
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Label-free quantification of single-stranded DNA utilizing enzymatic digestion and an off-the-shelf glucose test strip†
Faisal
Hossain
 ab and
Michael J.
Serpe
*a
ab and
Michael J.
Serpe
*a
aDepartment of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada. E-mail: serpe@ualberta.ca
bDepartment of Chemistry, Faculty of Science, University of Chittagong, Chattogram 4331, Bangladesh
First published on 5th February 2025
Abstract
A method was developed for quantifying single-stranded DNA (ssDNA) through enzymatic digestion and using commercially available glucose test strips. The process involves the initial digestion of ssDNA using a combination of exonuclease 1 and alkaline phosphatase enzymes, leading to the liberation of phosphates from the ssDNA backbone as free orthophosphate. Subsequently, the orthophosphates react with maltose and maltose phosphorylase, producing equivalent amounts of glucose to orthophosphate. The resulting glucose, which can be related to the ssDNA concentration, can be measured amperometrically with an off-the-shelf glucose test strip connected to a mini potentiostat. This method offers versatility, allowing the determination of ssDNA, regardless of nucleotide-count or sequence, with increased sensitivity as the number of nucleotides (NT) in the DNA increases. The method exhibits a limit of detection of 780 nM for 22-NT, 527 nM for 53-NT, 422 nM for 75-NT, and 329 nM for 87-NT ssDNA, and a linear range of 0–2 μM. To selectively quantify a specific ssDNA target, a magnetic microparticle-based isolation step was incorporated, demonstrating high selectivity for quantifying a particular ssDNA target from a mixture. The method holds potential for label-free quantification of ssDNA that can have an impact in myriad fields.
Introduction
Deoxyribonucleic acid (DNA) is a biopolymer that encodes genetic information in the linear sequence of its nucleotides (NT). NTs, the “building blocks” of DNA, consist of a nitrogenous base, a deoxyribose sugar, and a phosphate group. In the double helix structure of DNA, two polyNT chains with complementary sequences hybridize together through hydrogen bonding between nucleobases, e.g., guanine binds to cytosine, and adenine binds to thymine. To say DNA is important is a tremendous understatement as it is what defines who we are as individuals. That being said, DNA analysis is important in a number of areas.1,2 For example, in forensic science, DNA is vital for analyzing crime scene samples to aid in the identification of individuals. Additionally, the detection of DNA with specific sequences holds importance in the food industry, where certain DNA sequences can be indicative of bacterial contamination or cross contamination (e.g., Halal certification).3,4 Furthermore, DNA quantification plays a pivotal role in clinical diagnostics. It allows for the detection and quantification of specific DNA sequences, DNA sequences indicative of the presence of viruses or bacteria. This capability is instrumental in the diagnosis and treatment of various health conditions.5–8Numerous analytical methods are available for quantification of DNA in solution, and the choice depends on the purpose of the analysis, required sensitivity, and resource availability. The most basic method that can be used for DNA quantification is UV-visible spectroscopy which utilizes the absorbance of DNA at 260 and 280 nm.9 However, this method is susceptible to errors due to potential interfering species in the solution that absorb at the same wavelengths and suffer from low sensitivity due to the measurement method itself. Quantitative polymerase chain reaction (qPCR) is a powerful tool for DNA quantification, relying on enzymatic DNA amplification and subsequent fluorescence detection.10 While qPCR offers high sensitivity, it necessitates the use of expensive equipment and qualified personnel for experimental execution. To overcome these problems, researchers have developed less resource-intensive and quantitative biosensors to quantify DNA in solution.5,7,11,12 For example, Kirimli et al. developed a lead magnesium niobate–lead titanate piezoelectric sensor to quantify single-stranded DNA (ssDNA) by utilizing a complementary probe DNA (pDNA) immobilized on a sensor plate.13 Upon addition of a sample, the target DNA (tDNA) binds to the immobilized probe DNA that causes a frequency change of the sensor that can be monitored in real-time. In another example, Can et al. developed an electrochemical hybridization biosensor to quantify target tDNA found in E. coli K12 using immobilized pDNA on a gold electrode and finally measuring amperometric signal changes upon target addition.14 Although most of the recent methods provide very good sensitivity for DNA detection, they mostly require sophisticated sensor fabrication processes, which are sometimes expensive and difficult to use in a resource-limited environment.15,16 Conventional colorimetric assays need laboratory-based spectrophotometers to quantify analytes and some field applications are semi-quantitative in nature and any background solution color can interfere with the analysis.
In this study, we present a method for quantifying ssDNA that eliminates the need for complicated sensor design and fabrication processes while not requiring sophisticated laboratory equipment. Instead, an off-the-shelf glucose test strip can be employed for ssDNA quantification. Specifically, the ssDNA that is being detected is digested using a combination of exonuclease 1 (EXO 1) and alkaline phosphatase (AP) to release most of the phosphates from its backbone. EXO 1 degrades the phosphodiester bonds in the DNA backbone, while AP cleaves any remaining phosphomonoester bonds, resulting in the generation of free orthophosphate in the solution. Previously, we developed an orthophosphate sensor utilizing an enzymatic reaction that involves maltose phosphorylase and maltose and the reaction generates glucose and beta-D-glucose 1-phosphate as the final products.17 The amount of glucose has been found to be equivalent to the concentration of orthophosphate and the resultant glucose was quantified utilizing off-the-shelf glucose test strips. Based on the previously optimized orthophosphate sensor, in this study, we utilized the sensor to generate glucose from the orthophosphate obtained from DNA digestion. We found that the concentration of the produced glucose is directly proportional to the initial concentration of ssDNA in the solution and its NT count. Subsequently, the produced glucose can be measured amperometrically using a commercially available Accu-Chek Guide glucose test strip from Roche Diabetic Care, connected to a handheld mini-potentiostat (BDTminiSTAT100, from Biodevice Technology, Ltd, Ishikawa, Japan). Commercially available electrochemical glucose test strips use glucose oxidase or glucose dehydrogenase enzyme to selectively oxidize glucose from blood samples. This oxidation reaction generates 2 electrons for each glucose molecule that is carried by the redox mediators to the working electrode of the test strip.18 The voltage applied across the working and counter electrode (450 mV) is large enough so that the reaction at the electrode is instantaneous and the generated amperometric signal is limited by diffusion process. The amperometric current generated by the test strip follows the Cottrell equation:18
| i = (nFAD1/2C)/(πt)1/2 | (1) |
Experimental
Materials
EXO 1 (E. coli) and nuclease buffer were from New England Bio Lab Ltd. (Whitby, Ontario). AP from bovine intestinal mucosa and MP from Enterococcus sp., as well as boric acid and ammonium persulfate (APS), were purchased from Millipore Sigma (Milwaukee, Wisconsin). D-Maltose monohydrate, Dynabead streptavidin M280 magnetic microparticles, sodium citrate, and bromophenol blue were purchased from Sigma-Aldrich (Oakville, Ontario). Citric acid monohydrate was obtained from Caledon Laboratories Ltd (Edmonton, Alberta). Tetramethylethylenediamine (TEMED) was from Bio-Rad Laboratories, Inc. (Hercules, California). Stains-all, 95% powder, and formamide were from Fisher Chemical (Schwerte, Germany). Tris-HCl was from EMD Chemical Inc. (Darmstadt, Germany). Sodium phosphate monobasic monohydrate was purchased from EMD Chemicals Inc (Oakville, Ontario). Accu-Chek Guide and Accu-Chek Aviva test strips were purchased from a local pharmacy.Target ssDNA and probe DNA
All ssDNAs were purchased from Integrated DNA Technologies Inc. (Coralville, Iowa) with HPLC purification. Several ssDNAs were chosen with varying NT counts and different sequences: 22, 53, 75, and 87-NT, which were used to assess the sensitivity of this method. The 22, 75, and 53-NT ssDNA were previously reported as ssDNA aptamers.19–21 A biotin-trimethylene glycol (TEG)-pDNA (14-NT) immobilized on streptavidin magnetic microparticles was used to capture the 53-NT tDNA to show the selectivity of this method. All the sequences of the ssDNAs and their corresponding melting temperatures are shown in Table 1.| Type | Nucleotide count | Nucleotide sequence | Melting temperature, Tm (°C) in 50 mM NaCl |
|---|---|---|---|
| — | 22 | 5′-GCC GTT TGG GCC CAA GTT CGG C-3′ | 66.2 |
| Model target ssDNA | 53 | 5′-GCA GAA CTT ACG ACC CAG GGG GGT GGA CAG GCG GGG GTT AGG GGG GTC GTA AG-3′ | 75.1 |
| — | 75 | 5′-ATA CGA GCT TGT TCA ATA CGA AGG GAT GCC GTT TGG GCC CAA GTT CGG CAT AGT GTG GTG ATA GTA AGA GCA ATC-3′ | 71.2 |
| — | 87 | 5′-TAT CGC ATA GCG ATA CGA GCT TGT TCA ATA CGA AGG GAT GCC GTT TGG GCC CAA GTT CGG CAT AGT GTG GTG ATA GTA AGA GCA ATC-3′ | 71.6 |
| Probe DNA | 14 | 5′-GTC GTA AGT TCT GC/3 Bio-TEG/-3′ | 42.3 |
Immobilization of probe DNA on magnetic microparticles
50 μL of M280 streptavidin magnetic microparticles (10 mg mL−1) were washed once with 200 μL of hybridization buffer (20 mM Tris-HCl, 0.5 mM MgCl2, 1.5 M NaCl, and 5% DMSO, pH 7.4). The washing involved gentle vortexing for approximately 10 s, followed by magnetic precipitation for 2 min and discarding the supernatant. Subsequently, 100 μL of 5 μM pDNA was allowed to react with the washed magnetic microparticles overnight at room temperature. Afterward, the microparticles underwent a single wash with a hybridization buffer to remove any unbound pDNA, rendering them ready to selectively capture the corresponding tDNA from the solution.Procedure
The first step of this method involves the isolation of tDNA from its matrix. First, magnetic microparticles modified with pDNA were dispersed in 100 μL buffer and incubated at 50 °C for 30 min, followed by magnetic precipitation and discarding the supernatant solution. The Tm values are reported in Table 1 can be related to the corresponding incubation temperature needed to facilitate the unfolding of the probe/target ssDNA. This step was included to break any self-hybridization of the pDNA, allowing for the highest binding potential to tDNA. Simultaneously, a 100 μL tDNA solution in buffer was incubated at 100 °C for 10 min to facilitate the unfolding of any self-hybridized regions. Subsequently, the tDNA solution was added to the pDNA-modified magnetic microparticles, and the mixture was allowed to react for 1 h to enable hybridization between tDNA and pDNA. The tDNA can be captured by the magnetic particles through its hybridization with pDNA, and after magnetic separation for 2 min, the supernatant was decanted, successfully isolating the tDNA from the matrix. The magnetic microparticles were then dispersed in 50 μL deionized (DI) water and heated at 100 °C for 10 min to “melt” the double-stranded pDNA/tDNA into their respective single strands, and releasing the tDNA from the magnetic microparticles into solution while the magnetic microparticles can be separated from the supernatant using a magnet.The second step involved the determination of tDNA through enzyme treatment and subsequent detection using a glucose test strip. To accomplish this, 30 μL of the isolated tDNA solution in DI water (concentrations 0–4 μM) was reacted with an 8 μL digestion enzyme mix at 37 °C for 1 h. The digestion enzyme mix contains 4 μL of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1 EXO 1 enzyme, 2 μL of 10× nuclease buffer (10 mM 1,3-bis(tris(hydroxymethyl)methylamino)propane-HCl (Bis-Tris-Propane-HCl)), 10 mM MgCl2, 1 mM dithiothreitol, pH 7.0, and 2 μL of 1000 U mL−1 AP enzyme. During this reaction, nearly all of the single-stranded DNA breaks down into its nucleotides and generates free orthophosphate. The reaction product was then treated with 11 μL of a second reagent mixture (4 μL of 200 U mL−1 MP, 2 μL of 100 mM maltose, and 4 μL of 1 M citrate buffer, pH 4.6) at 30 °C for 90 min. In this step, orthophosphate in the solution reacts with maltose in the presence of MP to produce glucose. The solution containing glucose was introduced into the Accu-Chek Guide test strip connected to a potentiostat, and the amperometric signal was measured at 450 mV applied potential. The obtained current from 3 to 20 s was averaged. The ratio (relative current) of the averaged current of the sample to the corresponding averaged current obtained from the reagent blank was calculated and plotted against the sample concentration to generate calibration curves. The experimental steps are illustrated in Scheme 1.
000 U mL−1 EXO 1 enzyme, 2 μL of 10× nuclease buffer (10 mM 1,3-bis(tris(hydroxymethyl)methylamino)propane-HCl (Bis-Tris-Propane-HCl)), 10 mM MgCl2, 1 mM dithiothreitol, pH 7.0, and 2 μL of 1000 U mL−1 AP enzyme. During this reaction, nearly all of the single-stranded DNA breaks down into its nucleotides and generates free orthophosphate. The reaction product was then treated with 11 μL of a second reagent mixture (4 μL of 200 U mL−1 MP, 2 μL of 100 mM maltose, and 4 μL of 1 M citrate buffer, pH 4.6) at 30 °C for 90 min. In this step, orthophosphate in the solution reacts with maltose in the presence of MP to produce glucose. The solution containing glucose was introduced into the Accu-Chek Guide test strip connected to a potentiostat, and the amperometric signal was measured at 450 mV applied potential. The obtained current from 3 to 20 s was averaged. The ratio (relative current) of the averaged current of the sample to the corresponding averaged current obtained from the reagent blank was calculated and plotted against the sample concentration to generate calibration curves. The experimental steps are illustrated in Scheme 1.
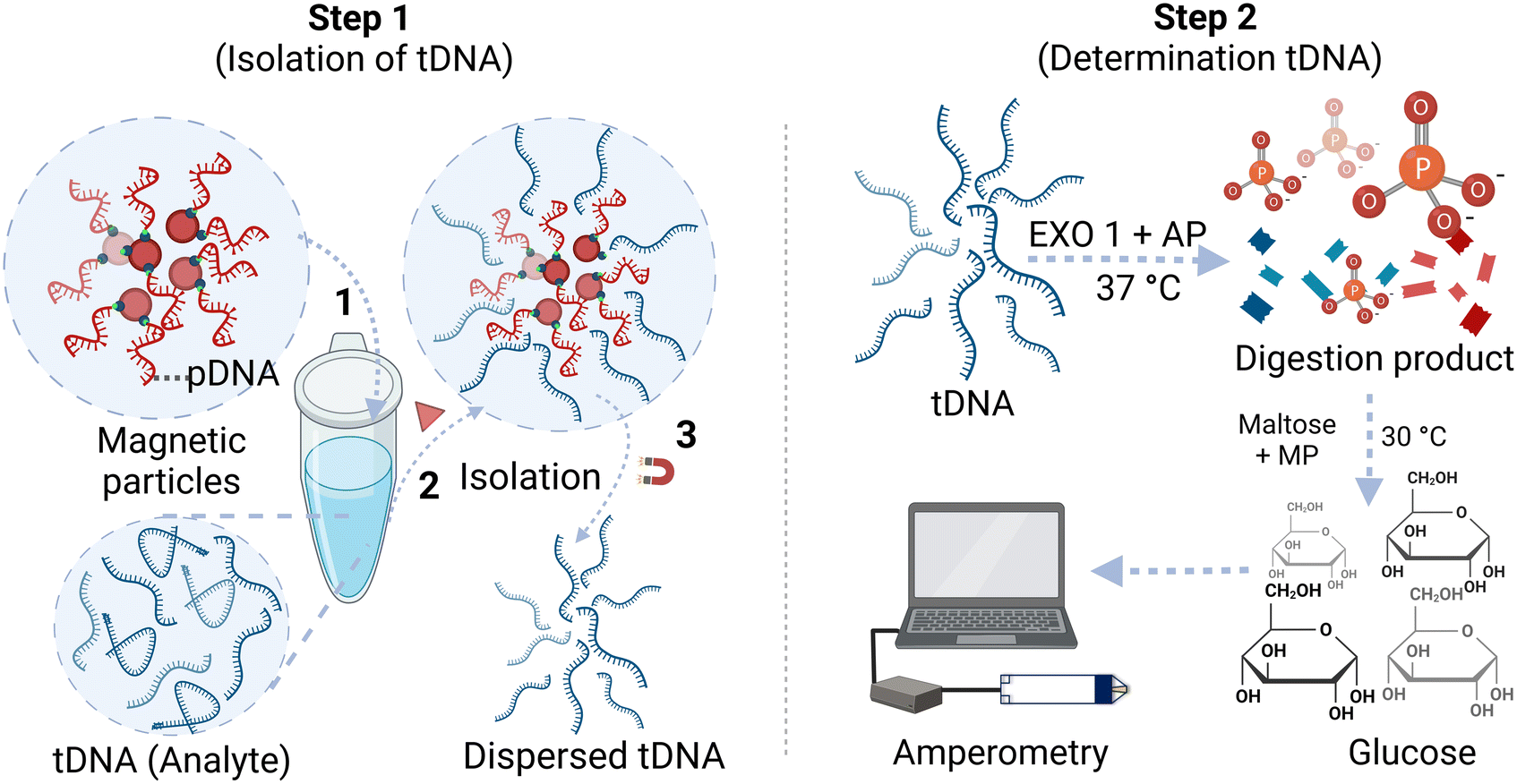 | ||
| Scheme 1 Experimental steps for determination of a target ssDNA using magnetic microparticle isolation and quantification using an off-the-shelf glucose test strip. | ||
Polyacrylamide gel electrophoresis
Non-denaturing polyacrylamide gel electrophoresis (PAGE) was used to assess the hybridization efficiency of the 14-NT pDNA and 53-NT model tDNA. A gel monomer solution was prepared by mixing 4.8 mL of 40% acrylamide:bis acrylamide (19:1) with 2.4 mL of 5× TBE buffer (0.45 M Tris base, 0.45 M boric acid, 0.5 M EDTA, pH 8.0) and 4.8 mL of DI water with continuous stirring. After that, 200 μL 10% (w/v) APS and 10 μL TEMED were added while stirring. The monomer solution was then poured into a 0.75 mm gel cassette assembly, ensuring the absence of any air bubbles. Polymerization was allowed to proceed for 30 min. Subsequently, the cassette was washed with 1× TBE buffer, and the assembly for the gel electrophoresis experiment was set up using a PowerPac basic power supply (Bio-Rad Laboratories Inc, California, USA). For the hybridization reaction, 20 μL of 4 μM pDNA and 20 μL of 4 μM tDNA were reacted at room temperature for 1 h. Controls consisted of individually incubating 40 μL of 2 μM pDNA and 40 μL of 2 μM tDNA under the same conditions. After the incubation period, 5 μL of 1 mg mL−1 sucrose was added to each solution to facilitate proper loading in the gel. 10 μL of each solution were loaded into the gel, which was run for 40 min at 170 V applied potential in 1× TBE running buffer. Following electrophoresis, the gel was washed with 1× TBE buffer and immersed in stains-all dye solution with continuous stirring for 10 min to ensure proper DNA staining. The stains-all solution was prepared by dissolving a small scoop of stains-all (95%) powder in a mixture of 40 mL formamide and H2O (1:1). After washing the gel with 1× TBE buffer, the image was captured using a Bio-Rad GelDoc Go imaging system (Bio-Rad Laboratories Inc, California, USA).
Results and discussion
Optimization of the enzymatic reactions
For this method, the choice of digestion enzymes that are used to cleave tDNA is critical to achieve optimal sensitivity for tDNA quantification. EXO 1 from E. coli is a nuclease selective to ssDNA, which catalyzes the removal of NTs from DNA in the 3′ to 5′ direction.22 Therefore, treatment of linear ssDNA with EXO 1 will yield the corresponding free NTs in solution through hydrolysis of the phosphodiester bonds of the ssDNA backbone. Furthermore, for equimolar amounts of ssDNA, it is expected that relatively long ssDNA will yield more free NTs in solution than shorter ssDNA. Importantly, each released NT contains one phosphate group bound to the ribose sugar through a phosphomonoester bond, which can be hydrolyzed using AP from bovine intestinal mucosa, liberating free orthophosphate into solution.23 The number of orthophosphates that can be generated is directly related to the NT count of the ssDNA. The recommended optimum solution pH for the EXO 1 reaction is 9.5 (supplier recommendation, New England Biolabs #M0568), and the optimum pH for AP is ∼9.7.24 Since both of these enzymes are active at similar pH, the enzymes were mixed together (as digestion enzyme mix) and reacted with the ssDNA to facilitate digestion in a single step. MP, in the presence of maltose, was used to generate glucose from orthophosphate in the digested solution.17 The optimum pH for MP is ∼4.6, much lower than the pH optimum for the digestion enzymes. Thus, the glucose generation reaction was done after the digestion reaction was complete with changing the solution pH to ∼4.6 using a high concentration of citrate buffer (1 M, pH 4.6).To investigate the impact of each digestion enzyme on a ssDNA, EXO 1 and AP were individually reacted with 4 μM 87-NT ssDNA following the procedure outlined in the Experimental section. Later, a mixture of both enzymes was reacted with 4 μM 87-NT ssDNA and detected with the Accu-Chek Guide test strip. When the mixture of EXO 1 (final concentration ∼1053 U mL−1) and AP (final concentration ∼106 U mL−1) was reacted with the 87-NT ssDNA, and subsequently reacted with MP and maltose, a significant increase in the amperometric signal was observed, indicating substantial breakdown of the ssDNA and the generation of a notable amount of orthophosphate/glucose (Fig. 1A). In contrast, individually reacting 4 μM 87-NT ssDNA with either AP (final concentration ∼106 U mL−1) or EXO 1 (final concentration ∼1053 U mL−1) resulted in very low relative currents, close to 1 (Fig. 1B), indicating that the cumulative action of EXO 1 and AP is essential for optimal digestion and the generation of free orthophosphate from the ssDNA backbone.
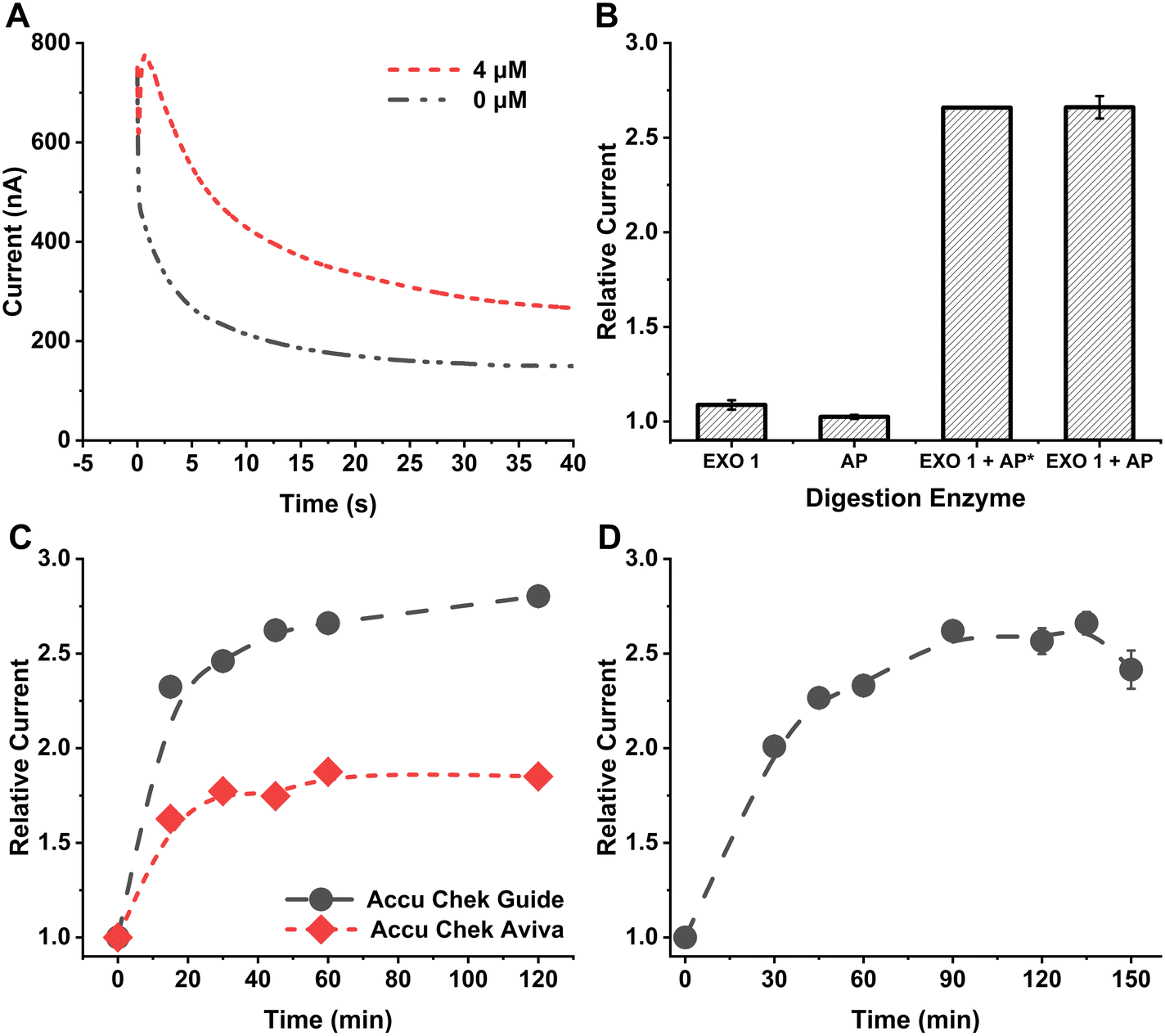 | ||
| Fig. 1 A. Amperometric response from 4 μM 87-NT ssDNA compared with the reagent blank using Accu-Chek Guide test strip at 450 mV applied potential. B. Effect of different digestion enzymes and their mixtures for detecting 4 μM 87-NT ssDNA using Accu-Chek Guide test strip at 450 mV applied potential. AP* has a final concentration of 53 U mL−1 while AP has a final concentration of 106 U mL−1. Error bars represent standard deviation of three replicates and may appear hidden as they are smaller than the size of the data points. C. Effect of digestion reaction time for 4 μM 87-NT ssDNA compared with Accu-Chek Guide and Accu-Chek Aviva test strips at 450 mV applied potential. The data points are connected with a line just to guide the eye. Error bars appear hidden as they are smaller than the size of the data points. D. Effect of reaction time for generation of glucose from maltose and orthophosphate in the presence of MP from 4 μM 87-NT ssDNA using Accu-Chek Guide test strip at 450 mV applied potential. The error bars represent the standard deviation of the three replicates of each data point. The data points are connected with a line as a guide to the eye. | ||
In further experiments, we investigated how AP concentration impacted the performance of the technique, i.e., we compared the signals obtained from ∼53 U mL−1 (AP*) to that obtained from ∼106 U mL−1 using the same amount of EXO 1 in the mixture (final concentration ∼1053 U mL−1). The relative currents for both AP concentrations were comparable (Fig. 1B), leading to the selection of ∼53 U mL−1 AP for subsequent experiments. To determine the optimal duration for the digestion reaction, 4 μM 87-NT ssDNA was exposed to the digestion enzyme mix for 0 to 120 min, followed by a subsequent reaction with maltose and MP for 120 min, following the procedure outlined in the Experimental section. The relative current exhibited an increase as reaction time increased, with the rise becoming less pronounced after 40 min of the digestion reaction (Fig. 1C). To ensure effective digestion with minimal time, a 60 min digestion reaction was selected. A comparison between the Accu-Chek Guide test strip, a newer version from Roche Diabetic Care, and Accu-Chek Aviva test strip used in our earlier reported methods17,25 revealed that the Accu-Chek Guide test strip provided significantly higher relative current values (Fig. 1C). The observed difference in current between the two test strips likely arises from a combination of different factors, including variations in enzyme type, mediator properties, and potentially electrode design or area. Our focus was on selecting a sensor based on experimental current outputs while maintaining a constant applied potential and without altering the test strips themselves. When selecting glucose test strips for this method, the primary considerations were selectivity and reproducibility. Since the method uses maltose as a substrate to generate glucose, it was crucial that the test strips demonstrate high selectivity for glucose over maltose. Both the Accu-Chek Aviva and Accu-Chek Guide strips use enzymes highly specific to glucose and exhibit a minimal response to maltose, enabling sensitive glucose detection in our approach. Of the two, the Accu-Chek Guide, being a newer version currently available on the market, provided higher sensitivity and was therefore chosen. The test strips were used in their original, unaltered form to ensure a user-friendly application. This method was designed so that users can purchase commercially available test strips and, without any modifications, perform DNA detection. By simply connecting the strips to a handheld mini-potentiostat, the process becomes accessible and straightforward. To assess the effect of reaction time on glucose generation from orthophosphate and maltose in the presence of MP, 4 μM 87-NT ssDNA was subjected to the digestion enzyme mix for 1 h, followed by reactions with maltose and MP for durations ranging from 0 to 150 min. The relative currents increased with reaction time and reached a plateau after 90 min (Fig. 1D). Hence, a 90 min duration for the glucose generation reaction was selected for further experiments.
Determination of ssDNA
Solutions with a range of concentrations, from 0 to 2 μM of 87-NT ssDNA, were prepared and subjected to a sequential reaction with the digestion enzyme mix, followed by a reaction with maltose and MP, as detailed in the Experimental section. We observed an increase in the amperometric signal with increasing concentration of ssDNA, as shown in Fig. 2A. These data can subsequently be used for generating calibration curves for quantification of ssDNA.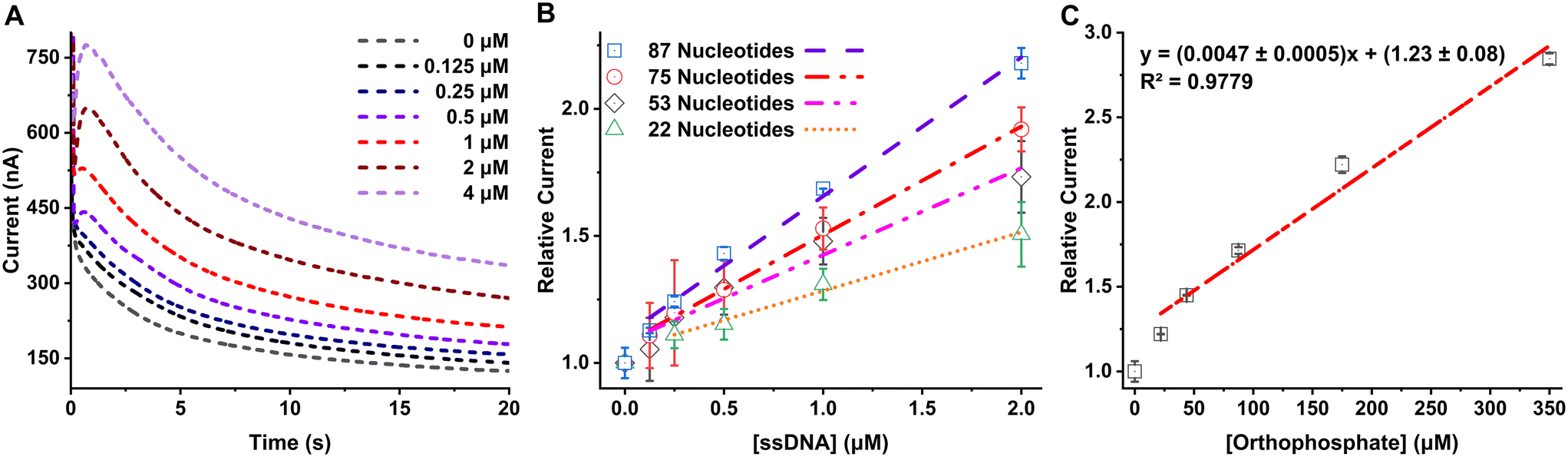 | ||
| Fig. 2 A. Amperometric response of 87-NT ssDNA from concentration 0–4 μM measured using Accu-Chek Guide test strip connected with a potentiostat with 450 mV applied potential. B. Response from ssDNA with different nucleotide counts for concentrations 0 to 2 μM measured using Accu-Chek Guide test strip connected with a potentiostat with 450 mV applied potential. C. Response from standard orthophosphate with concentrations 0 to 350 μM measured using Accu-Chek Guide test strip connected with a potentiostat with 450 mV applied potential. | ||
To evaluate the method's performance across ssDNAs with various NT counts, solutions of ssDNA with NTs ranging from 22 to 75-NT were prepared at concentrations varying from 0 to 2 μM. Each ssDNA solution underwent reactions with the digestion enzyme mix, followed by reactions with maltose and MP, utilizing the same procedure described in the Experimental section. The absolute current obtained was found to be dependent on the temperature, humidity, and potentially other factors and it was found to be different from day to day measurement. On the other hand, if the current is measured against the corresponding reagent blank and a ratio is taken it improves the day-to-day reproducibility of the signal and the obtained calibration curve is reproducible across experiments. The relative currents were calculated by taking the ratio of the averaged current (nA) from 3 s to 20 s for each ssDNA sample and the averaged current obtained from the reagent blank (0 μM ssDNA). The relative current increased proportionally with the concentration of the ssDNA samples. Notably, for the same concentration of ssDNAs, the signal increased as the length of the DNA (and the number of NTs present in each ssDNA) increased, as can be seen in Fig. 2B. This can be attributed to the increased availability of intrinsic orthophosphate in long/high NT count ssDNA (compared to short/low NT count ssDNA), leading to the generation of a higher amount of glucose and consequently yielding a larger signal in the amperometric measurements. The linear regression equations and limit of detection, calculated by taking 3 times the standard deviation of the blank divided by the slope of the calibration curve, for 0–2 μM linear range for all of the ssDNA analyzed are shown in Table 2. Blank data point (0, 1) was not included in the linear regression calculation since the method uses relative current (to the blank) for each concentration data for generating the calibration curves. Following the trend of improved LOD with longer DNA, it stands to reason that extremely long DNA (e.g., genomic DNA) would yield very low LODs. The LOD in ng μL−1 unit, which is independent of the length of the ssDNA, is also reported in Table 2 for comparison between other DNA detection methods.
| Nucleotide count | Regression equation | Regression coefficient, R2 | Limit of detection, LOD (nM) | Limit of detection, LOD (ng μL−1) |
|---|---|---|---|---|
| 22 | y = (0.23 ± 0.02)x + (1.05 ± 0.02) | 0.9599 | 780 | 5.26 |
| 53 | y = (0.34 ± 0.04)x + (1.08 ± 0.04) | 0.9966 | 527 | 8.79 |
| 75 | y = (0.43 ± 0.01)x + (1.08 ± 0.01) | 0.9911 | 422 | 9.84 |
| 87 | y = (0.55 ± 0.03)x + (1.11 ± 0.03) | 0.9899 | 329 | 8.89 |
We compared the performance and simplicity of the method with the recently published method in Table S1.† Although other DNA quantification method provides higher sensitivity, they require a complex sensor fabrication process which is somewhat difficult in a resource-limited scenario. On the other hand, this method provides a simple and label-free detection technique that does not require any sensor fabrication process and a user can simply buy off-the-shelf glucose test strips for sensitive measurement. Moreover, we anticipate getting significantly higher sensitivity when measuring genomic DNA/long DNA targets improving the performance of this method to several folds.
To determine the extent of the ssDNA digestion using this method, the relative currents obtained from ssDNA were compared with the relative current obtained from standard orthophosphate solutions. To facilitate this comparison, 0–350 μM standard orthophosphate solutions were subjected to all the steps that ssDNA would go through to generate the free NTs, and finally measured using the Accu-Chek Guide test strip. By employing the equation representing the best fit line in Fig. 2C, we can determine the theoretical relative current for the complete digestion of a ssDNA. That is, for a given concentration of ssDNA, with a known NT count, the theoretical relative current (“y”) can be calculated for 100% efficient digestion and phosphate generation (“x”). For example, after 100% efficient/complete digestion of 2 μM 87-NT ssDNA, the concentration of generated orthophosphate (“x”) would be 174 μM. By plugging this ideal orthophosphate concentration into the regression equation from Fig. 2C, the calculated relative current obtained is expected to be 2.0 ± 0.1. When the experiment was carried out, experimental relative current observed for 2 μM 87-NT was 2.18 ± 0.06. Using these values, we can calculate the % digestion using eqn (2), which is calculated as (109 ± 6)%. The calculated % digestion ranged from (97 ± 7) to (109 ± 6) across all ssDNA sequences investigated here, signifying this method can digest ssDNA with nearly 100% efficiency, allowing for maximal sensitivity.
| % Digestion = (Experimental relative current/Calculated relative current) × 100 | (2) |
| ssDNA nucleotide count | Concentration (1 μM) | Concentration (2 μM) | ||||
|---|---|---|---|---|---|---|
| Experimental relative current | Calculated relative current | % digestion | Experimental relative current | Calculated relative current | % digestion | |
| 22 | 1.30 ± 0.06 | 1.33 ± 0.08 | 98 ± 7 | 1.5 ± 0.1 | 1.44 ± 0.08 | 104 ± 9 |
| 53 | 1.48 ± 0.09 | 1.48 ± 0.08 | 100 ± 8 | 1.7 ± 0.1 | 1.73 ± 0.09 | 98 ± 8 |
| 75 | 1.53 ± 0.08 | 1.58 ± 0.09 | 97 ± 7 | 1.92 ± 0.09 | 1.9 ± 0.1 | 101 ± 7 |
| 87 | 1.686 ± 0.001 | 1.64 ± 0.09 | 103 ± 6 | 2.18 ± 0.06 | 2.0 ± 0.1 | 109 ± 6 |
Selective determination of a ssDNA target
The method employs pDNA-modified magnetic microparticles for selective capture, isolation and quantification of specific ssDNA targets (tDNA) within a mixture. The pDNA-modified magnetic microparticles were generated by exposing streptavidin-modified magnetic microparticles to biotin-TEG pDNA overnight at room temperature (see Experimental section). Then the difference in the concentrations of the initial pDNA and the final pDNA was measured. Specifically, 100 μL of 10 μM of the pDNA was reacted with 50 μL of 10 mg mL−1 of the streptavidin-modified magnetic microparticles overnight at room temperature. Then, the particles were precipitated with a magnet, and the concentration of the unbound pDNA in the supernatant solution was measured using a P360 NanoPhotometer (München, Germany) using lid10 (1 mm). The supernatant pDNA concentration was found to be 7.44 μM. From the difference of the initial pDNA concentration (10 μM) and the supernatant pDNA concentration (7.44 μM), we calculated the binding capacity: 512 pmol of pDNA mg−1 of magnetic microparticles, which exceeds the manufacturer-reported binding affinity: ∼200 pmol mg−1 of biotinylated single-stranded oligo NT. ssDNA are negatively charged and when approaching the surface of the particle, they experience repulsive force due to the negatively charged ssDNA concentrating onto the surface, which can reduce loading efficiency and affect the sensitivity of the method. Including the TEG chain minimizes repulsion while ssDNA approaches the particles and increases the loading efficiency on the particles. The presence of high salt concentration also helps loading by screening the charge of the approaching ssDNA molecules.To determine the efficiency of hybridization of pDNA and 53-NT tDNA, 4 μM of pDNA was reacted with 4 μM of tDNA for 1 h at room temperature with gentle shaking without any magnetic microparticles. The final solution was analyzed in 14% non-denaturing PAGE as described in the Experimental section with two control solutions: ‘only pDNA’ (4 μM) and ‘only tDNA’ (4 μM). As expected pDNA band was further down in the gel while tDNA band was slower due to its larger size and shape as shown in Fig. 3A. In a non-denaturing PAGE, a smaller size and shape DNA is able to move faster than a large DNA with more complex secondary structures.
 | ||
| Fig. 3 A. 14% polyacrylamide gel electrophoresis of 4 μM pDNA, 4 μM tDNA, bromophenol blue dye, and the mixture of the pDNA and tDNA (4 μM final concentration each). B. Response from 53-NT tDNA, 75-NT ssDNA, and mixtures of 53 and 75-NT ssDNA (1:1) from a final concentration of 0.25–2 μM. Error bars represent standard deviation of three replicates and may appear hidden as they are smaller than the size of the data points. | ||
The hybridized dsDNA band migrated slower than the tDNA band, signifying hybridization between the pDNA and tDNA. Importantly, the pDNA band was not observed for the mixture DNA solution, signifying almost no free pDNA was left in the solution. Hence, the pDNA chosen can efficiently bind to the tDNA and is able to capture and isolate the tDNA from the mixture for downstream analysis.
Next, solutions of 0–4 μM tDNA were prepared and individually exposed to pDNA-modified magnetic microparticles following the procedure outlined in the Experimental section. After isolation of the particles, they were redispersed in water and heated at 100 °C for 10 min to “melt” the hybridized double stranded DNA and release the tDNA back into solution. The tDNA solution was then reacted with the digestion enzyme mix and reagent to generate glucose. The relative current of the 53-NT tDNA increased linearly with increasing concentration, which allowed a calibration curve to be constructed, and the concentration of a tDNA to be determined. The calibration curve obtained is shown in Fig. 3B. To check for any non-specific binding on the magnetic microparticles, 75-NT ssDNA was exposed to the same pDNA-modified magnetic microparticles specific for the 53-NT tDNA. As expected, the relative current obtained from the 75-NT ssDNA was very low and did not increase with increasing concentration (Fig. 3B). This implies that the method is selective for specific tDNA sequences (in this case, 53-NT tDNA) with minimum non-specific binding. Then both 53 and 75-NT ssDNA were mixed together in a 1:1 ratio from concentration 0.25 μM to 2 μM and measured using the procedure described in the Experimental section. To check whether a mismatched DNA can interfere with the analysis of tDNA, we checked for any significant differences in each data points for a certain concentrations of tDNA to the mixture (53 and 75-NT). Paired t-tests were done on relative currents of the isolated 53-NT ssDNA and the mixture of 53-NT tDNA and 75-NT ssDNA for concentrations from 0.25 to 2 μM. The p values were: 0.43 for concentration 0.25 μM, 0.25 for concentration 0.5 μM, 0.16 for concentration 1 μM, and 0.14 for concentration 2 μM. Hence, the signals from the mixture were not significantly different (p > 0.01) from the signal that was obtained from the only 53-NT tDNA (Fig. 3B). Hence, this method can selectively isolate a certain tDNA from a mixture and allows for accurate determination.
Conclusions
We developed a method to quantify ssDNA in solution using commercially available glucose test strips. This method does not require any sophisticated laboratory-based instrument. The sensitivity of the method increases with increasing NT number of the ssDNA. The digestion efficiency of this method for all the ssDNA tested in this study was close to 100%. Utilization of magnetic microparticles facilitates the isolation of a specific tDNA from the solution, which provides the selectivity of this method to quantify a certain ssDNA target in the presence of other ssDNA in the mixture. This methodology holds promise for the quantification of viral or bacterial genomic ssDNA, offering a simple and cost-effective solution that is particularly valuable in resource-limited scenarios.Data availability
The datasets generated during and/or analysed during the current study are not publicly available due to potential patent filings, but are available from the authors on reasonable request.Conflicts of interest
There are no conflicts to declare.References
- E. A. Alacs, A. Georges, N. N. FitzSimmons and J. Robertson, Forensic Sci., Med., Pathol., 2010, 6, 180 CrossRef CAS PubMed.
- B. R. McCord, Q. Gauthier, S. Cho, M. N. Roig, G. C. Gibson-Daw, B. Young, F. Taglia, S. C. Zapico, R. F. Mariot and S. B. Lee, Anal. Chem., 2018, 91, 673 CrossRef PubMed.
- M. Woolfe and S. Primrose, Trends Biotechnol., 2004, 22, 222 CrossRef CAS PubMed.
- A. Prachugsorn, P. Thanakiatkrai, K. Phooplub, S. Ouiganon, Y. Sriaead, P. Thavarungkul, P. Kanatharana, C. Buranachai and T. Kitpipit, Food Control, 2022, 139, 108989 CrossRef CAS.
- X. Li, K. Scida and R. M. Crooks, Anal. Chem., 2015, 87, 9009 CrossRef CAS PubMed.
- B. S. Ferguson, S. F. Buchsbaum, T.-T. Wu, K. Hsieh, Y. Xiao, R. Sun and H. T. Soh, J. Am. Chem. Soc., 2011, 133, 9129 CrossRef CAS PubMed.
- F. Azek, C. Grossiord, M. Joannes, B. Limoges and P. Brossier, Anal. Biochem., 2000, 284, 107 CrossRef CAS PubMed.
- S. Nguyen, P. Vu and M. Tran, Sci. Rep., 2023, 13, 10235 CrossRef CAS PubMed.
- M. J. Holden, R. J. Haynes, S. A. Rabb, N. Satija, K. Yang and J. R. Blasic Jr, J. Agric. Food Chem., 2009, 57, 7221 CrossRef CAS PubMed.
- L. A. Shokere, M. J. Holden and G. R. Jenkins, Food Control, 2009, 20, 391 CrossRef CAS.
- L. Kékedy-Nagy, S. Shipovskov and E. E. Ferapontova, Anal. Chem., 2016, 88, 7984 CrossRef PubMed.
- J. Wang, G. Liu and M. R. Jan, J. Am. Chem. Soc., 2004, 126, 3010 CrossRef CAS PubMed.
- C. E. Kirimli, W.-H. Shih and W. Y. Shih, Analyst, 2014, 139, 2754 RSC.
- F. Can, H. E. Ökten, T. Ergön-Can, P. Ergenekon, M. Özkan and E. Erhan, Bioelectrochemistry, 2020, 135, 107553 CrossRef CAS PubMed.
- R. Gasparac, B. J. Taft, M. A. Lapierre-Devlin, A. D. Lazareck, J. M. Xu and S. O. Kelley, J. Am. Chem. Soc., 2004, 126, 12270 CrossRef CAS PubMed.
- G. F. Schneider, S. W. Kowalczyk, V. E. Calado, G. Pandraud, H. W. Zandbergen, L. M. Vandersypen and C. Dekker, Nano Lett., 2010, 10, 3163 CrossRef CAS PubMed.
- F. Hossain, N. Balasuriya, M. M. Hossain and M. J. Serpe, Anal. Chem., 2022, 94, 2056 CrossRef CAS PubMed.
- J. Hoenes, P. Müller and N. Surridge, Diabetes Technol. Ther., 2008, 10, S10 CrossRef.
- O. A. Alsager, S. Kumar, B. Zhu, J. Travas-Sejdic, K. P. McNatty and J. M. Hodgkiss, Anal. Chem., 2015, 87, 4201 CrossRef CAS PubMed.
- O. A. Alsager, S. Kumar, G. R. Willmott, K. P. McNatty and J. M. Hodgkiss, Biosens. Bioelectron., 2014, 57, 262 CrossRef CAS PubMed.
- H. Yu, Y. Luo, O. Alkhamis, J. Canoura, B. Yu and Y. Xiao, Anal. Chem., 2021, 93, 3172 CrossRef CAS PubMed.
- M. Viswanathan and S. T. Lovett, Genetics, 1998, 149, 7 CrossRef CAS PubMed.
- J. G. Zalatan, T. D. Fenn, A. T. Brunger and D. Herschlag, Biochemistry, 2006, 45, 9788 CrossRef CAS PubMed.
- O. Metwalli and F. Mourand, Z. Ernahrungswiss., 1980, 19, 154 CrossRef CAS PubMed.
- F. Hossain, Q. Shen, N. Balasuriya, J. L. M. Law, M. Logan, M. Houghton, D. L. Tyrrell, M. A. Joyce and M. J. Serpe, Anal. Chem., 2023, 95, 7620–7629 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00318g |
| This journal is © The Royal Society of Chemistry 2025 |