
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of asymmetric anhydrous quaternary ammonium fluoride electrolytes for fluoride ion batteries†
Tiancheng
Tan
a,
Richard
Murdey
 a,
Shunsuke
Sumitomo
b and
Atsushi
Wakamiya
*a
a,
Shunsuke
Sumitomo
b and
Atsushi
Wakamiya
*a
aInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: wakamiya@scl.kyoto-u.ac.jp
bOffice of Society-Academia Collaboration for Innovation, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
First published on 30th January 2025
Abstract
Fluoride ion batteries using quaternary ammonium fluoride-based liquid electrolytes have recently demonstrated promising performance. While liquid electrolytes are desirable for their high ionic conductivity at room temperature, finding solvent-fluoride salt systems with both high solubility and stability has been challenging. In this study, we synthesize novel asymmetric anhydrous quaternary ammonium fluoride salts with varying structural bulkiness to reveal how structural variations influence chemical properties such as solubility, ionic conductivity, and stability. The new fluoride salt electrolytes were successfully employed in a half-cell with BiF3 electrodes, achieving 80% of the 302 mA h g−1 theoretical capacity. We find that increasing the size of the cation led to higher conductivities for fluoride salts in bis(2,2,2-trifluoroethyl) ether solution. However, we also observe a loss of thermostability with increasing cation size. Optimizing this trade-off between ionic conductivity and thermostability will likely be critical for the future development of quaternary ammonium fluoride electrolytes.
Introduction
Due to their high theoretical capacities of more than 800 mA h g−1, fluoride ion batteries (FIBs) are promising alternatives to lithium-ion technologies.1–8 Their development can be traced back to early studies on ionic transport in solid materials. Researchers found that fluoride anions can move freely in fluoride salts, including PbF2, CaF2, LaF3, La1−xBaxF3−x, or BaSnF4 when heated, laying the foundation for FIB research. More recently, reversible all-solid-state FIBs were developed based on metal fluoride electrodes (CuF2, BiF3, SnF2, and KBiF4) and solid electrolytes such as La0.9Ba0.1F2.9, delivering a charge/discharge capacity of over 100 mA h g−1. In FIBs, the following electrochemical reactions occur during discharge:Anode:
| M′ + xF− → M′Fx + xe− |
Cathode:
| MFx + xe− → M + xF− |
Overall:
| MFx + M′ → M′Fx + M |
These all-solid-state FIBs require elevated temperatures to work efficiently due to the low ionic conductivities of the solid electrolytes at room temperature.9–16 One attractive option for reducing the operation temperatures is to replace the solid-state electrolyte with a liquid electrolyte, which generally retains high ionic conductivity at room temperature. However, solvent-fluoride salt systems that combine high solubility and stability have proven elusive.17–26 For example, although liquid electrolytes such as ammonium bifluoride and metal bifluoride can be readily prepared in high concentrations, their electrochemical stability falls below the threshold for good battery performance.17–21 To overcome this problem, researchers have focused on metal fluorides (CsF, KF, etc.), enhancing their poor solubility in aprotic solvents through the use of solvating additives such as fluorobis(2,4,6-trimethylphenyl)borane.1,27,28 While this method increases the solubility of fluoride salts in organic solvents, the formation energies of the B–F bond in the tetraborate species remained too high for optimal cell performance.1,27,28 One alternative, unprotonated fluoride electrolytes based on quaternary ammonium fluoride salts, has recently shown promise. V. K. Davis et al. reported two such quaternary ammonium fluoride salts, N,N,N-trimethyl-N-neopentylammonium fluoride (Np1F) and N,N-dimethyl-N,N-dineopentylammonium fluoride (Np2F), which showed good solubility (>2 M) and high ionic conductivity (>2 mS cm−1).22 Using Np1F/bis(2,2,2-trifluoroethyl) ether (BTFE) as the liquid electrolyte, the authors demonstrated rechargeable FIBs operating at room temperature.22
In our previous work, we developed a high-yield, rapid, and safe synthetic route for anhydrous Np2F through anion exchange.29 This new approach greatly simplifies the development of fluoride salts for use in FIBs. The resulting products have extremely low water content, enabling a wide voltage window of 3.5 V for Np2F/BTFE (0.7–4.2 V vs. Li+/Li). The performance demonstrates the inherent potential of Np2F, unaffected by HF2− species that could be introduced by water contamination.29 Our previous research confirmed that quaternary ammonium fluorides are well-placed to emerge as the electrolyte of choice for FIB applications. Despite the progress made in the field, there is a strong demand for electrolyte salts with superior characteristics, including improved solubility in organic solvents, higher ionic conductivity, and enhanced thermodynamic and electrochemical stability.
To make a wider selection of materials accessible for research, in the present work we develop three asymmetric quaternary ammonium fluorides with different alkyl substituents. The design strategy of the ammonium cations is illustrated in Fig. 1. Since fluoride is a strong base, quaternary ammonium fluorides like tetrabutylammonium fluoride (TBAF) are known for self-decomposition through Hofmann elimination.30 Therefore, removing the reactive β protons on the quaternary ammonium cation is an essential part of the molecular design. The reference compound Np2F features a symmetric cation with two tert-butyl groups on the tetramethyl ammonium backbone and is a benchmark for comparison. Compound N,N,2,2-tetramethyl-N-neopentylbutan-1-aminium fluoride (NpDMBF) replaces one tert-butyl group with a 1,1-dimethyl propyl group on Np2F, resulting in an asymmetrical cation with increased size. Compound N-((adamantan-1-yl)methyl)-N,N,2,2-tetramethylpropan-1-aminium fluoride (NpADMF) incorporates a bulky, rigid adamantyl group, which increases the overall size even further. Meanwhile, compound N,N,N,2,2-pentamethylbutan-1-aminium fluoride (MeDMBF) substitutes a hydrogen atom for the tert-butyl group on NpDMBF, resulting in an asymmetrical cation that is significantly smaller than both Np2F and NpDMBF. Theoretical calculations estimate the volumes of the MeDMB+, Np2+, NpDMB+, and NpADM+ cations to be 172, 233, 246, and 263 Å3, respectively. With these four fluoride salts, we can study how asymmetric substituents influence solubility, conductivity, and chemical stability. The fundamental properties of these three new asymmetrical quaternary ammonium fluorides were characterized together with the reference compound Np2F. Additionally, the charge/discharge performance of MeDMBF/BTFE, Np2F/BTFE, and NpDMBF/BTFE electrolytes was evaluated in half-cell devices with BiF3 electrodes.
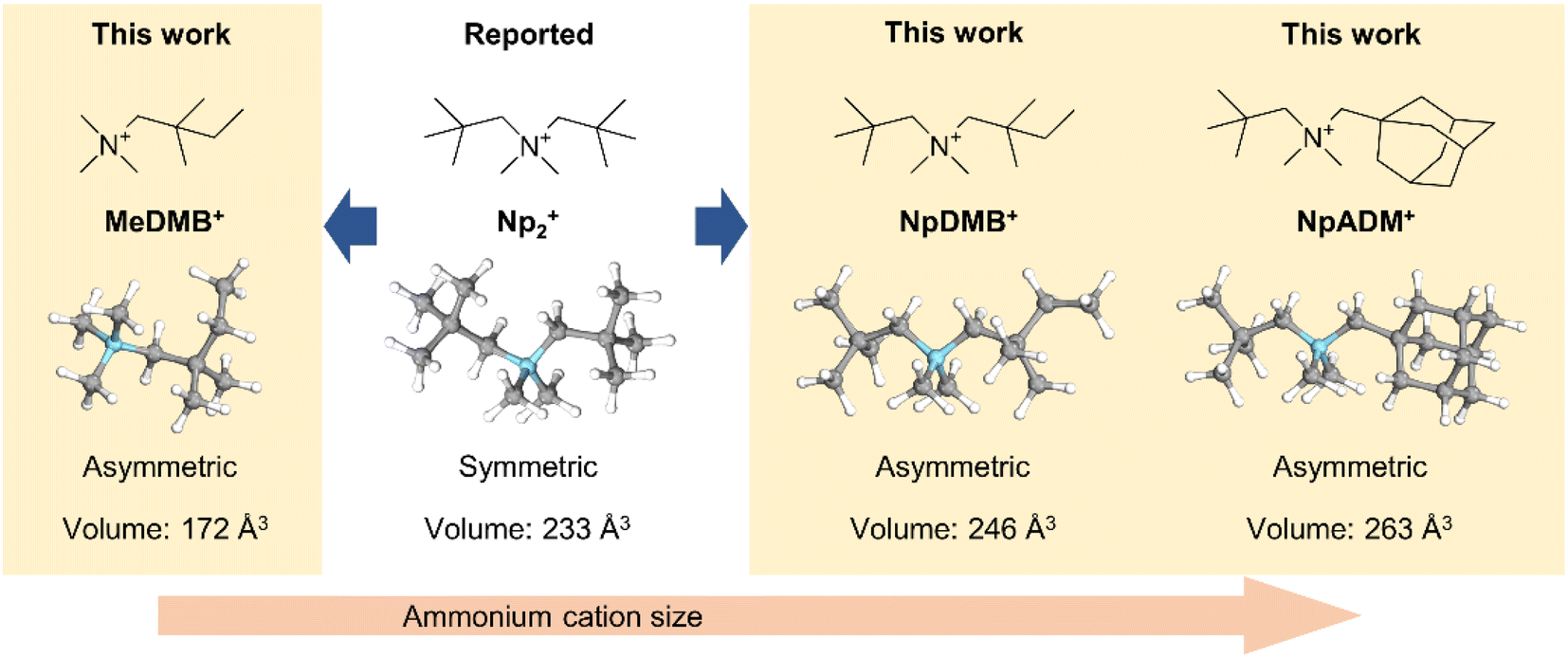 | ||
| Fig. 1 Molecule design of the quaternary ammonium cations studied in the present work for fluoride salts. | ||
Results and discussion
Synthesis
The synthesis of anhydrous quaternary ammonium fluorides, MeDMBF, NpDMBF, and NpADMF is shown in Scheme 1. As starting materials, tertiary amines were synthesized from commercially available compounds (Schemes S1, S3 and S5†). We employed a simple and safe anion exchange reaction with KF in methanol using the corresponding ammonium tetrafluoroborates as key intermediate compounds to obtain the anhydrous fluoride salts. Ammonium tetrafluoroborates, MeDMBBF4, NpDMBBF4, and NpADMBF4, were synthesized from the corresponding tertiary amines by oxonium borate methylation with Meerwein's salt (trimethyloxonium tetrafluoroborate) in dichloromethane. With these three borate salts in hand, anion exchange between the borate salts and KF in dry methanol produced anhydrous fluoride salts. After removing the KBF4 by filtration, the resulting solutions were further dried under vacuum at 90 °C to give anhydrous MeDMBF, NpDMBF, and NpADMF solid in 76%, 43%, and 44% yield, respectively.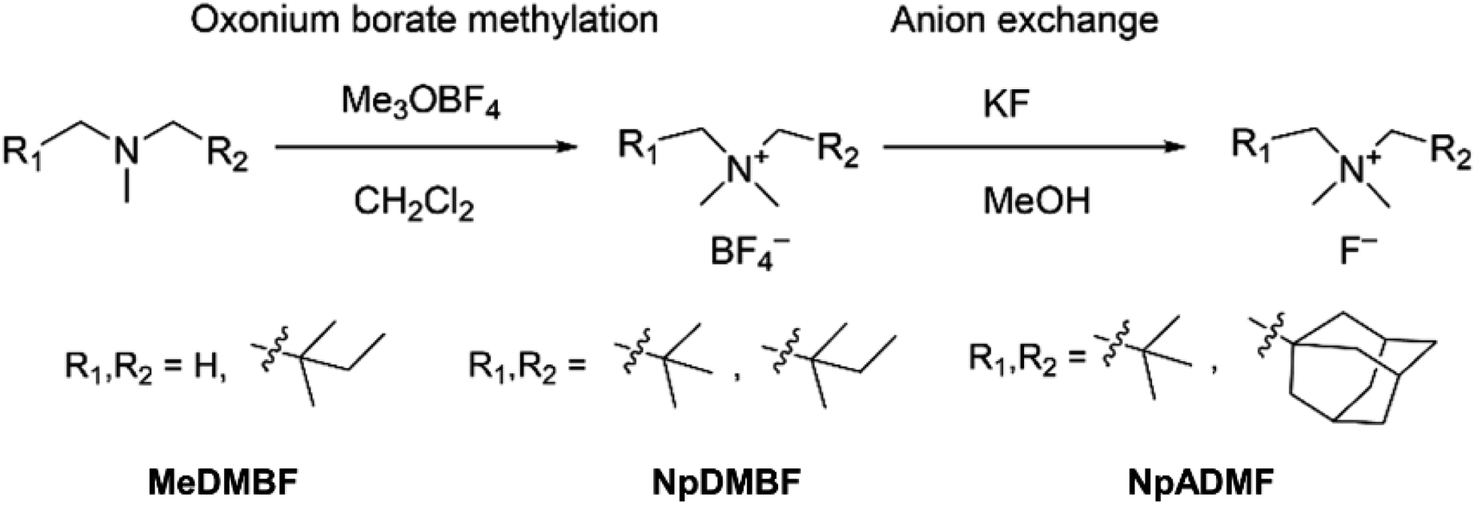 | ||
| Scheme 1 | ||
Detailed synthesis procedures are provided in the ESI (Schemes S2, S4 and S6†). The purity of the products was confirmed by 1H, 13C, and 19F NMR in deuterated organic solvents, and no HF2− species was observed. The water content of MeDMBF and NpDMBF was 15 and 16 ppm in 0.1 M BTFE, and 34 ppm for NpADMF in 0.1 M dichloromethane (DCM), respectively. The fluoride salts are highly hygroscopic. The solubility of MeDMBF, NpDMBF, and NpADMF in water exceeds 24 M, 28 M, and 5 M, respectively.
Properties and characterization
The solubility of MeDMBF, Np2F,29 NpDMBF, and NpADMF in 17 different solvents is summarized in Table S1,† with the residual water content of the dry solvents provided in Table S2.† MeDMBF, Np2F, and NpDMBF exhibit high solubility (>1 M) in bis(2,2,2-trifluoroethyl) ether (BTFE), dimethylsulfoxide (DMSO), acetonitrile (MeCN), γ-butyrolactone (GBL), and dichloromethane (DCM). Np2F and NpDMBF are also highly soluble (>1 M) in propionitrile (PN). MeDMBF, Np2F, and NpDMBF show poor solubility (<0.3 M) in N,N-dimethylformamide (DMF), and other hydrocarbon ethers like 1,2-dimethoxyethane (DME). MeDMBF also has low solubility (<0.3 M) in PN. NpDMBF is more soluble in DMF compared to MeDMBF or Np2F, likely due to the increased flexibility of its methyl-substituted alkyl chain. In contrast, NpADMF has very low solubility (<0.1 M) in all the aprotic solvents tested, a result we ascribe to the large, rigid structure of the adamantyl substituent.Since anhydrous quaternary ammonium fluorides are strongly basic due to the small ionic radius of F− and its high electronegativity of 3.98, the deprotonation of the solvent is a concern. Furthermore, quaternary ammonium fluorides can undergo halogen exchange reactions in alkyl halides.31 Therefore, evaluating the stability of MeDMBF, Np2F, NpDMBF, and NpADMF in different solvents is important. In DCM, the F− signal from 19F NMR was observed at around −100 ppm for all compounds, along with the signals for HF2−, CH2ClF, and CH2F2 at −150, −172, and −145 ppm, respectively. The presence of CH2ClF and CH2F2 suggests that halogen exchange has occurred (refer to ESI Fig. S9† for NMR charts and Fig. S14† for the single crystal structure of the chloride salt obtained from the NMR sample). For all the compounds in MeCN, DMSO, GBL, and PN, the F− signal was observed downfield (−73 to −80 ppm), while a signal from HF2− appeared upfield (−144 to −152 ppm), confirming that the F− anions could deprotonate these solvents. In PN and GBL, HF2− predominates, indicating a nearly complete conversion of F− to HF2−. In BTFE, quantitative analysis of the HF2− by 19F NMR is hindered by the strong signal from the fluorinated solvent. However, only trace amounts of HF2− were detected by 1H NMR (Fig. S10†). Therefore, while MeDMBF and NpDMBF may react with BTFE to some extent, their potential use for broader applications remains promising.
Thermogravimetric analysis (TGA), conducted in an argon-filled glove box, was employed to investigate the thermal stability of the ammonium fluoride salts. The mass loss for compounds MeDMBF, Np2F, NpDMBF, and NpADMF up to 70 °C was less than 1 wt%, confirming that the salts are stable at room temperature. The stability of MeDMBF was exceptional, with a mass loss of less than 1 wt% up to 108 °C compared to 2%, 3%, and 4% for Np2F, NpDMBF, and NpADMF, respectively (Fig. S5†). The trend in thermostability indicates that the reactivity of the fluorides becomes higher as the cation size increases. The long-term stability of these fluoride salts in BTFE was monitored by 1H NMR over 11 months. As shown in Fig. S6–S8,† the fluorides partially react with the quaternary cations to form tertiary amines and fluoromethane rather than deprotonate the BTFE solvent in all cases.
Theoretical calculations were performed using the ORCA package to gain further insight into the relative stability of the different compounds. The gas-phase geometries of the ground states of the initial salts, their decomposition products, and the corresponding probable transition states were fully optimized at the M06-2X/Def2-TZVPP level of theory (Fig. S13†). The overall decomposition reaction was exothermic for all the compounds, with a decomposition energy (ΔG) of −125, −144, −149, and −152 kJ mol−1 for MeDMBF, Np2F, NpDMBF, and NpADMF, respectively. The activation energy barrier (ΔG‡) was calculated to be 123, 112, 109, and 107 kJ mol−1 for MeDMBF, Np2F, NpDMBF, and NpADMF, respectively. Increasing the size of the ammonium cation reduces the activation energy, leading to an increase in the reactivity of the fluoride. These results are consistent with the experimental observations in TGA and NMR.
Due to the strong interaction between the fluoride anion and the ammonium cation, properties such as conductivity and diffusivity are significantly influenced by the solvent structure surrounding the fluoride anions. To gain further insight into this solvent structure, 19F and 1H heteronuclear Overhauser effect spectroscopy (HOESY) of MeDMBF and NpDMBF in 0.9 M BTFE (Fig. 2) was performed to characterize the interactions between the fluoride anion, cations, and solvents. Strong contacts were observed from the F− anions to the alpha protons (Ha and Hb) of MeDMB+ and the alpha proton (Hs) of BTFE. Strong contacts were also observed from the F− anions to the alpha protons (He, Hf, and Hg) of NpDMB+ and the alpha proton (Hs) of BTFE, indicating the presence of a contact ion pair or aggregate structures involving F−, ammonium cations, and BTFE.
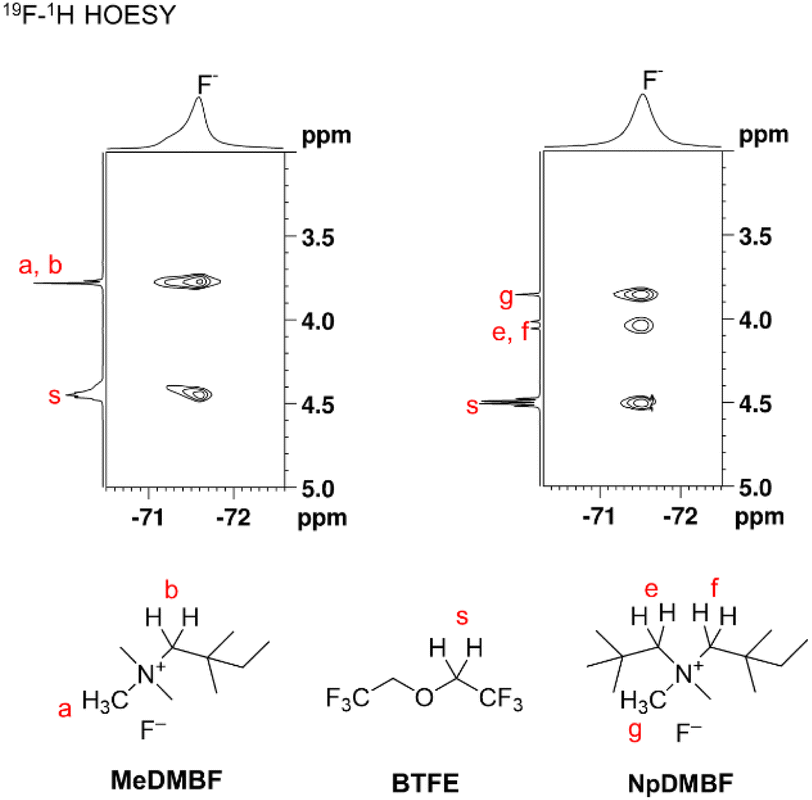 | ||
| Fig. 2 19F and 1H HOESY spectra of 0.9 M MeDMBF and NpDMBF in BTFE. | ||
To learn the details of how the F− anions interact with different sizes of cation, diffusion-ordered spectroscopy (DOSY) measurements were conducted on 0.9 M solutions of MeDMBF, Np2F, and NpDMBF BTFE respectively (Fig. S33–S37† DOSY charts). The diffusion coefficients, D, of the ions at room temperature were determined from the DOSY measurements, which are given in Fig. S3.† The ionic conductivities for MeDMBF, Np2F, and NpDMBF were determined from the diffusion coefficients using the Nernst–Einstein relation to be 14.9, 17.5, and 19.4 mS cm−1, respectively. This trend is consistent with the electrochemical measurements, where the ionic conductivities are 2.8, 3.0, and 4.0 mS cm−1, respectively – values comparable to those of the electrolytes used in lithium-ion cells.32–34 Regardless of the method used, the ionic conductivity of the electrolyte is observed to increase with the chemical size of the cation.
The hydrodynamic radii of the ions can be estimated from the diffusion coefficients using the Stokes–Einstein relation (rH = kBT/6πηD), as shown in Table 1. The hydrodynamic radius of the fluorides in MeDMBF/BTFE is 8.75 Å, whereas the radii of the fluorides of Np2F and NpDMBF are smaller, at 7.02 and 6.56 Å, respectively. Interestingly, increasing the chemical size of the cation (Fig. 1) effectively reduces the hydrodynamic radii of the corresponding fluoride anions. However, the hydrodynamic radius of F− in all three quaternary ammonium salts is much larger than the ionic radius of the naked F− anion (1.2 Å), confirming significant ion pair formation. Theoretical calculations were performed to estimate the binding energy between the anion and cation in the fluoride salts, which was determined to be in the order of NpDMBF < Np2F < MeDMBF. The smallest cation exhibits the highest binding energy. In contrast, the largest cation has the lowest binding energy, due to a reduced interaction between the ammonium cation and fluoride anion through hydrogen bonding. The lower binding energy (biggest cation) should lead to a smaller solvent structure around the fluoride anion and a smaller hydrodynamic radius.
| Salt | Solvent | D (10−10 m2 s−1) | r H (Å) | |
|---|---|---|---|---|
| a Results from ref. 29. | ||||
| MeDMBF | BTFE | MeDMB+ | 2.28 | 8.25 |
| F− | 2.15 | 8.75 | ||
| Np2F | BTFE | Np2+ | 2.52a | 7.46a |
| F− | 2.68a | 7.02a | ||
| NpDMBF | BTFE | NpDMB+ | 2.89 | 6.51 |
| F− | 2.87 | 6.56 | ||
| MeDMBF | CD3OD | MeDMB+ | 8.05 | 5.02 |
| F− | 6.91 | 5.84 | ||
| Np2F | CD3OD | Np2+ | 6.96 | 5.92 |
| F− | 6.89 | 5.86 | ||
| NpDMBF | CD3OD | NpDMB+ | 7.78 | 5.20 |
| F− | 6.97 | 5.80 | ||
Building on the observation of significant ion-pair formation of fluoride in BTFE solution, we further investigated the solvent structure of these fluoride salts in the protic organic solvent methanol, which typically does not promote ion pairing in solution. The diffusion coefficients of 0.9 M solutions of MeDMBF, Np2F, and NpDMBF in CD3OD were determined by DOSY measurement. As shown in Table 1, the corresponding hydrodynamic radii in CD3OD decreased only slightly compared with that in BTFE. At the same time, strong contacts of the F− anions to the ammonium cations were also observed by HOESY NMR (Fig. S4†). These results confirm the existence of strong interactions between the ammonium cations and the F− anions. We conclude that ion pairs still exist as a significant fraction in CD3OD even though methanol is a protic polar solvent that does not usually promote ion-pairing in solution.
The probable structures of the contact ion pairs of MeDMBF and NpDMBF in BTFE solutions are explored with molecular dynamics (MD) simulations. As depicted in Fig. 3a, MeDMBF has mainly a pentacoordinate contact ion pair structure around the F− anion with four BTFE solvent molecules and one MeDMB+ cation, whereas NpDMBF has a tetracoordinate contact ion pair structure with three BTFE solvent molecules due to the bulkiness of the NpDMB+ cation. The radial distribution functions (RDFs) are given in Fig. 3b and c. The radii of the coordination shells of F−–cations and BTFE–F− are 2.5 Å and 2.6 Å respectively. The small radii are consistent with the strong contacts observed by 19F and 1H HOESY NMR.
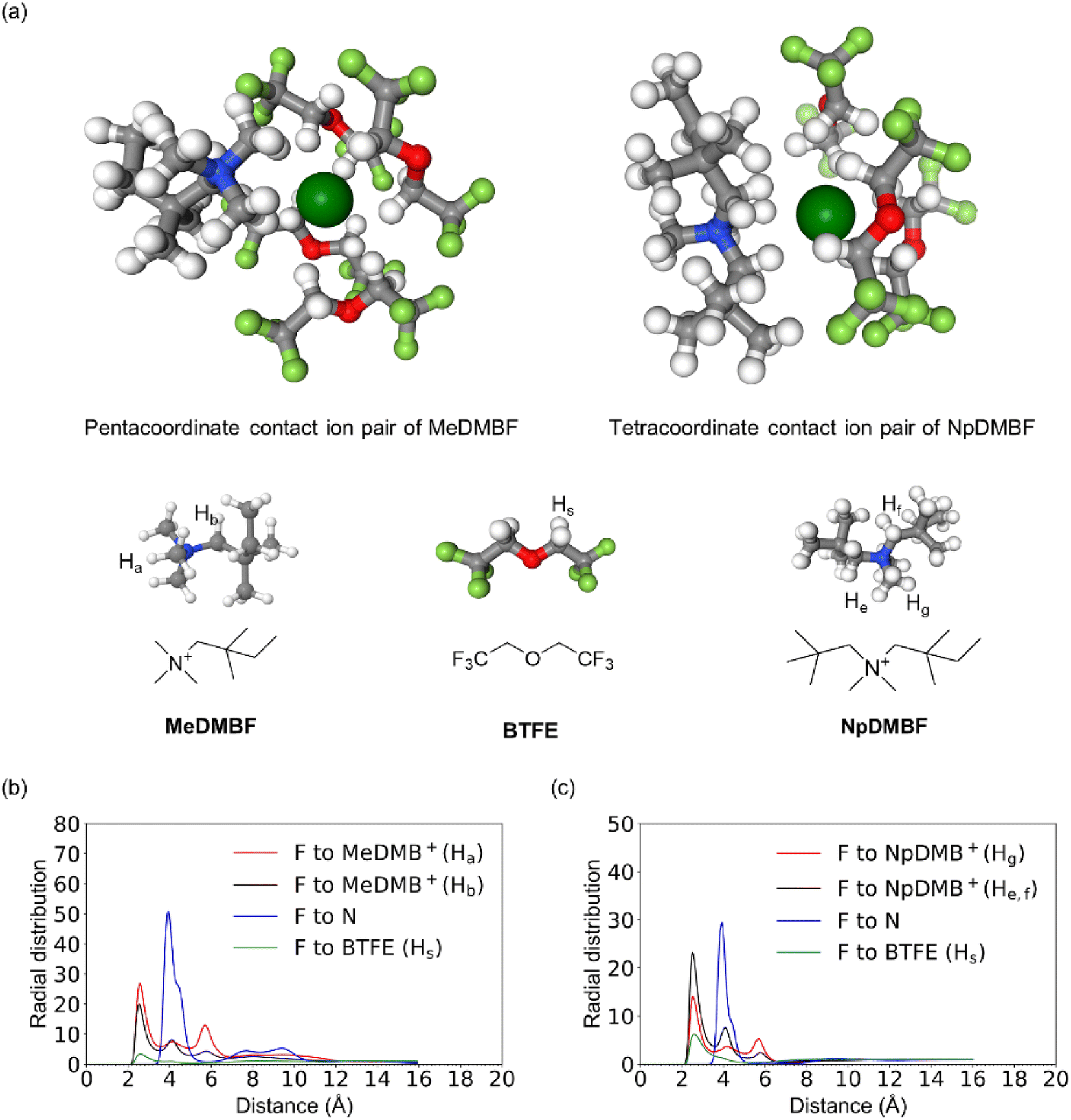 | ||
| Fig. 3 MD simulation results for a 0.9 M solution of MeDMBF and NpDMBF in BTFE at room temperature. (a) Snapshot view of the contact ion pair configuration, (b) radial distribution functions for the four chemically distinct hydrogens (Ha, Hb, Hs, and N) and the F− anion of MeDMB/BTFE, and (c) radial distribution functions for the four chemically distinct hydrogens (Ha, Hb, Hs, and N) and the F− anion of NpDMBF/BTFE. | ||
Electrochemical stability
Turning to the electrochemical stability, the voltage window of these anhydrous fluoride salts in GBL and BTFE was investigated by cyclic voltammetry, with the window defined based on an arbitrary current density cutoff, Jcutoff, of 100 μA cm−2. The voltage windows for MeDMBF and NpDMBF in BTFE were relatively wide, 3.3 V (0.9–4.2 V vs. Li+/Li) and 3.5 V (0.7–4.2 V vs. Li+/Li), respectively, comparable to that of the Np2F reference [3.5 V (0.7–4.2 V vs. Li+/Li)]. Next, we changed the solvent to GBL, which makes these fluoride salts highly soluble. However, the voltage window was reduced to 2.3 V (1.2–3.5 V vs. Li+/Li) for MeDMBF, 2.4 V for NpDMBF and 2.4 V for Np2F, due to the high concentration of HF2− species originating from the deprotonation of GBL by the F− anion (Fig. 4a, b and S1†). The oxidation peak observed in GBL probably originates from the oxidation of the deprotonated GBL anions. These results emphasize how a low HF2 concentration is essential for achieving a wide voltage window.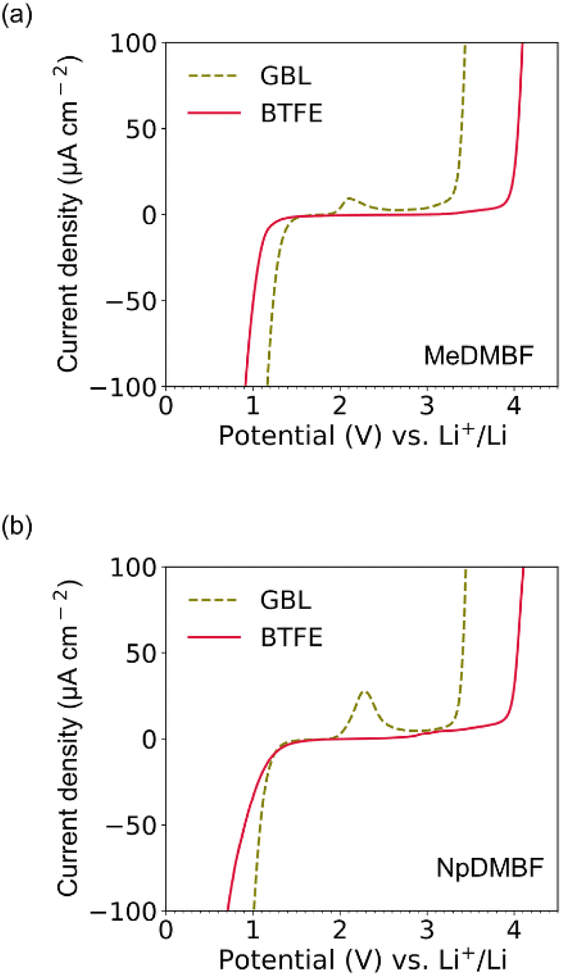 | ||
| Fig. 4 Cyclic voltammograms of (a) 0.1 M MeDMBF and (b) 0.1 M NpDMBF in GBL and BTFE. The scan rate was 1 mV s−1. | ||
Half-cell performance
A half-cell device with bismuth fluoride and carbon electrodes was fabricated to evaluate the charge/discharge performance in FIBs using 0.9 M Np2F/BTFE, MeDMBF/BTFE, and NpDMBF/BTFE electrolytes, respectively. (See the Experimental section for details.) The measurements were performed at 298 K. The double junction reference electrode was constructed from a silver (Ag) wire immersed in a mixture of 1-methyl-1-propylpyrrolidinium bis(fluorosulfonyl)imide (MPPy-FSI) and 0.1 M silver trifluoromethanesulfonate (AgOTf). The electrode was placed in a small compartment filled with MPPy-FSI and immersed in electrolyte. As a typical example, the cyclic voltammograms for this half-cell using 0.9 M Np2F/BTFE electrolyte are shown in Fig. 5a. In the first cycle, starting from −2.3 V (vs. ref.), the current rapidly increases during the cathodic sweep, corresponding to the reduction (defluorination) reaction of BiF3 to metallic bismuth. An oxidation peak is observed in the following scan toward a positive potential at −1.8 V, corresponding to the oxidation (fluorination) of metallic bismuth. Subsequent cyclic voltammograms overlap to a significant extent, confirming stable defluorination/fluorination reactions.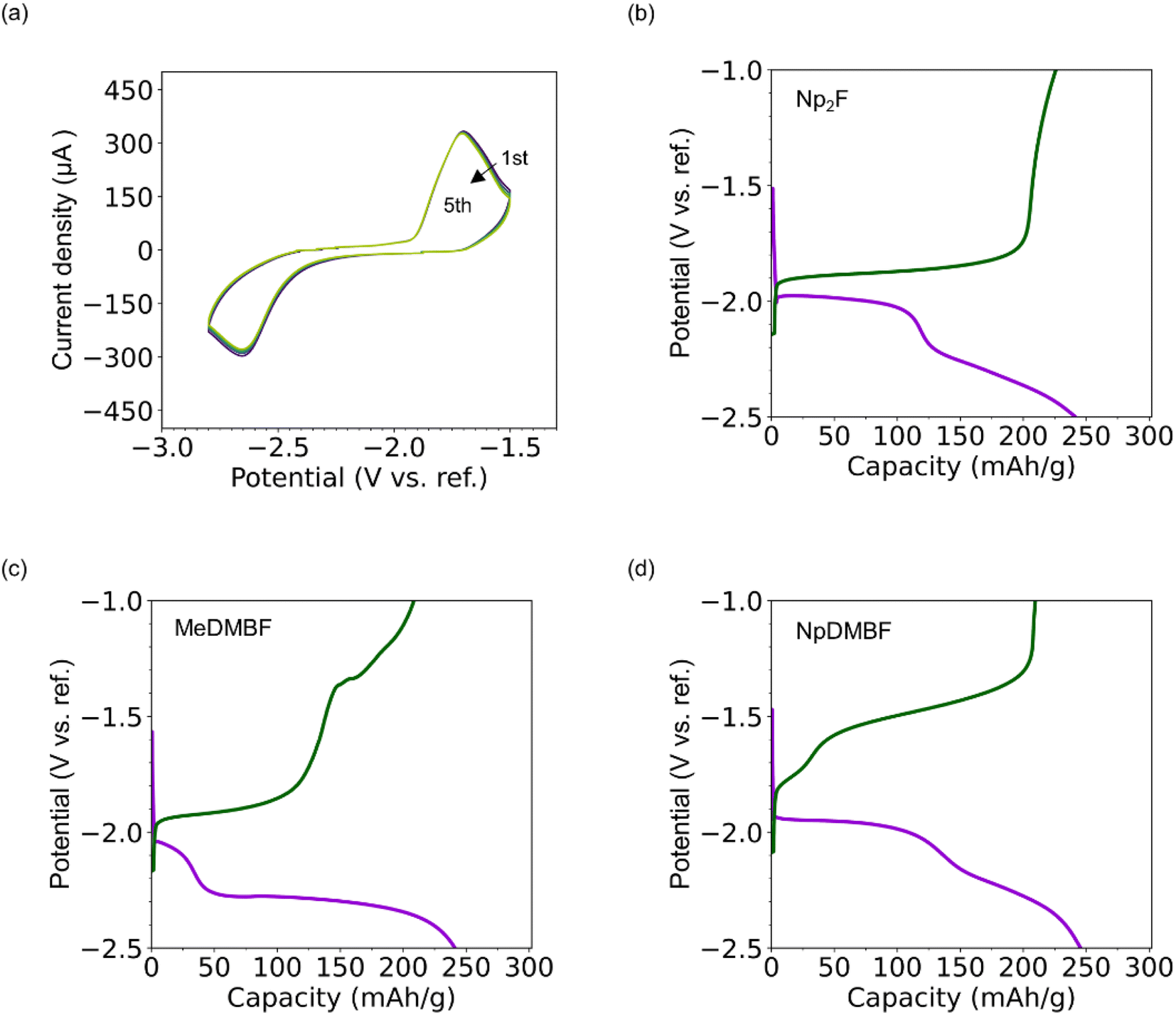 | ||
| Fig. 5 Half-cell performance. (a) Cyclic voltammograms (CVs) of the bismuth fluoride and carbon electrodes in 0.9 M Np2F/BTFE electrolyte at 298 K with a 1 mV s−1 scan rate, (b) charge/discharge curve (0.1C) for the BiF3 electrode in 0.9 M Np2F/BTFE, (c) charge/discharge curve (0.1C) for the BiF3 electrode in 0.9 M MeDMBF/BTFE, and (d) charge/discharge curve (0.1C) for the BiF3 electrode in 0.9 M NpDMBF/BTFE. The voltages are referenced to a double junction electrode based on Ag/AgOTf in MPPy-FSI (see the text). | ||
The total charge/discharge performance (at 0.1C) for 0.9 M Np2F/BTFE, 0.9 M MeDMBF/BTFE, and 0.9 M NpDMBF/BTFE is given in Fig. 5b, c and d, respectively. For Np2F/BTFE, the open-circuit voltage (OCV) before charge/discharge was −1.5 V. On the initial reduction side, the potential remained at −2.0 V until the capacity reached 100 mA h g−1, and then fell gradually before reaching the lower limit of −2.5 V. Subsequently, on the initial oxidation side, the potential remained stable at around −1.9 V before increasing dramatically. The discharge capacity was 244 mA h g−1 (81% of the theoretical capacity of 302 mA h g−1), and the charge capacity was 227 mA h g−1 (75% theoretical capacity). The discharge and charge capacities of MeDMBF and NpDMBF are comparable, at 241 mA h g−1 and 246 mA h g−1, and 208 mA h g−1 and 210 mA h g−1, respectively. Based on these results, three quaternary ammonium fluorides, MeDMBF, NpDMBF, and the reference compound Np2F, are promising as fluoride-ion battery electrolytes.
Conclusions
In this work, we successfully synthesized novel quaternary ammonium fluoride salts with varying alkyl substituents. Two salts, MeDMBF and NpDMBF, demonstrated excellent solubility (>1.7 M) and high conductivity (>2 mS cm−1) in the aprotic solvent BTFE, making them promising candidates for room-temperature FIB electrolytes. NMR spectroscopic studies and theoretical calculations revealed a correlation between cation size and fluoride ion diffusivity, with larger cations exhibiting higher diffusion coefficients. However, this trend was accompanied by a decrease in the thermal stability of the salts. This highlights the crucial trade-off between ionic conductivity and thermostability in these electrolyte systems. Despite this, MeDMBF and NpDMBF exhibited a wide electrochemical window exceeding 3 V in BTFE. Furthermore, preliminary electrochemical testing using BiF3 electrodes demonstrated promising charge/discharge behavior, with over 80% of the theoretical capacity achieved in the first cycle. These findings provide valuable insights into the design principles for high-performance fluoride-ion electrolytes, emphasizing the importance of balancing cation size and structural features to optimize conductivity and stability.Experimental
Materials
Neopentylamine, 1-(adamantan-1-yl)methanamine, pivalaldehyde, sodium triacetoxyborohydride, MeI, formaldehyde, trimethyloxonium tetrafluoroborate, LiAlH4, BTFE, PN, DMA, DME, DEC, TEGDME and DMI were purchased from Tokyo Chemical Industry Co., Ltd (TCI). Hydrochloric acid and anhydrous magnesium sulfate were purchased from Nacalai Tesque, Inc. Formic acid, sodium hydroxide, Et3N, dibutyl ether, super dehydrated MeOH, DMSO, GBL, NMP, EtOAc, and MeCN were purchased from FUJIFILM Wako Pure Chemicals Corporation. Dehydrated solvents, dichloromethane, diethyl ether, DMF, Et2O, and THF were purchased from Kanto Chemical Co., Inc. 2,2-Dimethylbutanoyl chloride, dimethylamine hydrochloride, and KF were purchased from Sigma-Aldrich. All chemicals were used as received, except PN, DMA, DME, DEC, TEGDME, DMI, and BTFE, which were dried over 4 Å molecular sieves, and KF, which was dried at 250 °C for 8 hours. Deuterated solvents were purchased from Eurisotop or Cambridge Isotope Laboratories, Inc. All deuterated solvents were used as received, except CD3CN, which was dried over 4 Å molecular sieves.Working electrodes
BiF3 and acetylene black (AB) were mixed and milled in a 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12.5 weight ratio. This mixture was then suspended in N-methylpyrrolidone (NMP) to form a smooth slurry with a 75:12:5:12.5 weight ratio of BiF3, AB, and polyvinylidene difluoride (PVDF). The slurry was coated onto a stainless current collector. The mixture was heated at under 80 °C for 0.5 hours, then dried overnight under vacuum at 90 °C and subsequently pressed. Electrodes with a 7.95 mm diameter were punched out from the dried mixture. The average weight of active material (BiF3) per cell is 0.7 mg.
:12.5 weight ratio. This mixture was then suspended in N-methylpyrrolidone (NMP) to form a smooth slurry with a 75:12:5:12.5 weight ratio of BiF3, AB, and polyvinylidene difluoride (PVDF). The slurry was coated onto a stainless current collector. The mixture was heated at under 80 °C for 0.5 hours, then dried overnight under vacuum at 90 °C and subsequently pressed. Electrodes with a 7.95 mm diameter were punched out from the dried mixture. The average weight of active material (BiF3) per cell is 0.7 mg.
Counter electrodes
A mixture of activated carbon and polytetrafluoroethylene (PTFE) (70:30 wt%) was prepared by a dry process. Electrodes with a 12 mm diameter were punched out from the mixture.
NMR measurements
The 1H, 13C, and 19F NMR spectra were recorded with a Bruker Advance III 400, 600 MHz spectrometer equipped with a BBFO probe. Chemical shifts are reported in δ ppm using a residual protonated solvent in the deuterated solvents for 1H as an internal standard. Deuterated solvent peaks were used as the internal standard for 13C NMR, whereas CFCl3 (0 ppm) was used as the external standard for 19F NMR. Anhydrous sample solutions were prepared in an argon gas-filled glove box (H2O < 0.1 ppm, O2 < 0.1 ppm). Wilmad low pressure/vacuum NMR tubes (LPV) were used.Electrochemical measurements
Cyclic voltammetry (CV) was performed in a glove box (H2O < 0.1 ppm, O2 < 0.1 ppm) using an HZ-Pro analyzer (Hokuto Denko). A three-electrode cell was constructed using Pt as the working electrode and the counter electrode, A silver wire immersed in 1-methyl-1-propylpyrrolidinium bis(fluorosulfonyl)imide (MPPy-FSI) containing 0.1 M silver trifluoromethanesulfonate then placed in a compartment that was filled with MPPy-FSI and immersed in 0.9 M Np2F/BTFE electrolyte was used as the reference electrode (vs. ref). The potential of Fc/Fc+ in a 0.2 M LiTFSI/DMSO solution was determined to be −0.42 V vs. ref. This value was subsequently converted to Li+/Li based on the literature report.35 Water content was measured by using a Karl Fischer moisture titrator (MKH-710M, Kyoto Electronics Manufacturing Co., Ltd).Battery performance
The charge/discharge performance of the BiF3 working electrodes was measured using a three-electrode cell (SB9 cells purchased from EC Frontier Co., Ltd), where 0.5 mL of a 0.9 M fluoride salt solution in BTFE electrolyte was used with a PTFE separator. The entire cell assembly process was conducted inside a glovebox. Active carbon was used as the counter electrode. A silver wire immersed in 1-methyl-1-propylpyrrolidinium bis(fluorosulfonyl)imide (MPPy-FSI) containing 0.1 M silver trifluoromethanesulfonate then placed in a compartment that was filled with MPPy-FSI and immersed in 0.9 M electrolyte was used as the reference electrode (vs. ref.). The charge/discharge rate was measured at 0.1C (theoretical capacity of BiF3: 302 mA h g−1, 1C = 302 mA g−1). The potential range of the charge/discharge cycle was set from −3.0 V to −0.5 V (vs. ref.).Theoretical calculations
DFT calculations were carried out using the ORCA 5.0.3 software package.36 The molecular geometries for the ground states were optimized by DFT at the M062X/Def2-TZVPP level of theory with the RIJCOSX approximation, and TightSCF convergence.37–40 Vibration analysis of all the stationary point structures confirmed that no imaginary or negative frequencies were present. Visualizations were generated by IboView or Avogadro.41–43 Molecular dynamics simulations were carried out at 300 K and 1 atm with the Gromacs 2022.4 software package.44–51 Molecular forces were calculated using the Optimized Potentials for Liquid Simulations All Atom (OPLSAA) force field.52 Topology files and bonded and Lennard-Jones parameters were generated with the LigParGen server.53 The initial dimensions of the simulation, generated by the Packmol program, were set to 33 Å × 33 Å × 33 Å.54 Visualizations were generated by VMD, MDAnalysis, and NGL viewer.55–60Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. T. conceived the idea and designed the experiment; synthesis procedures were designed and developed by T. T.; NMR, CV, and CDC measurements, and theoretical calculations were conducted by T. T.; S. S. prepared the electrodes for CDC measurements; T. T., R. M., and A. W. prepared the manuscript; A. W. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Research and Development Initiative for Scientific Innovation of New Generation Batteries 2 and 3 (RISING2 and RISING3, project codes JPNP16001 and JPNP21006, respectively) and commissioned by the New Energy and Industrial Technology Development Organization (NEDO). Tiancheng Tan gratefully acknowledges funding from JST through the establishment of university fellowships towards the creation of science and technology innovation, grant number JPMJFS2123. We thank Dr Minh Anh Truong (Kyoto University) and Dr Tomoya Nakamura (Kyoto University) for fruitful discussions. The NMR measurements were partly supported by the Joint Usage/Research Center (JURC) at the Kyoto University Institute for Chemical Research.Notes and references
- A. W. Xiao, G. Galatolo and M. Pasta, Joule, 2021, 5, 2823–2844 CrossRef CAS.
- F. Gschwind, G. Rodriguez-Garcia, D. J. S. Sandbeck, A. Gross, M. Weil, M. Fichtner and N. Hörmann, J. Fluorine Chem., 2016, 182, 76–90 CrossRef CAS.
- M. A. Nowroozi, I. Mohammad, P. Molaiyan, K. Wissel, A. R. Munnangi and O. Clemens, J. Mater. Chem. A, 2021, 9, 5980–6012 RSC.
- S. V. Gopinadh, P. V. R. L. Phanendra, B. John and T. D. Mercy, Sustainable Mater. Technol., 2022, 32, e00436 CrossRef CAS.
- M. A. Reddy and M. Fichtner, J. Mater. Chem., 2011, 21, 17059–17062 RSC.
- D. T. Thieu, M. H. Fawey, H. Bhatia, T. Diemant, V. S. K. Chakravadhanula, R. J. Behm, C. Kübel and M. Fichtner, Adv. Funct. Mater., 2017, 27, 1701051 CrossRef.
- Y. Wang, X. Yang, Y. Meng, Z. Wen, R. Han, X. Hu, B. Sun, F. Kang, B. Li, D. Zhou, C. Wang and G. Wang, Chem. Rev., 2024, 124, 3494–3589 CrossRef CAS PubMed.
- M. Zhang, X. Cao, Y. Hao, H. Wang, J. Pu, B. Chi and Z. Shen, Energy Rev., 2024, 3, 100083 CrossRef.
- C. Rongeat, M. Anji Reddy, R. Witter and M. Fichtner, ACS Appl. Mater. Interfaces, 2014, 6, 2103–2110 CrossRef CAS PubMed.
- Y. Yu, M. Lei, D. Li and C. Li, Adv. Energy Mater., 2023, 13, 2203168 CrossRef CAS.
- I. Mohammad, R. Witter, M. Fichtner and M. Anji Reddy, ACS Appl. Energy Mater., 2018, 1, 4766–4775 CrossRef CAS.
- K. Shimoda, Y. Morita, K. Noi, T. Fukunaga, Z. Ogumi and T. Abe, ACS Energy Lett., 2023, 8, 2570–2575 CrossRef CAS.
- H. Miki, K. Yamamoto, H. Nakaki, T. Yoshinari, K. Nakanishi, S. Nakanishi, H. Iba, J. Miyawaki, Y. Harada, A. Kuwabara, Y. Wang, T. Watanabe, T. Matsunaga, K. Maeda, H. Kageyama and Y. Uchimoto, J. Am. Chem. Soc., 2024, 146, 3844–3853 CrossRef CAS PubMed.
- V. Vanita, A. Iqbal Waidha, S. Vasala, P. Puphal, R. Schoch, P. Glatzel, M. Bauer and O. Clemens, J. Mater. Chem. A, 2024, 12, 8769–8784 RSC.
- Q. Nie, Y. Hao, L. Cheng, Y. Fu, G. Wang, M. Zhang and Z. Shen, Solid State Ionics, 2024, 405, 116454 CrossRef CAS.
- J. Liu, L. Yi, X. Chen, D. Li, S. Ni, J. Xia, L. Yang and X. Wang, Sustainable Mater. Technol., 2024, 39, e00810 CrossRef CAS.
- F. Gschwind, Z. Zao-Karger and M. Fichtner, J. Mater. Chem. A, 2013, 2, 1214–1218 RSC.
- O. Alshangiti, G. Galatolo, G. J. Rees, H. Guo, J. A. Quirk, J. A. Dawson and M. Pasta, ACS Energy Lett., 2023, 8, 2668–2673 CrossRef CAS PubMed.
- S. Kawauchi, H. Nakamoto, R. Takekawa, T. Kobayashi and T. Abe, ACS Appl. Energy Mater., 2022, 5, 2096–2103 CrossRef CAS.
- K. Okazaki, Y. Uchimoto, T. Abe and Z. Ogumi, ACS Energy Lett., 2017, 2, 1460–1464 CrossRef CAS.
- T. Yamamoto, K. Matsumoto, R. Hagiwara and T. Nohira, ACS Appl. Energy Mater., 2019, 2, 6153–6157 CrossRef CAS.
- V. K. Davis, C. M. Bates, K. Omichi, B. M. Savoie, N. Momčilović, Q. Xu, W. J. Wolf, M. A. Webb, K. J. Billings, N. H. Chou, S. Alayoglu, R. K. McKenney, I. M. Darolles, N. G. Nair, A. Hightower, D. Rosenberg, M. Ahmed, C. J. Brooks, T. F. Miller, R. H. Grubbs and S. C. Jones, Science, 2018, 362, 1144–1148 CrossRef CAS PubMed.
- G. Galatolo, O. Alshangiti, C. Di Mino, G. Matthews, A. W. Xiao, G. J. Rees, M. Schart, Y. A. Chart, L. F. Olbrich and M. Pasta, ACS Energy Lett., 2024, 9, 85–92 CrossRef CAS PubMed.
- Y. Yu, M. Lei and C. Li, Mater. Horiz., 2024, 11, 480–489 RSC.
- R. Yaokawa, T. Shiga, S. Moribe and K. Mukai, RSC Adv., 2022, 12, 31786–31791 RSC.
- Y. Yu, A. Lin, M. Lei, C. Lai, C. Wu, Y.-Y. Sun and C. Li, ACS Energy Lett., 2024, 9, 1008–1016 CrossRef CAS.
- A. C. Kucuk, T. Yamanaka and T. Abe, J. Mater. Chem. A, 2020, 8, 22134–22142 RSC.
- A. Celik Kucuk and T. Abe, J. Fluorine Chem., 2020, 240, 109672 CrossRef CAS.
- T. Tan, R. Murdey, S. Sumitomo, T. Nakamura, M. A. Truong and A. Wakamiya, Chem. Mater., 2024, 36, 4553–4560 CrossRef CAS.
- H. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050–2051 CrossRef CAS PubMed.
- K. O. Christe, W. W. Wilson, R. D. Wilson, R. Bau and J. A. Feng, J. Am. Chem. Soc., 1990, 112, 7619–7625 CrossRef CAS.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS.
- W. Xue, M. Huang, Y. Li, Y. G. Zhu, R. Gao, X. Xiao, W. Zhang, S. Li, G. Xu, Y. Yu, P. Li, J. Lopez, D. Yu, Y. Dong, W. Fan, Z. Shi, R. Xiong, C.-J. Sun, I. Hwang, W.-K. Lee, Y. Shao-Horn, J. A. Johnson and J. Li, Nat. Energy, 2021, 6, 495–505 CrossRef CAS.
- P. K. R. Kottam, D. Kalkan, M. Wohlfahrt-Mehrens and M. Marinaro, J. Electrochem. Soc., 2019, 166, A1574 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- P. Bauer, B. Hess and E. Lindahl, GROMACS 2022.4 Source Code, Zenodo, 2022 Search PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. Van Der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- S. Páll, M. J. Abraham, C. Kutzner, B. Hess and E. Lindahl, Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS, in Solving Software Challenges for Exascale (EASC 2014), Lecture Notes in Computer Science, ed. S. Markidis and E. Laure, Springer, Cham, 2015, vol. 8759, DOI:10.1007/978-3-319-15976-8_1.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110, 1657–1666 CrossRef CAS PubMed.
- L. S. Dodda, I. Cabeza de Vaca, J. Tirado-Rives and W. L. Jorgensen, Nucleic Acids Res., 2017, 45, W331–W336 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, J. Domański, D. L. Dotson, S. Buchoux, I. M. Kenney and O. Beckstein, Proc. 15th Python Sci. Conf., 2016, pp. 98–105 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- A. S. Rose and P. W. Hildebrand, Nucleic Acids Res., 2015, 43, W576–W579 CrossRef CAS PubMed.
- H. Nguyen, D. A. Case and A. S. Rose, Bioinformatics, 2018, 34, 1241–1242 CrossRef CAS PubMed.
- A. S. Rose, A. R. Bradley, Y. Valasatava, J. M. Duarte, A. Prlić and P. W. Rose, Bioinformatics, 2018, 34, 3755–3758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2385359. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4se01432d |
| This journal is © The Royal Society of Chemistry 2025 |