
Harnessing lattice oxygens in a high-entropy perovskite oxide for enhanced oxygen evolution reaction †
Sujan
Sen
 a and
Tapas Kumar
Mandal
*ab
a and
Tapas Kumar
Mandal
*ab
aDepartment of Chemistry, Indian Institute of Technology Roorkee, Roorkee – 247667, India. E-mail: tapas.mandal@cy.iitr.ac.in
bCenter for Nanotechnology, Indian Institute of Technology Roorkee, Roorkee – 247667, India
First published on 2nd October 2024
Abstract
The development of highly active and stable electrocatalysts for the oxygen evolution reaction (OER) is the main challenge in water electrolysis for green hydrogen production. Although Ru-based electrocatalysts have been in use for the past few decades, their stability in the reaction medium remains a major concern. Herein, a high-entropy simple perovskite oxide Ba0.33Sr0.67Co0.33Ti0.165Ru0.165Sb0.33O3 (BSCTRS) is designed and synthesized by introducing Ru at 16.5 mol% B-site positions of Ba0.33Sr0.67Co0.33Ti0.33Sb0.33O3 (BSCTS) to achieve enhanced lattice oxygen participation. The BSCTRS perovskite electrocatalyst exhibits an OER overpotential similar to RuO2 at 10 mA cm−2 and a far superior OER overpotential (340 mV) compared to the benchmark RuO2 at 100 mA cm−2. Moreover, BSCTRS shows ∼20% lower Tafel slope and ∼120% higher TOF than Ba0.33Sr0.67Co0.33Ti0.33Sb0.33O3. This results in a significant enhancement of current density to 263 mA (at 1.58 V vs. RHE) for BSCTRS compared to only 99 mA for the parent BSCTS. The enhanced activity of the catalyst stems from optimal filling of eg orbitals of the active metals and greater lattice oxygen participation. The work demonstrates the ability of high-entropy stabilized simple perovskite compositions with low concentrations of active noble metals to significantly enhance OER activity.
Introduction
Rising global energy demand and its associated environmental problems can only be addressed by transitioning from non-renewable to renewable energy sources.1,2 Water electrolysis is one of the clean and sustainable ways of energy production.3 A typical water electrolysis process involves two steps: the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) at the anode and cathode, respectively.4 The OER being a four-electron transfer reaction, is kinetically sluggish and requires a much higher overpotential than the HER.4,5 So, the development of efficient OER electrocatalysts is of paramount importance. Precious metal oxides like RuO2 and IrO2 have been widely accepted as the most efficient OER electrocatalysts.6 However, the low abundance and high cost of RuO2 and IrO2, in addition to their low stability in the reaction medium greatly hindered the industrial use of these catalysts.6,7 Synthesis of complex oxides using small amounts of noble metals is one of the promising strategies to reduce the cost while improving both stability and intrinsic activity.8 Complex oxides like perovskites, pyrochlores, etc., with small percentages of noble metal content have been reported to show excellent activity, even superior to that of binary metal oxides like RuO2 and IrO2.8,9It is reported that the inherent synergistic effect of different active metals in high-entropy oxides helps to improve the electrocatalytic activity.10,11 Perovskite oxides having higher compositional and structural flexibility can serve as a suitable host structure for multiple active metal based high-entropy compositions with low Ru content. In general, the A-site of the perovskite is occupied by s- or f-block elements while the B-site is occupied by transition elements and p-block metals.12 Although the nature of active sites remains elusive in perovskite oxides, researchers have suggested different origins for their activity.6,12 Matsumoto and co-workers first studied the La1−xSrxCoO3 perovskite system and proposed that the OER takes place at the surface transition metal active site and catalytic activity increases with the increase in the charge density of the active-site metal atom.13 Later, Bockris and Otagawa suggested that the activity of the perovskite based catalyst depends on the binding strength of oxygenated active species with the active-metal site (Mz). They further correlated the number of d-electrons with OER activity, suggesting that with an increase in the number of d-electrons, the occupancy of the Mz–OH anti-bonding orbital would also increase, resulting in enhanced OER activity.14 Yang Shao-Horn's group extensively studied the OER mechanism in different perovskite systems.15–17 They observed a volcano shaped dependence of activity on the eg-orbital filling of the surface transition metal, with near unity filling of the eg-orbital showing the best performance.15 Furthermore, they statistically evaluated various OER descriptors for perovskite oxide based OER electrocatalysts and found that optimal eg-orbital filling, covalency, and the number of d-electrons play a crucial role in OER activity.16
Conventionally, the mechanism of the OER for perovskite based catalysts is widely accepted to follow a concerted process involving four proton-electron transfer steps, commonly known as the adsorbate evolution mechanism (AEM).18–20 Recently, a non-concerted proton-electron transfer mechanism has been proposed by Grimaud et al.17In situ mass spectroscopy of the 18O isotope labelled catalyst confirmed the participation of lattice oxygens in the reaction. Based on this direct evidence, they suggested a new mechanism known as the lattice oxygen modulated (LOM) mechanism. It should be noted that the LOM mechanism is not universally followed by perovskite-based catalysts. During their study, they found that the highly active compounds La0.5Sr0.5CoO3−δ, Pr0.5Ba0.5CoO3−δ, and SrCoO3−δ show lattice oxygen participation, whereas LaCoO3 shows only a surface OER process that follows the AEM.17 However, to date, very few stable perovskite oxide based catalysts have been reported to follow the LOM mechanism.20 Stable electrocatalysts following the LOM mechanism can have enhanced OER activity and should be explored further. The enhancement of lattice oxygen participation in order to improve the OER activity always comes at the expense of stability.20 Herein, we have employed a strategy of partial substitution of OER-inactive B-site metal with Ru in a previously known active and stable OER electrocatalyst that follows the LOM mechanism. It is envisaged that even a low concentration of Ru will further improve the OER activity by enhancing lattice oxygen participation while maintaining the phase stability in the reaction medium. Although Ru-based simple perovskite oxide electrocatalysts, such as SrRuO3, Ca1−xSrxRuO3, Sr0.9Na0.1RuO3,etc., have been reported to show OER activity in basic media,21–23 their high overpotential and the requirement of a higher percentage of Ru in the composition make them unsuitable for practical applications. A perovskite with a small percentage of Ru, such as, SrTi0.7Ru0.3O3, is also known but it shows kinetically sluggish OER activity.21 We have designed a new high-entropy composition Ba0.33Sr0.67Co0.33Ti0.165Ru0.165Sb0.33O3 (BSCTRS) with much lower Ru content based on our previously studied OER active compound Ba0.33Sr0.67Co0.33Ti0.33Sb0.33O3.24 The compound has been synthesized by the conventional solid-state method. The compound shows lower overpotentials at both 10 and 100 mA cm−2 in comparison to the benchmark RuO2 electrocatalyst. Moreover, ∼120% enhanced turnover frequency (TOF), faster kinetics and substantially higher overall OER activity compared to Ba0.33Sr0.67Co0.33Ti0.33Sb0.33O3 is demonstrated. The kinetic results and enhanced oxygen participation through the LOM mechanism are confirmed by operando EIS and pH dependent OER studies, respectively. Our study shows the role of critical compositional design, using low Ru contents in the low-cost non-noble metal based electrocatalyst Ba0.33Sr0.67Co0.33Ti0.33Sb0.33O3, to significantly impact the activity by enhancing the lattice oxygen participation while maintaining phase stability in the reaction medium.
Experimental
Synthesis
A polycrystalline powder sample of Ba0.33Sr0.67Co0.33Ti0.165Ru0.165Sb0.33O3 (BSCTRS) was prepared by the conventional solid-state method. Stoichiometric amounts of BaCO3 (≥99%, Sigma-Aldrich), SrCO3 (≥99.9%, Aldrich), CoC2O4·2H2O (prepared from Co(NO3)2·6H2O, Sigma-Aldrich), TiO2 (99.8%, Aldrich), RuO2 (≥99.9%, Aldrich), and Sb2O3 (≥99.0%, Fluka) were mixed in a mortar and pestle to get a homogeneous mixture. The finely ground powder was then heated in a muffle furnace, first at 850 °C for 12 h followed by 1000 °C, 1025 °C, 1100 °C and 1150 °C for 24 h each, with intermediate grinding after each heating step.Characterization
The as prepared polycrystalline powder was characterized with powder X-ray diffraction (P-XRD) using a Rigaku automated X-ray diffractometer, SmartLab 3 kW, with a Cu anode as an X-ray source. The XRD pattern was analyzed with Rietveld refinement using the FullProf program to get the structural information.25,26 The microstructure and elemental composition of the catalyst were investigated by FE-SEM and EDS, respectively, using a Carl-Zeiss-Ultra Plus field-emission scanning electron microscope operating at 20 kV. Brunauer–Emmett–Teller (BET) analysis of the N2-adsorption–desorption isotherm was performed to obtain the specific surface area of the catalyst using a NOVA 2200e, Quantachrome. The chemical state of the surface elements was analysed by X-ray photoelectron spectroscopy (XPS) using a ULVAC PHI 5000 VersaProbe III with Al K-α as the X-ray source. The deconvolution of XPS peaks was performed using the PHI MultiPak program. The percentage of element leaching during the OER was evaluated by analyzing the electrolyte using a Microwave Plasma-Atomic Emission Spectrometer (MP-AES), Agilent (4210-MP-AES).Electrode fabrication and electrochemical measurements
The working electrodes were prepared on a nickel foam (NF) substrate by drop casting catalyst ink. The nickel foam sheet was cut into a 1 cm × 2 cm piece and sonicated in 3 M HCl solution, de-ionized water, and acetone for 15 minutes each, respectively, and dried overnight. The catalyst ink was prepared by mixing 20 mg of the catalyst with 2 mg poly(vinylidene fluoride) (PVDF) (Alfa-Aesar), and 2 mg carbon black (CB) (Alfa-Aesar) in 300 μL N-methyl-2-pyrrolidone (NMP) (≥99.00%, HIMEDIA) solvent. The suspension was then sonicated in an ultrasonic bath for 45 minutes followed by 12 h of magnetic stirring at 1000 rpm to get a homogeneous ink. Finally, the catalyst ink was carefully drop-cast onto a 1 cm × 1 cm area of the previously cleaned NF substrate and dried in a hot oven. The RuO2–NF working electrode was also prepared to compare and assess the catalytic performance using the method described above. The mass loading was optimized for both BSCTRS and RuO2 and a mass loading of 4 mg cm−2 was found to show the best activity for BSCTRS, while for RuO2, it was 2 mg cm−2.All the electrochemical measurements were performed in a standard three electrode setup comprising Ag/AgCl (3 M KCl) as the reference electrode, and Pt wire as the counter electrode, using a Metrohm Autolab M204 electrochemical workstation operating with Nova 2.1 software. A 1 M KOH solution was used as the electrolyte. Cyclic voltammetry (CV) was performed at a scan rate of 10 mV s−1 for 10 cycles to activate the working electrodes prior to other measurements. The linear sweep voltammetry (LSV) was recorded at a scan rate of 5 mV s−1 in the potential range of 0 to 0.8 V vs. Ag/AgCl. The electrochemical impedance spectroscopy (EIS) measurements were performed at open circuit potential (OCP) in the frequency range of 105 Hz to 0.1 Hz for iR correction. The iR-correction of the LSV curves was performed using the following formula:27
| ECorrected = ERaw − iRs |
Furthermore, operando EIS was also performed in the potential range of 1.20 V vs. RHE to 1.65 V vs. RHE. The EIS circuit fitting was conducted using NOVA 2.1 software. CV was performed in the non-faradaic region at different scan rates (20, 40, 60, 80, 100, and 120 mV s−1) to obtain the electrochemically active surface area (ECSA) (Fig. S2†). The current difference Δ j (=ja − jc) at 1.06 V (vs. RHE) vs. scan rate was plotted first to get the double layer capacitance (Cdl), which is half of the slope of the plot of Δj vs. scan rate. The Cdl value was then converted to ECSA by normalizing the Cdl of the working electrode. The normalization was performed by dividing the Cdl of the working electrode by the standard specific capacitance of a 1 cm2 substrate area. Since NF was used as the substrate, and the specific capacitance of a 1 cm2 area of NF is generally higher than the standard accepted value of 0.02–0.06 mF cm−2, Cdl of NF was also measured and used as the standard to calculate the ECSA as mentioned in the literature.28
All the potentials were standardized with respect to the reversible hydrogen electrode (RHE) using the following equation:11
| ERHE = EAg/AgCl + E0Ag/AgCl + 0.059 × pH |
The turnover frequency (TOF) was calculated at an overpotential of 350 mV according to the following equation:
| TOF = (j × A)/(4 × F × m) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485C mol−1) and m is the number of moles of catalyst deposited.29 The OER activity of benchmark RuO2 and bare Ni-foam (NF) was evaluated under the same environment. To check the stability of the catalyst, chronopotentiometry was performed for 16 h at a constant current density of 10 mA cm−2. A pH dependent OER study was also performed to gain mechanistic insight into the catalyst. LSV were recorded at varying pH (12.5, 13, 13.5, and 14) at a scan rate of 5 mV s−1.
485C mol−1) and m is the number of moles of catalyst deposited.29 The OER activity of benchmark RuO2 and bare Ni-foam (NF) was evaluated under the same environment. To check the stability of the catalyst, chronopotentiometry was performed for 16 h at a constant current density of 10 mA cm−2. A pH dependent OER study was also performed to gain mechanistic insight into the catalyst. LSV were recorded at varying pH (12.5, 13, 13.5, and 14) at a scan rate of 5 mV s−1.
Results and discussion
Material characterization
The entropy stabilization in compounds is based on the idea of enhancing configurational entropy by incorporating multiple atoms at a single site. The configurational entropy (Sconfig), is defined by the following equation:where, A and B are the number of elements present at the A- and B-site of a perovskite, xa and xb are the mole fractions and R is the gas constant.30 The compound Ba0.33Sr0.67Co0.33Ti0.165Ru0.165Sb0.33O3 has an Sconfig value of 1.96 R, making it a high-entropy perovskite oxide.
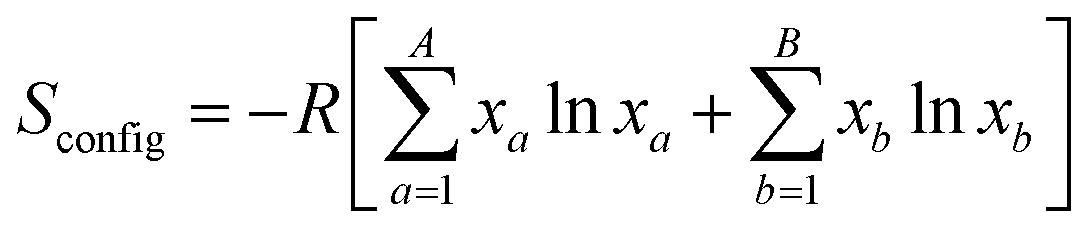
The PXRD pattern of the compound shows diffraction peaks corresponding to the simple cubic perovskite phase of the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group with no detectable impurity phase. The Rietveld refinement performed adopting the Pmm space group using slow scan P-XRD data has resulted in a refined lattice parameter of 3.9850(1) Å. The refinement profile is shown in Fig. 1 along with the crystal structure drawn using the diamond program using the refined structural parameters (inset of Fig. 1). The refined lattice parameters and reliability factors are given in Table S1,† whereas the atomic position, occupancies, and thermal parameter are given in Table S2.†
m space group with no detectable impurity phase. The Rietveld refinement performed adopting the Pmm space group using slow scan P-XRD data has resulted in a refined lattice parameter of 3.9850(1) Å. The refinement profile is shown in Fig. 1 along with the crystal structure drawn using the diamond program using the refined structural parameters (inset of Fig. 1). The refined lattice parameters and reliability factors are given in Table S1,† whereas the atomic position, occupancies, and thermal parameter are given in Table S2.†
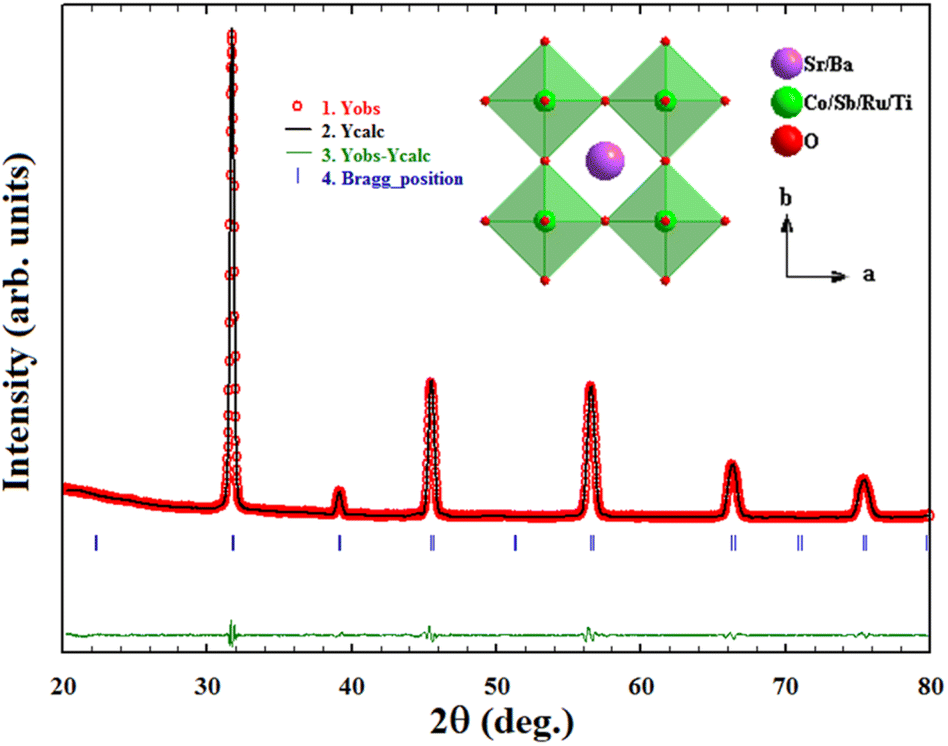 | ||
| Fig. 1 Rietveld refinement profile fitting and the crystal structure (inset) of BSCTRS. | ||
The microscopic morphology of the compound analyzed with FE-SEM shows an agglomeration of faceted irregular shaped submicron sized particles throughout the area under observation (Fig. 2a and b). The agglomeration of the particles is expected as the compound was synthesized at an elevated temperature. Consequently, the BET analysis also shows a relatively low surface area of 5 m2 g−1 and an average pore size distribution of 5.3 nm (Fig. 3). The EDS data show that the observed average elemental composition aligns with the nominal composition of the compound (Fig. 2c). The elemental mapping of the compound displays an even distribution of Ba, Sr, Co, Ti, Ru, and Sb elements throughout the whole region under observation (Fig. 2d).
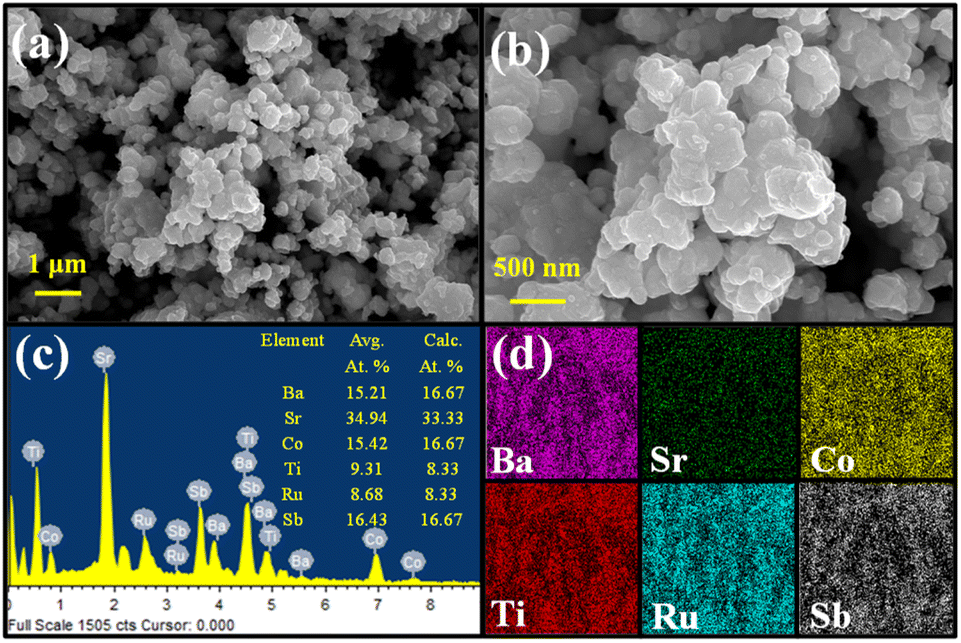 | ||
| Fig. 2 (a) and (b) FE-SEM images; (c) EDS and (d) elemental mapping of BSCTRS. | ||
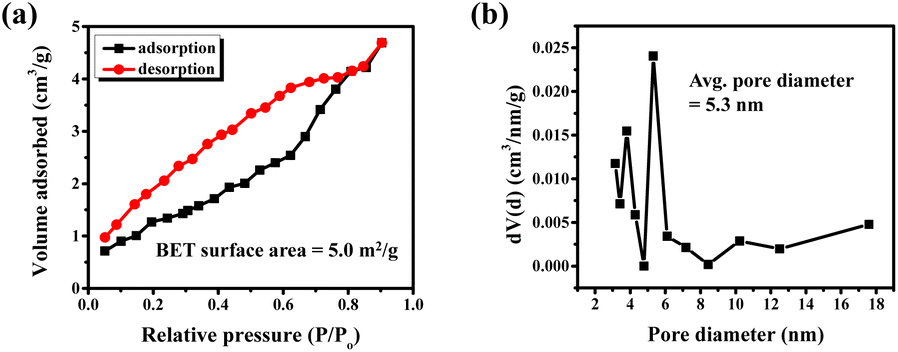 | ||
| Fig. 3 (a) BET isotherm and (b) pore size distribution of BSCTRS. | ||
XPS was performed to study the oxidation state of the elements at the surface of the catalyst. The XPS survey spectra confirm the presence of all the elements (Fig. S3†). Fig. 4a shows the deconvoluted XPS peaks of Ru 3p. Since the Ti 2p1/2 XPS peaks overlap in the same region (460–470 eV) with Ru 3p3/2, to deconvolute the Ru 3p and Ti 2p peaks, Ti 2p3/2 is indexed first, followed by the identification of the Ti 2p1/2 peak with the help of the spin–orbit coupling value (∼5.54 eV). Once the Ti 2p peaks were identified, Ru 3p peaks were deconvoluted. The peak positions of Ru 3p3/2 and 3p1/2 at 464.4 eV and 486.6 eV, respectively, indicate the oxidation state of Ru to be +4.7 Also, it is observed that the Co 2p and Ba 3d peaks appear in the same region of 770 to 820 eV and their spin–orbit coupling value is also similar (∼15 eV).31 In our earlier observation, it was noted that the Co 2p XPS peak for Co3+ appears at a higher binding energy as compared to the 3d peak of Ba2+.24 Additionally, two satellite peaks are also observed at 789 and 804 eV, which indicates the presence of Co in the +2 oxidation state along with the expected +3 oxidation state.32–35 Moreover, 2p binding energy of Co2+ is higher than that of Co3+.36 Considering all these, the peaks at ∼780 and ∼795 eV are deconvoluted into three peaks corresponding to Ba 3d, Co3+ 2p and Co2+ 2p (Fig. 4b). The ratio of Co3+ to Co2+ is also calculated based on the area of the peaks and is given in Table 1.
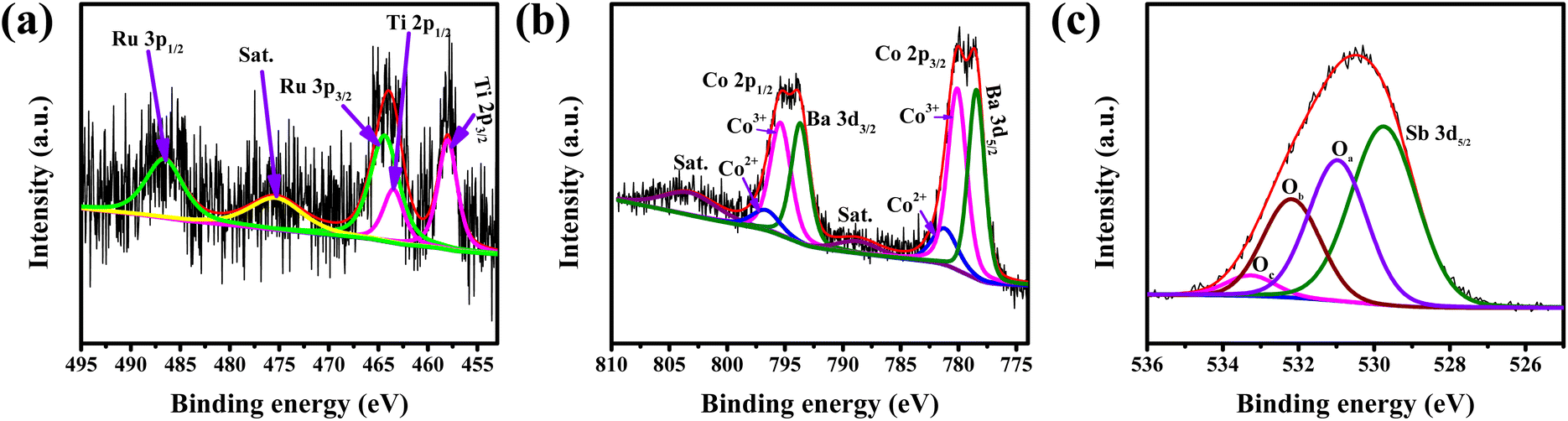 | ||
| Fig. 4 Deconvoluted XPS spectra of (a) Ru 3p, (b) Co 2p and (c) O 1s. | ||
| Element | Percentage (%) | |
|---|---|---|
| Co 2p | Co3+ | 80.94 |
| Co2+ | 19.06 | |
| O 1s | Oa | 56.35 |
| Ob | 36.65 | |
| Oc | 7.00 | |
The presence of low coordinated surface oxygens enhances the OER activity in perovskite oxides.21 Therefore, identification of different oxygen species from XPS becomes important to understand the lattice oxygen mediated OER activity. The O 1s XPS spectrum is deconvoluted into four peaks (Fig. 4c). Since the Sb 3d5/2 peaks also appear in the same region as O 1s (520–550 eV), the peak corresponding to Sb 3d5/2 is identified first with the help of the Sb 3d3/2 peak and the spin orbit coupling value. The rest three oxygen peaks are marked as Oa, Ob, and Oc corresponding to lattice oxygens, low coordinated surface oxygens, and adsorbed hydroxyl group oxygens, respectively.21
The peak area of Oa, Ob, and Oc is used to calculate the percentage of different oxygen species and tabulated in Table 1. The presence of a significant percentage (36.65%) of low coordinated surface oxygen is expected to help in the OER activity of the catalyst.
Electrocatalytic oxygen evolution reaction (OER)
The electrocatalytic OER activity of the catalyst was studied on a standard three electrode setup with 1 M KOH as the electrolyte. Fig. 5a shows the iR-corrected polarization curves of BSCTRS, RuO2 and NF. The iR correction was performed using the Rs value from the first intersection of the EIS curve with the real axis of the Nyquist plot (Fig. S1†). As shown in Fig. 5a, BSCTRS required a lower overpotential (286 mV) at a current density of 10 mA cm−2 (η10) and a significantly lower overpotential (340 mV) at a current density of 100 mA cm−2 (η100) as compared to the benchmark RuO2 (η10 = 292 mV and η100 = 444 mV). The NF shows much lower activity, with an η10 value of 405 mV, which is consistent with previous reports.21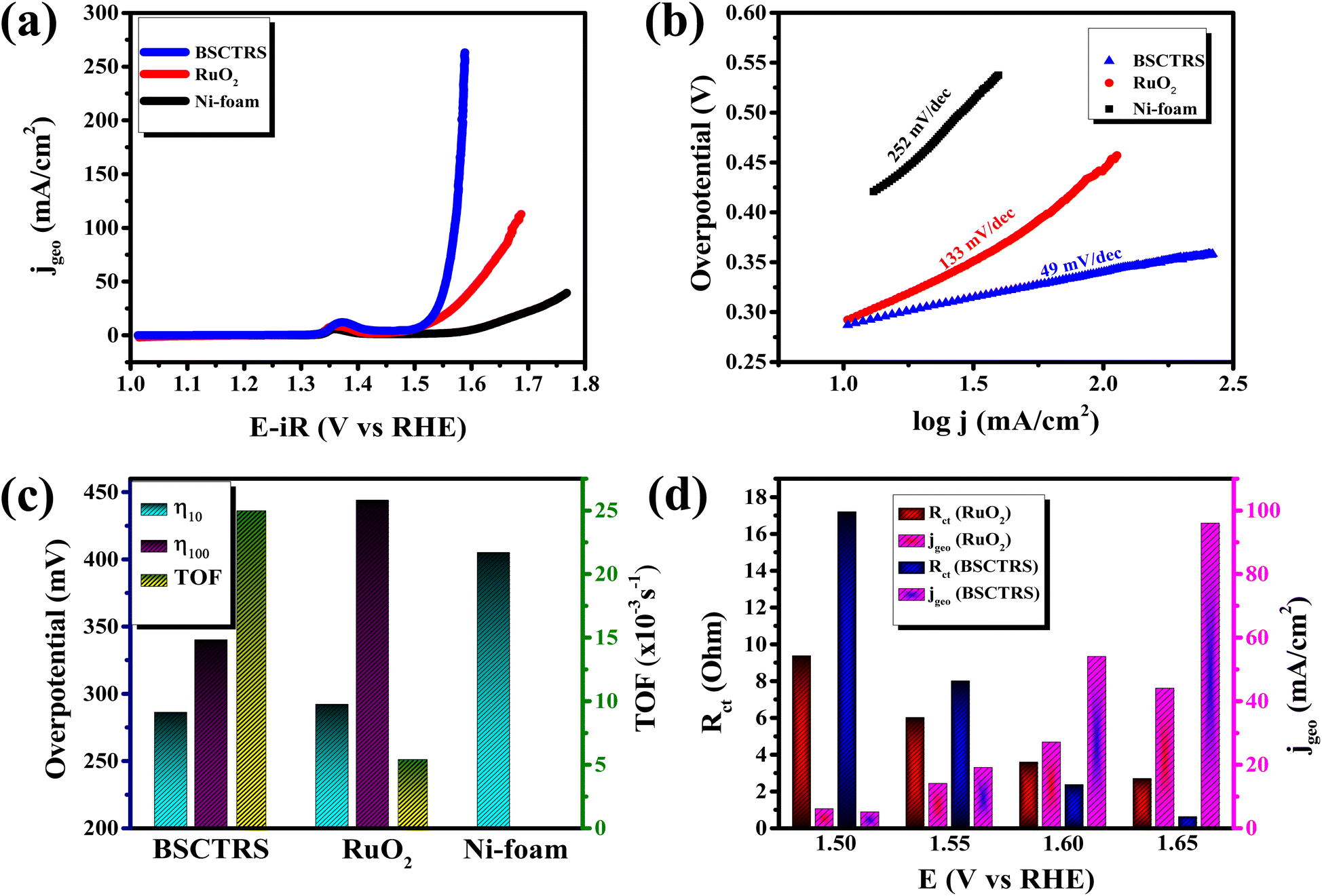 | ||
| Fig. 5 (a) Polarization curves, (b) Tafel plots, (c) overpotential and TOF of BSCTRS, RuO2 and NF and (d) correlation of charge transfer resistance and current density at different potentials. | ||
The kinetic relationship of OER can be expressed with the Butler–Volmer equation:37
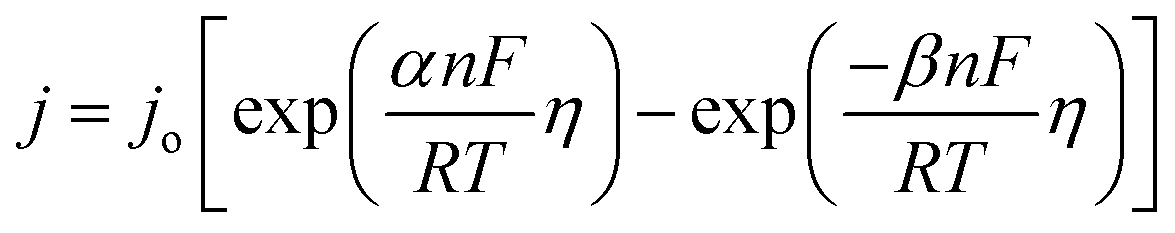
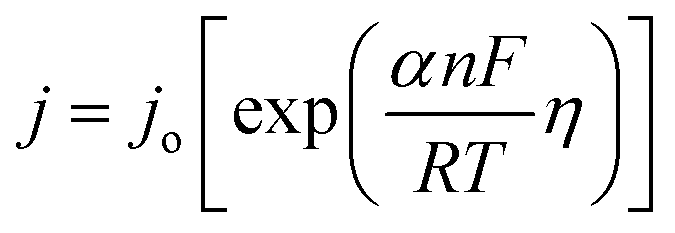
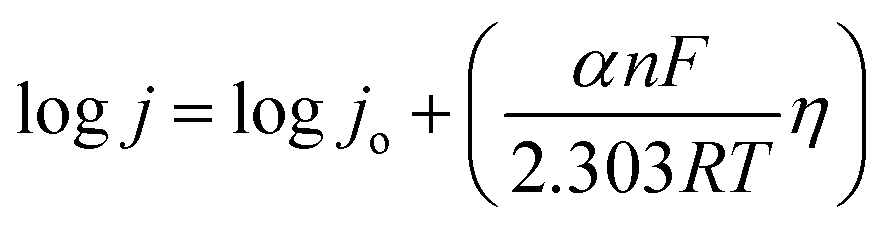
which is generalized as,
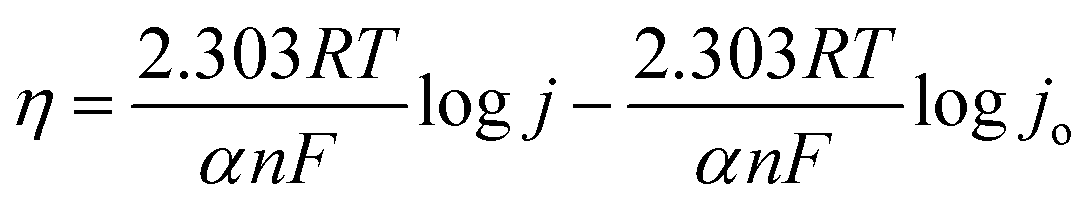
| η = blogj + a |
j vs. η plot, b values are calculated. The OER kinetics of the catalyst was evaluated with the Tafel plots derived from the iR-corrected polarization curves. The BSCTRS catalyst shows a Tafel slope that is nearly 2.7 times lower than that of the benchmark RuO2 electrocatalyst (Fig. 5b). Furthermore, the Tafel slope is also 20% lower than that of the unsubstituted BSCTS.24 The much lower Tafel slope for BSCTRS indicates that the compound is kinetically superior to RuO2 and BSCTS. In addition, to understand the intrinsic activity of the catalyst, turnover frequency (TOF) has also been calculated at 350 mV overpotential using the equation mentioned above. The TOF of the BSCTRS catalyst (0.02497 s−1) is nearly five-fold and more than two-fold higher than that of the RuO2 (0.00538 s−1) and BSCTS (0.01140 s−1),24 respectively (Fig. 5c). The catalyst's notably high TOF value suggests that the intrinsic activity of the catalyst surpasses that of RuO2 and BSCTS. The kinetic superiority and higher intrinsic activity of BSCTRS compared to BSCTS results in a significantly higher current density of 263 mA at 1.58 V (vs. RHE) compared to 99 mA for BSCTS at the same potential.
Operando EIS was performed at different potentials ranging from 1.20 to 1.65 V vs. RHE to study the internal charge transfer resistance behavior of the compound under OER conditions. The EIS circuit fitting was carried out with different circuit elements, viz., series resistance (Rs), two charge-transfer resistance (Rct) and two constant phase elements (CPE); the equivalent circuit diagram is given in Fig. S4c.† The fitted circuit consists of two Rct elements. The first Rct (Rct1) is consistent for both the catalyst and RuO2, indicating that it is due to the resistance of the NF substrate as reported in the literature,19 whereas the second Rct (Rct2) is associated with the catalyst. As shown in Fig. 5d, the Rct of RuO2 is much lower as compared to that of BSCTRS at 1.50 V vs. RHE, and the corresponding current density of BSCTRS is slightly lower than that of RuO2. As we increased the potential to 1.55 V vs. RHE, the Rct of BSCTRS remained marginally higher than that of RuO2, but the current density of BSCTRS became higher than that of RuO2. At even higher potentials (1.60 and 1.65 V vs. RHE), the Rct of BSCTRS becomes lower than that of RuO2, as evident from the semicircle of the Nyquist plot in Fig. S4d and e,† while the corresponding current density becomes much higher than that of RuO2. A rapid decrease in Rct and an increase in current density are observed for BSCTRS, whereas a slow decrease in Rct and a much slower increase in current density is observed for RuO2 with increasing applied potential. This observation corroborates well with the Tafel slope and further establishes the faster OER kinetics for BSCTRS. Bode plots were also analyzed to study the changes in the interface during the OER study. Fig. 6 shows the Bode plots of BSCTRS and RuO2 in the low frequency region; the corresponding Nyquist plots are shown in Fig. S4.† The change in the phase angle in the low frequency region at potentials of 1.45 and 1.475 V vs. RHE for RuO2 and BSCTRS, respectively, indicates the OER activity of the catalysts, which matches with the onset potential observed for the catalysts in the LSV curves. Additionally, when the potential is increased from 1.50 to 1.65 V vs. RHE, a greater phase angle change and a larger shift of peaks toward the higher frequency region are observed in BSCTRS as compared to RuO2. The higher change in the phase angle value indicates a faster decrease in charge transfer resistance across the interface with increasing potential for BSCTRS as compared to RuO2. The phase angle data for both BSCTRS and RuO2 align well with the observation that, although the onset of OER potential is slightly lower for RuO2 compared to BSCTRS, the latter exhibits significantly higher current density at higher overpotentials. It is well established that the ECSA plays an important role in the OER activity.32 The estimated values of electrical double layer capacitance (Cdl) and ECSA are given in Table 2. The plot of scan rate vs. Δj (Fig. 7a) depicts the steepest slope for BSCTRS indicating enhanced Cdl and consequently a larger ECSA as compared to the benchmark RuO2. The higher ECSA of the catalyst indicates the presence of more exposed catalytically active sites compared to RuO2, supporting the superior OER activity of the catalyst. To further evaluate the intrinsic activity of the catalyst, LSV curves were normalized with respect to the ECSA (Fig. 7b). The ECSA normalized LSV data resulted in an overpotential of 326 mV as compared to 362 mV for RuO2, demonstrating superior OER performance and high intrinsic activity of BSCTRS compared to RuO2.
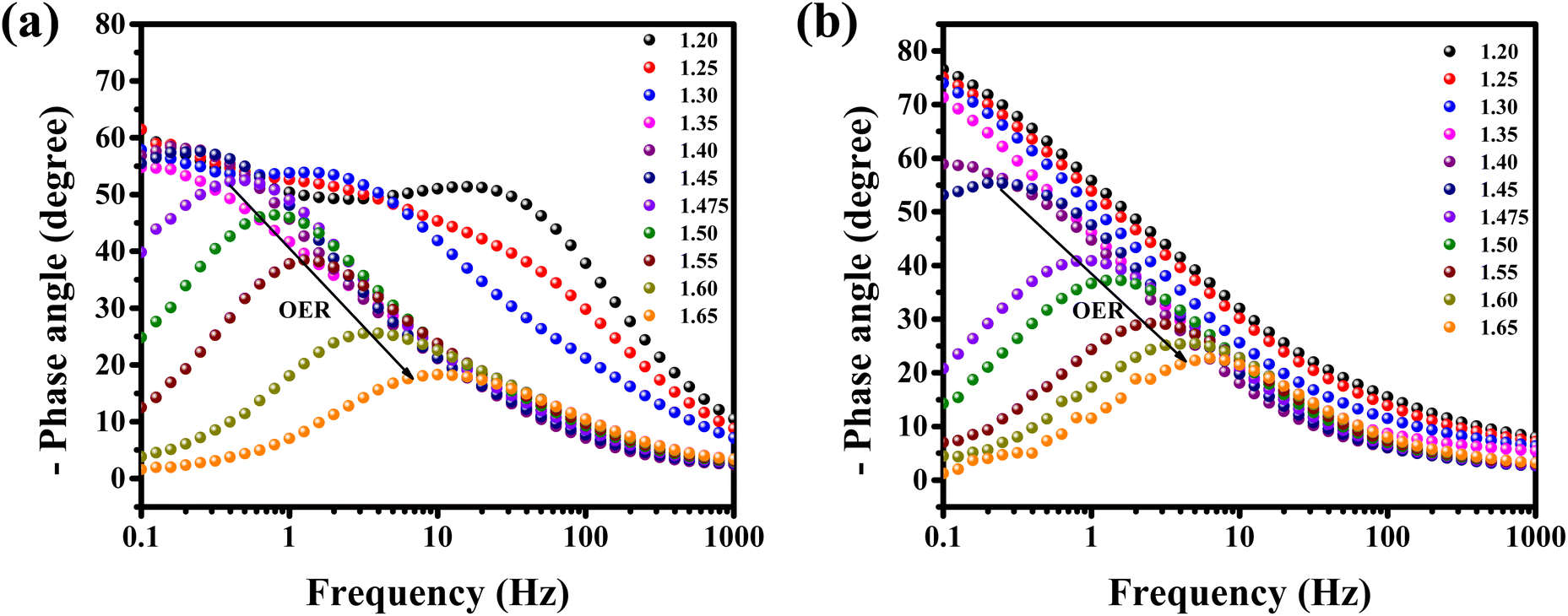 | ||
| Fig. 6 Bode plots of (a) BSCTRS and (b) RuO2 in the low frequency region at different potentials vs. RHE. | ||
| Sample | Slope (mF cm−2) | C dl (mF cm−2) | ECSA (cm2) |
|---|---|---|---|
| BSCTRS | 6.61 | 3.305 | 5.046 |
| RuO2 | 4.82 | 2.410 | 3.679 |
| NF | 1.31 | 0.655 | — |
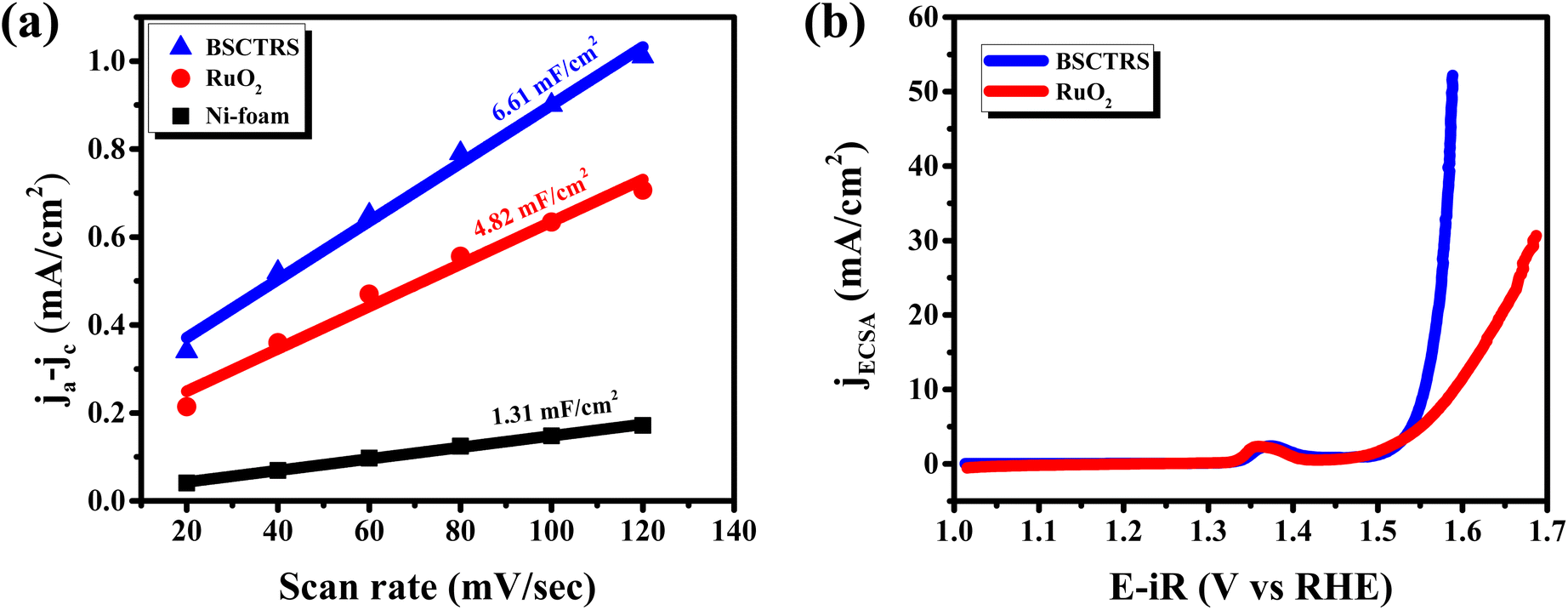 | ||
| Fig. 7 (a) Plot of scan rate vs. Δj, and (b) ESCA normalized polarization curves. | ||
Since the catalyst contains the precious element Ru in its composition, albeit in smaller amounts, the mass activity (MA) is an important parameter to evaluate its activity and efficiency. Fig. 8a and b show the enhanced mass activity of BSCTRS per gram of Ru as well as per gram of the oxide, respectively. The MA for BSCTRS turns out to be 37 times higher than that of RuO2, reaching 932 A/gRu at 1.59 V vs. RHE in comparison to 25 A/gRu for RuO2.
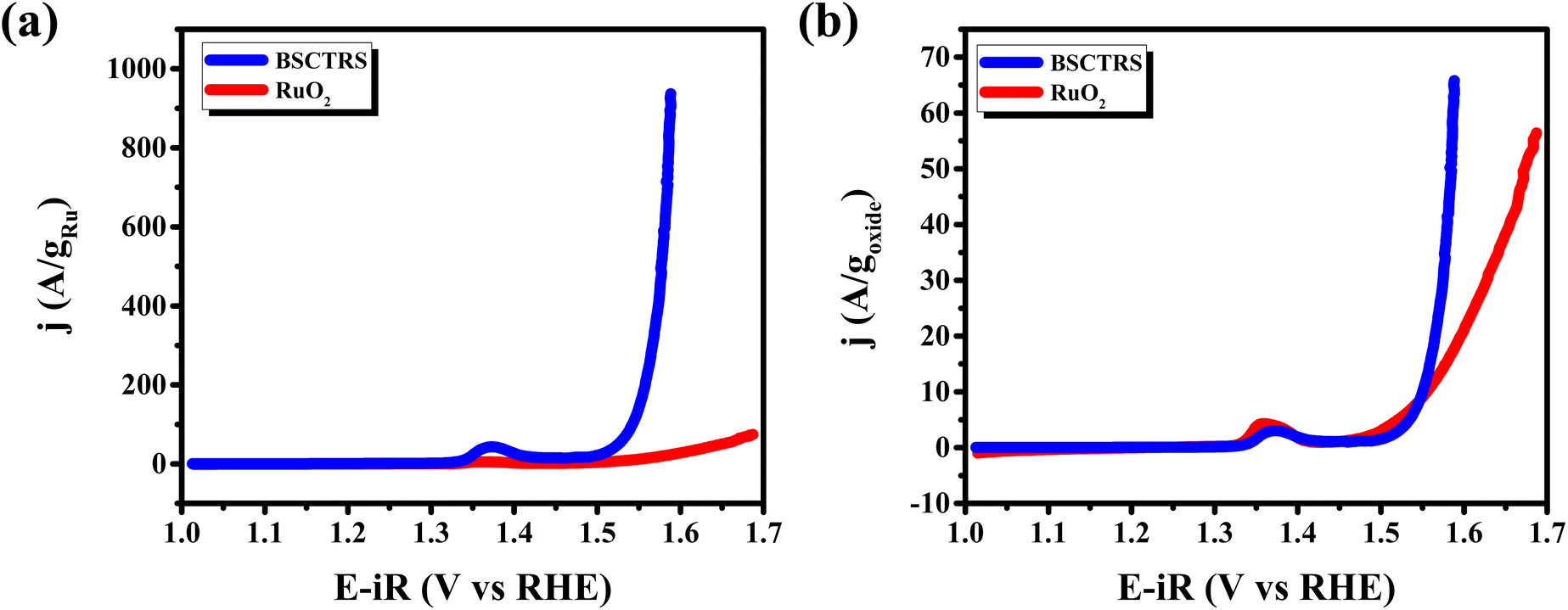 | ||
| Fig. 8 (a) Mass activity per gram of Ru, and (b) mass activity per gram of oxide. | ||
The compound shows low η10 and η100 values and one of the lowest reported Tafel slope values among the various perovskite ruthenate-based OER electrocatalysts reported recently (Table 3). Although, Sr0.9Ag0.1RuO3 possesses a lower η10 value than BSCTRS (220 vs. 286 mV), a much higher Tafel slope of the former (156 vs. 49 mV dec−1) indicates its kinetic sluggishness, making it a less efficient OER electrocatalyst than BSCTRS.
| Catalyst/substrate | Electrolyte | η 10 (mV) | η 100 (mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|
| a Abbreviations for substrates: GCE: glassy carbon electrode and NF: nickel foam. | |||||
| SrRuO3/NF | 0.1 M KOH | 483 | — | 169 | 21 |
| CaLaScRuO6+δ/GCE | 1 M KOH | 478 | — | — | 38 |
| La1.5Sr0.5Ni0.5Mn0.5Ni0.5Ru0.5O6/GCE | 0.1 M KOH | 430 | — | — | 39 |
| RuO2/La0.9Fe0.92Ru0.08O3/GCE | 0.1 M KOH | 420 | — | 116 | 7 |
| Ca2FeRuO6/GCE | 1 M KOH | 400 | — | 102 | 40 |
| SrTi0.7Ru0.3O3/NF | 0.1 M KOH | 375 | — | 224 | 21 |
| Ca0.9Sr0.1RuO3/GCE | 0.1 M KOH | 370@17 mA cm−2 | — | 43 | 22 |
| Ca2ScRuO6/GCE | 1 M KOH | 353 | — | 105 | 41 |
| (La0.8Sr0.2)0.9Co0.1Fe0.8Ru0.1O3−δ/GCE | 1 M KOH | 347 | 411 | 54.65 | 42 |
| CaSrScRuO6/GCE | 1 M KOH | 323 | — | 98 | 41 |
| Sr0.9Na0.1RuO3/GCE | 1 M KOH | 305 | — | 300 | 23 |
| Sr0.9Ag0.1RuO3/GCE | 1 M KOH | 220 | — | 156 | 23 |
| Ba 0.33 Sr 0.67 Co 0.33 Ti 0.165 Ru 0.165 Sb 0.33 O 3 /NF | 1 M KOH | 286 | 340 | 49 | This Work |
The chronopotentiometry data (Fig. 9a), show stable potential over a 16 h test period. The slight gradual increase in potential is due to the consumption and evaporation of electrolyte during the experiment, as no fresh electrolyte was replenished to compensate for the losses. Post chronopotentiometry LSV measurements conducted after replenishing the electrolyte show no observable change in the activity of the catalyst, demonstrating high stability of the catalyst under OER conditions (Fig. 9b).
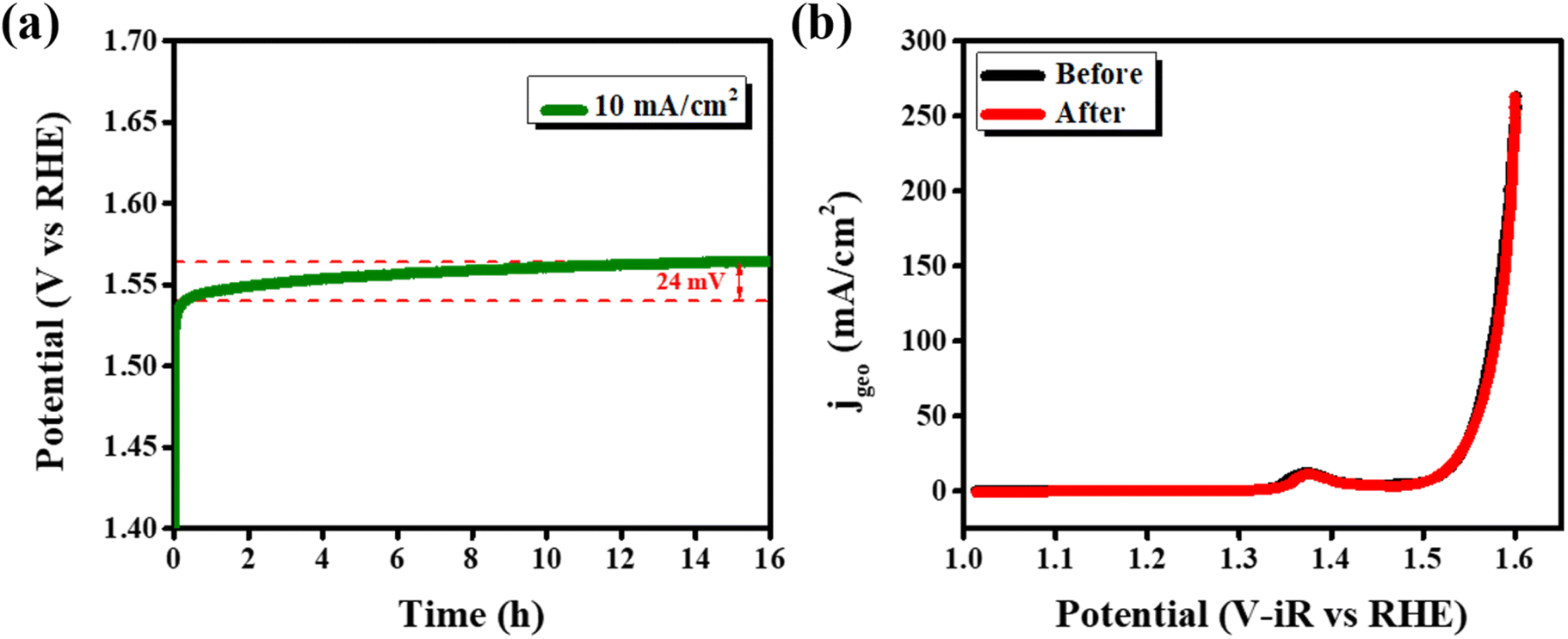 | ||
| Fig. 9 (a) Chronopotentiometry, and (b) polarization curves after chronopotentiometry. | ||
The post-catalytic P-XRD (Fig. S5†) of the compound shows an intact crystal structure of the catalyst with a lattice parameter of 3.9851(1) Å, similar to the pristine sample. Previous studies reported the dissolution of Sr and Ru for A-site Sr based perovskite ruthenates.21,39 Moreover, the SEM-EDS study also indicated patch formation with missing elemental signals in the EDS due to metal dissolution. FE-SEM elemental mapping of post-catalytic BSCTRS (Fig. 10b) shows uniform distribution of all the elements, demonstrating no patch formation or loss of elements due to dissolution during the OER process, further validating the high stability of the catalyst under OER conditions. The post catalytic EDS spectra (Fig. S6†) also confirmed the retention of original elemental composition. Furthermore, the MP-AES study of the electrolyte after chronopotentiometry also shows no significant presence of dissolved elements in the electrolyte (Table S3†).
 | ||
| Fig. 10 Post-catalytic (a) FE-SEM image, and (b) elemental mapping of BSCTRS. | ||
Origin of high intrinsic OER activity
Among the various factors affecting the OER activity, eg-orbital filling of the active surface metal site plays an important role.15 Suntivich et al. found that near unity filling of the eg orbital of the surface active metal site significantly improves the OER activity.15 The valence state of Co and Ru is analyzed with XPS technique. As stated earlier, we observed that Co exists in +2 and +3 oxidation states in BSCTRS. From the deconvoluted XPS peaks it was found that the ratio of Co3+/Co2+ is 4.26, which indicates that the average valence state of surface Co is ∼+2.8. A similar oxidation state has also been observed for Co based perovskite oxides in many previous studies.15,31,43 Furthermore, it is well established that the Co ion exists in an intermediate spin state in the BO6 octahedral environment.15,44 An electronic configuration of t2g5eg∼1.2 is expected with an eg-orbital filling of ∼1.2 based on the above observations and this configuration is reported to be most favorable for OER activity.15,31 Furthermore, the presence of Ru in the +4 oxidation state also suggests the t2g3eg1 electronic configuration with unity eg-filling of surface Ru ions. The presence of near unity filling of both the active metal sites of BSCTRS plays a significant role in the high activity of the catalyst.The involvement of lattice oxygens during the OER is reported to significantly improve the OER activity.45 It is well documented that oxides showing participation of lattice oxygens during the OER also show pH dependent OER kinetics on the RHE scale.17 Consequently, a pH dependent OER study was performed for BSCTRS (Fig. 11). The OER kinetics depends on the current, potential and pH according to the following equations:
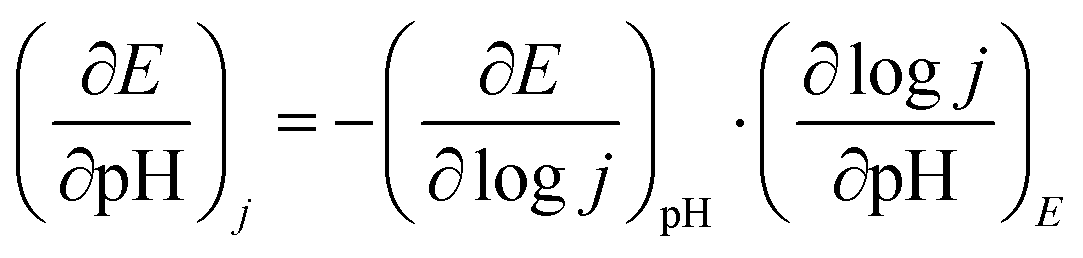
where, ρRHE is the reaction order on the RHE scale.46 Furthermore, for a pH dependent OER, current can be expressed as:
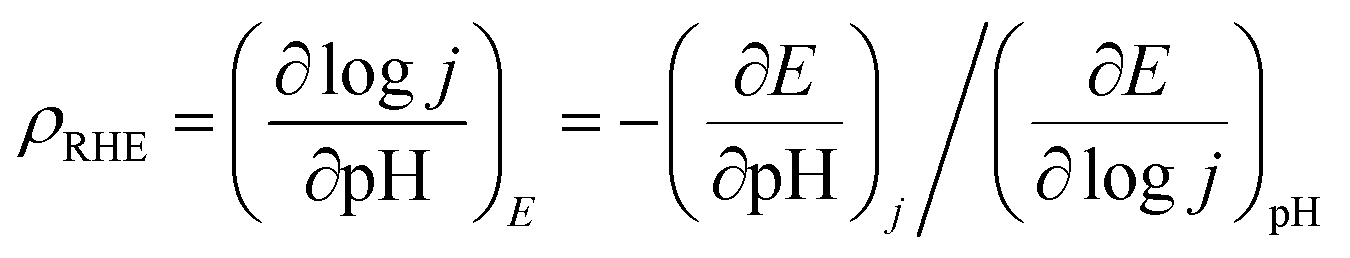
| j = θ·COH·e−ΔG/RT |
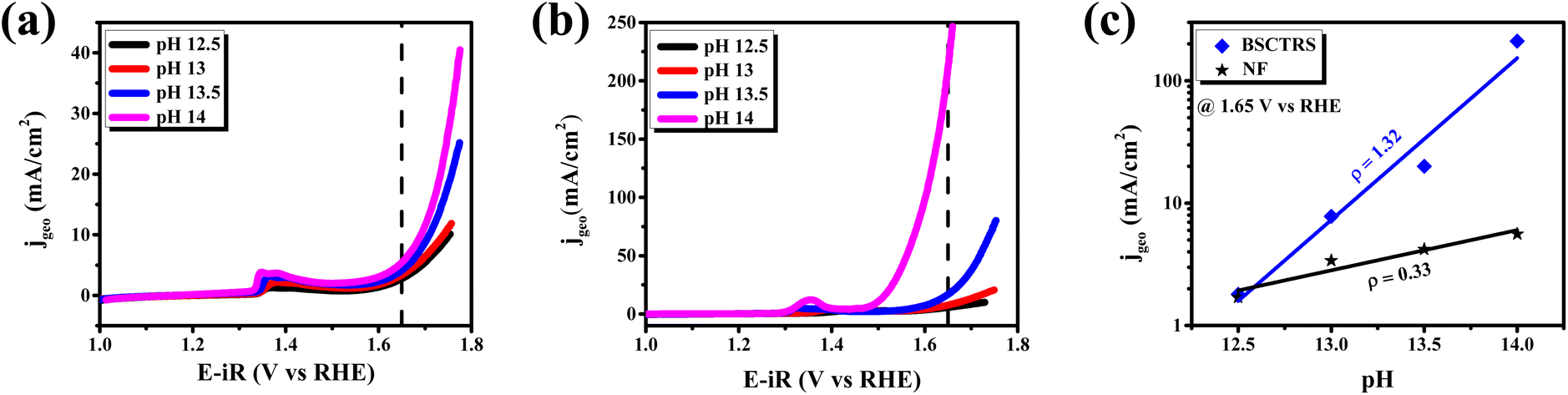 | ||
| Fig. 11 pH dependent polarization curves of (a) NF, and (b) BSCTRS. (c) OER specific activities at 1.65 V vs. pH of the electrolyte. | ||
Activation of the oxygen ligand is accepted as a prerequisite for an electrocatalyst to follow the LOM pathway.48 To confirm the oxygen activation, O 1s XPS of BSCTRS was recorded after treating it under different conditions, viz., after treating in 1 M KOH, after catalyst activation by 10 CV cycles, and after the chronopotentiometry test. As shown in the deconvoluted O 1s XPS spectra, after treating the catalyst in 1 M KOH solution, the percentage of adsorbed hydroxyl groups (corresponding to the Oc peak feature) increases significantly due to the higher OH− concentration in the solution (Fig. S7 and Table S4†). As can be seen in Fig. S7b,† a sharp increase in the reactive oxygen species or the low coordinated surface oxygen species (Ob peak feature) is observed upon activation of the catalyst by performing CV in the range of 1 to 1.6 V vs. RHE. This increase in reactive low coordinated lattice oxygen species confirms the activation of the catalyst and paves the way for the LOM OER mechanism to take place. The post chronopotentiometry deconvoluted O 1s XPS spectra also shows an enhanced presence of Ob and Oc features at the expense of the Oa feature. This also supports the constant lattice oxygen participation in BSCTRS during the OER process.
Conclusions
Ba0.33Sr0.67Co0.33Ti0.165Ru0.165Sb0.33O3 as a high-entropy perovskite oxide was synthesized by solid-state reactions. The compound exhibited comparable η10 and far superior η100 and Tafel slope compared to the benchmark RuO2 for the OER. A substantial reduction in the Tafel slope and increased TOF of BSCTRS as compared to Ba0.33Sr0.67Co0.33Ti0.33Sb0.33O3 demonstrated the successful adaptation of strategic incorporation of Ru in the composition. Moreover, the electrocatalyst also showed much higher mass activity in comparison to the benchmark RuO2. The enhanced OER of BSCTRS is primarily due to near unity filling of the eg-orbital for both Co and Ru, a higher percentage of low coordinated surface oxygens, and increased lattice oxygen participation. Enhanced lattice oxygen participation and the LOM mechanistic pathway were confirmed through the pH dependent OER study. The high stability of the compound despite enhanced lattice oxygen participation demonstrated the significance of high-entropy perovskites in the quest for highly active perovskite based electrocatalysts for the OER. With its low Ru content along with high activity and stability, BSCTRS shows great promise as a viable and affordable alternative to commercial precious metal oxide based OER electrocatalysts.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.† The raw data that support the findings of this study are available from the corresponding author, [TKM], upon reasonable request.Author contributions
Sujan Sen: conceptualization, methodology, data curation, formal analysis, investigation, and writing – original draft. Tapas Kumar Mandal: conceptualization, formal analysis, resources, supervision, validation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Sen acknowledges the Ministry of Education (MoE), Govt. of India, New Delhi, for providing a research fellowship. The authors also acknowledge the Department of Chemistry, and Department of Metallurgical and Materials Engineering, IIT Roorkee, for providing various instrumental facilities.References
- S. Chu and A. Majumdar, Opportunities and Challenges for a Sustainable Energy Future, Nature, 2012, 488, 294–303, DOI:10.1038/nature11475
.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Perovskites in Catalysis and Electrocatalysis, Science, 2017, 358, 751–756, DOI:10.1126/science.aam7092
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Energy and Fuels from Electrochemical Interfaces, Nat. Mater., 2017, 16, 57–69, DOI:10.1038/nmat4738
- M. Yu, E. Budiyanto and H. Tuysuz, Principles of Water Electrolysis and Recent Progress in Cobalt-, Nickel-, and Iron-Based Oxides for the Oxygen Evolution Reaction, Angew. Chem., Int. Ed., 2022, 61, e202103824, DOI:10.1002/anie.202103824
- Y. Wei, C.-H. Shin, E. B. Tetteh, B.-J. Lee and J.-S. Yu, Insight into the Boosted Electrocatalytic Oxygen Evolution Performance of Highly Hydrophilic Nickel−Iron Hydroxide, ACS Appl. Energy Mater., 2020, 3, 822–830, DOI:10.1021/acsaem.9b01952
- B.-J. Kim, D. F. Abbott, X. Cheng, E. Fabbri, M. Nachtegaal, F. Bozza, I. E. Castelli, D. Lebedev, R. Schäublin, C. Copéret, T. Graule, N. Marzari and T. J. Schmidt, Unraveling Thermodynamics, Stability, and Oxygen Evolution Activity of Strontium Ruthenium Perovskite Oxide, ACS Catal., 2017, 7, 3245–3256, DOI:10.1021/acscatal.6b03171
- Y. Jiang, Z. Geng, L. Yuan, Y. Sun, Y. Cong, K. Huang, L. Wang and W. Zhang, Nanoscale Architecture of RuO2/La0.9Fe0.92Ru0.08−xO3−δ Composite via Manipulating the Exsolution of Low Ru-Substituted A-Site Deficient Perovskite, ACS Sustain. Chem. Eng., 2018, 6, 11999–12005, DOI:10.1021/acssuschemeng.8b02288
- X. Miao, L. Zhang, L. Wu, Z. Hu, L. Shi and S. Zhou, Quadruple Perovskite Ruthenate as a Highly Efficient Catalyst for Acidic Water Oxidation, Nat. Commun., 2019, 10, 3809, DOI:10.1038/s41467-019-11789-3
- D. F. Abbott, R. K. Pittkowski, K. Macounová, R. Nebel, E. Marelli, E. Fabbri, I. E. Castelli, P. Krtil and T. J. Schmidt, Design and Synthesis of Ir/Ru Pyrochlore Catalysts for the Oxygen Evolution Reaction Based on Their Bulk Thermodynamic Properties, ACS Appl. Mater. Interfaces, 2019, 11, 37748–37760, DOI:10.1021/acsami.9b13220
- M. V. Kante, M. L. Weber, S. Ni, I. C. G. van den Bosch, E. van der Minne, L. Heymann, L. J. Falling, N. Gauquelin, M. Tsvetanova, D. M. Cunha, G. Koster, F. Gunkel, S. Nemšák, H. Hahn, L. Velasco Estrada and C. Baeumer, A High-Entropy Oxide as High-Activity Electrocatalyst for Water Oxidation, ACS Nano, 2023, 17(6), 5329–5339, DOI:10.1021/acsnano.2c08096
- T. X. Nguyen, Y.-C. Liao, C.-C. Lin, Y.-H. Su and J.-M. Ting, Advanced High Entropy Perovskite Oxide Electrocatalyst for Oxygen Evolution Reaction, Adv. Funct. Mater., 2021, 31, 2101632, DOI:10.1002/adfm.202101632
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. F. B. D. Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, Dynamically Stable Active Sites from Surface Evolution of Perovskite Materials during the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2021, 143, 2741–2750, DOI:10.1021/jacs.0c08959
- Y. Matsumoto, H. Manabe and E. Sato, Oxygen Evolution
on La1−xSrxCoO3 Electrodes in Alkaline Solutions, J. Electrochem. Soc., 1980, 127, 811–814, DOI:10.1149/1.2129762
- J. O. Bockris and T. Otagawa, The Electrocatalysis of Oxygen Evolution on Perovskites, J. Electrochem. Soc., 1984, 131, 290–302, DOI:10.1149/1.2115565
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles, Science, 2011, 334, 1383–1385, DOI:10.1126/science.1212858
- W. T. Hong, R. E. Welsch and Y. Shao-Horn, Descriptors of Oxygen-Evolution Activity for Oxides: A Statistical Evaluation, J. Phys. Chem. C, 2016, 120, 78–86, DOI:10.1021/acs.jpcc.5b10071
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Activating Lattice Oxygen Redox Reactions in Metal Oxides to Catalyse Oxygen Evolution, Nat. Chem., 2017, 9, 457–465, DOI:10.1038/nchem.2695
- C. Feng, M. B. Faheem, J. Fu, Y. Xiao, C. Li and Y. Li, Fe-Based Electrocatalysts for Oxygen Evolution Reaction: Progress and Perspectives, ACS Catal., 2020, 10, 4019–4047, DOI:10.1021/acscatal.9b05445
- A. K. Tomar, U. N. Pan, N. H. Kim and J. H. Lee, Enabling Lattice Oxygen Participation in a Triple Perovskite Oxide Electrocatalyst for the Oxygen Evolution Reaction, ACS Energy Lett., 2023, 8, 565–573, DOI:10.1021/acsenergylett.2c02617
- C. E. Beall, E. Fabbri and T. J. Schmidt, Perovskite Oxide Based Electrodes for the Oxygen Reduction and Evolution Reactions: The Underlying Mechanism, ACS Catal., 2021, 11, 3094–3114, DOI:10.1021/acscatal.0c04473
- H.-J. Liu, C.-Y. Chiang, Y.-S. Wu, L.-R. Lin, Y.-C. Ye, Y.-H. Huang, J.-L. Tsai, Y.-C. Lai and R. Munprom, Breaking the Relation between Activity and Stability of the Oxygen-Evolution Reaction by Highly Doping Ru in Wide-Band-Gap SrTiO3 as Electrocatalyst, ACS Catal., 2022, 12, 6132–6142, DOI:10.1021/acscatal.1c05539
- S. Hirai, T. Ohno, R. Uemura, T. Maruyama, M. Furunaka, R. Fukunaga, W.-T. Chen, H. Suzuki, T. Matsuda and S. Yagi, Ca1-xSrxRuO3 Perovskite at the Metal–Insulator Boundary as a Highly Active Oxygen Evolution Catalyst, J. Mater. Chem. A, 2019, 7, 15387–15394, 10.1039/c9ta03789f
- Y. Wang, J. Wu, X. Lu, Y. Guo, H. Zhao and X. Tang, A-Site Doped Ruthenium Perovskite Bifunctional Electrocatalysts with High OER and ORR Activity, J. Alloys Compd., 2022, 920, 165770, DOI:10.1016/j.jallcom.2022.165770
- S. Sen, M. Goyal, L. Kumar and T. K. Mandal, Excellent Electrocatalytic Oxygen Evolution Reaction by Non-Noble Metal Based 3D Perovskite Oxides Ba3-xSrxMTiSbO9 (x = 1, 1.5 for M = Co and x = 2 for Mn/Co), ACS Appl. Energy Mater., 2024, 7(4), 1495–1507, DOI:10.1021/acsaem.3c02750
- H. M. Rietveld, A Profile Refinement Method for Nuclear and Magnetic Structures, J. Appl. Crystallogr., 1969, 2(2), 65–71, DOI:10.1107/S0021889869006558
- J. Rodríguez-Carvajal, Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction, Phys. B, 1993, 192(1), 55–69, DOI:10.1016/0921-4526(93)90108-I
- H. Liu, J. Yu, J. Sunarso, C. Zhou, B. Liu, Y. Shen, W. Zhou and Z. Shao, Mixed Protonic-Electronic Conducting Perovskite Oxide as a Robust Oxygen Evolution Reaction Catalyst, Electrochim. Acta, 2018, 282, 324–330, DOI:10.1016/j.electacta.2018.06.073
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlögl and H.
N. Alshareef, Plasma-Assisted Synthesis of NiCoP for Efficient Overall Water Splitting, Nano Lett., 2016, 16, 7718–7725, DOI:10.1021/acs.nanolett.6b03803
- Y. Xu, L. Xie, D. Li, R. Yang, D. Jiang and M. Chen, Engineering Ni(OH)2 Nanosheet on CoMoO4 Nanoplate Array as Efficient Electrocatalyst for Oxygen Evolution Reaction, ACS Sustain. Chem. Eng., 2018, 6, 16086–16095, DOI:10.1021/acssuschemeng.8b02663
- T. Erdil and C. Toparli, B-Site Effect on High-Entropy Perovskite Oxide as a Bifunctional Electrocatalyst for Rechargeable Zinc–Air Batteries, ACS Appl. Energy Mater., 2023, 6(21), 11255–11267, DOI:10.1021/acsaem.3c02149
- S. Wu, J. Li, J. Hu, Y. Huang, H.-S. Xu and K. Tang, Perovskite Oxide Pr0.5Sr0.5Co0.8Fe0.2O3−δ as an Efficient Catalyst for Oxygen Evolution Reaction in Alkaline Media, ACS Appl. Energy Mater., 2023, 6, 6289–6298, DOI:10.1021/acsaem.3c00807
- M. Yang, W. Lu, R. Jin, X.-C. Liu, S. Song and Y. Xing, Superior Oxygen Evolution Reaction Performance of Co3O4/NiCo2O4/Ni Foam Composite with Hierarchical Structure, ACS Sustain. Chem. Eng., 2019, 7, 12214–12221, DOI:10.1021/acssuschemeng.9b01535
- D. Ji, C. Liu, Y. Yao, L. Luo, W. Wang and Z. Chen, Cerium Substitution in LaCoO3 Perovskite Oxide as Bifunctional Electrocatalysts for Hydrogen and Oxygen Evolution Reactions, Nanoscale, 2021, 13, 9952–9959, 10.1039/d1nr00069a
- T. Zhao, Y. Wang, X. Chen, Y. Li, Z. Su and C. Zhao, Vertical Growth of Porous Perovskite Nanoarrays on Nickel Foam for Efficient Oxygen Evolution Reaction, ACS Sustain. Chem. Eng., 2020, 8, 4863–4870, DOI:10.1021/acssuschemeng.0c00060
- Q. Luo, D. Lin, W. Zhan, W. Zhang, L. Tang, J. Luo, Z. Gao, P. Jiang, M. Wang, L. Hao and K. Tang, Hexagonal Perovskite Ba0.9Sr0.1Co0.8Fe0.1Ir0.1O3−δ as an Efficient Electrocatalyst towards the Oxygen Evolution Reaction, ACS Appl. Energy Mater., 2020, 3, 7149–7158, DOI:10.1021/acsaem.0c01192
- H. Chen, S. Ouyang, M. Zhao, Y. Li and J. Ye, Synergistic Activity of Co and Fe in Amorphous Cox−Fe−B Catalyst for Efficient Oxygen Evolution Reaction, ACS Appl. Mater. Interfaces, 2017, 9, 40333–40343, DOI:10.1021/acsami.7b13939
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Insight on Tafel Slopes from a Microkinetic Analysis of Aqueous Electrocatalysis for Energy Conversion, Sci. Rep., 2015, 5, 13801, DOI:10.1038/srep13801
- N. Kumar, M. Kumar, T. C. Nagaiah, V. Siruguri, S. Rayaprol, A. K. Yadav, S. N. Jha, D. Bhattacharyya and A. K. Paul, Investigation of New B-Site-Disordered Perovskite Oxide CaLaScRuO6+δ: An Efficient Oxygen Bifunctional Electrocatalyst in a Highly Alkaline Medium, ACS Appl. Mater. Interfaces, 2020, 12, 9190–9200, DOI:10.1021/acsami.9b20199
- M. Retuerto, F. Calle-Vallejo, L. Pascual, G. Lumbeeck, M. T. Fernandez-Diaz, M. Croft, J. Gopalakrishnan, M. A. Peña, J. Hadermann, M. Greenblatt and S. Rojas, La1.5Sr0.5NiMn0.5Ru0.5O6 Double Perovskite with Enhanced ORR/OER Bifunctional Catalytic Activity, ACS Appl. Mater. Interfaces, 2019, 11, 21454–21464, DOI:10.1021/acsami.9b02077
- N. Kumar, K. Naveen, M. Kumar, T. C. Nagaiah, R. Sakla, A. Ghosh, V. Siruguri, S. Sadhukhan, S. Kanungo and A. K. Paul, Multifunctionality Exploration of Ca2FeRuO6: An Efficient Trifunctional Electrocatalyst toward OER/ORR/HER and Photocatalyst for Water Splitting, ACS Appl. Energy Mater., 2021, 4, 1323–1334, DOI:10.1021/acsaem.0c02579
- N. Kumar, T. Rom, M. Kumar, T. C. Nagaiah, E. Lee, H. C. Ham, S. H. Choi, S. Rayaprol, V. Siruguri, T. K. Mandal, B. J. Kennedy and A. K. Paul, Unraveling the Effect of A-Site Sr-Doping in Double Perovskites Ca2−xSrxScRuO6 (x = 0 and 1): Structural Interpretation and Mechanistic Investigations of Trifunctional Electrocatalytic Effects, ACS Appl. Energy Mater., 2022, 5, 11632–11645, DOI:10.1021/acsaem.2c02101
- Y. Liang, Y. Cui, Y. Chao, N. Han, J. Sunarso, P. Liang, X. He, C. Zhang and S. Liu, Exsolution of CoFe(Ru) Nanoparticles in Ru-Doped (La0.8Sr0.2)0.9Co0.1Fe0.8Ru0.1O3−δ for Efficient Oxygen Evolution Reaction, Nano Res., 2022, 15, 6977–6986, DOI:10.1007/s12274-022-4328-0
- Y. Zhu, W. Zhou, Z.-G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, SrNb0.1Co0.7Fe0.2O3−δ Perovskite as a Next-Generation Electrocatalyst for Oxygen Evolution in Alkaline Solution, Angew. Chem., Int. Ed., 2015, 54, 3897–3901, DOI:10.1002/anie.201408998
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Double Perovskites as a Family of Highly Active Catalysts for Oxygen Evolution in Alkaline Solution, Nat. Commun., 2013, 4, 2439, DOI:10.1038/ncomms3439
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J.-P. M. Veder, D. Guan, R. O'Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Direct Evidence of Boosted Oxygen Evolution over Perovskite by Enhanced Lattice Oxygen Participation, Nat. Commun., 2020, 11, 2002, DOI:10.1038/s41467-020-15873-x
- L. Giordano, B. Han, M. Rischc, W. T. Hong, R. R. Rao, K. A. Stoerzinger and Y. Shao-Horn, pH Dependence of OER Activity of Oxides: Current and Future Perspectives, Catal. Today, 2016, 262, 2–10, DOI:10.1016/j.cattod.2015.10.006
- C. Yang, C. Laberty-Robert, D. Batuk, G. Cibin, A. V. Chadwick, V. Pimenta, W. Yin, L. Zhang, J.-M. Tarascon and A. Grimaud, Phosphate Ion Functionalization of Perovskite Surfaces for Enhanced Oxygen Evolution Reaction, J. Phys. Chem. Lett., 2017, 8, 3466–3472, DOI:10.1021/acs.jpclett.7b01504
- N. Zhang and Y. Chai, Lattice Oxygen Redox Chemistry in Solid-State Electrocatalysts for Water Oxidation, Energy Environ. Sci., 2021, 14(9), 4647–4671, 10.1039/D1EE01277K
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01204f |
| This journal is © The Royal Society of Chemistry 2025 |