminescence and chemiluminescence imaging suffer from signal decay resulting from continuous substrate consumption, which constrains their effectiveness for longitudinal studies of biological processes. In contrast, afterglow imaging captures persistent luminescence after excitation ceases and permits repeated reactivation, offering superior capabilities for longitudinal biological imaging. Despite these advantages, afterglow imaging remains limited by luminescent attenuation over time, which compromises quantitative accuracy, and the inert structure of many existing afterglow materials, which restricts the development of activatable probes for specific detection. To address these issues, Zhang et al. developed an innovative ratiometric afterglow nanoplatform (RAN) based on an afterglow resonance energy transfer (ARFT) strategy, enabling the customization of activatable afterglow probes for the specific and quantitative detection of biological analytes.92 The RAN system was constructed via self-assembly of four components: responsive molecules, an afterglow matrix (poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene], MEHPPV), afterglow initiators, and surfactants (F127) (Fig. 8a). Under light excitation, the afterglow initiator generates singlet oxygen, which initiates the afterglow matrix (acting as the energy donor) to form an unstable PPV-dioxetane intermediate, resulting in an initial afterglow emission (AF1). The energy from AF1 is then transferred to responsive molecules (energy acceptors), generating a longer-wavelength afterglow signal (AF2) (Fig. 8b). This design enables quantitative detection of analytes by calculating the afterglow intensity ratio (AF2/AF1). As a proof-of-concept for NO detection, a NO-activated ratiometric afterglow nanoprobe (RAN1) was developed by employing a typical benzo[c][1,2,5]thiadiazole-5,6-diamine-based semiconducting compound (NRM, Fig. 8c) as the NO-responsive molecule and a brominated boron dipyrromethene photosensitizer (BDP) as the afterglow initiator. In the presence of NO, the NRM undergoes oxidative cyclization to generate a 5H-[1,2,3]triazolo[4,5-f]-2,1,3-benzothiadiazole-based luminophore (NRM-NO), improving the ICT effect and resulting in a red-shifted emission at 830 nm (Fig. 8c). According to the sensing mechanism (Fig. 8a), RAN1 could emit two corresponding afterglow signals (AF1 at 600 nm from MEHPPV and AF2 at 830 nm from NRM-NO), allowing for quantitative NO detection via the AF2/AF1 ratio. The experiment results demonstrated that RAN1 exhibited brighter AF2 and weaker AF1 with increasing NO concentration after 660 nm laser pre-irradiation (Fig. 8d). The afterglow intensity ratio (AF2/AF1) showed a linear correlation with NO concentration, with a detection limit of 0.21 μM. Importantly, the ratiometric signal was independent of laser power, irradiation duration, and acquisition time, showing the sensing reliability. Finally, RAN1 successfully enabled visualization of endogenous NO in inflammatory regions and tumor-associated M1 macrophages through ratiometric afterglow imaging (Fig. 8e).
 | ||
| Fig. 8 (a) Schematic illustration of ARET-based ratiometric nanoplatform. (b) Schematic diagram of the probable responsive mechanism of afterglow probes. AF1 and AF2 represent the afterglow for afterglow substrate (MEHPPV) and responsive molecules, respectively. A12RET represents the afterglow resonance energy transfer from AF1 to AF2. (c) Structural change of the responsive molecule NRM before and after response to NO. (d) Afterglow images of RAN1 treated with different NO concentrations. (e) Representative afterglow images, as a function of post-injection time of RAN1 (10 μg mL−1) in LPS or PBS-pretreated mice. Reproduced from ref. 92 and with permission from Springer Nature, copyright 2022. | ||
5. Organic probes for NO-activatable photoacoustic imaging
NIR photoacoustic (PA) imaging is an advanced hybrid technique that integrates optical excitation with ultrasonic detection, offering substantial advantages in biomedical imaging. Upon absorption of NIR light (700–1700 nm), PA probes undergo localized thermoelastic expansion, generating high-frequency ultrasound signals.93 This unique mechanism effectively bypasses the inherent limitations of conventional optical imaging, such as light scattering and tissue autofluorescence, enabling high-resolution imaging at depths of several centimeters.94,95 Unlike fluorescence imaging, where signal intensity can be significantly influenced by environmental factors, PA signal intensity depends primarily on the probe's optical absorption coefficient and photothermal conversion efficiency, ensuring superior stability and reliability.96,97 In particular, ratiometric PA imaging with self-calibration enables accurate and quantitative detection in complex biological environments.5.1 NIR-I photoacoustic imaging
Due to its exceptional photochemical and chemical stability, Chan et al. utilized the boron-azadipyrromethene (aza-BODIPY) dye platform to design NO-responsive photoacoustic (PA) probes.98 Five candidate probes (APNOs, Scheme 6) were created by incorporating different NO-responsive units into the aza-BODIPY platform and systematically evaluated for performance. APNO-1 responded to NO via cyclization of o-phenylenediamine, resulting in an adsorption blue-shift from 731 to 676 nm and enabling ratiometric PA detection of NO. However, its practical application was limited by instrumentation constraints and low synthetic yield. APNO-2, featuring an o-aminophenol group, underwent NO-mediated deamination to induce an adsorption blue-shift from 735 to 676 nm, achieving a 13.1-fold ratiometric PA enhancement (750/680 nm excitation). Despite its strong signal response, the slow reaction kinetics reduced its practicality. APNO-3, a structural isomer of APNO-2, showed minimal change in adsorption wavelength after reaction with NO and was affected by pH within the biological range. To enhance NO reactivity and prevent unwanted side reactions, APNO-4 was designed with an N-methylaniline trigger and a methoxy substituent in place of the hydroxyl group to avoid pH sensitivity. In the presence of NO, it underwent rapid N-nitrosation, resulting in an 86 nm blue shift in absorption from 764 to 678 nm. However, the poor aqueous solubility of the nitrosated product led to aggregation and inconsistent performance. To address this issue, APNO-5 was developed as an analogue of APNO-4, featuring a PEGylated tetraalkylammonium group to enhance water solubility. APNO-5 exhibited an 18.6-fold ratiometric PA turn-on response to NO and was able to non-invasively detect endogenous NO in a mouse model of LPS-induced inflammation, producing stable and robust ratiometric PA signals. However, APNO-5 suffers from low sensitivity, which is insufficient to detect low NO levels in cancer. To improve the sensitivity, Chan et al. proposed a new design strategy based on steric relaxation to planarize the aza-BODIPY dye platform to further enhance PA performance.99 By replacing bulky phenyl rings with smaller thiophene groups, three probes (SR-APNO-1, SR-APNO-2, and SR-APNO-3, Scheme 6) were synthesized. Among them, SR-APNO-3, featuring two thiophenes, exhibited the most favourable properties, including the highest molar absorptivity and a red-shifted absorbance ideal for PA imaging windows. After N-nitrosation with NO, SR-APNO-3 exhibited a maximal absorbance shift from 790 to 704 nm and a 4.4-fold increase in ratiometric PA response compared to APNO-5. Owing to the high sensitivity, SR-APNO-3 enabled noninvasive, real-time detection of endogenous NO in both LPS-induced inflammation and murine breast cancer models. Due to its deep tissue penetration and high spatial resolution, PA imaging has emerged as a promising tool for brain imaging. However, achieving effective PA brain imaging of NO remains challenging because traditional PA probes suffer from poor blood–brain barrier (BBB) permeability. To conquer this obstacle, Liu et al. designed a series of NO-activated ratiometric PA probes (PANO–1–3, Scheme 6) based on a meso-trifluoromethyl BODIPY (CF3-BODIPY) core.100 The introduction of phenyl groups bearing various substituents at the 5-position of the CF3-BODIPY scaffold not only redshifts the probes' absorption wavelengths to the NIR region but also facilitates the modulation of their lipophilicity (logP value). Among these probes, PANO–2 exhibited an optimal logP value (2.1), allowing it to effectively penetrate the blood–brain barrier. Upon reacting with NO, the PANO–2 probe displayed a maximum absorption redshift from 785 nm to 705 nm (Fig. 9a). Dual-channel PA imaging experiments (790/690 nm excitation) confirmed that the PA signal at 790 nm gradually decreased, while the PA signal at 690 nm increased after adding NO to the PANO–2 probe solution (Fig. 9b). The PA ratio (PA690/PA790) displayed a linear correlation with NO concentration (Fig. 9c). These properties enabled PANO–2 to achieve real-time, ratiometric three-dimensional (3D) PA imaging of endogenous NO in a Parkinson's disease (PD) mouse model. Furthermore, PANO–2 was successfully used for screening natural polyphenols with potential neuroprotective effects against PD by monitoring NO levels.
 | ||
| Scheme 6 Chemical structures of NO-activated photoacoustic probes. | ||
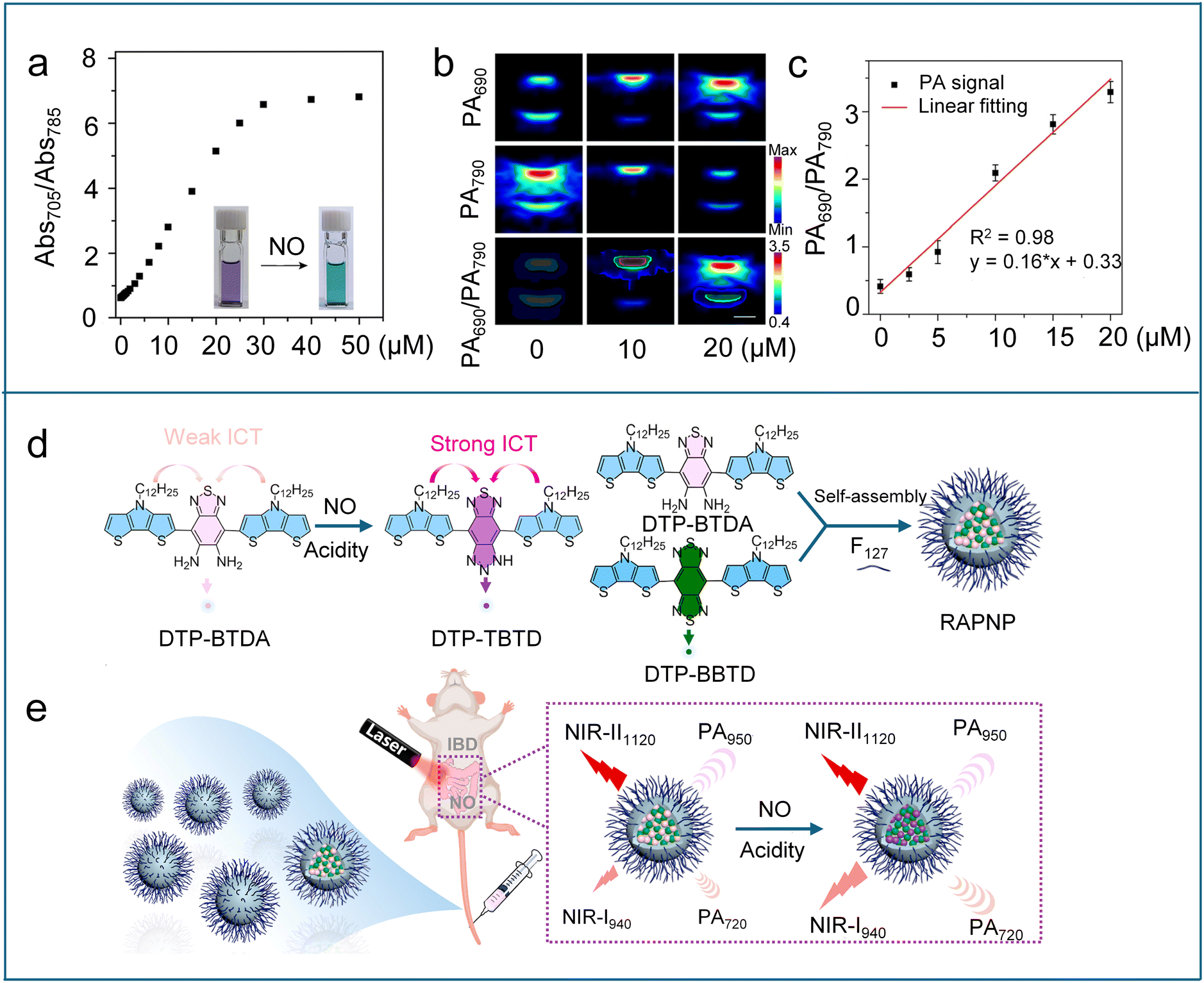 | ||
| Fig. 9 (a) Absorbance ratio (Abs705/Abs785) of PANO–2 at different NO concentrations in PBS buffer. Insets: photographs of PANO–2 solutions in the absence and presence of NO. (b) PA images of the PANO–2 solution recorded at different NO concentrations upon excitation at 690 and 790 nm, respectively, and the corresponding ratiometric PA images (PA690/PA790). (c) The PA ratios (PA690/PA790) of PANO–2 at different NO concentrations and the linear fitting (red). Reproduced from ref. 100 with permission from American Chemical Society, copyright 2024. (d) NO-reactive mechanism of DTP-BTDA and construction of RAPNP. (e) Schematic showing the activation of the fluorescence signal at 940 nm and the PA signal at 720 nm of RAPNP in IBD mice by the endogenous NO. Reproduced from ref. 102 with permission from Elsevier, copyright 2023. | ||
Most PA probes are activated by a single stimulus, which often limits the specificity for in vivo imaging. To achieve accurate and specific detection of cancer in vivo, Zhang et al. reported a “dual-key–one-lock” PA nanoprobe (DATN) that responded exclusively to the coexistence of NO and tumour acidity.101 DATN was constructed by co-encapsulating a typical NO-responsive molecule (NRM, Scheme 6) and NO-insensitive internal reference (NIM) within F127 nanoparticles. Upon simultaneous exposure to NO and an acidic environment, DATN exhibited a marked increase in absorbance at 680 nm, while the reference signal at 950 nm from NIM remained unchanged, enabling reliable ratiometric PA measurements (PA680/PA950). Compared to stimulation by either NO alone or acidity alone (pH 5.4), dual stimulation produced a significantly higher PA680/PA950 ratio, highlighting the superiority of the dual-activatable design. Benefiting from the elevated NO levels and acidic conditions typical of the tumour microenvironment (TME), DATN enabled accurate and specific ratiometric photoacoustic imaging of cancer in vivo.
Recently, Wu et al. adopted a similar strategy to develop a NO/acid-responsive nanoprobe RAPNP by using DTP-BTDA as the responsive molecule and DTP-BBTD as the nonresponsive internal reference (Fig. 9d).102 Under weakly acidic environments, DTP-BTDA underwent NO-mediated cyclization to generate DTP-TBTD with a strong ICT effect. This transformation induced a marked enhancement in both the absorption at 720 nm and the fluorescence emission at 940 nm for the RAPNP probe. In contrast, the maximal absorption and emission wavelengths of DTP-BBTD (950 nm and 1120 nm, respectively) remained unchanged under NO/acidity stimuli. The immutable long-wavelength NIR absorption and emission of DTP-BBTD make it very suitable as an internal reference for the ratiometric NIR fluorescence/PA imaging. The ratiometric NIR fluorescence/PA dual-modal imaging approach, which combines the strengths of both imaging modalities, enables more accurate, reliable, and sensitive molecular imaging and detection. Leveraging the elevated NO levels and the weakly acidic environment characteristic of inflammatory bowel disease (IBD), RAPNP successfully enabled detection of endogenous NO through NIR fluorescence/PA dual-modal ratiometric imaging (Fig. 9e).
5.2 NIR-II photoacoustic imaging
Most reported PA imaging agents use NIR-I light (650–900 nm) to generate ultrasound signals. However, this wavelength range suffers from significant interference from endogenous PA-active biomolecules like hemoglobin. Shifting to longer excitation wavelengths, specifically NIR-II (1000–1700 nm), reduces background noise and enables enhanced sensitivity for detecting low-abundance targets, such as NO. Towards this end, Chan et al. utilized a two-phase optimization strategy to successfully construct the first NIR-II PA probe, APNO-1080, for deep-tissue NO imaging.103 APNO-1080 integrated an optimized and highly reactive p-anisidine trigger onto a low-aggregation NIR-II cyanine platform (IR-1048) (Scheme 6). The probe could rapidly react with NO to form the N-nitrosated product, which had a 1080 nm absorption maximum with no spectral overlap with APNO-1080 and an enhanced extinction coefficient (Fig. 10a). In vitro tests using a tissue-mimicking phantom showed that APNO-1080 was 17.7-fold more sensitive than an NIR-I analogue. To evaluate the in vivo imaging capability, PA imaging of APNO-1080 was performed in both an orthotopic breast cancer model and a heterotopic lung cancer model (Fig. 10b and c). Compared to control mice without tumours, the normalized turn-on response was 1.3 ± 0.12 for the breast cancer model and 1.65 ± 0.07 for the lung cancer model. Recently, Scott designed a series of xanthene-based shortwave infrared (SWIR, 900–1400 nm) dyes (SCR).104 By capping the carboxylate group of a SCR dye with o-phenylenediamine, a NO-responsive PA probe (SCR-NO, Scheme 6) was constructed with PA-active “open” form. After reaction with NO, an acyl triazole intermediate was generated and spontaneously underwent rate-limiting hydrolysis to release a free dye in PA-inactive lactone form. To enable ratiometric PA imaging, they formulated a ratiometric nanoprobe (rNP-NO) through precise co-encapsulation of SCR-NO with the reference dye IR-1061 in phospholipid-polymer (DSPE-PEG) nanoparticles. The nanoprobe demonstrated high specificity and sensitivity for monitoring NO levels in a drug-induced liver injury model through deep-seated PA imaging.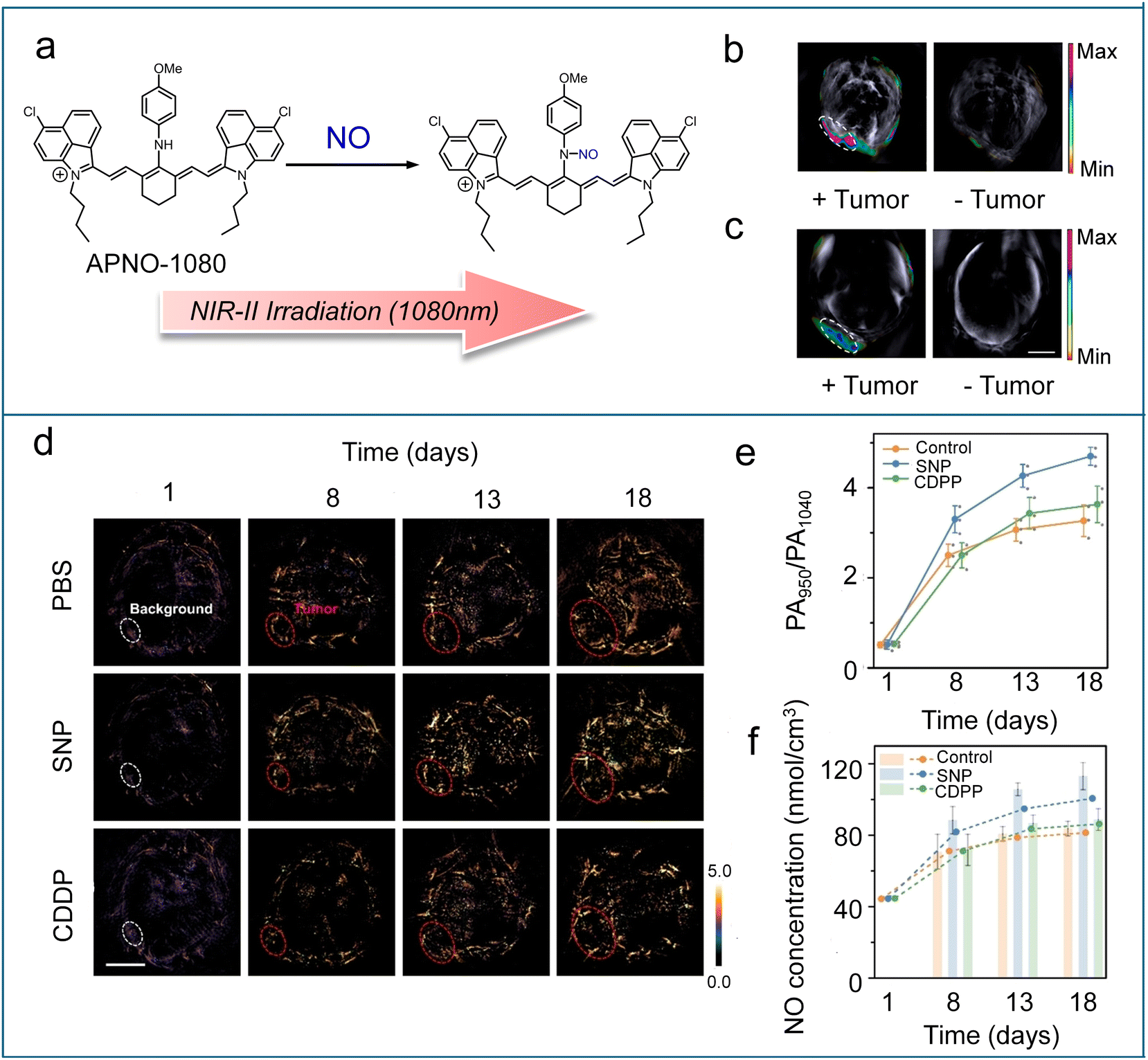 | ||
| Fig. 10 (a) Schematic showing the reaction between APNO-1080 and NO. (b) Representative PA images of a 4T1-Luc tumour and the tumour-less control after treatment with APNO-1080 (50 μM). (c) Representative PA images of a heterotopic A549-Luc2 tumour and the tumour-less control after treatment with APNO-1080. Reproduced from ref. 103 with permission from American Chemical Society, copyright 2021. (d) Ratiometric PA images of tumours in different treatment groups (SNP, sodium nitroprusside; CDDP, cisplatin). (e) PA ratios of tumours from (d). (f) Line graph: NO concentration levels of tumours were calculated from the average PA ratio in (e). Reproduced from ref. 105 and with permission from Wiley, copyright 2024. | ||
However, the aforementioned NIR-II photoacoustic probes for NO detection are constrained by poor photostability (particularly with cyanine dyes), difficulties in achieving ratiometric responses, and reproducibility and leakage issues associated with nanoparticles. In 2024, Liu et al. developed a novel class of NIR-II PA probes (SPANO–1–2, Scheme 6) based on an ethylene-bridged BODIPY platform (BisBDP1).105 The probes featured an N-methylaniline group for NO-specific nitrosation, which induced a hypsochromic shift from 1010 nm to 950 nm, thereby enabling ratiometric detection towards NO. SPANO–1–2 demonstrated superior photostability, rapid reaction kinetics, low detection limitation (nanometer level), and high selectivity for NO. Thanks to the incorporation of PEG and biotin, SPANO–2 had excellent water solubility and tumor targeting capability, making it suitable for in vivo studies. SPANO–2 enabled high spatial resolution ratiometric imaging and quantitative analysis of tumour NO levels in living mice treated with the drug (Fig. 10d and e). The imaging data revealed a critical NO concentration threshold of approximately 80 nmol cm−3, playing a key role in mediating the “dual-edged sword” effects of NO within tumours (Fig. 10f). Additionally, the results uncovered the possibility of NO-mediated communication between the tumour and the liver during both tumour progression and drug therapy.
6. Conclusions and outlook
Significant progress has been made in the development of organic molecular probes for NO detection, leveraging advanced optical modalities including near-infrared fluorescence, self-luminescence, and photoacoustic imaging. Near-infrared probes offer reduced light scattering and low background interference; self-luminescent probes eliminate the need for real-time light excitation, thereby enhancing signal-to-noise ratios; photoacoustic probes combine the merits of optical and ultrasonic imaging, affording high-resolution imaging at centimeter-level depths. These probes capitalize on the unique chemical reactivity of NO with specific recognition groups to achieve highly selective and sensitive detection. Moreover, the emergence of ratiometric and dual-activatable platforms has further improved the accuracy and reliability of NO imaging in complex biological systems. These innovations have greatly expanded the biomedical applications of NO probes, ranging from disease diagnostics to drug screening and therapeutic monitoring.Despite these advances, several critical challenges remain to be addressed. First, the limited diversity of NO-responsive recognition units restricts probe design flexibility and may affect the specificity of NO detection, particularly in low-concentration or complex biological environments. Integrating artificial intelligence into molecular engineering could accelerate the discovery of novel recognition groups with enhanced performance. Second, the development of multi-responsive probes that simultaneously detect NO and other biomarkers is still in its infancy and warrants further exploration. Such probes could provide deeper insights into disease mechanisms by capturing the interplay between NO and other pathological factors. Third, self-luminescent NO probes, particularly chemiluminescence and afterglow systems, remain extremely scarce. Innovative strategies are urgently needed to enhance the performance of self-luminescent NO probes, including extending signal half-life, achieving red-shifted emission (transitioning from NIR-I to NIR-II), and improving selectivity under physiological conditions. In addition, considering future clinical translation, probe molecules should be designed with optimal water solubility, high biocompatibility, excellent in vivo stability, good reproducibility, low immunogenicity, predictable pharmacokinetic behaviour, and the ability to selectively accumulate in target tissues. Overall, the continuous evolution of molecular probe design will contribute significantly to the precise and real-time visualization of NO in complex physiological and pathological processes, ultimately accelerating the development of next-generation diagnostic and therapeutic technologies.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Q. M. and Q. L. conceived the outline of the manuscript. W. A. and Z. W. wrote the original draft of the manuscript. Q. M. and Q. L. revised and supervised the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (32471428 and 22274107) and the Outstanding Youth Fund of Jiangsu Province (BK20230009).References
- S. Panthi, S. Manandhar and K. Gautam, Transl. Neurodegener., 2018, 7, 3 CrossRef.
- J. Kim and S. N. Thomas, Pharmacol. Rev., 2022, 74, 1146–1175 CrossRef CAS.
- C. Farah, L. Y. M. Michel and J. L. Balligand, Nat. Rev. Cardiol., 2018, 15, 292–316 CrossRef CAS PubMed.
- A. de Mel, F. Murad and A. M. Seifalian, Chem. Rev., 2011, 111, 5742–5767 CrossRef CAS.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- H. Hong, J. Sun and W. Cai, Free Radical Biol. Med., 2009, 47, 684–698 CrossRef CAS PubMed.
- Y. Chen, Nitric Oxide, 2020, 98, 1–19 CrossRef CAS.
- J. Garthwaite, Eur. J. Neurosci., 2008, 27, 2783–2802 CrossRef.
- C. Szabo, Nat. Rev. Drug Discovery, 2015, 15, 185–203 CrossRef PubMed.
- B. N. Gantner, K. M. LaFond and M. G. Bonini, Redox Biol., 2020, 34, 101550 CrossRef CAS.
- D. A. Wink, H. B. Hines, R. Y. Cheng, C. H. Switzer, W. Flores-Santana, M. P. Vitek, L. A. Ridnour and C. A. Colton, J. Leukocyte Biol., 2011, 89, 873–891 CrossRef CAS.
- M. Weng, X. Yang, Y. Ni, C. Xu, H. Zhang, J. Shao, N. Shi, C. Zhang, Q. Wu, L. Li and W. Huang, Sens. Actuators, B, 2019, 283, 769–775 CrossRef CAS.
- L. Wang, J. Zhang, X. An and H. Duan, Org. Biomol. Chem., 2020, 18, 1522–1549 RSC.
- F. L. Kiechle and T. Malinski, Am. J. Clin. Pathol., 1993, 100, 567–575 CrossRef CAS PubMed.
- T. Yang, A. N. Zelikin and R. Chandrawati, Small, 2020, 16, 1906744 CrossRef CAS.
- D. D. Thomas, L. A. Ridnour, J. S. Isenberg, W. Flores-Santana, C. H. Switzer, S. Donzelli, P. Hussain, C. Vecoli, N. Paolocci, S. Ambs, C. A. Colton, C. C. Harris, D. D. Roberts and D. A. Wink, Free Radical Biol. Med., 2008, 45, 18–31 CrossRef CAS PubMed.
- H. Ding, L. Luo, L. Su, J. Chen, Y. Li, L. Hu, K. Luo and X. Tian, Biosens. Bioelectron., 2024, 247, 115939 CrossRef CAS PubMed.
- M. Zhang and C. Tong, J. Hazard. Mater., 2024, 472, 134558 CrossRef CAS PubMed.
- Z. Dai, L. Tian, B. Song, X. Liu and J. Yuan, Chem. Sci., 2017, 8, 1969–1976 RSC.
- X. Wu, W. Shi, X. Li and H. Ma, Acc. Chem. Res., 2019, 52, 1892–1904 CrossRef CAS PubMed.
- M. Yuan, X. Fang, J. Liu, K. Yang, S. Xiao, S. Yang, W. Du and J. Song, Small, 2022, 19, 2206666 CrossRef.
- Z. Jiang, Z. Yang and W. Li, Adv. Healthcare Mater., 2023, 12, 2302089 CrossRef CAS.
- J. Chen, R. Chen, C. V. Chau, A. C. Sedgwick, Q. Xue, T. Chen, S. Zeng, N. Chen, K. K. Y. Wong, L. Song, Y. Ren, J. Yang, J. L. Sessler and C. Liu, J. Am. Chem. Soc., 2024, 146, 4620–4631 CrossRef CAS.
- W. Zhang, C. Chan, K. Zhang, H. Qin, B. Y. Yu, Z. Xue, X. Zheng and J. Tian, Adv. Mater., 2024, 36, e2311397 CrossRef.
- Y. Liu, L. Teng, B. Yin, H. Meng, X. Yin, S. Huan, G. Song and X. B. Zhang, Chem. Rev., 2022, 122, 6850–6918 CrossRef CAS PubMed.
- M. Y. Lucero and J. Chan, Nat. Chem., 2021, 13, 1248–1256 CrossRef CAS PubMed.
- Z. Gong, Z. Liu, Z. Zhang, Y. Mei and Y. Tian, CCS Chem., 2022, 4, 2020–2030 CrossRef CAS.
- J. Shang, Y. Cui, P. Wu, W. Chi, P. Yu, K. Fu, D. L. Phillips, Z. Sun, Y. Liu, L. Du and Y. Zhou, Sens. Actuators, B, 2024, 405, 135287 CrossRef CAS.
- L. Zhou, C. Liu, Y. Zheng, Z. Huang, X. Zhang and Y. Xiao, Anal. Chem., 2022, 94, 15678–15685 CrossRef CAS PubMed.
- Y. Zhou, X. Kuang, X. Yang, J. Li, X. Wei, W. J. Jang, S. S. Zhang, M. Yan and J. Yoon, Chem. Sci., 2024, 15, 19669–19697 RSC.
- G. Gan, T. Ma, G. Zhang, K. He and J. Hu, TrAC, Trends Anal. Chem., 2023, 168, 117340 CrossRef CAS.
- T. Han, Y. Sun, C. Zhao, H. Y. Wang, H. Yu and Y. Liu, J. Med. Chem., 2024, 67, 4026–4035 CrossRef CAS PubMed.
- J. Hu, R. Wang, W. Liao, J. Hu, L. Li, Z. Cheng and W. H. Chen, Anal. Chim. Acta, 2024, 1296, 342333 CrossRef CAS PubMed.
- R. Huang, Z. Zhang, Z. Shi, Y. Yang, J. Sun and F. Gao, Chem. Commun., 2024, 60, 6793–6796 RSC.
- J. Li, T. Xu, J. Chen, X. He, R. Ma, X. Lu, J. Yuan, M. Yao, Y. Tang and J. Li, Adv. Healthcare Mater., 2024, 13, 2402333 CrossRef CAS.
- Z. Wang, W. Wang, P. Wang, X. Song, Z. Mao and Z. Liu, Anal. Chem., 2021, 93, 3035–3041 CrossRef CAS PubMed.
- Q. Zhao, H. Zhao, Y. Guo, Z. Zhang, Y. Hu and Y. Tang, Anal. Chem., 2018, 90, 12663–12669 CrossRef CAS PubMed.
- R. Borlan, M. Focsan, D. Maniu and S. Astilean, Int. J. Nanomed., 2021, 16, 2147–2171 CrossRef.
- T. Pu, Y. Liu, Y. Pei, J. Peng, Z. Wang, M. Du, Q. Liu, F. Zhong, M. Zhang, F. Li, C. Xu and X. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 32226–32239 CrossRef CAS.
- S. J. Li, D. Y. Zhou, Y. Li, H. W. Liu, P. Wu, J. Ou-Yang, W. L. Jiang and C. Y. Li, ACS Sens., 2018, 3, 2311–2319 CrossRef CAS PubMed.
- Z. Mao, W. Feng, Z. Li, L. Zeng, W. Lv and Z. Liu, Chem. Sci., 2016, 7, 5230–5235 RSC.
- N. G. Zhegalova, G. Gonzales and M. Y. Berezin, Org. Biomol. Chem., 2013, 11, 8228–8234 RSC.
- X. Zhang, B. Wang, Y. Xiao, C. Wang and L. He, Analyst, 2018, 143, 4180–4188 RSC.
- Y.-H. Pan, X.-X. Chen, L. Dong, N. Shao, L.-Y. Niu and Q.-Z. Yang, Chin. Chem. Lett., 2021, 32, 3895–3898 CrossRef CAS.
- Y. Dong, X. R. Li, Q. Chen, R. Y. Guo, B. X. Tang, W. J. Kan, W. Zhang, Y. Hu, J. Li, Y. Zang and X. Li, Anal. Chem., 2020, 92, 699–706 CrossRef CAS PubMed.
- Z. Mao, H. Jiang, X. Song, W. Hu and Z. Liu, Anal. Chem., 2017, 89, 9620–9624 CrossRef CAS.
- Y. U. Yu Gabe, K. Kikuchi, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2004, 126, 3357–3367 CrossRef PubMed.
- E. Sasaki, H. Kojima, H. Nishimatsu, Y. Urano, K. Kikuchi, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2005, 127, 3685 Search PubMed.
- W. Liu, C. Fan, R. Sun, Y. J. Xu and J. F. Ge, Org. Biomol. Chem., 2015, 13, 4532–4538 RSC.
- J. Tang, Z. Guo, Y. Zhang, B. Bai and W. H. Zhu, Chem. Commun., 2017, 53, 10520–10523 RSC.
- Y. Huo, J. Miao, L. Han, Y. Li, Z. Li, Y. Shi and W. Guo, Chem. Sci., 2017, 8, 6857–6864 RSC.
- B. Wang, S. Yu, X. Chai, T. Li, Q. Wu and T. Wang, Chemistry, 2016, 22, 5649–5656 CrossRef CAS PubMed.
- S. Liu, Y. Zhu, P. Wu and H. Xiong, Anal. Chem., 2021, 93, 4975–4983 CrossRef CAS PubMed.
- S. He, P. Cheng and K. Pu, Nat. Biomed. Eng., 2023, 7, 281–297 CrossRef CAS.
- X. Zou, S. Yang, S. Jiao and Z. Zhang, Sens. Actuators, B, 2024, 421, 136491 CrossRef CAS.
- Y. Liu, H. Fan, Y. Wen, T. Jia, Q. Su and F. Li, Dyes Pigm., 2019, 166, 211–216 CrossRef CAS.
- Y. Huo, J. Miao, Y. Li, Y. Shi, H. Shi and W. Guo, J. Mater. Chem. B, 2017, 5, 2483–2490 RSC.
- S. Jiao, S. Yang, Y. Wang and A. Ma, Sens. Actuators, B, 2023, 374, 132790 CrossRef CAS.
- F. Xu, Q. Wang, L. Jiang, F. Zhu, L. Yang, S. Zhang and X. Song, Anal. Chem., 2022, 94, 4072–4077 CrossRef CAS.
- Y. Xi, S. Bai, Y. Tian, Y. Lv, L. Ji, W. Li, G. He and L. Yang, Bioorg. Chem., 2024, 148, 107476 CrossRef CAS.
- X. Wang, W. Wu, B. Yun, L. Huang, Z.-H. Chen, J. Ming, F. Zhai, H. Zhang and F. Zhang, J. Am. Chem. Soc., 2025, 147, 2182–2192 CrossRef CAS.
- X. Zhong, A. Patel, Y. Sun, A. M. Saeboe and A. M. Dennis, Angew. Chem., Int. Ed., 2024, 63, 2410936 Search PubMed.
- Y. Zhang, M. Zhao, D. Cheng, J. Zhu, B. Cheng, M. Miao, Q. Li and Q. Miao, ACS Appl. Nano Mater., 2024, 7, 22117–22129 CrossRef CAS.
- M. Zhao, W. Lai, B. Li, T. Bai, C. Liu, Y. Lin, S. An, L. Guo, L. Li, J. Wang and F. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202403968 CrossRef CAS PubMed.
- N. Gill, I. Srivastava and J. Tropp, Adv. Healthcare Mater., 2024, 13, 2401297 CrossRef CAS.
- J. Miao, M. Miao, Y. Jiang, M. Zhao, Q. Li, Y. Zhang, Y. An, K. Pu and Q. Miao, Angew. Chem., Int. Ed., 2023, 62, e202216351 CrossRef CAS PubMed.
- L. Xu, X. Wang, L. Huang, L. Dai, D. Tan and W. Lin, Small, 2025, 21, 2407918 CrossRef CAS.
- D. Hu, M. Zha, H. Zheng, D. Gao and Z. Sheng, Research, 2025, 8, 0583 CrossRef CAS PubMed.
- Y. Zhang, D. Liu, W. Chen, Y. Tao, W. Li and J. Qi, Adv. Mater., 2024, 36, e2409661 CrossRef PubMed.
- L. Guo, Q. Wang, F. Gao, Y. Liang, H. Ma, D. Chen, Y. Zhang, H. Ju and X. Zhang, Small, 2024, 20, e2405320 CrossRef PubMed.
- Y. Tang, Y. Li, Z. Wang, F. Pei, X. Hu, Y. Ji, X. Li, H. Zhao, W. Hu, X. Lu, Q. Fan and W. Huang, Chem. Commun., 2018, 55, 27–30 RSC.
- P. Yuan, X. Xu, D. Hu, Y. Chen, J. Fan, S. Yao, Y. Piao, Z. Zhou, S. Shao, N. K. H. Slater, Y. Shen and J. Tang, J. Am. Chem. Soc., 2023, 145, 7941–7951 CrossRef CAS PubMed.
- J. Chen, L. Chen, Y. Fang, F. Zeng and S. Wu, Cell Rep. Phys. Sci., 2022, 3, 100570 CrossRef CAS.
- Q. Xu, Y. Zhang, M. Zhu, C. Yan, W. Mao, W. H. Zhu and Z. Guo, Chem. Sci., 2023, 14, 4091–4101 RSC.
- Z. She, J. Chen, L. Sun, F. Zeng and S. Wu, Chem. Commun., 2022, 58, 13123–13126 RSC.
- Z. Zheng, J. Gao, R. Wang, C. Dong, X. Dong, J. Sun, L. Sun, X. Gu and C. Zhao, ACS Nano, 2023, 17, 22060–22070 CrossRef.
- Q. Fu, X. Yang, M. Wang, K. Zhu, Y. Wang and J. Song, ACS Nano, 2024, 18, 3916–3968 CrossRef CAS PubMed.
- B. Hu, Q. Liu, Y. Jiang, Y. Huang, H. Ji, J. Zhang, X. Wang, X. C. Shen and H. Chen, Angew. Chem., Int. Ed., 2024, 64, 2418378 Search PubMed.
- C. Wu, F. Zhang, Y. Mao, X. Qi, X. Wang, W. Zhang, B. Tang and P. Li, Chem. Biomed. Imaging, 2023, 1, 372–379 CrossRef CAS.
- S. Han, L. Yang, M. Liu, H. Li and X. Song, Chem. Eng. J., 2023, 463, e202301689 Search PubMed.
- X. Ren, S. Han, Y. Li, Y. Zeng, H. Li, C. Yao, L. Yang and X. Song, Anal. Chem., 2024, 96, 8689–8695 CrossRef CAS PubMed.
- F. Bai, W. Du, X. Liu, L. Su, Z. Li, T. Chen, X. Ge, Q. Li, H. Yang and J. Song, Anal. Chem., 2021, 93, 15279–15287 CrossRef CAS.
- H. Wu, Y. Fang, L. Tian, X. Liu, X. Zhou, X. Chen, H. Gao, H. Qin and Y. Liu, ACS Sens., 2023, 8, 3205–3214 CrossRef CAS PubMed.
- J. Zhu, W. Chen, L. Yang, Y. Zhang, B. Cheng, W. Gu, Q. Li and Q. Miao, Angew. Chem., Int. Ed., 2024, 63, e202318545 CrossRef CAS PubMed.
- R. Ding, D. Liu, Y. Feng, H. Liu, H. Ji, L. He and S. Liu, Chem.–Eur. J., 2023, 29, e202301689 CrossRef CAS PubMed.
- J. Zhu, L. Zhao, W. An and Q. Miao, Chem. Soc. Rev., 2025, 54, 1429–1452 RSC.
- C. Chen, H. Gao, H. Ou, R. T. K. Kwok, Y. Tang, D. Zheng and D. Ding, J. Am. Chem. Soc., 2022, 144, 3429–3441 CrossRef CAS PubMed.
- Y. Zhang, W. Xu, D. Cheng, M. Zhao, J. Xiong, Q. Li and Q. Miao, Angew. Chem., Int. Ed., 2025, e202500801 CAS.
- H. Takakura, R. Kojima, M. Kamiya, E. Kobayashi, T. Komatsu, T. Ueno, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2015, 137, 4010–4013 CrossRef CAS PubMed.
- A. K. Yadav, M. C. Lee, M. Y. Lucero, S. Su, C. J. Reinhardt and J. Chan, ACS Cent. Sci., 2022, 8, 461–472 CrossRef CAS PubMed.
- J. Huang, M. Xu, P. Cheng, J. Yu, J. Wu and K. Pu, Angew. Chem., Int. Ed., 2024, 63, e202319780 CrossRef CAS PubMed.
- Y. Liu, L. Teng, Y. Lyu, G. Song, X. B. Zhang and W. Tan, Nat. Commun., 2022, 13, 2216 CrossRef CAS.
- Y. Pan, J. Chen, Y. Zhang, Y. Ren, Z. Wu, Q. Xue, S. Zeng, C. Fang, H. Zhang, L. Zhang, C. Liu and J. Zeng, Mol. Pharm., 2024, 21, 1804–1816 CrossRef CAS.
- H. Chen, K. Li, L. Yuan and X.-B. Zhang, Chem. Commun., 2023, 59, 10520–10523 RSC.
- Y. Zhang, S. He, C. Xu, Y. Jiang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2022, 61, e202203184 CrossRef CAS.
- H. Mu, K. Miki, H. Harada, K. Tanaka, K. Nogita and K. Ohe, ACS Sens., 2020, 6, 123–129 CrossRef PubMed.
- J. Wu, L. You, S. T. Chaudhry, J. He, J.-X. Cheng and J. Mei, Anal. Chem., 2021, 93, 3189–3195 CrossRef CAS.
- C. J. Reinhardt, E. Y. Zhou, M. D. Jorgensen, G. Partipilo and J. Chan, J. Am. Chem. Soc., 2018, 140, 1011–1018 CrossRef CAS.
- C. J. Reinhardt, R. Xu and J. Chan, Chem. Sci., 2020, 11, 1587–1592 RSC.
- Z. Jiang, Z. Liang, Y. Cui, C. Zhang, J. Wang, H. Wang, T. Wang, Y. Chen, W. He, Z. Liu and Z. Guo, J. Am. Chem. Soc., 2023, 145, 7952–7961 CrossRef CAS.
- L. Teng, G. Song, Y. Liu, X. Han, Z. Li, Y. Wang, S. Huan, X. B. Zhang and W. Tan, J. Am. Chem. Soc., 2019, 141, 13572–13581 CrossRef CAS.
- P. Xiao, M. Liang, S. Yang, Y. Sun, J. Li, Z. Gu, L. Zhang, Q. Fan, X. Jiang and W. Wu, Biomaterials, 2023, 294, 121993 CrossRef CAS PubMed.
- M. Y. Lucero, A. K. East, C. J. Reinhardt, A. C. Sedgwick, S. Su, M. C. Lee and J. Chan, J. Am. Chem. Soc., 2021, 143, 7196–7202 CrossRef CAS PubMed.
- C. S. L. Rathnamalala, S. Hernandez, M. Y. Lucero, C. B. Swartchick, A. Kalam Shaik, N. I. Hammer, A. K. East, S. R. Gwaltney, J. Chan and C. N. Scott, Angew. Chem., Int. Ed., 2023, 62, e202214855 CrossRef CAS.
- Z. Jiang, C. Zhang, Q. Sun, X. Wang, Y. Chen, W. He, Z. Guo and Z. Liu, Angew. Chem., Int. Ed., 2024, 63, e202320072 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |