
Fluorine-rich Schiff base ligand derived Fe/N–C–F and Co/N–C–F catalysts for the oxygen reduction reaction: synthesis, experimental validation, and DFT insights†
Sumanta Kumar
Das‡
 ab,
Shaik Gouse
Peera‡
c,
Aiswarya
Kesh
a,
Prabakaran
Varathan
ab and
Akhila Kumar
Sahu
*ab
ab,
Shaik Gouse
Peera‡
c,
Aiswarya
Kesh
a,
Prabakaran
Varathan
ab and
Akhila Kumar
Sahu
*ab
aCSIR – Central Electrochemical Research Institute-Madras Unit, CSIR Madras Complex, Taramani, Chennai – 600 113, India. E-mail: aksahu@cecri.res.in; Fax: +91-44-22544554; Tel: +91-44-22544554
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
cNatural Science Research Institute, College of Natural Sciences, Keimyung University, 1095, Dalseo-gu, Daegu 42601, Republic of Korea
First published on 28th November 2024
Abstract
The development of cost effective and durable catalysts for the electrochemical reduction of O2 to H2O is paramount for energy conversion devices such as fuel cells and Zn–air batteries. In this research work, we have developed a unique strategy for the synthesis of active and stable electrocatalysts comprising Fe and Co transition metals in combination with N and F dopants in the carbon matrix. This research also introduces an innovative approach for synthesizing Fe/N–C–F and Co/N–C–F electrocatalysts utilizing organic Schiff base ligands and their coordination complexes with Fe and Co transition metals. The synthesized Fe/N–C–F and Co/N–C–F catalysts have been systematically evaluated for their physicochemical properties and electronic states by using HR-TEM, XPS analysis and electrochemical characterization in 0.1 M aqueous KOH electrolyte. The optimized Fe/N–C–F catalyst shows a half-wave potential of 0.88 V vs. RHE and superior durability evaluated up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles with only a marginal potential drop of ∼27 mV in its E1/2 potential value compared to the Pt/C catalyst. Furthermore, the reaction pathway and Gibbs free energy of the ORR intermediates in Fe/N–C–F and Co/N–C–F catalysts have been evaluated by DFT analysis.
000 cycles with only a marginal potential drop of ∼27 mV in its E1/2 potential value compared to the Pt/C catalyst. Furthermore, the reaction pathway and Gibbs free energy of the ORR intermediates in Fe/N–C–F and Co/N–C–F catalysts have been evaluated by DFT analysis.
1 Introduction
The multi-step, sluggish, cathodic electrocatalytic oxygen reduction reaction (ORR) is considered as a key reaction in several energy conversion devices such as chemical and biological fuel cells and Zn–air batteries, requiring efficient, noble metal based electrocatalysts to perform a direct 4e− reduction of O2 to H2O.1 To date, Pt supported on high surface area carbon (Pt/C) has been considered as the state-of-the-art catalyst for the ORR, due to its ability to activate and cleave the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond efficiently.2 In addition, due to its high exchange current density, reduced overpotential, and reduced activation energy towards the ORR, Pt/C is considered as the most efficient cathode catalyst.3 However, the successful usage of Pt/C catalysts in several energy conversion devices is halted due to their high cost, limited economic viability, scarcity and uneven geographical distribution of Pt, which cause supply-chain issues and volatility in the price.4 In addition, the poor stability of the Pt/C catalyst under harsh acidic and alkaline conditions, due to carbon corrosion, Pt nanoparticle dissolution, redeposition, Pt nanoparticle agglomeration and coalescence, further necessitates the search for alternative electrocatalysts that are potentially active for the ORR and stable under long term operating conditions.5–7 In this regard, several alternative catalysts made of cheap, earth abundant transition metals such as Fe, Co, Ni, Mn, etc., in combination with heteroatom (N, F, P, B, S, etc.) doped carbons, have been explored as electrocatalysts for the ORR in both acidic and alkaline electrolytes.8–13 A special geometrical arrangement, in which transition metals form strong co-ordination bonds with nitrogen atoms, is known as an M–N4–C (M = transition metal) active site, and it has been the subject of over twenty years of research by researchers from around the world.13,14 Among several types of transition metal-based M–N4–C catalysts, Fe and Co based catalysts have particularly attracted greater attention, due to the fact that Fe–N4–C and Co–N4–C active sites mimic the active centers of Pt, balanced O2 adsorption/desorption energies and the strong co-ordination environment around the metal centers enhances the activity and stability of the catalyst.14,15 In most cases, it is found that transition metal atoms in combination with N/F dopants in the carbon matrix are found to be particularly active catalysts, while second heteroatoms such as S, P and B act as ancillary dopants that help in modulating the charge/spin density distribution and hence alter the electronic conductivity around the Fe–N4–C and Co–N4–C active sites, synergistically improving the overall catalytic activity.16
O bond efficiently.2 In addition, due to its high exchange current density, reduced overpotential, and reduced activation energy towards the ORR, Pt/C is considered as the most efficient cathode catalyst.3 However, the successful usage of Pt/C catalysts in several energy conversion devices is halted due to their high cost, limited economic viability, scarcity and uneven geographical distribution of Pt, which cause supply-chain issues and volatility in the price.4 In addition, the poor stability of the Pt/C catalyst under harsh acidic and alkaline conditions, due to carbon corrosion, Pt nanoparticle dissolution, redeposition, Pt nanoparticle agglomeration and coalescence, further necessitates the search for alternative electrocatalysts that are potentially active for the ORR and stable under long term operating conditions.5–7 In this regard, several alternative catalysts made of cheap, earth abundant transition metals such as Fe, Co, Ni, Mn, etc., in combination with heteroatom (N, F, P, B, S, etc.) doped carbons, have been explored as electrocatalysts for the ORR in both acidic and alkaline electrolytes.8–13 A special geometrical arrangement, in which transition metals form strong co-ordination bonds with nitrogen atoms, is known as an M–N4–C (M = transition metal) active site, and it has been the subject of over twenty years of research by researchers from around the world.13,14 Among several types of transition metal-based M–N4–C catalysts, Fe and Co based catalysts have particularly attracted greater attention, due to the fact that Fe–N4–C and Co–N4–C active sites mimic the active centers of Pt, balanced O2 adsorption/desorption energies and the strong co-ordination environment around the metal centers enhances the activity and stability of the catalyst.14,15 In most cases, it is found that transition metal atoms in combination with N/F dopants in the carbon matrix are found to be particularly active catalysts, while second heteroatoms such as S, P and B act as ancillary dopants that help in modulating the charge/spin density distribution and hence alter the electronic conductivity around the Fe–N4–C and Co–N4–C active sites, synergistically improving the overall catalytic activity.16
Several researchers highlighted the effect of heteroatom dopants on the ORR activity and stability of Fe/Co–N–C type catalysts.17–19 For instance, Jin et al.20 demonstrated the effect of N species in the Fe–N–C catalyst, with a particular focus on Fe–pyridinic-N4–C and Fe–pyrrolic-N4–C structures, contributing to the enhanced ORR kinetics and the stability of Fe in the catalyst. While Fe–pyridinic-N4–C contributes to the ORR activity, the activity and stability of Fe–pyrrolic-N4–C structures were found to be poor. This is due to the facile protonation of Fe–pyrrolic-N4–C structures, whereas the protonation occurs on the central Fe metal atom in Fe–pyridinic-N4–C structures. S optimizes charge distribution around the M–N–C active sites and induces structural defects that could aid O2 adsorption due to its larger atomic radius and high polarizability compared to N atoms. In a short conclusion, it is well accepted that the ORR activity of transition metal-based catalysts originates from the presence of Fe–N4–C and Co–N4–C active sites, primarily due to the presence of heteroatom dopants that alter the electronic and structural properties of the catalyst due to their difference in electronegativity, atomic size, and ability to polarize the C–C bonds by varying the charge and spin density.21 In this regard, the presence of N atoms is found to be a “must-be” dopant, in order to form M–N4–C structures, while other secondary heteroatoms such as S, P and B help in modifying the surface properties to further assist in elevating the overall activity and stability.22
In this regard, the combination of the N dopant with halogens, such as F, Cl, Br and I, is very interesting to explore towards synergistic ORR catalysis. The ORR catalysis of halogen doped carbons not only depends on the electronegativity, but also on dopant atomic size.23,24 Among the investigated halogen doped carbon catalysts, F doped carbons in combination with the N dopant have recently gained tremendous interest, due to the fact that F is the highest electronegative element not only among the halogen group elements, but also in the entire periodic table with an electronegativity value of 3.98 on the Pauling electronegativity scale; therefore, F doping could provide an ultimate charge/spin distribution in the carbon matrix, even more than the N dopant.25 Several studies have shown that semi-ionic and ionic C–F bonding structures are potential ORR active sites in F-doped carbons.26,27 Most importantly, it has recently been postulated that –F doping provides extraordinary stability to the ORR catalyst by mitigating carbon corrosion, much higher than currently proposed N-doped carbons.28 Particularly, the edge positioned –F atoms are found to strongly protect the carbon against corrosion under harsh fuel cell operating conditions.29 Based on these observations, several research studies have emerged, in which N and F dopants have been introduced into the carbon matrix to gain the supreme polarizability and modulation of the electronic structure of Fe–N4–C and Co–N4–C active sites to further maximize the ORR activity.30,31
In a traditional synthesis strategy of Fe–N4–C/N–F or Co–N4–C/N–F doped carbons, the N and F precursors are mixed with carbon and metal precursors and the final mixtures are pyrolyzed in an inert atmosphere to obtain catalysts with M–N4–C structures and C and F doped carbons.32 Several types of precursors have been used in the literature such as PTFE, PVDF, NH4F, trifluoroacetic acid, ammonium tetrafluoroborate, NaF, HF and NH4BF4 as F precursors, while PANI, melamine, polyacrylonitrile (PAN), urea, hydrazine, and 2-methyl imidazole are the common N precursors.33 In several studies, a secondary carbon source such as Ketjen-black (KB), graphene oxide (GO), graphitic carbon nanofibers, phloroglucinol–formaldehyde resin, etc., have been used.34 The doping of N and F from the precursors into the secondary carbon matrix has always been challenging in terms of dopant content and their even distribution due to poor surface functionalities, heterogeneity and different pore geometries of different carbon supports.35 Therefore, the catalysts with a single precursor that serves as the C and dopant source ( e.g., N) together with transition metal atoms are always advantages as this approach combines all components in a single source that leads to the possibility of high dopant concentration and potential M–N4–C structures. This is the reason why the catalysts with M–N4–C structures derived from metal–organic frameworks (MOFs) are so popular and extremely active and stable catalysts synthesized so far in the literature.36,37 In this regard, studies which involve the use of a single precursor as the N and F source are extremely rare, except for a few studies with NH4F. The use of a single precursor simplifies the synthesis process in an environmentally friendly manner and reduces the use of toxic chemicals of environmental toxicity such as HF, trifluoroacetic acid, and ammonium tetrafluoroborate.38,39 In this work, a novel single precursor is synthesized from a Schiff base ligand, intrinsically contained N and F in their organic structures together with the metallic precursors, guarantees the formation of potential M–N4–C type active sites, and thus avoids the use of two different precursors for N and F and an additional carbon source/matrix, as it is the case in previous studies.40–43
Similar to MOFs, the Schiff bases, resulting from a condensation reaction between a primary amine and a carbonyl compound, usually an aldehyde or ketone, are a class of co-ordination organic compounds with imine (–CN–) functional groups that have attracted considerable interest in organic and medicinal chemistry and are extremely rarely explored as precursors for electrocatalyst synthesis.44,45 The presence of imine functional groups that act as ligands and their ability to form co-ordinate complexes with a variety of metal ions could be very interesting precursors to synthesize electrocatalysts.46 To the best of our knowledge, we have not come across literature exploring the synthesis of transition metal coordinated Schiff base derived electrocatalysts for the ORR. In this work, we attempted to synthesize Schiff base ligands followed by their complexation with Fe and Co metallic ions with a single compound acting as the N, C, F and Fe/Co precursor. The pyrolysis of the metal/coordinated Schiff base complex yields Fe/N–C–F and Co/N–C–F catalysts. The optimized Fe/N–C–F catalyst with a half-wave potential of 0.88 V vs. RHE and superior durability evaluated up to 20000 cycles with only a marginal potential drop of ∼27 mV in its E1/2 value compared to the Pt/C catalyst. Furthermore, the reaction pathway and Gibbs free energy of the ORR intermediates have been evaluated by DFT analysis.
2 Experimental section
2.1. Synthesis of the Schiff base ligand
A Schiff base ligand is synthesized with an ∼80% yield by refluxing salicylaldehyde and pentafluorophenylhydrazine in ethanol solvent under acidic conditions. In a typical synthesis, 2.44 g salicylaldehyde and 3.96 g of pentafluorophenylhydrazine were dissolved in 50 mL of ethanol and magnetically agitated until the organic ligands dissolved completely. The pH of the mixtures is adjusted to ∼5 using glacial acetic acid. The resulting pH adjusted mixture is subjected to refluxing conditions at 80 °C under continuous magnetic agitation for 24 h. A pale-yellow colored precipitate appeared after 24 h reaction time, and was collected, filtered and washed several times with ethanol and ether solvents to remove any unreacted molecules. The obtained precipitation powder, termed Schiff base ligand (SL), is then dried at room temperature. The precipitation yield obtained is close to ∼80%. The reaction mechanism is explained in Scheme 1.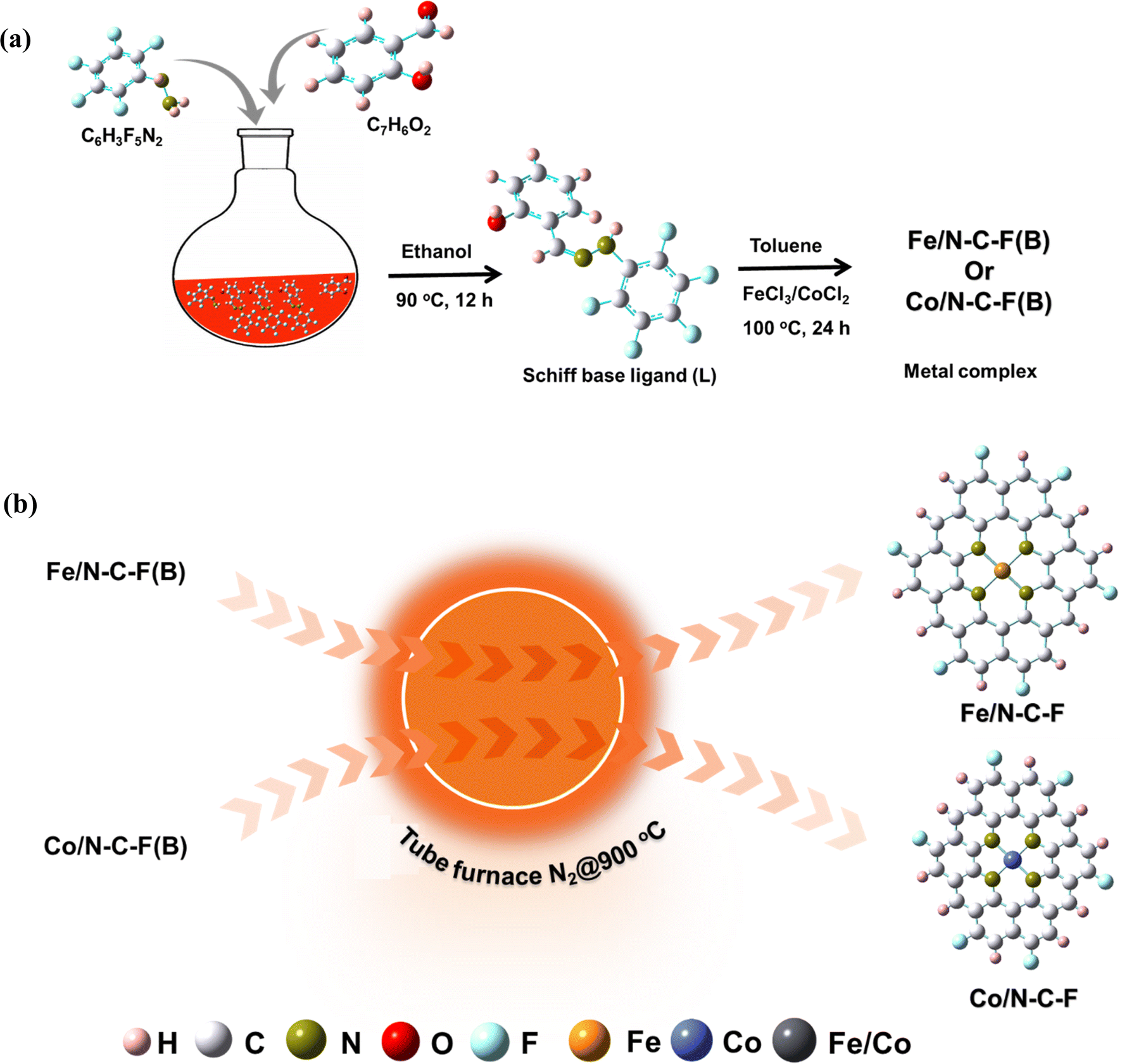 | ||
| Scheme 1 (a) Representation of the synthesis of the Schiff base ligand, and the synthesis of metal complexes (Fe/N–C–F(B) and Co/N–C–F(B)) (B – before pyrolysis); (b) synthesis of Fe/N–C–F and Co/N–C–F. | ||
2.2. Synthesis of Fe/SL and Co/SL co-ordination complexes
The Fe and Co coordinated/SL complexes are synthesized by a complexation reaction between preciously obtained SL and Fe3+/Co2+ ions. In a typical synthesis, 100 mg of SL was added to a mixture of aqueous acetic acid (25 mL, CH3–COOH/H2O = 1/50, v/v), followed by the addition of toluene (10 mL) and FeCl3 (ferric chloride anhydrous) in sequence. After the addition of the ligands and Fe3+ precursor, the resulting solution is then refluxed at 100 °C for 24 h, for a complexation reaction to occur and to obtain the co-ordination compound. After the complexation reaction, the reaction mixture is filtered to remove any unreacted precursors. The obtained product is then dried in a hot-air oven, to obtain the Fe/SL co-ordination complex. The resulting powder is then ground into a fine powder in a mortar and pestle. The SL:metal ratio was varied (SL:Fe, i.e., 1:1, 2:1, and 3:1) accounting for the increased ligand concentration for optimizing the complexation reaction between the SL and Fe. For the synthesis of the Co/SL co-ordination complex, a similar procedure is repeated, in which the CoCl2 precursor is used. The details of chemicals used can be accessed in ESI Section S1.†
2.3. Synthesis of Fe/N–C–F and Co/N–C–F catalysts from Fe/SL and Co/SL co-ordination complexes
For the synthesis of Fe/N–C–F and Co/N–C–F catalysts from Fe/SL and Co/SL co-ordination complexes, the finely ground powders of Fe/SL and Co/SL co-ordination complexes are transferred into a graphite boat and then placed in a tube furnace and then subjected to a high temperature pyrolysis at 800 °C, 900 °C and 1000 °C with a heating rate of 3 °C min−1, for 1 hour under a continuous flow of N2 inert gas. The black powder that is collected is then ground into a fine powder in a mortar and pestle and is designated as Fe/SL → Fe/N–C–F and Co/SL → Co/N–C–F.2.4. Physicochemical characterization
The successful synthesis proof of the Schiff base ligand has been obtained from 1H NMR analysis. The pyrolyzed Fe/SL and Co/SL co-ordination complexes resulting in the Fe/N–C–F and Co/N–C–F catalysts have been characterized comprehensively for their morphological and structural properties by SEM, TEM measurements, and crystallographic analysis, phase properties and defect chemistry by XRD and Raman spectra analysis, and the bonding structure of N, F, C, Fe and Co by XPS analysis. The detailed characterization procedures and additional results can be accessed in ESI Section S2.†2.5. Electrochemical characterization
Electrochemical ORR analysis of Fe/N–C–F and Co/N–C–F catalysts has been comprehensively performed using several traditional electrochemical techniques that include cyclic voltammetry (CV), linear sweep voltammetry (LSV), and rotating disk and rotating ring disk electrode (RDE and RRDE) techniques. For the electrochemical analysis, a traditional three electrode system has been used with Hg/HgO, a graphite rod, and a glassy carbon (GC) electrode with a thin layer of the catalyst deposited on top of it as reference, counter and working electrodes, respectively. The catalyst ink made of Fe/N–C–F and Co/N–C–F catalysts is prepared by dispersing a definite amount of the catalyst in a mixture of ethanol and water, and Nafion ionomer as the binder, which is then deposited on the previously polished GC electrode. The catalyst deposited GC electrode is then used as the working electrode for the electrochemical measurements, for the ORR, in 0.1 M KOH electrolyte. For the stability test, the catalyst deposited GC electrode is subjected to repetitive potential cycling for 20000 potential cycles and then LSVs were recorded to assess the stability of the Fe/N–C–F and Co/N–C–F catalysts. The detailed electrochemical measurements are given in ESI Section S3† of this article.
2.6. MEA fabrication and AEM fuel cell performance analysis of Fe/N–C–F and Co/N–C–F catalysts
Anion exchange membrane fuel cells (AEMFCs) were evaluated using optimized Fe/N–C–F and Co/N–C–F as cathode catalysts in a membrane electrode assembly (MEA) configuration along with commercial Pt/C on the anode side. MEA fabrication was achieved using a brush coating technique, and performance tests were conducted at 45 °C by using a fuel cell test station (Fuel Cell Technologies Inc., N3300A). Detailed experimental procedures and additional results are available in the ESI Section S4.†3 Results and discussion
Schiff bases and their derivatives have played a significant role in the development of coordination chemistry, inorganic chemistry and bioinorganic chemistry research. Schiff base chemistry has been popular in the fields of organic chemistry and medical chemistry, in which Schiff base derived compounds have been used in diverse applications such as antibacterial, antiviral, antifungal, autoinhibitory and analysis applications.47 For the first time, in this study, we have explored the synthesis of electrocatalysts from metal-coordinated Schiff base ligands for the ORR in fuel cells. The versatility of the traditional Schiff base reaction between the amine and the aldehyde or ketone carbonyl group allows for the selection of ligands with a variety of atoms in the side chain that includes S, P, N and F.48 Therefore, organic ligands with a side chain of atoms of interest can be chosen to derive the Schiff bases of our interest, and this is possible because the condensation reaction occurs between the amine and the aldehyde or ketone carbonyl group, leaving the atoms attached to the ligands intact. Furthermore, choosing a suitable organic ligand with metal co-ordinate sites (for example, –N) helps to further form co-ordination complexes with the d-block elements.49 This unique ability of Schiff bases allowed us to formulate Fe/N–C–F and Co/N–C–F as cathode catalysts in a unique way, in contrast to the traditional way of mixing precursors of each containing C, N, F and metal precursors. At first a condensation reaction is achieved between salicylaldehyde and pentafluorophenylhydrazine in ethanol solvent under acidic conditions, to obtain the Schiff base ligand (SL). In a second step, a co-ordination reaction is performed with the previously synthesized SL with Fe3+/Co2+ metallic ions to obtain Fe/SL and Co/SL. The pyrolysis of Fe/SL and Co/SL gives Fe/N–C–F and Co/N–C–F as cathode catalysts. During the pyrolysis, the decomposition and carbonization of the salicylaldehyde and pentafluorophenylhydrazine ligands lead to the formation of carbon, along with the N and F dopants incorporated into it. During the pyrolysis, the salicylaldehyde ligand decomposes into phenolic derivatives, CO2 and CO gases and other smaller fragments of hydrocarbons and then eventually into carbon residue by a thermal carbonization process. Parallelly, pentafluorophenylhydrazine primarily decomposes into pentafluorobenzene, N2 gas, and other fluorinated fragments. With further increase in temperature, thermal cleavage of C–F bonds in fluorocarbon fragments leads to the generation of F radicals, which form C–F bonds, potentially generating C–F bonds in the carbon. Collectively, the decomposition and pyrolysis of Fe/SL and Co/SL give Fe/N–C–F and Co/N–C–F as cathode catalysts. The pre-existing co-ordination bonds between transition metals and N atoms in Schiff's base (SL) can be directly transformed into Fe–Nx–C and Co–Nx–C active sites in Fe/N–C–F and Co/N–C–F as cathode catalysts.The successful formation of Schiff's base (SL) is analyzed through the quantitative 1H-NMR spectroscopic technique as shown in Fig. 1. The SL is characterized by the presence of a unique imine linkage, specifically –CHN– that is generated from the condensation reaction. The chemical shift of the imine linkage in general appears in the range of 7.5–8.5 ppm, which is due to the presence of high electronegativity of N in the imine carbon (–CHN–). In addition, since the imine carbon has no protons directly attached to it that could split, the imine linkage –CHN– generally appears to be a singlet peak. So direct evidence of SL formation can be assessed through the appearance of a singlet peak around 7.5–8.5 ppm. Evidently, the 1H-NMR spectra of SL show a clear singlet peak at 7.9 ppm (marked as “b”), suggesting the presence of the imine linkage –CHN–, suggesting the successful formation of SL. Further, to prove the presence of an aromatic ring containing pentafluorophenyl moieties that is attached to the imine linkage –CHN–, one can look at the splitting of the aromatic proton's peaks in the region of 6.8–8.5 ppm, which exhibits multiplex peaks arising due to the coupling of the protons adjacent to the aromatic ring and the –F chemical environment around the carbons. Fig. 1 clearly shows the presence of multiple peaks in the 6.5–8.0 ppm (marked as “c, e, f and g”), clearly suggesting that the pentafluorophenyl moieties are chemically bonded to the salicylaldehyde moieties by the imine linkage –CHN–, indicating the presence of aryl protons in the formed Schiff base ligand. Furthermore, the signal at 10.37 ppm (marked as “a”) suggests the presence of the C–OH group in the salicylaldehyde ligand. The peak “d” is attributed to the solvent. The presence of a singlet peak that corresponds to the imine linkage –CHN– and multiplex peaks that correspond to aryl protons clearly suggests the successful formation of the Schiff base (SL). When SL is complexed with Fe/Co, the colored products obtained during the complexation reaction further confirmed the successful formation of the Fe/SL and Co/SL co-ordination compounds, which upon pyrolysis yield Fe/N–C–F and Co/N–C–F as catalysts.
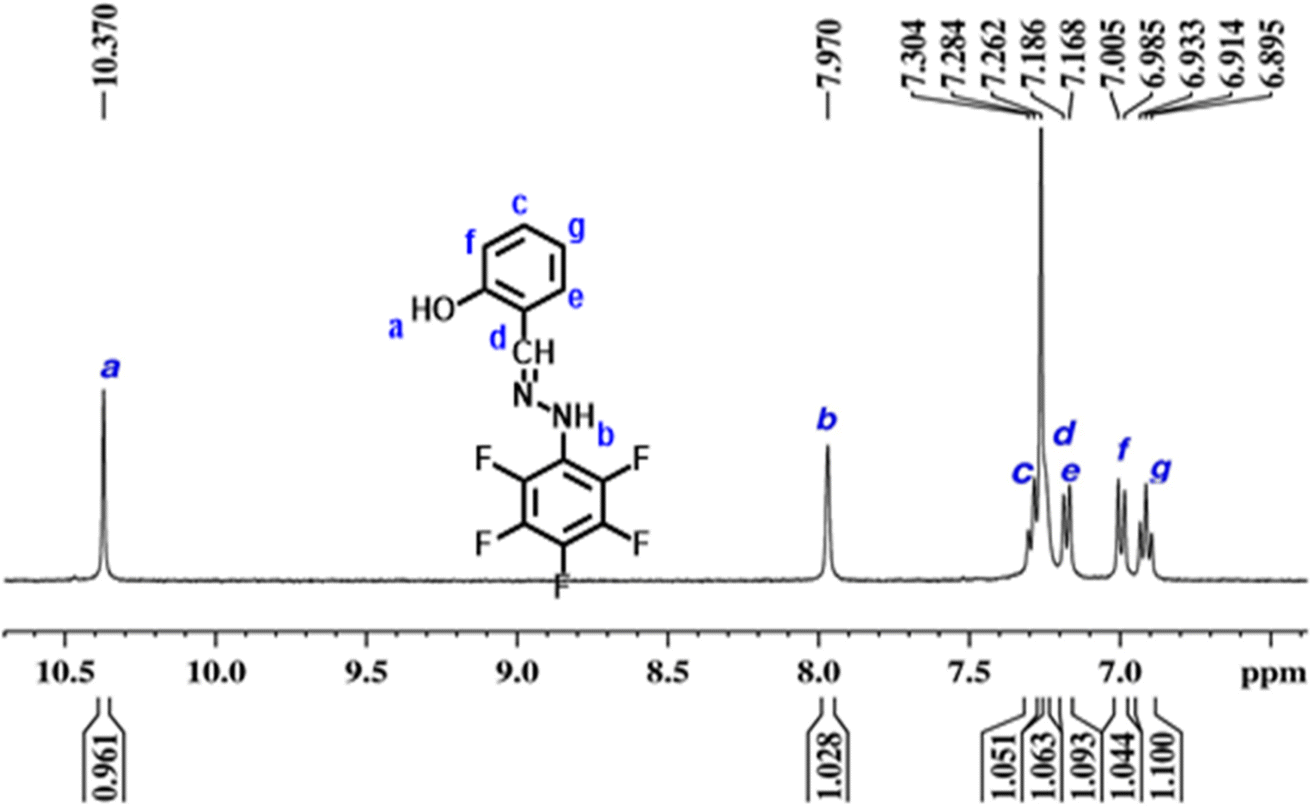 | ||
| Fig. 1 1H NMR of ligand-SL1. | ||
Fig. 2a shows the XRD patterns of Fe/N–C–F and Co/N–C–F catalysts, which show the crystallographic and phase characteristics of the catalysts. Both the catalysts show a diffraction peak around a 2-theta angle of 26.8°, originating from the presence of graphitic carbon (0 0 2) peaks. Interestingly, the diffraction patterns of Fe/N–C–F and Co/N–C–F as catalysts show well resolved peaks associated with the various phases of the metal crystal particles in the catalysts. Both Fe/N–C–F and Co/N–C–F catalysts have been found to contain both Fe and Co species in the mixed phases of metal oxides, metal nitrides and metallic species of Fe and Co nanoparticles. Though the ORR activity of each of these phases might differ, it is generally accepted that metal nitrides in which transition metals are coordinated with N, which form potential M–N–C type active centers, are primarily responsible for the ORR activity, followed by the metal oxides and metallic components of transition metals in the catalysts.50 The XRD of Fe/N–C–F and Co/N–C–F catalysts shows the possible presence of metal nitrides and therefore hints at the presence of M–N–C type active centers in the catalysts. Furthermore, the metal nitrides of Fe are found to be more active than the Co nitrides, influencing their density and distribution within the catalyst layer.51 It is generally assumed that Fe–Nx sites possess increasingly intrinsic catalytic properties than Co–Nx; therefore, we believe that Fe/N–C–F catalysts might present higher ORR activity than Co/N–C–F catalysts as indicated by the ORR studies, which will be discussed in the later section of this article. It is worth noting that the XRD patterns do not show any diffraction peaks associated with the formation of transition metal fluorides, in consistent with the literature reported earlier on non-precious metal catalysts along with N and F doped carbons.52
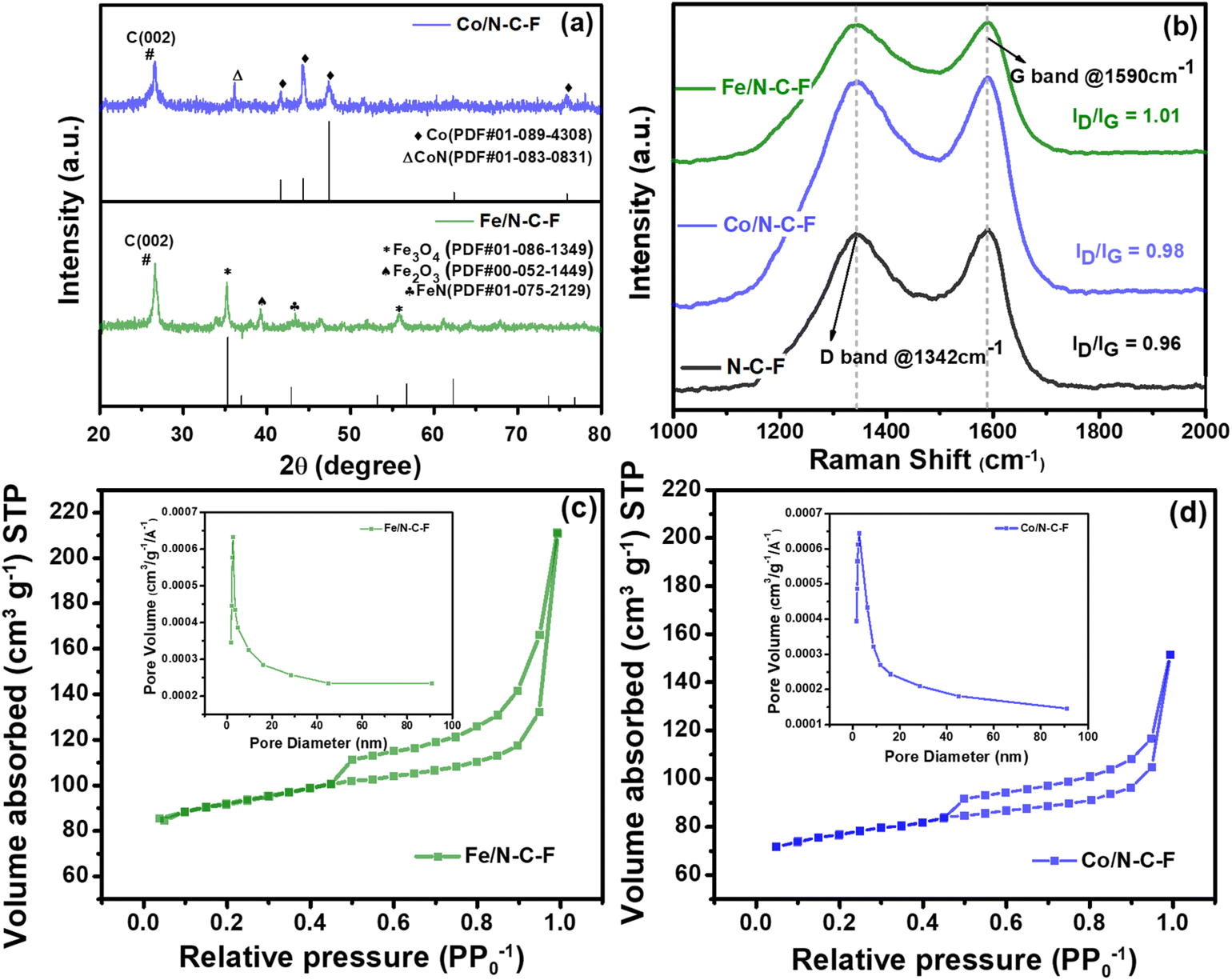 | ||
| Fig. 2 (a) XRD patterns; (b) Raman spectra for the Schiff base ligand (SL), Fe/N–C–F and Co/N–C–F catalysts; N2 absorption–desorption isotherm of (c) Fe/N–C–F and (d) Co/N–C–F catalysts. Equivalent PSDs are presented in the insets of the panels. | ||
The disorder and defect analysis of the Fe/N–C–F and the Co/N–C–F catalysts are performed through Raman spectroscopy through an analysis of defect and graphitic bands, D-band, and G-band, respectively (Fig. 2b). The D band originates from sp3 type carbon, representing the defects and disorder structures in the carbon lattice, with A1g symmetry and arises from the carbon atoms on the lattice edge due to the presence of oxygen functionalities and N/F doping.53 The G band originates from the sp2 type of graphitic carbon lattice mode with E2g symmetry, representing the presence of graphitic carbon.54 The relative intensity of these two bands (ID/IG) generally indicates the presence of defective and graphitic carbon in the catalysts. The Raman spectra of the simple SL pyrolyzed carbon, which gives rise to N–C–F carbon, show a lowest ID/IG ratio of 0.96, whereas Fe/N–C–F and Co/N–C–F catalysts showed marginally increased ID/IG ratios (1.01 and 0.98), indicating slightly increased content of graphitized carbon, possibly due to the influence of the Fe/Co metallic component on the graphitization of carbon at high temperatures during the pyrolysis process. Increased content of graphitic carbon is advantageous in terms of enhanced electronic conductivity of the catalysts, which in turn influences the ORR kinetics.
The textural and substantial characteristics of the catalyst are precisely determined by analyzing the N2 physisorption isotherms and the corresponding pore size distribution (PSD) curves derived from the N2 adsorption data, as shown in Fig. 2c and d. It is clearly evident that the N2 physisorption isotherms of both Fe/N–C–F and Co/N–C–F catalysts presented typical type-II isotherms, which are characteristic of materials composed of nanostructures. These isotherms display a pronounced hysteresis loop at relative pressure intervals of 0.5 and 1.0, indicating their mesoporous nature. The obtained BET surface area values were found to be 245 and 222 m2 g−1 for Fe/N–C–F and Co/N–C–F catalysts, respectively. The obtained surface area values are in the typical range of commercial Pt/C catalysts.55
The obtained average pore diameter for Fe/N–C–F catalysts is found to be 2.8 nm, while for Co/N–C–F catalysts it is 2.7 nm. Furthermore, the area under the hysteresis loop for Fe/N–C–F catalysts is observed to be larger than that for the Co/N–C–F catalyst, possibly due to the opening of secondary pores/cavities at a relatively higher pressure. The adequate surface area and mesoporous nature of the Fe/N–C–F and Co/N–C–F catalysts indicate that the catalyst surface provides sufficient surface area to host Fe/Co active sites and enhance the mass transfer of reactants and quick removal of the products from the ORR.
The morphological analysis of the catalysts is performed by TEM measurements. Fig. 3 shows the TEM images and the elemental mapping of the Fe/N–C–F catalyst. The Fig. 3a shows the assembled, aggregated nanorod morphology of the Fe/N–C–F catalyst. Further, the elemental mapping suggests the presence of C, O, N, F and Fe atoms; all are integrated into the assembled, aggregated, thick nanorod morphology of the Fe/N–C–F catalyst, suggesting the possibility of Fe atoms being bonded to the N-atoms of the carbon derived from the pyrolysis of the SL. Furthermore, the higher resolution image reveals a distinguished porous pattern with elevated lattice fringes corresponding to an interplanar distance of 0.532 nm of the (012) plane of hematite (α-Fe2O3), in consistent with the XRD results, which show the presence of the Fe-oxide phase (Fig. 2a). Similar morphological observations are also found for the Co/N–C–F catalyst (Fig. S1†). The Co/N–C–F catalyst also shows a thick sheet morphology with evenly distributed C, O, N, F and Co atoms in a selected area of elemental mapping. Furthermore, the higher resolution image reveals a distinguished porous pattern with elevated lattice fringes corresponding to an interplanar distance of 0.378 nm of the (311) plane of Co3O4, in consistent with the XRD results, which show the presence of the Co-oxide phase (Fig. 2a).
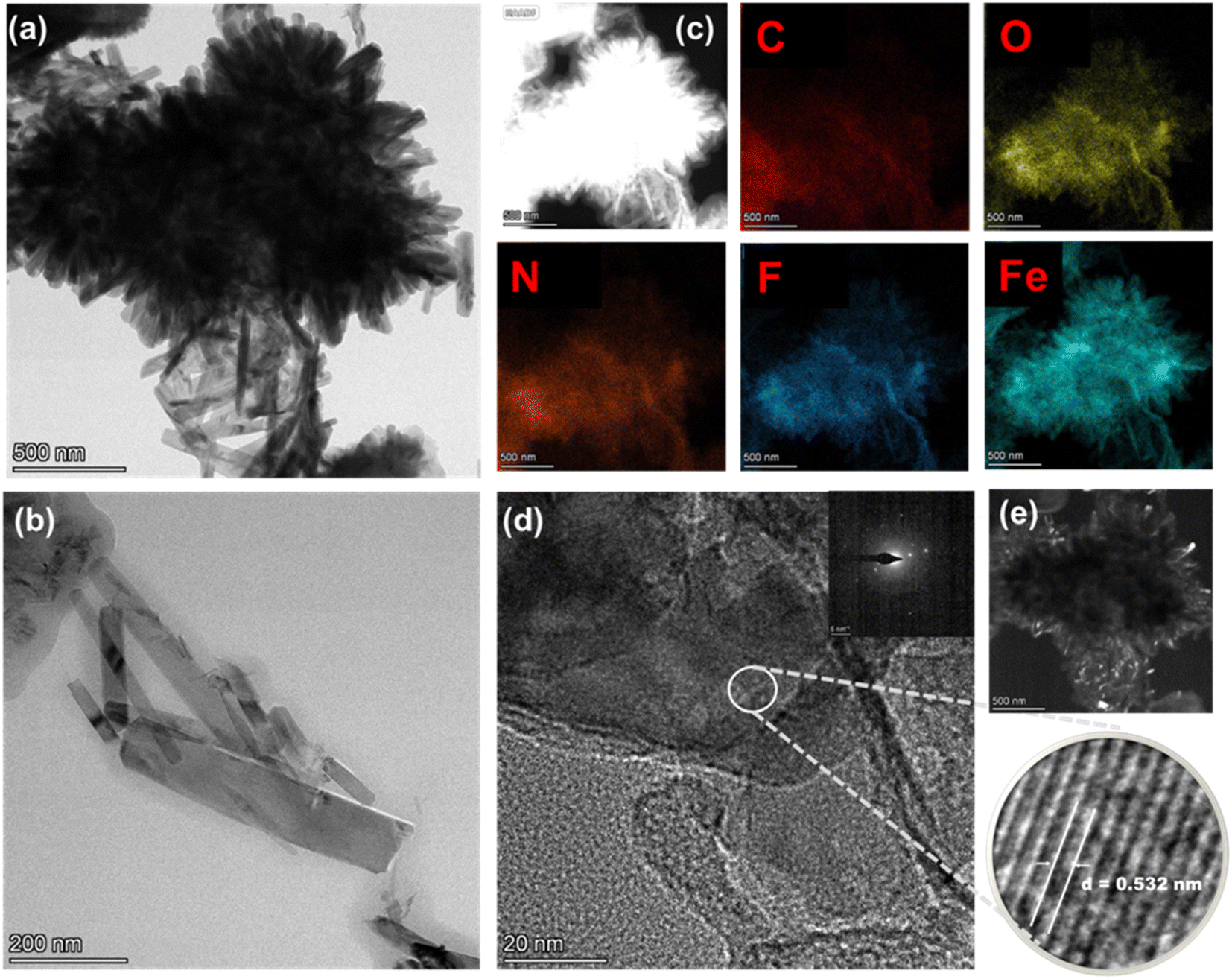 | ||
| Fig. 3 TEM images of (a) and (b) Fe/N–C–F; (c) EDS-mapping images of Fe/N–C–F; (d) high magnified image of Fe/N–C–F, and the corresponding selected area diffraction patterns are shown in insets with lattice fringes; (e) dark field image of Fe/N–C–F. | ||
XPS analysis of the Fe/N–C–F and Co/N–C–F catalysts is performed to determine the electronic states of the catalysts. Table 1 shows the detailed chemical states and their binding energy values of the various elements in Fe/N–C–F and Co/N–C–F catalysts acquired from XPS analysis. Fig. 4a shows the survey spectra of Fe/N–C–F catalysts representing the presence of various elements in the catalysts that include C, N, O, F and Fe. Furthermore, all the existing elements are further recorded with a high resolution scan and deconvoluted into their chemical states to understand the active sites and their distribution in the catalyst layer. Fig. 4b shows the N1s spectra of Fe/N–C–F catalysts, which can be further deconvoluted into five different peaks associated with five different chemical functionalities in the catalyst layer, pyridinic-N, pyrrolic-N, pyridinic-N–Fe, graphitic-N and oxidized-N–O peaks. It is well known that doping of N into the carbon species generates these types of bonding states and the presence of N in the carbon matrix alters the charge/spin density of surrounding carbon atoms, which helps in the ORR.56 Though all three major bonding states, namely, pyridinic-N, pyrrolic-N and graphitic-N species, are responsible for the intrinsic ORR, their order of reactivity is considered as a debatable perspective. Several researchers believe that the order of ORR activity is pyridinic-N > graphitic-N > pyrrolic-N species.57 Among these three major peaks, especially, the pyridinic-N species is found to be very important in terms of its ability to co-ordinate with a variety of transition metals including Fe and Co; therefore, it tends to form M–Nx–C active sites, which are assumed to be the highly active ORR sites in the catalyst layer. Fig. 4c shows the F1s spectra of the Fe/N–C–F catalysts, which can be further deconvoluted into two distinct electronic arrangements of F atoms in the catalyst, namely ionic C–F and semi-ionic C–F bonds. The carbon matrix with sp3 hybridization carbon atoms, specially formed on buckled, defected carbons, forms covalent C–F bonds, while planar carbon sheets form semi-ionic bonds.58 Since semi-ionic C–F bonds increase carbon's electronic conductivity, they are crucial for electrochemical reactions. Semi-ionic C–F (sp2 hybridization) bonds are more polar than covalent C–F bonds (sp3 hybridization of C), leading to more positive charge on adjacent carbon atoms.59 Because of this, F-doped carbon with semi-ionic C–F bonds should have higher ORR activity than covalent carbon.60Fig. 4d shows the C1s deconvoluted spectra which can be classified into three distinct major peaks associated with sp2, sp3 and carbon linked oxygen derivatives. The presence of sp3 carbon arises due to the defects induced by the –N and F doping and the presence of oxygen functionalities. Fig. 4e shows the deconvoluted Fe2p spectra, which show several peaks of Fe associated Fe2p3/2 and Fe2p1/2 along with their satellite peaks. The various binding energies of Fe2+/Fe3+ of Fe2p3/2 and Fe2p1/2 peak splitting are given in Table 1. The appearance and co-existence of Fe2+/Fe3+ help in catalyzing the ORR due to their ease of interconversion between Fe2+ and Fe3+ during the electron exchange in the ORR. Further, the Fe3+ ionic species are particularly beneficial in absorbing and activating the O2 molecules, during the ORR, due to their high positive charge, which can polarize the adsorbed O2 molecule. In conclusion, the Fe/N–C–F catalyst ORR activity is assumed to originate from the presence of various N and F-doped functionalities and the possible formation of M–Nx–C active sites along with the appearance and co-existence of Fe2+/Fe3+.
| Catalyst | ‘N’ configuration@eV | ‘F’ configuration@eV | Catalyst ‘Fe/Co’ configuration@eV | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pyridinic | Fe/Co–N | Pyrrolic | Graphitic | Oxidised N | Ionic | Semi-ionic | Fe/Co 2p3/2 | Fe/Co 2p1/2 | |||
| Fe/Co3+ | Fe/Co2+ | Fe/Co3+ | Fe/Co2+ | ||||||||
| Fe/N–C–F | 397.7 | 398.6 | 399.5 | 400.7 | 401.9 | 684.5 | 688.1 | 712.7 | 710.8 | 724.5 | 727.1 |
| Co/N–C–F | 398.3 | 399.7 | 400.9 | 402.5 | 404.5 | 684.6 | 687.3 | 784.4 | 789.1 | 800.1 | 805.2 |
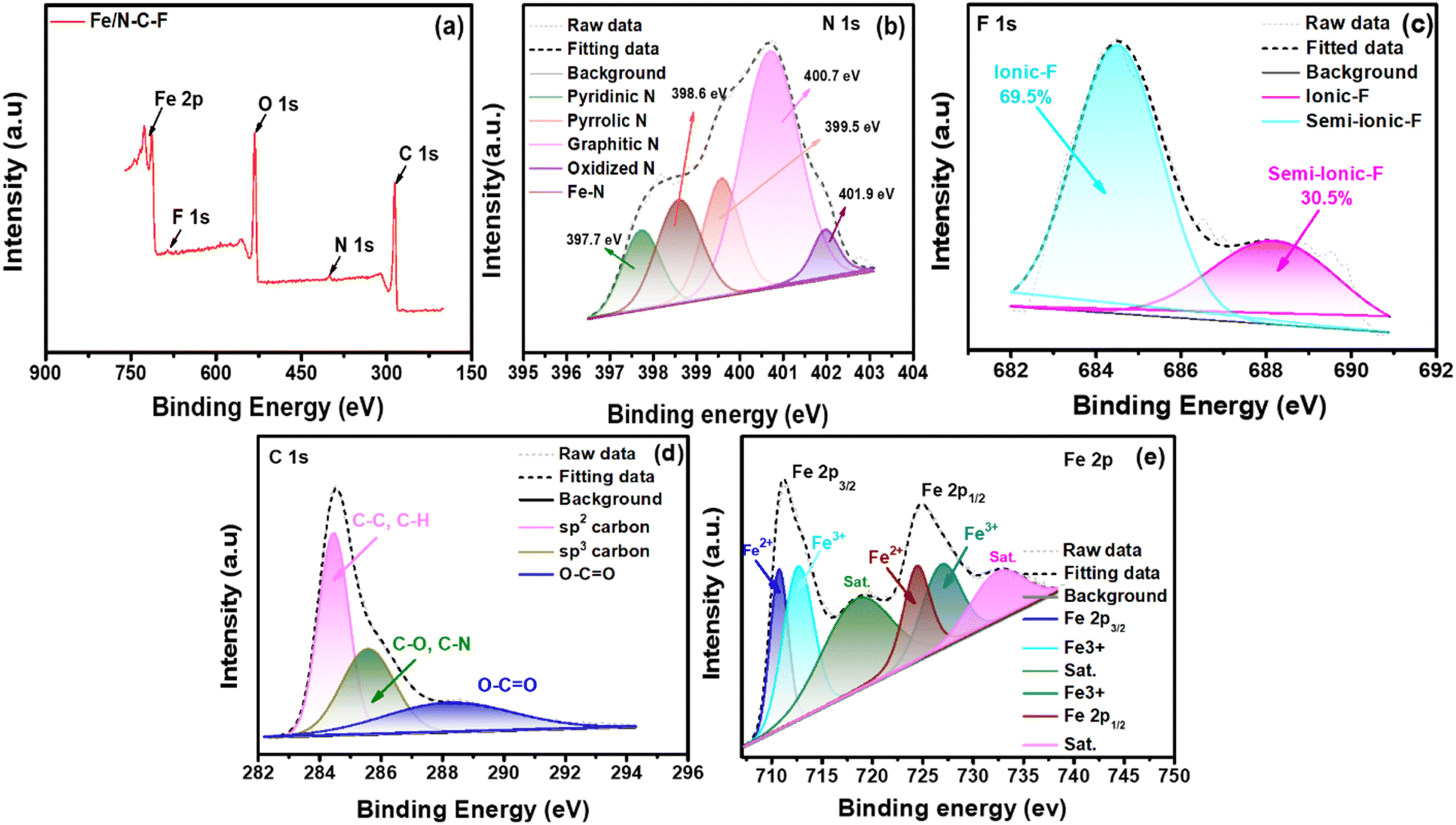 | ||
| Fig. 4 (a) XPS survey spectra of Fe/N–C–F; (b)–(e) deconvoluted XPS spectra of N1s, F1s, C1s, and Fe2p. | ||
Similar ORR active sites can also be deduced from the Co/N–C–F catalyst as shown in Fig. 5. Fig. 5a shows the survey spectra of the Co/N–C–F catalyst, indicating the presence of various elements, including C, N, O, F, and Co. Additionally, all the present elements are further analyzed with high-resolution scans and deconvoluted into their chemical states to understand the active sites and their distribution in the catalyst layer. Fig. 5b shows the N1s spectra of Co/N–C–F catalysts, which can be deconvoluted into five different peaks corresponding to five distinct chemical functionalities in the catalyst layer: pyridinic-N, pyrrolic-N, pyridinic-N–Co, graphitic-N, and oxidized-N–O peaks. However, the distinct chemical environment of N and F dopants differs from that of the Co/N–C–F catalysts. Especially, the abundance of graphitic-N is found to be higher in Fe/N–C–F catalysts than in Co/N–C–F catalysts (Fig. 5b). In addition, the abundance of semi-ionic-C–F bonds is almost double in the Fe/N–C–F catalysts compared to the Co/N–C–F catalysts (Fig. 5c). As explained above, several literature studies also suggest that the ORR activity and stability of the F-doped carbons are directly linked to the content of semi-ionic-C–F bonds. Therefore, it is reasonably concluding that the presence of a high abundance of graphitic-N and semi-ionic-C–F bonds might strongly influence the ORR activity in Fe/N–C–F catalysts compared to the Co/N–C–F catalysts, as deduced from the experimental observations in RDE studies. Furthermore, Fig. 5d shows the C1s deconvoluted spectra, which can be classified into three distinct major peaks associated with sp2, sp3 and carbon linked oxygen derivatives. The presence of sp3 carbon arises due to the defects induced by the N and F doping and the presence of oxygen functionalities. Fig. 5e shows the deconvoluted Co2p spectra, which show several peaks of Co associated Co2p3/2 and Co2p1/2 along with their satellite peaks. The various binding energies of Co2+/Co3+ of Co2p3/2 and Co2p1/2 peak splitting are given in Table 1. The presence of Co2+/Co3+ facilitates the catalysis of the ORR due to their easy inter-conversion between Co2+ and Co3+ during electron exchange in the ORR. Additionally, Co3+ ionic species are particularly effective in absorbing and activating O2 molecules during the ORR due to their high positive charge, which can polarize the adsorbed O2 molecules. In summary, the ORR activity of the Co/N–C–F catalyst is attributed to the presence of various N and F-doped functionalities, and the potential formation of M–Nx–C active sites, along with the coexistence of Co2+/Co3+. The comparison of the elemental composition of Fe/N–C–F and Co/N–C–F catalysts can be accessed in ESI Table S2.†
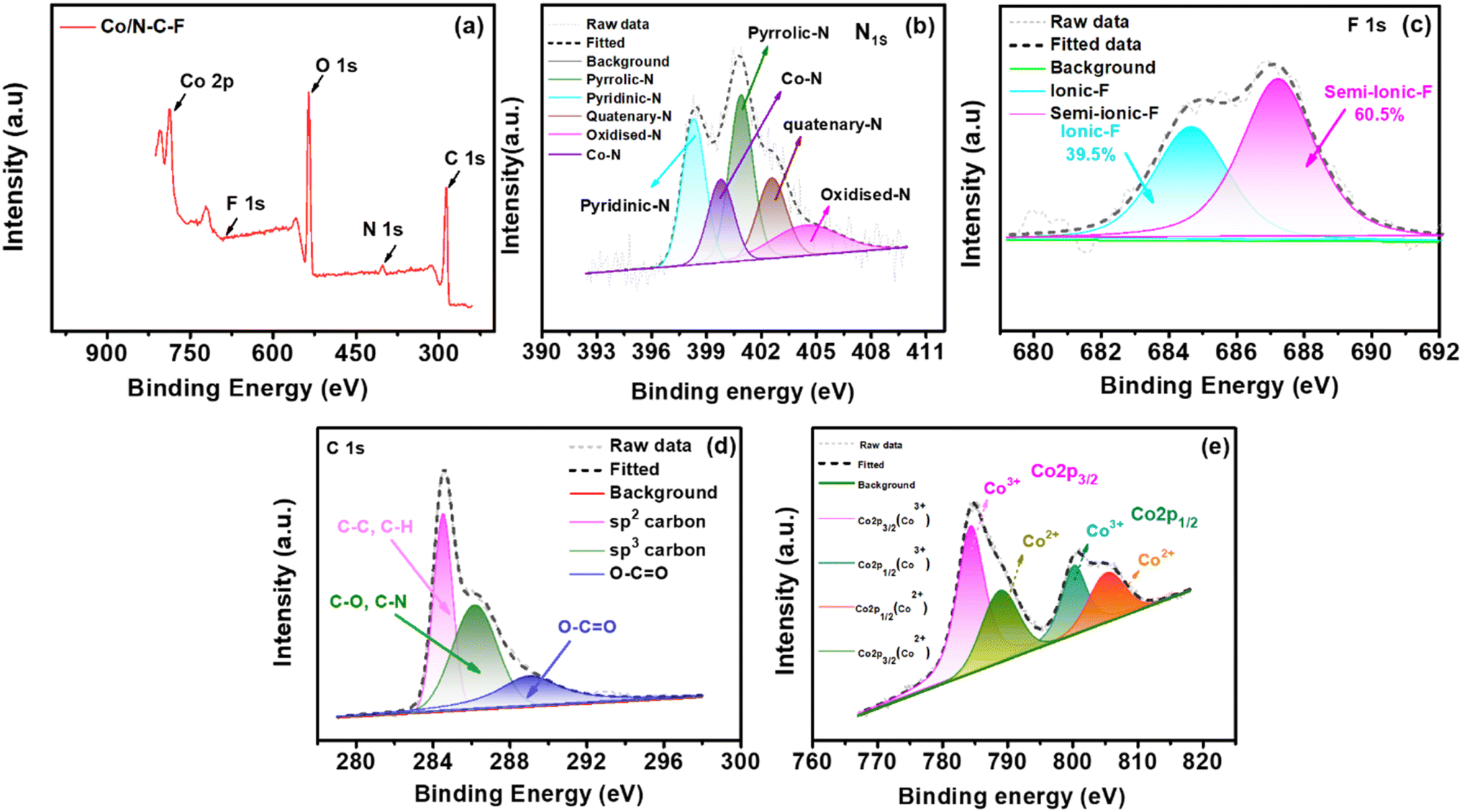 | ||
| Fig. 5 (a) XPS survey spectra of Co/N–C–F. (b)–(e) Deconvoluted XPS spectra of N1s, F1s, C1s, and Co2p. | ||
After a detailed microstructural analysis of Fe/N–C–F and Co/N–C–F catalysts through several physicochemical characterization studies, the electrocatalytic ORR activity of the catalysts was further examined in O2 saturated 0.1 M aqueous KOH electrolyte on a thin layer deposited glassy carbon electrode. The optimum ORR performance of Fe/N–C–F and Co/N–C–F catalysts was assessed by varying the Schiff base ligand:metal ratio, aiming to obtain ideal catalysts with a high density of N and F dopants and metal active sites in the catalyst. For this reason, the Schiff base ligand:metal ratio has been varied at 1:1, 2:1 and 3:1 by keeping the metal concentration constant and varying the Schiff base ligand concentration. We choose to vary the Schiff base ligand, due to the fact that pyrolysis of the Schiff base ligand leads to the formation of a carbon support with N and F dopants, which acts as a base for even distribution of metallic active sites. The catalysts synthesized in this way are then pyrolyzed to obtain the Fe/N–C–F and Co/N–C–F catalysts. Fig. 6a and d show the cyclic voltammograms of the Fe/N–C–F and Co/N–C–F catalysts, respectively. Both catalysts exhibited typical CV curves with capacitance and oxygen reduction regions. Interestingly, all the catalysts also showed a definite Eredox peak associated with O2 reduction in the potential range of 0.8–0.9 V vs. RHE, indicating that all the catalysts possess intrinsic ORR activity. Furthermore, the Eredox peaks were observed to shift to higher potentials with an increase in the SL concentration, in the case of Fe/N–C–F catalysts. The Eredox peak potentials were found to be 0.81, 0.86 and 0.83 V vs. RHE, respectively for 1:1, 2:1 and 3:1 molar ratios of SL:metal precursor (Fe) (Fig. 6a). The LSV curves further confirm the trend observed from the CV curves and their Eredox potentials. The half-wave potentials obtained for the Fe/N–C–F catalyst, for 1:1, 2:1 and 3:1 molar ratios of SL:metal precursor, were calculated to be 0.79, 0.88 and 0.83 V vs. RHE, respectively (Fig. 6b). The highest Eredox peak potentials and half-wave potentials for Fe/N–C–F catalysts with a 2:1 SL:metal precursor ratio were identified as indicative of the best and optimum catalysts. When similar catalysts were obtained with Co precursors, the optimum catalysts was found to be that with a 1:1 SL:metal precursor ratio. The CV curves obtained show that the Eredox peak potential constantly decreases with increasing SL concentration. The Eredox peak potentials were found to be 0.84, 0.79 and 0.76 V vs. RHE, respectively, for 1:1, 2:1 and 3:1 molar ratios of SL:metal (Co) precursor (Fig. 6d). The LSV curves further confirm the trend observed from the CV curves and their Eredox potentials. The half-wave potentials obtained for the Co/N–C–F catalyst, for 1:1, 2:1 and 3:1 molar ratios of SL:metal precursor, were calculated to be 0.80, 0.79 and 0.75 V vs. RHE, respectively (Fig. 6e). The highest Eredox peak potentials and half-wave potentials for the Co/N–C–F catalyst with 1:1 SL:metal precursor were identified as indicative of the best and optimum catalysts. Among the investigated Fe/N–C–F and Co/N–C–F catalysts, with the highest half-wave potentials of 0.88 V vs. RHE, Fe/N–C–F is found to be the best catalyst, compared to 0.80 V vs. RHE for Co/N–C–F catalysts. We believe that the high ORR activity of Fe/N–C–F catalysts is credited to the unique nanorod morphology and relatively higher surface area of 245 m2 g−1, compared to Co/N–C–F catalysts. The number of electrons transferred per O2 molecule is a measure of the desired direct 4e− reduction of electrocatalysts. The K–L plots (inset of Fig. 6c and f) derived from the LSV curves recorded at different rotations per minute for the optimized Fe/N–C–F and Co/N–C–F catalysts are shown in Fig. 6c and f. It is observed that the limiting current density constantly increases with the increase in the rotations per minute, with almost no change in the kinetic and mixed kinetic diffusion-controlled regions of the LSV curves indicating that the ORR is a diffusion-controlled process, where gaseous O2 needs to be diffused from the bulk solution to the electrocatalyst surface. The K–L plots derived from the LSV curved show a linear plot at different potentials indicating that the ORR follows a similar reaction trend with a similar number of electrons transferred per O2 molecules at different potentials. The average number of electrons transferred calculated from the K–L plots were found to be 3.91 and 3.87 for Fe/N–C–F and Co/N–C–F catalysts, respectively.
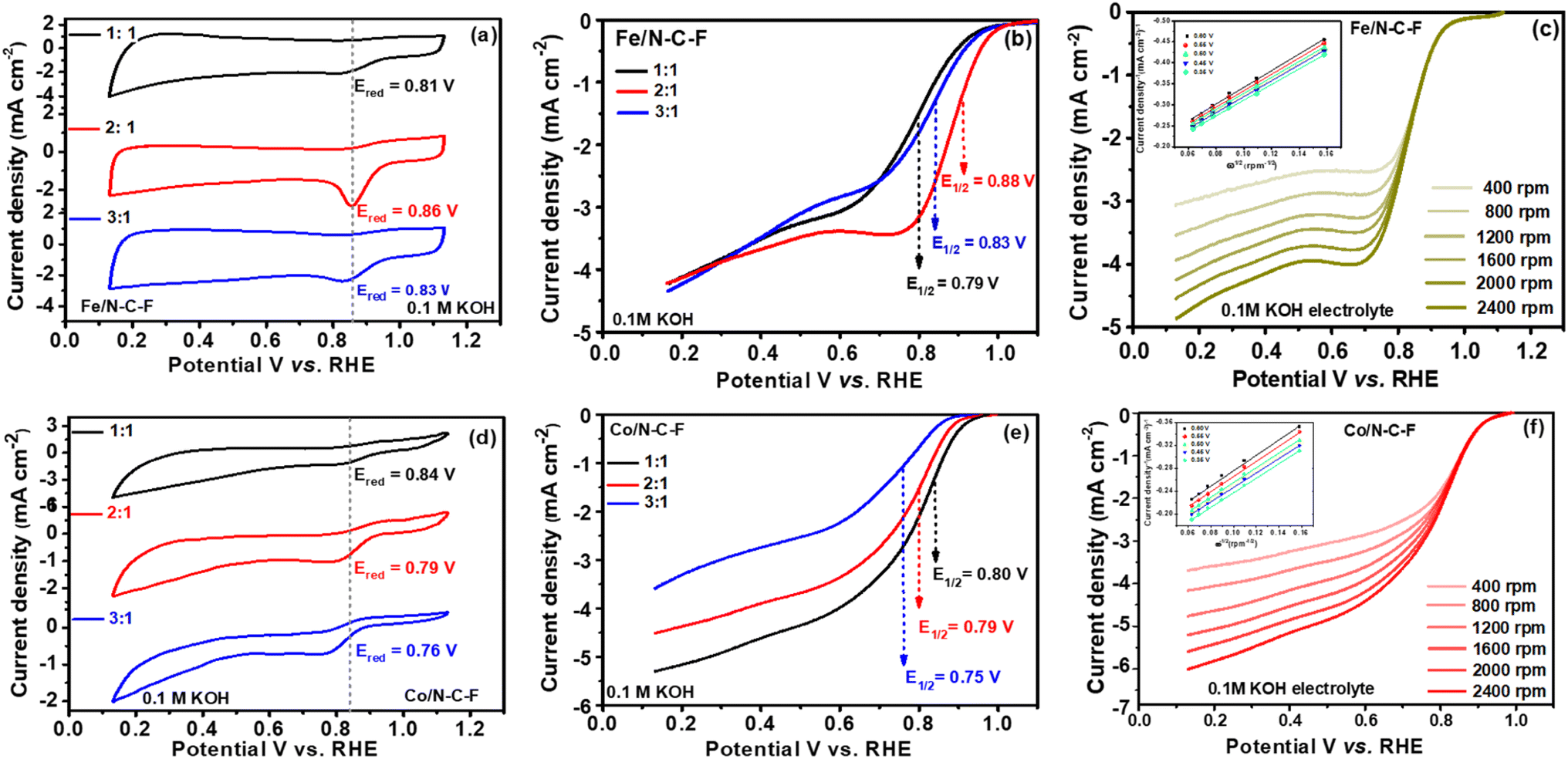 | ||
| Fig. 6 (a) CV for Fe/N–C–F with varied molar ratios at a scan rate of 20 mV s−1 and (b) comparison of LSV for Fe/N–C–F with varied molar ratios at a scan rate of 5 mV s−1, recorded in O2–saturated aq. 0.1 M KOH electrolyte at a rotation rate of 1600 rpm. K–L plots for (c) Fe/N–C–F; (d) CV for Co/N–C–F with varied molar ratios at a scan rate of 20 mV s−1 and (e) comparison of LSV for Co/N–C–F with varied molar ratios at a scan rate of 5 mV s−1, recorded at a rotation rate of 1600 rpm in O2–saturated 0.1 M KOH; K–L plots for (f) Co/N–C–F. | ||
The ORR activity of the optimized Fe/N–C–F and Co/N–C–F catalysts is finally compared with that of the state-of-the-art commercial Pt20%/C catalyst, as shown in Fig. 7a, along with the RRDE curves. The half-wave potential of the Pt20%/C catalyst is 0.90 V vs. RHE, which is 20 mV higher than that of Fe/N–C–F and 100 mV lower than that of Co/N–C–F catalysts. Therefore, the ORR activity of Fe/N–C–F is found to be closer to that of the commercial Pt20%/C catalyst, with just 20 mV lower half-wave potential values. However, the obtained ORR activity of the Fe/N–C–F catalyst can still be considered significant and admirable in terms of the catalyst price, as the catalyst price of Fe/N–C–F could be much lower than that of the noble metal based Pt20%/C catalyst. The RRDE curves shown in Fig. 7a indicate that Pt20%/C and Fe/N–C–F catalysts show almost similar ORR kinetics. The calculated hydroperoxide yield is found to be 3.1, 6.5 and 4.5% and the average number of electrons transferred per O2 molecule calculated from RRDE studies was obtained to be 3.91, 3.87 and 3.94 for Fe/N–C–F, Co/N–C–F and Pt20%/C catalysts, respectively, as shown in Fig. 7b. The Tafel plots derived from the LSV curves are shown in Fig. 7c, where the obtained Tafel values are 87, 89 and 77 mV dec−1 for Fe/N–C–F, Co/N–C–F and Pt20%/C catalysts, respectively, suggesting almost similar O2 adsorption and reduction kinetics of Fe/N–C–F and Co/N–C–F, in relation to the commercial Pt20%/C catalyst. Though the overpotential for the Pt20%/C catalyst is slightly lower than those of the Fe/N–C–F and Co/N–C–F catalysts, the ORR activity is still considered appreciable considering the cost and abundance of Fe and Co metallic precursors compared to Pt and therefore serves as a competitive alternative to Pt20%/C catalysts for the ORR. In order to truly compare the competitiveness of Fe/N–C–F and Co/N–C–F catalysts with Pt20%/C catalysts, the mass and specific activities are determined as shown in Fig. 7d. The mass activity of the Fe/N–C–F catalyst is 0.46 A mg−1, which is close to 85% of the mass activity of the Pt20%/C catalyst (0.54 A mg−1). A comparison of the iK, MA and Tafel slope of Fe/N–C–F, Co/N–C–F and commercial Pt20%/C catalysts can be accessed in ESI Table S1.†
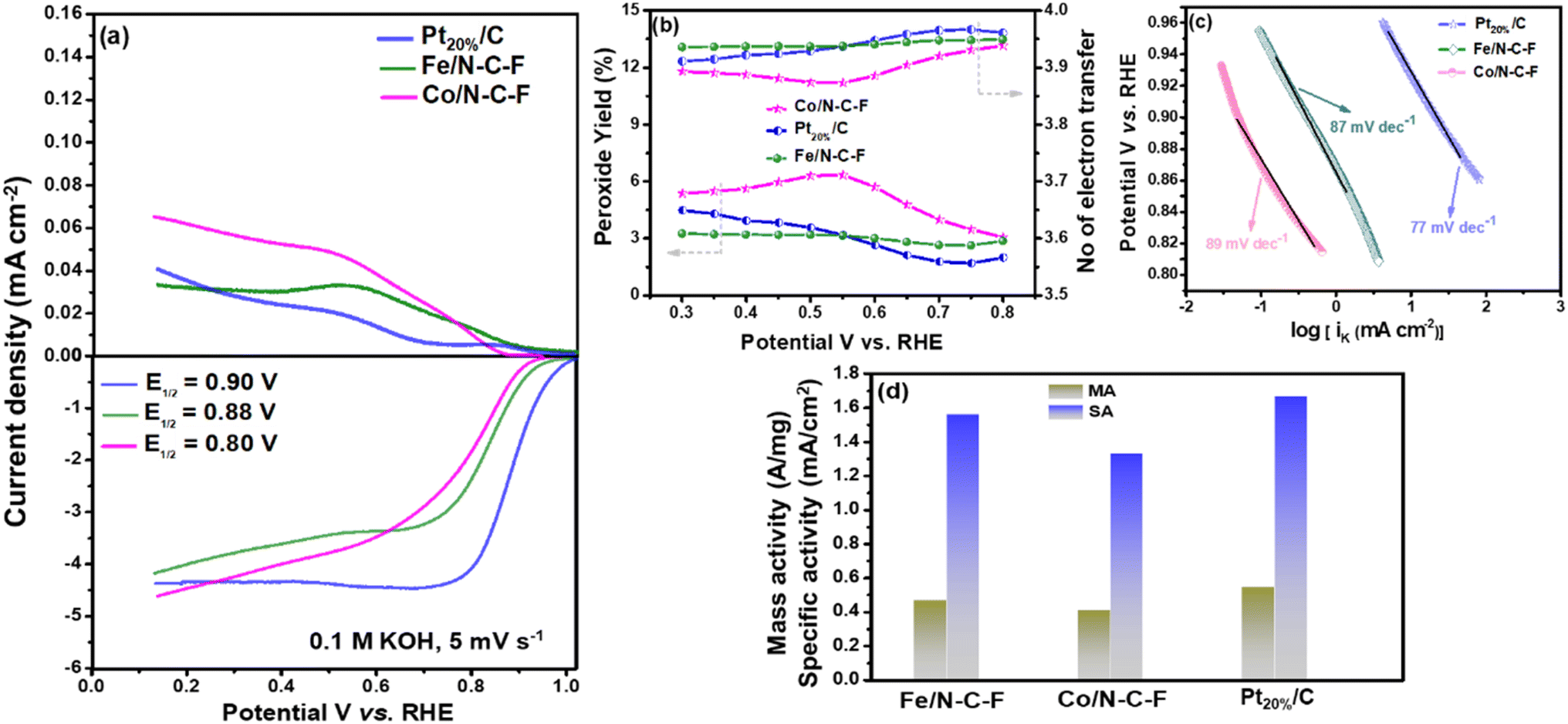 | ||
| Fig. 7 (a) RRDE curves of Fe/N–C–F and Co/N–C–F catalysts and Pt20%/C at a scan rate of 5 mV s−1; (b) number of electrons transferred and HO2− yield of Fe/N–C–F and Co/N–C–F catalysts and Pt20%/C during the ORR process; (c) Tafel slope and (d) MA and SA of Fe/N–C–F, Co/N–C–F and Pt20%/C, respectively. | ||
After a detailed analysis of the ORR activity, the stability of Fe/N–C–F, Co/N–C–F and Pt20%/C catalysts has been evaluated by potential cycling using CV curves for 20000 potential cycles, in the potential range of 0 to 1.2 V vs. RHE, in 0.1 M KOH electrolyte. The stability of the Fe/N–C–F, Co/N–C–F and Pt20%/C catalysts is assessed by recording LSV curves after 20000 potential cycles at 1600 rpm and the half-wave potentials have been deduced, as shown in Fig. 8a–c. The loss of half-wave potentials for Fe/N–C–F and Co/N–C–F catalysts was found to be ∼27 and ∼30 mV, respectively. In contrast, under similar experimental conditions, the Pt20%/C catalyst lost ∼70 mV of its half-wave potentials after just 1000 cycles. These results indicate the extremely poor stability of the Pt20%/C catalyst in 0.1 M KOH electrolyte. It is well known that the poor stability of the Pt20%/C catalyst is due to several reasons such as the instability of the carbon support that leads to carbon corrosion, Pt nanoparticle agglomeration, Pt dissolution and redeposition and Pt nanoparticle detachment from the support due to weak metal–support interactions.61 In contrast, the excellent stability of the Fe/N–C–F catalyst is credited to several unique properties such as the stable nanorod morphology and stable N and F doped carbon support. It is well known that N doping enhances the metal nanoparticle interaction with the support and therefore enhances the metal–support interaction.62,63 In addition, the strong bonding between Fe and N in the carbon support forms Fe–Nx–C type coordination structures, which ensure the successful bonding of the metallic active sites with the carbon support and therefore help in the mitigation of metal nanoparticle detachment and resistance to dissolution under highly alkaline conditions.64 In addition to the effect of nitrogen (N) on catalyst stability, it has been recently reported that the presence of –F atoms in the carbon support outperforms –N doping in enhancing stability. –F doping provides exceptional thermal and electrochemical stability to the carbon due to the presence of –C–F bonds.65 The highly polarized C–F bonds in the carbon strengthen the carbon structure against corrosion. In addition, the hydrophobicity induced by F-doping helps in reducing the adsorption and interaction of O2 and H2O with the carbon support and hence alleviates the chances of carbon corrosion.29 In addition, the C–F bonds are mostly located in the defect and edge sites that are normally the start points of carbon corrosion. Due to edge occupation of –F the edge carbons are highly protected by highly polarized C–F bonds and therefore strongly enhance the ORR activity and catalyst stability.26,66 Therefore, the presence of N helps in enhancing the electronic conductivity and mitigating the detachment of Fe active sites through Fe–Nx–C type coordination structures, while –F doping helps in stabilizing the carbon against electrochemical corrosion, which are believed to be the factors effecting the high stability of the Fe/N–C–F catalyst.
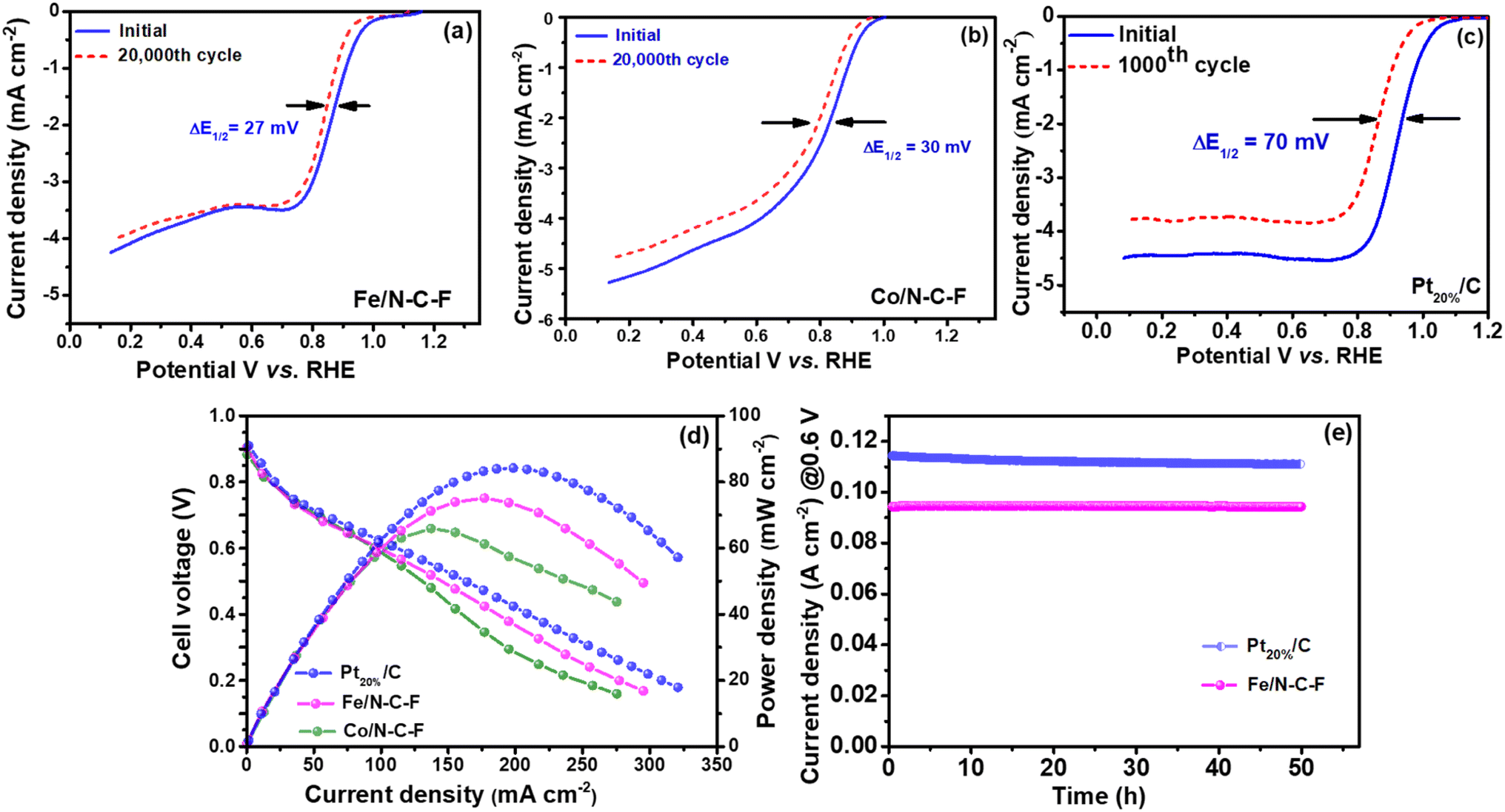 | ||
| Fig. 8 (a) and (b) LSVs of Fe/N–C–F and Co/N–C–F catalysts before and after 20000 recurring potential cycles and (c) Pt20%/C afore and after 1000 repeated potential cycles in 0.1 M KOH electrolyte. (d) Fuel cell polarization and power density data in AEMFCs with Fe/N–C–F and Co/N–C–F as cathode catalysts in comparison with the commercial Pt20%/C catalyst. (e) Fuel cell stability of Fe/N–C–F in comparison to Pt20%/C. | ||
The stability of the Fe/N–C–F catalyst is further assessed by recording post-structural and morphological changes that occur in the Fe/N–C–F catalyst through TEM analysis. Fig. S3 of the ESI† shows the TEM images of the Fe/N–C–F catalyst that are collected after 20000 potential cycles. It is clearly seen that the Fe/N–C–F catalyst morphology hardly changed when compared to the TEM images recorded before the stability test, indicating the extraordinary stability of the Fe/N–C–F catalyst. Table S3 of ESI† shows the ORR kinetic data extracted from the literature that are based on the electrocatalysts of transition metals supported on N and F doped carbons. It is interesting to note that the electrocatalytic performance of the Fe/N–C–F catalyst developed in this study showed the best ORR performance among the existing literature, suggesting the promising results of Fe/N–C–F synthesized by a unique Schiff base synthesis strategy.
After the detailed morphological, structural and electrochemical analysis of the Fe/N–C–F and Co/N–C–F catalysts, finally, the i–v characterization of the catalysts is performed in a membrane electrode assembly (MEA) configuration in an AEMFC as shown in Fig. 8d. To evaluate the power density in a single cell configuration, MEAs are fabricated with Fe/N–C–F, Co/N–C–F and Pt20%/C catalysts as cathodes and Pt20%/C as anodes, with H2 as the fuel and O2 as the oxidant. The AEMFCs comprising Fe/N–C–F and Co/N–C–F catalysts deliver peak power densities of ∼75 and ∼67 mW cm−2 at load current densities of 175 and 137 mA cm−2 under atmospheric pressure and a temperature of ∼30 °C, while Pt20%/C catalysts deliver a power density of ∼85 mW cm−2 and a current density of ∼200 mA cm−2. The obtained power density values are further clearly reflected in RDE measurements, in which the ORR activity trend is Pt20%/C > Fe/N–C–F > Co/N–C–F catalysts. In order to assess the stability of the catalyst under real fuel cell operating conditions, the chronoamperometric analysis of the catalysts in a MEA configuration is performed at a constant voltage of 0.6 V for 50 h (Fig. 8e). The chronoamperometric results show that the Fe/N–C–F and Pt20%/C catalysts both show excellent stability in the MEA configuration.
4 Theoretical calculations
In order to gain a deeper understanding of the factors that contribute to the increased ORR activity of the Fe/N–C–F and Co/N–C–F catalysts, DFT simulations were carried out as shown in Fig. 9. Consistent with prior studies, all modelling and optimization tasks were executed using the DFT method, specifically employing the B3LYP function and 6-31G* basis set.67,68 The catalytic ORR mechanisms of Fe/N–C–F and Co/N–C–F catalysts were investigated by constructing a carbon matrix with conjugated aromatic rings incorporated with N and F, along with N-atoms coordinated with Fe/Co metal atoms by four N atoms, mimicking M–N4–C structures (M = Fe/Co), all situated within a system of 10 conjugated aromatic rings. Associative and dissociative mechanisms in accordance with the 2e− and 4e− pathways respectively are the two major reactions through which the electrochemical oxygen reduction reaction occurs and we have investigated both of these mechanisms on Fe/N–C–F and Co/N–C–F catalysts.61–66 Detailed theoretical procedures and additional results are available in ESI Section S5 (Table 2).†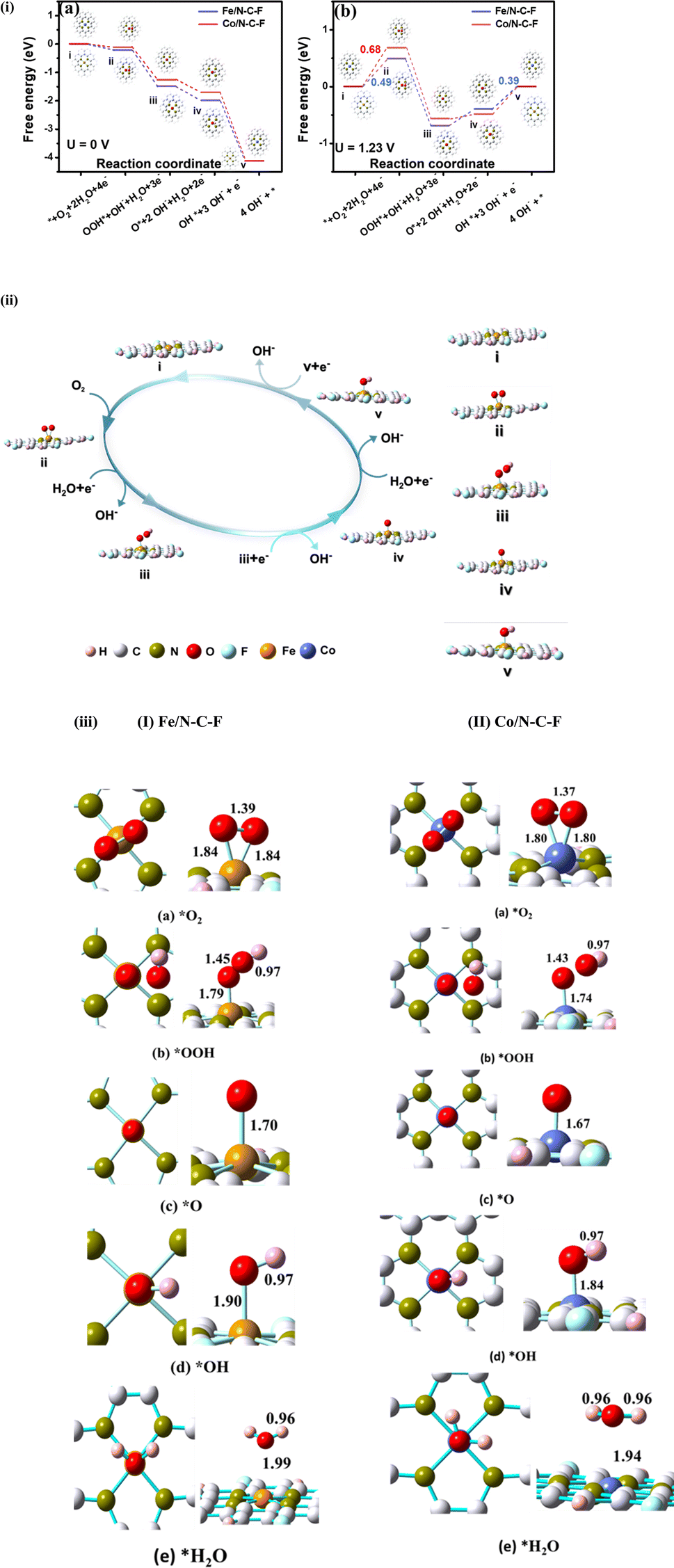 | ||
| Fig. 9 (i) The free energy diagram at (a) U = 0 V vs. NHE and (b) U = 1.23 V vs. NHE for the oxygen reduction reaction on the Fe/N–C–F and Co/N–C–F catalysts in alkaline medium (the corresponding energy differences between the adjacent steps are presented in Table 3). (ii) Contemplated reaction pathway of the oxygen reduction reaction on the Fe/N–C–F catalyst. (iii) Optimized geometries (top view and side view) of ORR species on Fe/N–C–F and Co/N–C–F. (I) For Fe/N–C–F: (a) O2, (b) OOH, (c) O, (d) OH and (e) H2O. (II) for Co/N–C–F: (a) O2, (b) OOH, (c) O, (d) OH and (e) H2O. Grey balls represent C atoms, green balls represent N atoms, pink balls represent H atoms, red balls represent O atoms, cyan balls represent F atoms, orange balls represent Fe atoms and blue balls represent Co. | ||
| Catalyst | Mulliken charges | |
|---|---|---|
| Fe | Co | |
| Cat. | 1.068 | 0.849 |
| Cat. + O2(ads) | 1.186 | 0.984 |
| Cat. + OOH(ads) | 1.214 | 1.021 |
| Cat. + O(ads) | 1.193 | 0.989 |
| Cat. + OH(ads) | 1.176 | 0.969 |
Gaussian 09 software was used to carry out all calculations and the stimulation results were envisaged using Gauss View 6 software. In the self-consistent reaction field (SCRF) model, the presence of the solvent (water) was factored into calculations and models were built keeping them in field of view.69 Models were built using fragments of graphene in order to investigate the catalytic parameters of the material. XPS analysis has confirmed the presence of Fe/Co–N bonds, as well as C–N bonds, within the carbon matrix. This discovery is essential for constructing the DFT model, offering valuable insights into the electronic structure and catalytic properties of the material. The surface of graphene having conjugated aromatic rings incorporated with a nitrogen atom surrounding a metal atom in the arrangement, i.e., a metal atom enclosed by four N atoms, all situated within a system of 10 conjugated aromatic rings, has been intensively studied. Phthalocyanines of metals corresponding to the metal precursors used play a vital role in determining the experimental structure explained above. Associative and dissociative mechanisms in accordance with the 2e− and 4e− pathways respectively are the two major reactions through which the electrochemical oxygen reduction reaction occurs.
In view of the optimized structures of intermediates used to simulate catalytic processes (Table S4†), it becomes evident that oxygen adsorption is limited on the surface. Consequently, catalytically active sites within the catalyst consist of metal atoms firmly rooted within the carbon matrix. The energy profiles of the explored electrochemical reaction of oxygen reduction both in its free form and on the studied carbon support were acquired as part of demonstration (as shown in Fig. 9). The catalytic cycle of Fe/N–C–F and Co/N–C–F shown in Fig. 9ii and the relevant calculations shown in Table S4† and Fig. 9i reveal the higher performance of Fe/N–C–F in comparison to Co/N–C–F. Adsorption, being the preliminary step of the catalytic cycle, plays an essential role in the entire process. In this study, various catalytic oxygenated intermediates of the ORR (such as O2, OOH, O, OH and OH−) that are being absorbed on Fe/N–C–F and Co/N–C–F surfaces are explored in order to determine the entire mechanism evidently. Further, to have a clear insight into the reaction pathway of Fe/N–C–F and Co/N–C–F in alkaline medium, the free energy diagrams (Fig. 9ii) for the ORR were computed and were also compared with that of pristine graphene to highlight the reaction coordinates and characteristics. The initial stage was the chemisorption of O2, followed by hydrogenation to form the *OOH intermediate. As evident, O2 binds the single metal Fe/Co surrounded by N atoms directly in a side-on orientation, featuring an O–O bond distance of 1.39 Å (Fig. 9iii-I) for the Fe/N–C–F catalyst and 1.37 Å (Fig. 9iii-II) for the Co/N–C–F catalyst, which are also tabulated in Table S4.† Further hydrogenation of absorbed O2 leads to the end-on configuration of the O2 molecule where one of the oxygen atoms adheres to the Fe/Co atom, while the other is positioned on hollow site of hexagonal (MCN4) or pentagonal (MN4) ring. This leads to a longer O–O bond length for O2 absorbed on the metal atom, indicating their easier breakage and leading to the formation of the *OOH intermediate. The *OOH intermediate on Fe/Co sites further stretches the O–O bond length to 1.45 Å (Fig. 9iii-I) for the Fe/N–C–F catalyst and 1.43 Å (Fig. 9iii-II) for the Co/N–C–F catalyst, giving rise to the dissociation of the O–O bond in consequential steps. Moreover, the additional hydrogenation of *OOH could lead to the creation of the H2O2 intermediate, or alternatively, the adsorbed *OOH species may undergo further reduction, yielding 2H2O. Importantly, the pivotal chemical event in the 4e− ORR pathway is the cleavage of the O–O bond within *OOH at the Fe/Co site. Finally, two more consequent hydrogenation steps of O* leading to the formation of *OH and the final product H2O are demonstrated in the catalytic cycle (Fig. 9ii). The structural insights into the formed ORR intermediate sites of Fe/Co corroborate the calculated bond distance between Fe–O or Co–O and OH of both as calculated and tabulated in Table S4.†Fig. 9i also provides the energy profile diagram for the proposed reaction mechanism on Fe/Co sites. The free energy associated with each reaction step is determined through DFT calculations, considering the total energy with Zero Point Energy (ZPE) and entropy adjustments. Open circuit conditions clarify that the calculated free energy of the proposed reaction of the ORR steps is exothermic. The data stated above elucidate that 4e− reduction of O2 is energetically favourable in the ORR. The most stable configuration for each absorbed species, which usually has various adsorption configurations and different adsorption sites, is considered. At an applied voltage of U = 0 V, the Fe/N–C–F and Co/N–C–F catalysts both exhibit energetically favorable pathways, indicating that each electron transfer event during the ORR process occurs spontaneously from a thermodynamic perspective.19 The relatively smaller magnitude of the negative free energy change (ΔG) observed on the Fe/N–C–F catalyst during the initial reaction step (O2 + H2O + e− → OOH + OH*) signifies a higher propensity for O2 activation on Fe/N–C–F compared to Co/N–C–F. When the applied voltage is U = 1.23 V, the computed overpotential for the ORR on Fe/N–C–F (0.49 eV) remains lower than that of the Co/N–C–F species (0.68 eV). This further implies a superior ORR performance of the Fe/N–C–F catalyst compared to Co/N–C–F. These computational findings provide a clear rationale for the activity disparity observed in M-based catalysts prepared through different methods, which is influenced not only by the exposure of M/N–C–F (M = Fe/Co) species but also by variations in M coordination.
According to our calculations, including hydrogenation steps, the *OH state is determined to be the least energetically favorable. Subsequently, we analyzed the subsequent steps of the ORR, specifically focusing on the addition of an additional H atom per step. During the second stage of the ORR, two distinct pathways were observed: a 2e− transfer pathway and a 4e− transfer pathway. Our analysis, as presented in Table 3, clearly demonstrates that the Fe/N–C–F catalyst is more energetically favorable in an alkaline medium compared to the Co/N–C–F catalyst. These findings are in good agreement with experimental data and provide valuable insights into the ORR mechanisms on Fe/N–C–F and Co/N–C–F catalysts in an alkaline medium. Recent experimental studies delved into the evolution of both the structural and performance characteristics of atomically dispersed FeN4 sites for oxygen reduction under thermal conditions. The findings revealed that Fe–N–C catalysts exhibited decent ORR performance via a 4e− pathway. The authors also attributed catalytic activity to the synergistic effects of metallic Fe and quaternary N. These experimental observations support our theoretical results on the ORR using Fe/N–C–F or Co/N–C–F catalysts, highlighting the effectiveness of the creation of ordered linkages, a strategy to enhance ORR activity. However, it is important to note that there are limited reports on heteroatom-doped M/N/F/C catalysts, and to the best of our knowledge, no experimental investigations have been conducted on the ORR using Fe/N–C–F or Co/N–C–F catalysts in an alkaline medium. Nonetheless, our DFT results provide valuable insights and can guide experimental researchers in exploring cost-effective and highly efficient Fe/N–C–F or Co/N–C–F catalysts in an alkaline medium as potential replacements for Pt catalysts.
| Steps | Energy barrier (eV) | |
|---|---|---|
| Fe | Co | |
| i → ii | 0.49 (uphill) | 0.68 (uphill) |
| ii → iii | 1.17 (downhill) | 1.14 (downhill) |
| iii → iv | 0.20 (uphill) | 0.16 (uphill) |
| iv → v | 0.39 (uphill) | 0.40 (uphill) |
5 Conclusions
In this work, a unique synthesis method of Fe/N–C–F and Co/N–C–F catalysts by an unconventional Schiff-base synthesis strategy, using fluorine-rich pentafluorophenylhydrazine and salicylaldehyde organic ligands, is presented. The successful synthesis of the Schiff-base ligand has been established by 1H NMR spectroscopy. The metal coordinated SL with Fe/Co leads to the formation of in situ M–Nx–C type active sites. The Fe/N–C–F and Co/N–C–F catalysts have been characterized by various physicochemical techniques. Especially the Fe/N–C–F catalyst shows a rod type morphology and exhibits exceptional electrocatalytic performance for the ORR. In addition, the optimized Fe/N–C–F catalyst shows a half-wave potential of 0.88 V vs. RHE and superior durability evaluated up to 20000 cycles with only a marginal potential drop of ∼27 mV in its E1/2 potential value compared to the Pt/C catalyst. Furthermore, the reaction pathway and Gibbs free energy of the ORR intermediates in Fe/N–C–F and Co/N–C–F catalysts have been evaluated by DFT analysis. The rate-determining step for the ORR on the Fe/N–C–F catalyst has been predicted to involve the conversion of adsorbed OH* species into OH− ions. Overall, our study provides valuable insights into the design and development of efficient catalysts for polymer electrolyte fuel cells, bridging the gap between theoretical investigations and experimental evaluations. The successful synthesis and characterization of Fe/N–C–F catalysts pave the way for further advancements in electrocatalysis and hold great promise for the realization of cost-effective and efficient fuel cells in the future.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.Author contributions
S. K. D.: conceptualization, methodology, data curation, and writing – original draft. S. G. P.: data curation and writing – original draft. A. K.: formal analysis and writing – original draft. P. V.: writing – review & editing. A. K. S.: supervision and review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
CSIR, New Delhi, India, is gratefully acknowledged for the financial support. We thank Dr K. Ramesha, Director, CSIR-CECRI, for his constant encouragement and support. S. D. thanks CSIR for awarding the Junior Research Fellowship (31/0068(0188)/2019-EMR-I) to pursue research at CSIR-CECRI. This work was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (Grant No. 2021R1F1A1046648), Republic of Korea.References
- J. Tao, X. Wang, M. Xu, C. Liu, J. Ge and W. Xing, Ind. Chem. Mater., 2023, 1, 388–409 RSC.
- A. Pedersen, J. Pandya, G. Leonzio, A. Serov, A. Bernardi, I. E. L. Stephens, M. M. Titirici, C. Petit and B. Chachuat, Green Chem., 2023, 25, 10458–10471 RSC.
- G. Xu, L. Yang, J. Li, C. Liu, W. Xing and J. Zhu, Advanced Sensor and Energy Materials, 2023, 2, 100058 CrossRef.
- X. Zhao and K. Sasaki, Acc. Chem. Res., 2022, 55, 1226–1236 CrossRef CAS PubMed.
- M. Janssen, P. Weber and M. Oezaslan, Curr. Opin. Electrochem., 2023, 40, 101337 CrossRef CAS.
- Y. Zeng, J. Liang, C. Li, Z. Qiao, B. Li, S. Hwang, N. N. Kariuki, C. W. Chang, M. Wang, M. Lyons, S. Lee, Z. Feng, G. Wang, J. Xie, D. A. Cullen, D. J. Myers and G. Wu, J. Am. Chem. Soc., 2023, 145, 17643–17655 CrossRef CAS.
- J. Zhang, L. Wang, A. Zhong, G. Huang, F. Wu, D. Li, M. Teng, J. Wang and D. Han, Dyes Pigm., 2019, 162, 590–598 CrossRef CAS.
- Y. Chen, S. Zhang, J. Chung-Yen Jung and J. Zhang, Prog. Energy Combust. Sci., 2023, 98, 101101 CrossRef.
- A. Sarapuu, J. Lilloja, S. Akula, J. H. Zagal, S. Specchia and K. Tammeveski, ChemCatChem, 2023, 15(22), e202300849 CrossRef CAS.
- X. Chen, D. D. Ma, B. Chen, K. Zhang, R. Zou, X. T. Wu and Q. L. Zhu, Appl. Catal., B, 2020, 267, 118720 CrossRef CAS.
- P. Varathan, P. Moni, S. Kumar Das and A. K. Sahu, J. Electrochem. Soc., 2023, 170, 050536 CrossRef.
- P. Mukherjee, I. Patil, B. Kakade, S. Kumar Das, A. K. Sahu and A. Swami, Catal. Today, 2022, 1–11 Search PubMed.
- P. Varathan, S. K. Das and A. K. Sahu, J. Energy Storage, 2024, 90, 111845 CrossRef.
- W. Zhang, J. K. Pan, Y. F. Yu, X. J. Zhang, J. H. Wang, W. X. Chen and G. L. Zhuang, Phys. Chem. Chem. Phys., 2023, 25, 11673–11683 RSC.
- L. Xie, W. Zhou, Y. Huang, Z. Qu, L. Li, C. Yang, Y. Ding, J. Li, X. Meng, F. Sun, J. Gao, G. Zhao and Y. Qin, Mater. Horiz., 2024, 11, 1719–1731 RSC.
- S. G. Peera and C. Liu, Coord. Chem. Rev., 2022, 463, 214554 CrossRef CAS.
- H. Chang, Y. F. Guo, X. Liu, P. F. Wang, Y. Xie and T. F. Yi, Appl. Catal., B, 2023, 327, 122469 CrossRef CAS.
- Q. Miao, Z. Chen, X. Li, M. Liu, G. Liu, X. Yang, Z. Guo, C. Yu, Q. Xu and G. Zeng, ACS Catal., 2023, 11127–11135 CrossRef CAS.
- J. Zhang, Z. Mei, L. Yi, J. Tian, K. Li, X. Hu, Y. Zhang, R. Wang, H. Guo and S. Zang, Appl. Catal., B, 2023, 338, 123044 CrossRef CAS.
- H. Jin, R. Yu, P. Ji, W. Zeng, Z. Li, D. He and S. Mu, Chem. Sci., 2024, 15, 7259–7268 RSC.
- S. Gouse Peera, H. J. Kwon, T. G. Lee and A. M. Hussain, Ionics, 2020, 26, 1563–1589 CrossRef CAS.
- C. Tang and Q. Zhang, Adv. Mater., 2017, 29(13), 1604103 CrossRef PubMed.
- G. Escobar, R. Venegas, I. Ponce, A. Toro-Labbé, J. H. Zagal, F. J. Recio and K. Muñoz-Becerra, Electrochim. Acta, 2023, 466, 143060 CrossRef CAS.
- L. Feng, L. Yang, Z. Huang, J. Luo, M. Li, D. Wang and Y. Chen, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- P. Zhu, J. Gao, X. Chen and S. Liu, Int. J. Hydrogen Energy, 2020, 45, 9512–9521 CrossRef CAS.
- Y. Chang, J. Chen, J. Jia, X. Hu, H. Yang, M. Jia and Z. Wen, Appl. Catal., B, 2021, 284, 119721 CrossRef CAS.
- G. Wang, S. Jia, H. Gao, Y. Shui, J. Fan, Y. Zhao, L. Li, W. Kang, N. Deng and B. Cheng, J. Energy Chem., 2023, 76, 377–397 CrossRef CAS.
- F. Forouzandeh, X. Li, D. W. Banham, F. Feng, A. Joseph Kakanat, S. Ye and V. Birss, J. Power Sources, 2018, 378, 732–741 CrossRef CAS.
- S. Jin, S. Y. Yang, J. M. Lee, M. S. Kang, S. M. Choi, W. Ahn, X. Fuku, R. M. Modibedi, B. Han and M. H. Seo, ACS Appl. Mater. Interfaces, 2021, 13, 26936–26947 CrossRef CAS.
- Y. Diao, H. Liu, Z. Yao, Y. Liu, G. Hu, Q. Zhang and Z. Li, Nanoscale, 2020, 12, 18826–18833 RSC.
- X. Tao, R. Lu, L. Ni, V. Gridin, S. H. Al-Hilfi, Z. Qiu, Y. Zhao, U. I. Kramm, Y. Zhou and K. Müllen, Mater. Horiz., 2022, 9, 417–424 RSC.
- L. Osmieri, ChemEngineering, 2019, 3, 1–32 CrossRef.
- S. G. Peera, R. S. Menon, S. K. Das, A. Alfantazi, K. Karuppasamy, C. Liu and A. K. Sahu, Coord. Chem. Rev., 2024, 500, 215491 CrossRef CAS.
- H. Huang, Z. Jiang, X. Tian, Y. Zheng and Z. J. Jiang, Int. J. Hydrogen Energy, 2023, 48, 3385–3401 CrossRef CAS.
- D. Wang, X. Pan, P. Yang, R. Li, H. Xu, Y. Li, F. Meng, J. Zhang and M. An, ChemSusChem, 2021, 14, 33–55 CrossRef CAS PubMed.
- S. Wu, X. Qu, J. Zhu, X. Liu, H. Mao, K. Wang, G. Zhou, J. Chi and L. Wang, J. Alloys Compd., 2024, 970, 172518 CrossRef CAS.
- Y. Qian, F. Zhang, S. Zhao, C. Bian, H. Mao, D. J. Kang and H. Pang, Nano Energy, 2023, 111, 108415 CrossRef CAS.
- A. Ghosh, K. Mukherjee, S. K. Ghosh and B. Saha, Res. Chem. Intermed., 2013, 39, 2881–2915 CrossRef CAS.
- M. d. l. A. Garavagno, R. Holland, M. A. H. Khan, A. J. Orr-Ewing and D. E. Shallcross, Sustainability, 2024, 16(6), 2382 CrossRef CAS.
- S. G. Peera, R. S. Menon, S. K. Das, A. Alfantazi, K. Karuppasamy, C. Liu and A. K. Sahu, Coord. Chem. Rev., 2024, 500, 215491 CrossRef CAS.
- Z. Chen, F. Jing, M. Luo, X. Wu, H. Fu, S. Xiao, B. Yu, D. Chen, X. Xiong and Y. Jin, Carbon Energy, 2024, 6, 1–12 Search PubMed.
- P. Luo, Z. Huang, T. Wang, H. Xiao, X. Ma, R. Yan and G. Zhao, Solid State Sci., 2024, 156, 107678 CrossRef CAS.
- X. Ding, Y. Fan, Z. Zhang, D. Chen and S. Yao, Int. Commun. Heat Mass Transfer, 2024, 159, 107942 CrossRef CAS.
- B. Musikavanhu, Y. Liang, Z. Xue, L. Feng and L. Zhao, Molecules, 2023, 28(19), 6960 CrossRef CAS.
- J. Zhang, A. Zhong, G. Huang, M. Yang, D. Li, M. Teng and D. Han, Sol. Energy, 2020, 209, 316–324 CrossRef CAS.
- Z. Ma, Y. Chu, C. Fu, H. Du, X. Huang and J. Zhao, Catalysts, 2018, 8(4), 156 CrossRef.
- C. Boulechfar, H. Ferkous, A. Delimi, A. Djedouani, A. Kahlouche, A. Boublia, A. S. Darwish, T. Lemaoui, R. Verma and Y. Benguerba, Inorg. Chem. Commun., 2023, 150, 110451 CrossRef CAS.
- S. Nagar, S. Raizada and N. Tripathee, Results Chem., 2023, 6, 101153 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- P. Xu, W. Chen, Q. Wang, T. Zhu, M. Wu, J. Qiao, Z. Chen and J. Zhang, RSC Adv., 2015, 5, 6195–6206 RSC.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 RSC.
- S. G. Peera, A. Arunchander and A. K. Sahu, Nanoscale, 2016, 8, 14650–14664 RSC.
- S. Gupta and N. Dimakis, J. Appl. Phys., 2021, 130, 084902 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- M. Kim, J. N. Park, H. Kim, S. Song and W. H. Lee, J. Power Sources, 2006, 163, 93–97 CrossRef CAS.
- J. Zhang, J. Zhang, F. He, Y. Chen, J. Zhu, D. Wang, S. Mu and H. Y. Yang, Nano-Micro Lett., 2021, 13, 1–30 CrossRef CAS PubMed.
- J. Liu, P. Song, M. Ruan and W. Xu, Chin. J. Catal., 2016, 37, 1119–1126 CrossRef CAS.
- W. Feng, P. Long, Y. Feng and Y. Li, Adv. Sci., 2016, 3, 1500413 CrossRef PubMed.
- Y. Lee and J. Fluor, Chem, 2007, 128, 392–403 CAS.
- J. Kim, R. Zhou, K. Murakoshi and S. Yasuda, RSC Adv., 2018, 8, 14152–14156 RSC.
- S. G. Peera, R. Koutavarapu, S. Akula, A. Asokan, P. Moni, M. Selvaraj, J. Balamurugan, S. O. Kim, C. Liu and A. K. Sahu, Energy Fuels, 2021, 35, 11761–11799 CrossRef CAS.
- Y. Li, J. Li, Y. G. Wang, X. Chen, M. Liu, Z. Zheng and X. Peng, Int. J. Hydrogen Energy, 2021, 46, 13273–13282 CrossRef CAS.
- W. Yoshimune, N. Kikkawa, H. Yoneyama, N. Takahashi, S. Minami, Y. Akimoto, T. Mitsuoka, H. Kawaura, M. Harada, N. L. Yamada and H. Aoki, ACS Appl. Mater. Interfaces, 2022, 14, 53744–53754 CrossRef CAS PubMed.
- S. Akula, M. Mooste, B. Zulevi, S. McKinney, A. Kikas, H. M. Piirsoo, M. Rähn, A. Tamm, V. Kisand, A. Serov, E. B. Creel, D. A. Cullen, K. C. Neyerlin, H. Wang, M. Odgaard, T. Reshetenko and K. Tammeveski, J. Power Sources, 2022, 577, 230819 CrossRef.
- G. Zhang, M. Colin, X. Yang, S. Sun, J. P. Dodelet and M. Dubois, Appl. Surf. Sci., 2022, 577, 151721 CrossRef CAS.
- Y. N. Sun, J. Yang, X. Ding, W. Ji, A. Jaworski, N. Hedin and B. H. Han, Appl. Catal., B, 2021, 297, 120448 CrossRef CAS.
- S. K. Das, A. Kesh, S. Akula, S. K. Panda, G. V. M. Kiruthika and A. K. Sahu, ACS Appl. Energy Mater., 2022, 5, 10240–10253 CrossRef CAS.
- S. K. Das, A. Kesh, S. Akula and A. K. Sahu, Energy Fuels, 2022, 36, 2108–2122 CrossRef CAS.
- S. K. Das, A. Kesh, D. Sujatha, S. K. Panda and A. K. Sahu, ACS Appl. Eng. Mater., 2024, 2, 1894–1907 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01370k |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |