
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
YFO photocathode fabricated via spray pyrolysis for unassisted solar water splitting for generation of hydrogen fuel†
Bandar Y.
Alfaifi
a,
Hameed
Ullah
 *b,
Xin
Jiang
*b and
Asif Ali
Tahir
*c
*b,
Xin
Jiang
*b and
Asif Ali
Tahir
*c
aDepartment of Electrical Engineering, College of Engineering and Information Technology, Onaizah Colleges, Qassim, Saudi Arabia
bChair of Surface and Materials Technology, Institute of Materials Engineering, University of Siegen, 57076 Siegen, Germany. E-mail: hameed.ullah@uni-siegen.de; xin.jiang@uni-siegen.de
cSolar Energy Research Group, Environment and Sustainability Institute, Department of Engineering, University of Exeter, Penryn Campus, Cornwall, UK. E-mail: A.Tahir@exeter.ac.uk
First published on 26th November 2024
Abstract
Efficient solar to fuel conversion technology is highly desirable to meet future global renewable energy demands as conventional energy resources are environmentally irresponsible and depleting rapidly. Photoelectrochemical (PEC) water splitting without the use of any external potential bias and/or assistance to produce hydrogen (a clean and renewable fuel) is a technology having the potential to fulfil this desire. However, the main bottleneck is the unavailability of cost-effective, efficient and stable photoelectrodes, which are used to conduct water splitting using light photons. YFeO3 (YFO) thin films with a small energy band gap (Eg), suitable band positions straddling water redox potential and high stability were fabricated using a simple, cost-effective and scalable synthesis technique i.e., spray pyrolysis. The optimum YFO film was applied, for the first time to the best of our knowledge, for generation of hydrogen fuel through water splitting without applying any external potential bias and/or assistance. Orthorhombic YFO (o-YFO) showed a maximum photocurrent of ∼0.65 mA cm−2 at 0.46 V vs. RHE, faradaic efficiency of ∼70%, and excellent stability spanning over 6 hours. UV-visible and electrochemical impedance spectroscopy (EIS) revealed the p-type characteristic, narrow Eg of 2.45 eV and suitable band positions, which encompassed the redox potential of water, of the o-YFO film. The o-YFO film generated 0.41 µmol cm−2 of hydrogen over 6 hours without any assistance in a spontaneous hydrogen evolution reaction (HER). In a subsequent cycle, it generated 0.35 µmol cm−2 of hydrogen, showing its potential as a reusable photoelectrode in the HER. Post HER characterizations did not show any visible/significant changes in the phase and morphology of the o-YFO film, indicating its stability under the applied HER conditions.
Introduction
The global energy demand is increasing almost exponentially, and for the most parts, the demand is met by fossil fuel-based energy resources, which include coal, liquid fuels and natural gas.1 However, such fuels are responsible for global warming as they emit greenhouse gases (GHGs) upon combustion, which affect the climate adversely.2 Moreover, these energy resources are limited, facing challenges of depletion with the growing demand, and are concentrated in specific regions, making their secure and continuous supply a challenge.3,4 Therefore, intensive efforts are underway to transform the economy completely from fossil fuel-based to renewable-based and with globally evenly distributed energy resources.5 Among the renewable energy resources, solar power is at the forefront as the earth receives more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TW of energy annually from the sun.6,7 However, there are still a number of challenges encountered in conversion of this huge amount of sun energy into safely storable and easily transportable fuel.8,9 One of the fuels that can be produced by exploiting sun power is hydrogen, which is renewable and environmentally benign.10 To achieve this goal, there is a need to explore and optimize solar-driven hydrogen-production methods that are economically viable and technologically efficient.9
000 TW of energy annually from the sun.6,7 However, there are still a number of challenges encountered in conversion of this huge amount of sun energy into safely storable and easily transportable fuel.8,9 One of the fuels that can be produced by exploiting sun power is hydrogen, which is renewable and environmentally benign.10 To achieve this goal, there is a need to explore and optimize solar-driven hydrogen-production methods that are economically viable and technologically efficient.9
Among the different solar-power-driven hydrogen-production technologies, photoelectrochemical (PEC) water splitting is highly promising owing to its lower overpotential needed to drive the water splitting reaction and use of cost-effective instrumentation constructed of simple components.7,11,12 Fujishima and Honda pioneered PEC technique for water splitting using a titanium dioxide (TiO2) photoanode in combination with a platinum (Pt) wire counter electrode.13 Since then, tremendous progress has been made in PEC technology for water splitting to generate hydrogen.9,14 One of the main and critical components of PEC devices is the photoelectrode, which determines the performance of PEC devices, and should be made of a material that is cost-effective, stable in aqueous media, and has high solar-to-hydrogen (STH) conversion efficiency.15 However, despite many investigations into diverse materials, from conventional metal oxide photoanodes, like TiO2,16 bismuth vanadate (BiVO4),17 ferric oxide (Fe2O3),18 and tungsten trioxide (WO3),19 to a number of photocathode materials, like cuprous oxide (Cu2O),20 indium gallium nitride (InGaN),21 copper iron oxides (CuFe2O422 and CuFeO223), and lanthanum ferrite (LaFeO3),24 an ideal semiconductor material to be employed as a photoelectrode in PEC has not yet been found. The lack of success of these materials as photoelectrodes in practical PEC devices could be due to their poor charge mobility, low stability, and high costs.7,15,25 It is pertinent to mention here that an ideal photoelectrode material for application in practical PEC devices must meet some essential requirements, which include a suitable energy band gap to allow absorption in the visible part of the solar spectrum and a potential that can straddle the redox potential of water. It must also have the ability to effectively separate the generated charges and migrate them to the surface without recombination. It must be capable of reducing and oxidizing the adsorbed species by using the generated electron–hole pairs to produce hydrogen and oxygen.26 Finally, the desired photoelectrode must be stable in aqueous electrolyte.27,28 Additionally, it would be exceptionally ideal if it could split water into hydrogen in the absence of any external potential bias and/or assistance.
The conventional metal oxide semiconductors mentioned above can meet the requirements up to some extent but they could not promise large-scale application in practical PEC devices. For example, metal oxides like TiO2, tin dioxide (SnO2), zinc oxide (ZnO), and tantalum pentoxide (Ta2O5) have shown excellent stability, but are limited by their wide energy band gap (Eg), which allows them to absorb only in the UV region of the visible spectrum.6 Also, while metal oxides like α-Fe2O3 and zinc ferrite (ZnFe3O4) are decorated with small Eg, making them suitable for absorption in the visible region, they can only split water under external potential bias/assistance because of their more positive conduction band potential than the reduction potential of water.6 Meanwhile photoelectrode materials that do not need assistance and/or external potential bias owing to their ideal band structure tend to suffer from extreme instability. For example, metal chalcogenides, nitrides, and oxynitrides suffer from photo-corrosion and photo-oxidation during hydrogen generation reactions.6 Although p-type semiconductors, for example Cu2O,29 nickel oxide (NiO),30 and calcium ferrite (CaFe2O4),31 have shown encouraging activities as photocatalysts for water splitting, they also face certain challenges that hinder their use for the large-scale production of hydrogen from water splitting under solar illumination. With a current density as high as 10 mA cm−2, Cu2O is considered one of the most highly promising photoelectrode materials for PEC devices. However, it is extremely unstable and degrades very quickly during the hydrogen evolution reaction (HER).32 To improve its stability, different coatings have been applied.33 However, these coating methods have their own problems and challenges. NiO and CaFe2O4 are highly stable narrow band gap materials, respectively, making them interesting as photoelectrodes in PEC devices. However, the former, i.e. NiO, is limited by its large Eg, absorbing only in the UV region, while the latter, i.e. CaFe2O4, requires extreme synthesis conditions, which make it not cost-effective at all.
Perovskite-type metal ferrite materials (MFeO3) have been found to meet numerous requirements of ideal photoelectrodes for unassisted water splitting,34 and they are therefore are at the centre of intensive research work towards the realization of practical PEC devices. The importance of ferrite photoelectrodes for unassisted water splitting is evident from the number of review articles published in the last couple of years alone.34–36 They are cost-effective, highly stable over a large pH range in aqueous media, and have a low band gap (2.3–2.4 eV).34,37 We fabricated a lanthanum ferrite (LaFeO3) photocathode by a scalable and cost-effective spray pyrolysis method, and found it was highly stable and efficient for the production of hydrogen from water splitting without the need for any external potential bias and/or assistance.12 This encouraged us to extend the method to one of the highly important metal ferrite photocathodes, i.e. yttrium orthoferrite (YFeO3), which is abbreviated henceforth as YFO. YFO crystallizes in two different phases, namely hexagonal (h) and orthorhombic (o), as a function of the annealing temperature.38 In h-YFO, the bond angles and distances are more uniform, allowing the orbitals to overlap more effectively. This overlap brings the energy levels of the valence and conduction bands closer together, which narrows the band gap. In contrast, o-YFO has a more distorted oxygen environment around the iron ions. This distortion reduces orbital overlap, causing a greater separation between the energy levels and resulting in a wider band gap.39 Therefore, the energy band gap of h-YFO (∼1.8 eV) is smaller than that of o-YFO (∼2.4 eV), enabling it to harvest higher sun light.36 However, the metastability of h-YFO due to its thermodynamically unstable crystal system makes it challenging to be realized in the correct calcination temperature window.40 Therefore, o-YFO has emerged as a stable photoelectrode with an energy band gap suitable to absorb in the visible region of the solar spectrum. Owing to this, o-YFO has found uses in photocatalytic water remediation41,42 and photochemical hydrogen evolution.43 Díez-García et al.37 applied o-YFO as a photocathode for hydrogen evolution. They sintered o-YFO nanoparticles at 1000 °C after preparation by the so-called ionic liquid method and then deposited these on FTO glass by the doctor blade technique to prepare YFO films. They also prepared films by spin-coating using a sol–gel precursor. However, the film they obtained was amorphous even though they heat treated it at 600 °C for 2 h. We have experienced in case of LaFeO3 that the quality of the nanostructured thin film has a profound effect on the PEC performance of photoelectrode.6 However, the methods reported so far lack control over the quality of the o-YFO thin film, and thus do not possess the flexibility to allow tailoring of photoelectrodes with the desired stability, low cost, and ideal band structure. We believe that spray pyrolysis as a novel, inexpensive, and scalable method has the potential to overcome these issues on the one hand, and on the other hand, could enable us to realize a high-quality nanostructured o-YFO film directly on FTO glass substrate at moderate temperature for unassisted PEC water splitting to hydrogen under visible light. Spray pyrolysis has been previously used for the fabrication of YFO films.44 However, detailed analysis of the films and their use as photoelectrodes is still missing, and needs to be explored.
Here in this work, we present the fabrication of nanostructured o-YFO thin films on FTO glass substrate by a simple, inexpensive, and scalable method i.e. spray pyrolysis. The resulting films were employed as photocathodes for PEC water splitting to generate hydrogen under alkaline conditions without the use of external potential bias and/or assistance. To the best of our knowledge, no spray pyrolyzed nanostructured o-YFO film has been used so far in this role. We investigated the key properties of the thin film materials using state-of-the-art techniques. The film exhibited p-type semiconductor properties and was stable under the applied HER conditions. The band structure was determined, and was found to straddle above and below the redox potential of water. Owing to this, the film showed excellent PEC performance.
Experimental
Materials
The chemicals, iron(III) acetylacetonate (Fe(acac)3), yttrium(III) nitrate hexahydrate (Y(NO3)3·6H2O), dodecyltrimethylammonium bromide (DTAB), and polyethylene glycol (PEG) were purchased from commercial suppliers and used as received without any further treatment and/or purification. The solvents methanol (CH3OH), iso-propanol ((CH3)2CHOH), and acetone (CH3COCH3) were purchased from Sigma-Aldrich and used as received without any further drying. Fluorine-doped tin oxide (FTO) glass was purchased from MSE Supplies LLC.Fabrication of the o-YFO thin film photocathode
Following a reported method,6 the o-YFO photocathodes were prepared by spray pyrolysis, for which the solution was prepared by dissolving 5 mmol each of Fe(acac)3, Y(NO3)3·6H2O, and DTAB in 200 mL methanol. A few drops of polyethylene glycol (PEG) were added to the prepared solution, which was then stirred at 60 °C for 5 min. After cooling down, the solution was used for the preparation of o-YFO by spray pyrolysis on an FTO glass substrate. Before spraying with the prepared solution, the FTO glass was cleaned ultrasonically in ethanol, iso-propanol, and acetone for 15 min successively with each liquid. The precursor solution was sprayed on to the FTO glass substrate maintained at 200 °C via a syringe pump (New Era Pump Systems NE-1000), with an ultrasonic atomizer nozzle (Sonozap) of 1 mm diameter and a vortex attachment. With a spray rate of 1 mL min−1, 30 mL of the prepared solution was sprayed on to the FTO glass substrate under compressed air assistance at a rate of 4 L min−1. Upon completion of the spray process, the prepared samples were annealed at different temperatures for 3 hours in air to obtain different samples, i.e. 500 °C (YFO-500), 550 °C (YFO-550), 600 °C (YFO-600), 650 °C, and (YFO-650).Materials characterization
X-Ray diffraction (XRD) patterns were acquired using a Siemens D5000 diffractometer equipped with a Cu target emitting Kα radiation. The diffractograms were recorded in the 2θ range of 20–80° with a step size of 0.02° and a scan time of 2.5 s per step. The surface morphology and elemental compositions of the fabricated films were studied by high-resolution scanning electron microscopy (SEM, TESCAN VEGA3). The thickness of the films was characterized by a focused ion beam (FIB, NOVA NanoLab FEI 600 dual beam FIB SEM). Optical absorption measurements were acquired using a spectrophotometer (PerkinElmer lambda 1050 with a 150 mm integrated InGaAs sphere). Photoelectrochemical (PEC) characterizations were performed using Metrohm Autolab. The Raman shift patterns were obtained using a Renishaw RM1000 instrument (Renishaw plc, Gloucestershire, UK) with a 532 nm Ar ion laser.PEC measurements of the o-FYO photoelectrodes
PEC studies of the fabricated o-YFO photoelectrodes were carried out in 0.1 M NaOH electrolyte (pH 13) under chopped light using a 100 W ozone-free xenon lamp equipped with an AM 1.5 filter (Oriel LCS-100, Newport). The light intensity was 1 sun illumination (100 mW cm−2). Linear sweep voltammetry (LSV) was performed, scanning in the positive to negative direction between the ranges of +0.3 V to −0.7 V vs. Ag/AgCl. Chronoamperometric (CA) measurements of o-YFO were performed over a period of 6 h with periodic light chopping. The sample was illuminated from the film side for 30 min and another 30 min in dark conditions. The PEC measurements were conducted in a standard three-electrode system at ambient temperature. All the potentials described in this study refer to the reversible hydrogen electrode (RHE) potential, which was calculated according to the following formula (eqn (1)).45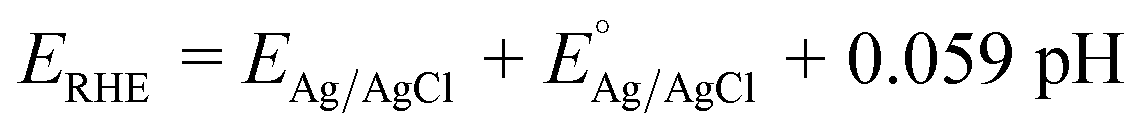 | (1) |
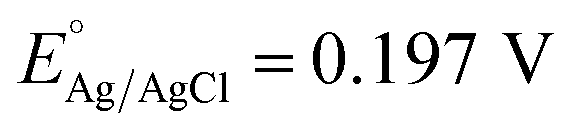 at 25 °C.
at 25 °C.
Hydrogen evolution measurements
Hydrogen evolution measurements were carried out using gas chromatography (GC) (PerkinElmer Clarus 580). A custom-made glass reactor vessel with an attached fused silica viewport containing 0.1 M NaOH (pH 13) with a dead space of 100 mL was purged with argon for 2 h with gentle stirring to remove atmospheric air from the system. The sealed vessel contained the working o-YFO electrode connected to a Pt mesh by a single outer wire was subjected to light illumination for the water splitting reaction. A digital image of the vessel is given in Fig. S1 in the ESI.† GC measurements were taken every 1 h. The faradaic efficiency of the HER was determined following eqn (S1) (ESI†).7Results and discussion
Powder XRD was used to study the phase and composition of the fabricated YFeO3 films, and the pertinent normalized diffractograms are presented in Fig. 1(a). As can be seen, the diffractogram of the YFeO3 film at 500 °C (YFO-500) showed no peaks other than those of the FTO glass substrate, indicating that the film was amorphous. By increasing the annealing temperature to 550 °C, peaks appeared for the YeFeO3 film besides those of the FTO glass substrate. The peaks were searched and matched using XertPro software and it was found that the diffractogram of YFO-550 matched with the standard diffraction patter of orthorhombic phase YFeO3 (o-YFO) given by PDF number [01-086-0171] in the joint committee for powder diffraction standards (JCPDS) database. Although the crystalline phase, i.e. orthorhombic, remained unchanged, the crystal quality was enhanced with the increasing annealing temperature, as shown by the increasing intensity of the peaks in Fig. 1(a). The crystallite size was estimated for all the samples by applying Scherrer's equation (eqn (2)) to the highest intensity reflection at 2θ = 33°.| τ = Kλ/βcosθ | (2) |
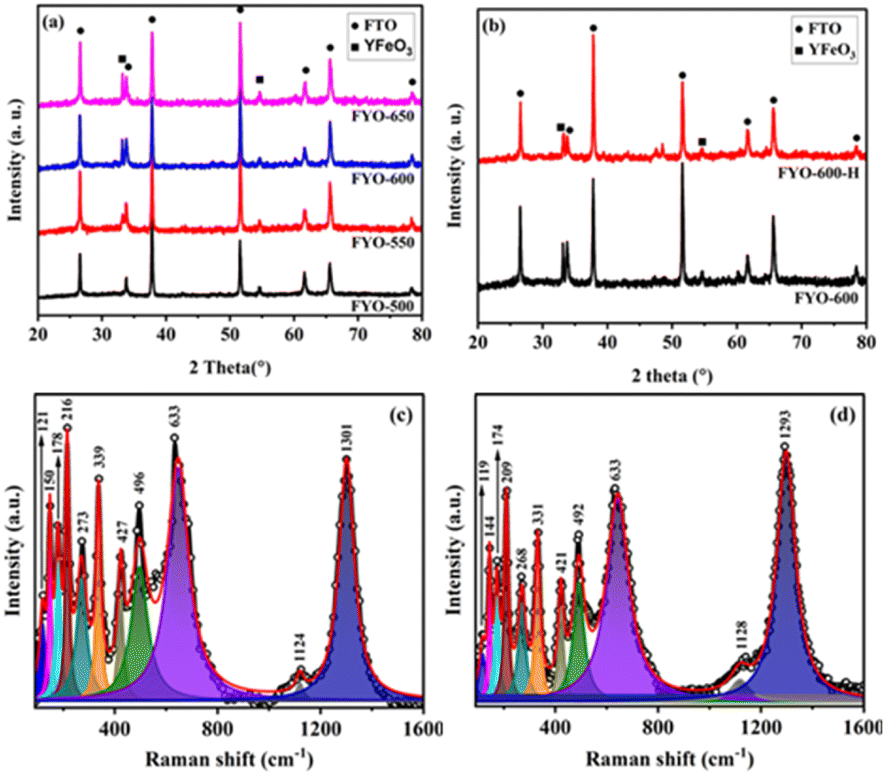 | ||
| Fig. 1 (a) XRD patterns of the o-YFO thin film at different annealing temperatures of 500 °C (FYO-500), 550 °C (FYO-550), 600 °C (FYO-600) and 650 °C (FYO-650). (b) XRD patterns of FYO-600 before (FYO-600) and after (FYO-600-H) the hydrogen evolution test, and Raman spectra of the o-YFO photoelectrode before (c) and after (d) the hydrogen evolution test. | ||
The Raman spectrum of the YFeO3 film (YFO-600) in the wavenumber range of 100–1600 cm−1 is presented in Fig. 1(c), and is in good agreement with the data reported in the literature.47–49 The spectrum was deconvoluted into a number of strong bands at wavenumbers of 150, 178, 216, 273, 339, 427, 496, 633, and 1301 cm−1. The bands at 150 and 178 cm−1 corresponded to the A1g modes of Y–O bond vibration.46,49 The band at 216 cm−1 corresponded to the A1g mode of FeO6 octahedra vibration.49 The peaks at 273, 339, 427, and 496 cm−1 corresponded to the B1g, B2g, A1g, and B3g vibrational modes of Fe–O bonds.46,49,50 The peak at 633 cm−1 corresponded to the B2g + B3g mode47,51 and the peak at 1301 cm−1 corresponded to the vibration of magnetic Fe3+ ions.49,50 The Raman data were in accordance with the XRD results presented above in Fig. 1(a) and consistent with the literature, confirming further that the YFeO3 film crystallized in orthorhombic phase with the Pnma symmetry.
The surface morphology, and adhesion as well as thickness of the film were followed by SEM analysis, and the pertinent data are presented in Fig. 2. The top-view SEM image of the o-YFO film (YFO-600) in Fig. 2(a) showed a uniform film with good interactions among its crystalline grains. The cross-section of the film was imaged by SEM and, as can be seen in Fig. 2(c), the film strongly adhered to the substrate, i.e. the FTO glass. The thickness of the film was found to be approximately 273 nm. The elemental composition from the EDX spectrum in Fig. 2(e) indicated that the film was without any chemical contaminations as it only displayed peaks for the elements that are an integral part of the YFeO3 film. As presented in the inset of Fig. 2(e), the weight percentage amounts of Y (44.6%), Fe (27.0%), and O (21.5%) were comparable to the theoretical values of these elements in stoichiometric YFeO3. The distributions of the two essential metallic elements Y and Fe were determined through their elemental mapping, as presented in Fig. 2(f) and (g), respectively. The uniform composition was evident through these elemental mappings, which showed the homogeneous distribution of the elements Fe and Y in the YFeO3 film.
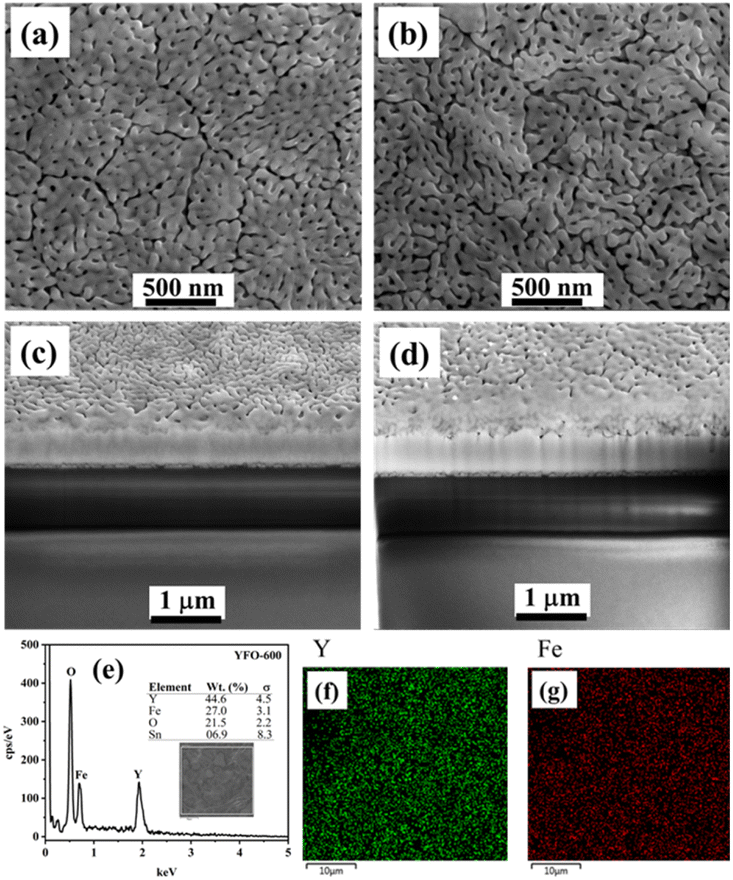 | ||
| Fig. 2 Top view (a) and cross-sectional (c) SEM images of the o-YFO film; SEM images of top view (b) and cross-section (d) of the o-YFO film after the hydrogen evolution reaction. EDX spectrum (e) and elemental mapping of Y (f) and Fe (g) of the o-YFO film. | ||
The PEC performance of the fabricated o-YFO photocathode was measured in a three-electrode cell. The photocathode was illuminated from the film side and the illuminating light was chopped every 0.1 V. The photocurrent response plot between the current density and biased potential vs. RHE is presented in Fig. 3(a). The photocurrent density at 0.5 V was −0.57 mA cm−2, indicating the significant sensitivity of the photocathode to light. The plot of the current density and potential further confirmed the absence of any dark current. This was evident from the flat curves of the current density at zero mA cm−2 during the absence of illumination of the photocathode. Only insignificant spikes during each chop appeared, indicating that the recombination of the generated transient electrons with their holes was insignificant under the applied illumination voltage window of 0.1 V. The photogenerated electrons had enough time to interact with the electrolyte before or after each chop. It is pertinent to mention here that the steady-state photocurrent onset was 1.30 V, as estimated from the current density vs. voltage plot given in Fig. S2 (ESI†).
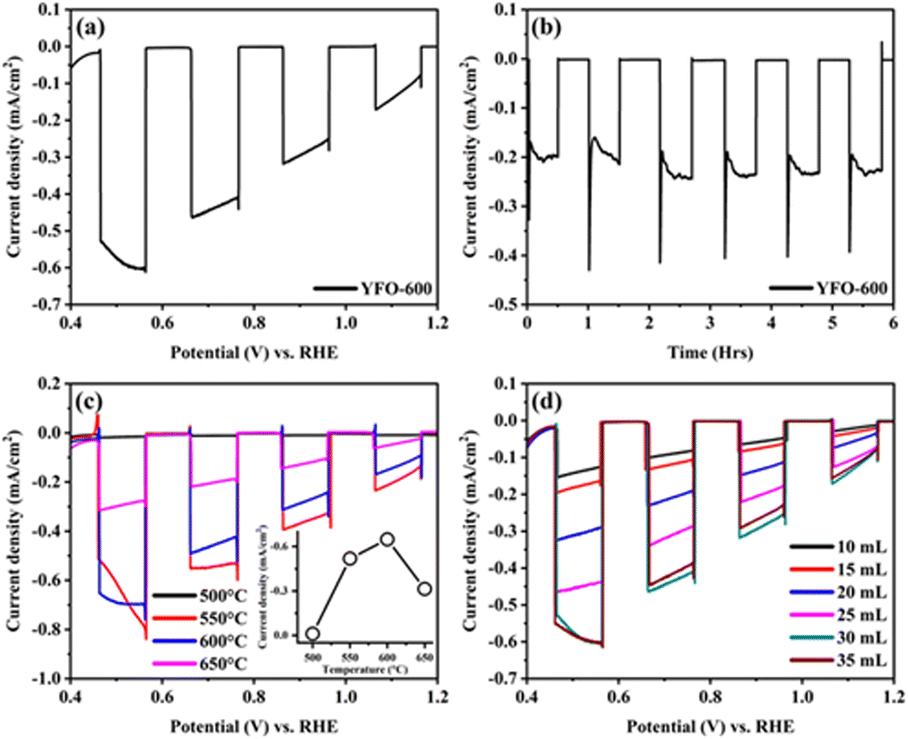 | ||
| Fig. 3 (a) Linear sweep voltammetry curves of YFeO3 obtained by plotting current density (mA cm−2) as a function of potential (V) vs. RHE in 0.1 M NaOH solution of pH 13 under chopped light. (b) Chronoamperometric curve of YFeO3 with chopped illumination from the backside at −0.3 V vs. Ag/AgCl. (c) Photocurrent as a function of potential (V) vs. RHE for YFeO3 annealed at various temperatures; inset shows the current density trend as a function of annealing temperature. (d) Plots of the photocurrent as a function of the potential (V) vs. RHE for YFeO3 deposited under varying amounts of spray solution at 1 mL min−1. | ||
A chronoamperometric test of the o-YFO photocathode was carried out to investigate its stability, and the pertinent plot of the current density vs. time in hours is presented in Fig. 3(b). The photocathode was illuminated for 30 min followed by a 30-min dark period at a constant potential of −0.3 V. From the graph in Fig. 3(b), it is evident that the photocathode was photosensitive till the end of the test and gave a stable response upon each illumination period. No significant decrease in current density was observed for the photocathode upon illumination for up to 6 h. Furthermore, the film was illuminated for 24 h, giving nearly constant current, as shown by the i–t curve in Fig. S3 (ESI†), corresponding to other materials reported for the HER.52 Based on these results, it is anticipated that the hydrogen evolution efficiency of the fabricated o-YFO photocathode will be appreciably stable over time.
The fabrication conditions were optimized to obtain a photoelectrode with optimum properties for PEC applications. Two of the vital fabrication parameters, namely the annealing temperature and spray amount, were optimized by varying them to get different photoelectrodes, which were then tested for their PEC characteristics. Fig. 3(c) shows the current density and potential vs. RHE plots of the o-YFO photoelectrodes annealed at different temperatures. The photocurrents at 0.46 V vs. RHE of the YFeO3 photoelectrodes annealed at 500 °C, 550 °C, 600 °C, and 650 °C were −0.010, −0.518, −0.654, and −0.316 mA cm−2, respectively. The plot in the inset of Fig. 3(c) shows the trend for the current density as a function of temperature. As shown by the plot, the photocurrent increased as the annealing temperature was increased up to 600 °C and then decreased. Previously, we have also observed the same trend in the case of LaFeO3 photoelectrodes, except the optimum temperature was 550 °C compared to 600 °C in the case of this sample (YFO-600).6 The lower photocurrent at low annealing temperature could correspond to the incomplete decomposition of the photoelectrode materials and thus the low crystallization, as was confirmed by the XRD analysis. As the annealing temperature increased, up to 600 °C in this case, the crystallinity improved and thus the photocurrent too. With further increasing the annealing temperature above the optimum, i.e. up to 650 °C, the photoelectrodes developed defects, dislocations, and kinks, which could act as recombination centres for the photogenerated electron–hole pairs.6 An increase in recombination of photogenerated charge carriers (electrons and holes) would directly lead to a decrease in the photocurrent.
Similarly, the effect of the amount of spray solution per given time on the photocurrent was studied by varying it over a range of 10 mL to 35 mL (spray rate was 1 mL min−1 for all the films). The current density was plotted against the potential vs. RHE in Fig. 3(d) and showed that the photocurrent increased with increasing the amount of spray solution. The maximum photocurrent was achieved with a film prepared by spraying 30 mL solution at a rate of 1 mL min−1. With further increasing the amount of spray solution to 35 mL, a decrease in photocurrent was observed (Fig. 3(d)). The photocurrent generated upon illumination of the different films could correspond to the thickness of the films.6 A lower amount of spray solution gave thinner films, which had insufficient sites for the generation of a large number of photoelectrons. As the spray solution amount increased, the film thickness also increased, also leading to an increasing number of photoelectron generation sites. However, increasing the amount of spray solution beyond the optimum amount led to films that were excessively thick. These films were thick enough that the photogenerated electrons needed to travel longer distances to reach the current collector, i.e. the FTO glass. The photogenerated electrons thus become extraordinary slow, leading to their excessive recombination with holes. The result of the slow transport and trapping of the photogenerated electrons was a reduction in the photocurrent. This charge-transport mechanism dominated by the slow transport of electrons and their trapping by recombination has previously been observed in the case of other metal oxide films.
To measure the band gap energy (Eg) of the fabricated o-YFO thin film (YFO-600), its UV-visible spectrum was measured in the range of 350 nm to 800 nm. As shown by the optical absorption spectrum of YFO-600 presented in Fig. 4(a), the sample absorbed strongly in the region of 350–600 nm. The Eg was determined by fitting the absorption data with the Tauc model using eqn (3).53
| (αhv) = A(hv − Eg)n | (3) |
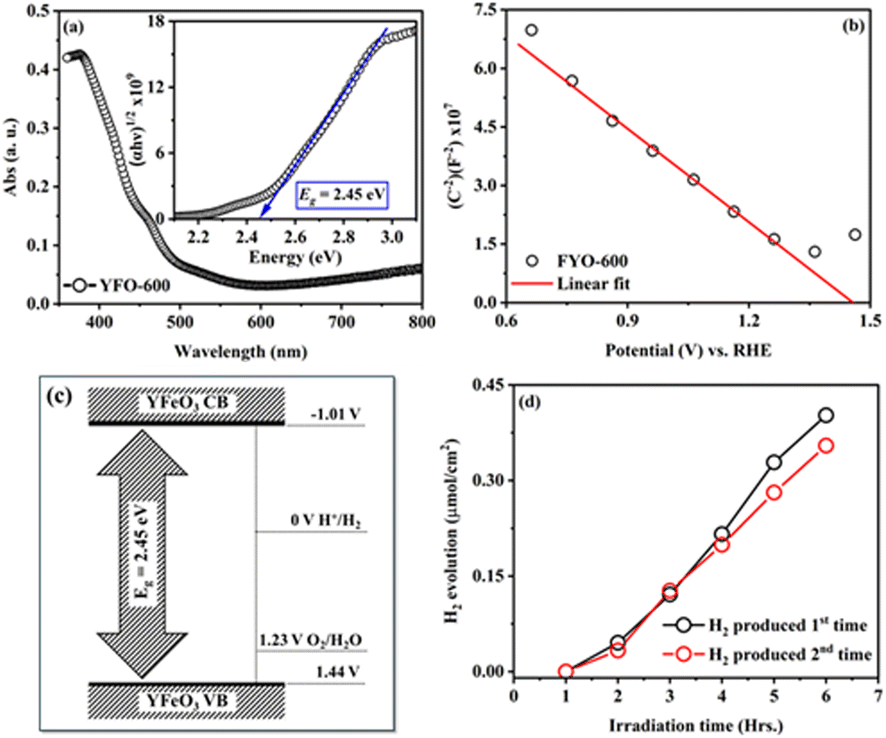 | ||
| Fig. 4 (a) Absorbance spectra and Tauc plot (in inset) of the o-YFO photoelectrode prepared as a thin film on an FTO glass substrate. (b) Mott–Schottky plot of the capacitance obtained from the EIS spectrum as a function of the potential vs. RHE. (c) Band alignment diagram of the fabricated o-YFO film vs. RHE. (d) Test of hydrogen evolution as a function of irradiation time in hours for the fabricated o-YFO photoelectrode (YFO-600). | ||
The flatband potential of the film YFO-600 (YFeO3) was determined from electrochemical impedance spectroscopy (ESI) measurements in a 0.1 M NaOH solution of pH 13. The EIS spectrum was fitted with Randles circuit using ZView software and the capacitance was extracted for drawing the Mott–Schottky plot between (1/C)2 and the potential (V) vs. RHE. The Mott–Schottky plot presented in Fig. 4(b) showed p-type characteristics for the fabricated o-YFO film. Therefore, the Mott–Schottky equation (eqn (4)) for p-type semiconductors was used to determine the flatband potential of the photoelectrode (YFeO3).6,55
 | (4) |
The fabricated o-YFO thin film (FYO-600) was used for the production of hydrogen from water splitting, and so its performance as a photoelectrode for the HER was tested under constant illumination in 0.1 M NaOH solution. A digital image of the custom-made reaction vessel used for testing the hydrogen-production capability of the fabricated photoelectrode is presented in Fig. S1 (ESI†). The HER was carried out without applying any external bias voltage. The photoelectrode (YFeO3-coated FTO glass) was connected to a counter electrode made up of Pt through an external single looped wire. Upon illumination without any external applied potential, the photoelectrode split water, and hydrogen was produced spontaneously during the 6 h cycle. As shown in Fig. 4(d), the amount of hydrogen produced after 6 h of illumination was 0.41 µmol cm−2 with a faradaic efficiency of 73%, which was comparable to the previously reported non-film-based systems.57 After the first cycle of 6 h, the photoelectrode was subjected to a second cycle to check its re-usability. The photoelectrode was illuminated again for 6 h, generating 0.35 µmol cm−2 of hydrogen (Fig. 4(d)). The amount of hydrogen produced in the second cycle was decreased by approximately 15% compared to the hydrogen produced in the first cycle. The decrease in hydrogen amount in the subsequent testing cycles could be correlated to certain factors, including the accumulation of gas bubbles and electrolyte particles on the surface of the photoelectrode (YFeO3 thin film), which may have had a shading effect.6,56
To follow the changes, if any, in the crystals of YFeO3 after the hydrogen evolution test, the YFO-600 electrode after the hydrogen evolution reaction was also analysed by XRD. As shown in Fig. 1(b), the diffractograms of YFO-600 before and after the hydrogen evolution test were identical in terms of the peak positions and intensities, indicating that there was no change in the basic crystal phase of the YFeO3 films. The crystallite size of the post-hydrogen-evolution test film was identical to that of the pre-hydrogen-evolution test. These observations indicate that the YeFeO3 films were stable under the applied reaction conditions for hydrogen production from the splitting of water. After the HER, the YFeO3 film was also analysed by Raman spectroscopy. The Raman spectrum (Fig. 1(d)) of the YFeO3 film after the hydrogen evolution test was almost identical to that of the pre-hydrogen-evolution reaction (Fig. 1(c)). This further confirmed that the film was stable under the applied hydrogen evolution reaction conditions. The effect of the HER on the morphology was followed by SEM, and the image of the YFeO3 film after the hydrogen evolution test is presented in Fig. 2(b), which showed an identical morphology to that before the hydrogen evolution reaction (Fig. 2(a)). A cross-sectional SEM image of the film after the HER was also acquired, and as shown in Fig. 2(d), the thickness of the YFeO3 film did not change under the applied test conditions. This indicates that the film was morphologically stable for the purposed reaction conditions.
Conclusions
We fabricated YFeO3 thin films by a simple, cost-effective, and scalable method, i.e. spray pyrolysis on an FTO glass substrate. The film was subjected to post-synthesis annealing temperatures of 500 °C to 650 °C for optimization to get the desired phase, namely an orthorhombic YFeO3 (o-YFO) film. XRD and Raman scattering data confirmed the attainment of the optimum o-YFO film at 600 °C, yielding a photocurrent density of ∼0.65 mA cm−2 at 0.46 V vs. RHE. The films annealed at lower and/or higher temperatures than 600 °C showed decreased photocurrent densities at 0.46 V vs. RHE. Besides the annealing temperature, it was found that the amount of spray solution per given time also affected the photocurrent density, and the optimum response was given by the o-YFO film prepared by spraying a 30 mL solution at a rate of 1 mL min−1. Furthermore, the o-YFO film (FYO-600) showed a stable photoresponse over 6 h. The UV-visible and EIS data confirmed the narrow Eg of 2.45 eV, showing it could absorb in the visible region of the solar spectrum, with the value straddling the redox potential of water. The valence and conduction bands were determined to be 1.44 V and −1.01 V, respectively, encompassing the potential of 1.23 V as the redox potential of water. The HER test was conducted in a home-made vessel without applying any external potential bias and/or assistance and the system generated 0.41 µmol cm−2 of hydrogen over a period of 6 h. The faradaic efficiency in the HER test was significant, amounting to 73%. The re-usability of the photocathode for the spontaneous production of hydrogen from water splitting was studied by repeating the HER test, and 0.35 µmol cm−2 hydrogen was produced in the second run of the experiment for the same illumination time of 6 h. This would be the first time, to the best of our knowledge, that a spray pyrolysis-fabricated o-YFO film has been used for the spontaneous production of hydrogen from water splitting without the use of any external applied potential as an assistance to the HER. The encouraging results have motivated us to continue work on the photocathode in our future investigations, focusing on the improvement of the photocurrent densities, charge carrier dynamics, etc.Author contributions
All authors contributed to the development of the research proposal and the writing of the manuscript. Dr Bandar Y. Alfaifi conducted the experiments, investigated the processes, and acquired data using various techniques. He analyzed the data to establish the research results and prepared the initial draft of the manuscript. Dr Hameed Ullah conducted research, validated the results and experiments, and secured funding through a post-doctoral research grant. He co-authored the initial draft and contributed to reviewing and editing the manuscript for publication. Prof. Dr Asif Ali Tahir, one of the co-principal investigators (PIs), conceptualized the research objectives and secured financial support for the project through an EPSRC grant. He supervised and mentored Dr Bandar, coordinated research activities, and assisted in reviewing and editing the final manuscript. Prof. Dr Xin Jiang was involved in conceptualizing the research, setting its aims, and securing financial resources through the European Union's Horizon 2020 research and innovation program. As one of the PIs, he managed and supervised the project, mentoring Dr Ullah. He also contributed to reviewing, editing, and approving the final manuscript for submission.Data availability
All relevant data supporting the findings of this study are included within the article and its ESI† files.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 26th November 2024, where Fig. 3 and 4 were incorrectly positioned.Acknowledgements
This work was funded by the Engineering and Physical Sciences Research Council (EPSRC), grant numbers EP/T025875/1 and EP/V049046/1. This project has also received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 945422. College of Engineering and Information Technology at Onaizah Colleges, Saudi Arabia, is also acknowledged for their support.References
- D. Raimi, Y. Zhu, R. G. Newell and B. C. Prest, Global Energy Outlook 2024: Peaks or Plateaus?, Resources for the Future, 2024, https://www.rff.org/publications/reports/global-energy-outlook-2024/ Search PubMed.
- M. Mathew, Prog. Nucl. Energy, 2022, 143, 104080 CrossRef CAS.
- M. Hoel and S. Kverndokk, Resour. Energy Econ., 1996, 18, 115–136 CrossRef.
- D. Ginley, M. A. Green and R. Collins, MRS Bull., 2008, 33, 355–364 CrossRef CAS.
- N. T. Mbungu, R. M. Naidoo, R. C. Bansal, M. W. Siti and D. H. Tungadio, J. Energy Storage, 2020, 29, 101385 CrossRef.
- G. S. Pawar and A. A. Tahir, Sci. Rep., 2018, 8, 3501 CrossRef PubMed.
- C. Jiang, S. J. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- M. T. Spitler, M. A. Modestino, T. G. Deutsch, C. X. Xiang, J. R. Durrant, D. V. Esposito, S. Haussener, S. Maldonado, I. D. Sharp and B. A. Parkinson, Sustainable Energy Fuels, 2020, 4, 985–995 RSC.
- H. Song, S. Luo, H. Huang, B. Deng and J. Ye, ACS Energy Lett., 2022, 7, 1043–1065 CrossRef CAS.
- S. E. Hosseini and M. A. Wahid, Int. J. Energy Res., 2020, 44, 4110–4131 CrossRef.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS PubMed.
- M. A. Marwat, M. Humayun, M. W. Afridi, H. Zhang, M. R. Abdul Karim, M. Ashtar, M. Usman, S. Waqar, H. Ullah and C. Wang, ACS Appl. Energy Mater., 2021, 4, 12007–12031 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- A. Vilanova, P. Dias, T. Lopes and A. Mendes, Chem. Soc. Rev., 2024, 53, 2388–2434 RSC.
- D. Hansora, J. W. Yoo, R. Mehrotra, W. J. Byun, D. Lim, Y. K. Kim, E. Noh, H. Lim, J.-W. Jang and S. I. Seok, Nat. Energy, 2024, 9, 272–284 CrossRef CAS.
- M. Jankulovska, I. Barcelo, T. Lana-Villarreal and R. Gomez, J. Phys. Chem. C, 2013, 117, 4024–4031 CrossRef CAS.
- J. Quinonero, T. Lana-Villarreal and R. Gómez, Appl. Catal., B, 2016, 194, 141–149 CrossRef CAS.
- M. Tallarida, C. Das, D. Cibrev, K. Kukli, A. Tamm, M. Ritala, T. Lana-Villarreal, R. Gómez, M. Leskelä and D. Schmeisser, J. Phys. Chem. Lett., 2014, 5, 3582–3587 CrossRef CAS PubMed.
- V. Cristino, S. Caramori, R. Argazzi, L. Meda, G. L. Marra and C. A. Bignozzi, Langmuir, 2011, 27, 7276–7284 CrossRef CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- K. Fujii, K. Kusakabe and K. Ohkawa, Jpn. J. Appl. Phys., 2005, 44, 7433 CrossRef CAS.
- M. I. Díez‐García, T. Lana‐Villarreal and R. Gómez, ChemSusChem, 2016, 9, 1504–1512 CrossRef.
- M. S. Prévot, N. Guijarro and K. Sivula, ChemSusChem, 2015, 8, 1359–1367 CrossRef PubMed.
- M. I. Díez‐García and R. Gómez, ChemSusChem, 2017, 10, 2457–2463 CrossRef PubMed.
- Y. Yang, S. Niu, D. Han, T. Liu, G. Wang and Y. Li, Adv. Energy Mater., 2017, 7, 1700555 CrossRef.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske and M. Sunkara, J. Mater. Res., 2010, 25, 3–16 CrossRef CAS.
- R. Zhang, A. Xie, L. Cheng, Z. Bai, Y. Tang and P. Wan, Chem. Commun., 2023, 59, 8205–8221 RSC.
- L. Pan, Y. Liu, L. Yao, D. Ren, K. Sivula, M. Grätzel and A. Hagfeldt, Nat. Commun., 2020, 11, 318 CrossRef CAS PubMed.
- P. Sahoo, A. Sharma, S. Padhan and R. Thangavel, Superlattices Microstruct., 2021, 159, 107050 CrossRef CAS.
- N. Yadav and M. Ahmaruzzaman, Energy Environ., 2024, 35, 458–490 CrossRef CAS.
- M.-K. Son, L. Pan, M. T. Mayer, A. Hagfeldt, M. Gratzel and J. Luo, ACS Appl. Mater. Interfaces, 2021, 13, 55080–55091 CrossRef CAS.
- H. V. Le, T. T. Ung, P. D. Tran, H. V. Mai, B. D. Do and L. Q. Nguyen, J. Phys. D: Appl. Phys., 2023, 56, 465502 CrossRef CAS.
- Y. Wang, J. Huang, L. Wang, H. She and Q. Wang, Chin. J. Struct. Chem., 2022, 41, 2201054–2201068 CAS.
- H. S. Jarusheh, A. Yusuf, F. Banat, M. A. Haija and G. Palmisano, J. Environ. Chem. Eng., 2022, 10, 108204 CrossRef CAS.
- J. H. Kim, H. E. Kim, J. H. Kim and J. S. Lee, J. Mater. Chem. A, 2020, 8, 9447–9482 RSC.
- M. I. Díez-García, V. Celorrio, L. Calvillo, D. Tiwari, R. Gómez and D. J. Fermín, Electrochim. Acta, 2017, 246, 365–371 CrossRef.
- Y. Guo, N. Zhang, H. Huang, Z. Li and Z. Zou, RSC Adv., 2017, 7, 18418–18420 RSC.
- M. Wang, T. Wang, S. Song and M. Tan, Materials, 2017, 10, 626 CrossRef.
- Y. Sui, F. Lu, X. Liu, Y. Zhang, X. Sun and C. Liu, Results Mater., 2021, 10, 100186 CrossRef CAS.
- P. Tang, H. Chen, F. Cao and G. Pan, Catal. Sci. Technol., 2011, 1, 1145–1148 RSC.
- Y. Chen, J. Yang, X. Wang, F. Feng, Y. Zhang and Y. Tang, J. Ceram. Soc. Jpn., 2014, 122, 146–150 CrossRef.
- M. Khraisheh, A. Khazndar and M. A. Al‐Ghouti, Int. J. Energy Res., 2015, 39, 1142–1152 CrossRef CAS.
- N. Mihailov, O. Vankov, N. Petrova and D. Kovacheva, Open Chem., 2004, 2, 188–195 CrossRef CAS.
- C. Gómez-Solís, J. Ballesteros, L. M. Torres-Martínez and I. Juárez-Ramírez, Fuel, 2016, 166, 36–41 CrossRef.
- D. Du Boulay, E. Maslen, V. Streltsov and N. Ishizawa, Acta Crystallogr., Sect. B:Struct. Sci., 1995, 51, 921–929 CrossRef.
- Z. Ž. Lazarević, Č. Jovalekić, M. Gilić, V. Ivanovski, A. Umićević, D. Sekulić and N. Ž. Romčević, Sci. Sintering, 2017, 49, 277–284 CrossRef.
- A. Vazquez-Olmos, M. Sánchez-Vergara, A. Fernández-Osorio, A. Hernández-García, R. Sato-Berrú and J. Alvarez-Bada, J. Cluster Sci., 2018, 29, 225–233 CrossRef CAS.
- M. Wang, T. Wang, S.-H. Song, M. Ravi, R.-C. Liu and S.-S. Ji, Ceram. Int., 2017, 43, 10270–10276 CrossRef CAS.
- A. Kuzmenko, P. Abakumov and M. Dobromyslov, J. Magn. Magn. Mater., 2012, 324, 1262–1264 CrossRef CAS.
- P. Mandal, V. S. Bhadram, Y. Sundarayya, C. Narayana, A. Sundaresan and C. Rao, Phys. Rev. Lett., 2011, 107, 137202 CrossRef CAS PubMed.
- W. Liu, W. Liu, T. Hou, J. Ding, Z. Wang, R. Yin, X. San, L. Feng, J. Luo and X. Liu, Nano Res., 2024, 17, 4797–4806 CrossRef CAS.
- R. Gakhar, K. Summers, R. Palaniappan, S. K. Pilli and D. Chidambaram, RSC Adv., 2014, 4, 49729–49736 RSC.
- H. Baqiah, M. M. A. Kechik, R. Al-Gaashani, A. A. Al-Zahrani, N. M. Al-Hada, N. Zhang, J. Liu and S. Xu, Ceram. Int., 2023, 49, 600–606 CrossRef CAS.
- K. Gelderman, L. Lee and S. Donne, J. Chem. Educ., 2007, 84, 685 CrossRef CAS.
- S. Hernández, G. Barbero, G. Saracco and A. Alexe-Ionescu, J. Phys. Chem. C, 2015, 119, 9916–9925 CrossRef.
- Z. Lu, H. Yang, Q. Liu, J. Luo, L. Feng, L. Chu and X. Liu, Small, 2024, 20, 2305434 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01276c |
| This journal is © The Royal Society of Chemistry 2025 |