
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding charge separation in CdS/Ce-UiO66-NH2 heterojunctions for enhanced photocatalytic hydrogen evolution†‡
Wenqing
Hou
 ab,
Sam A. J.
Hillman
a,
Soranyel
Gonzalez-Carrero
ad,
Shilin
Yao
a,
Huangtianzhi
Zhu
c and
James R.
Durrant
*a
ab,
Sam A. J.
Hillman
a,
Soranyel
Gonzalez-Carrero
ad,
Shilin
Yao
a,
Huangtianzhi
Zhu
c and
James R.
Durrant
*a
aDepartment of Chemistry and Centre for Processable Electronics, Imperial College London, White City Campus, London W12 0BZ, UK. E-mail: j.durrant@imperial.ac.uk
bState Key Laboratory of Silicon Materials and Department of Chemistry, Zhejiang University, Hangzhou 310027, China
cYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
dInstitute of Molecular Science, University of Valencia, Paterna, Valencia 46980, Spain
First published on 26th November 2024
Abstract
UiO metal–organic frameworks (MOFs) are regarded as promising photocatalysts due to their unique stability and band designability. We recently demonstrated that the cerium-based Ce-UiO-NH2 exhibited an enhanced hydrogen evolution relative to zirconium (Zr)-UiOs, when loaded with cadmium sulfide (CdS). However, the underlying charge separation dynamics of this system is unclear. In this work, we optimised the CdS loading and used transient absorption and electrochemical spectroscopy to investigate the charge separation dynamics and energetics in the CdS/Ce-UiO-NH2 heterojunction. The optimised heterojunction showed improved stability and achieved an external quantum efficiency (EQE) of 2.2% under 420 nm LED illumination whilst using methanol as a sacrificial agent. The heterojunction facilitates charge separation, generating long-lived (ms) holes on Ce-UiO-NH2 and electrons on the CdS. In contrast with electron-accepting Zr-UiOs, this study reveals a reversed charge separation direction in CdS/UiO heterojunctions with Ce-UiO-NH2 acting as the electron donor.
Introduction
In recent years, the world's energy shortage and environmental pollution have become urgent issues. It is therefore imperative to seek new technologies to relieve these pressures. Among them, photocatalytic hydrogen (H2) evolution from water under solar light irradiation is regarded as a promising clean energy technology and has developed rapidly in recent years.1,2 In particular, research has focused on exploring new, more efficient materials and heterojunctions and has achieved significant progress.3,4 Meanwhile, understanding such materials' underlying reaction mechanisms is attracting increasing attention, and is playing a significant role in the design of efficient photocatalysts.Among various photocatalysts, MOFs have received significant attention owing to their high surface areas, visible-light-response, and tunable functional structure.5 The large number of possible combinations of constituent metal clusters and bridging organic linkers offer considerable synthetic flexibility when targeting superior visible-light-driven photocatalysis.6,7 It has been demonstrated that MOFs can show charge transfer from the organic linkers to the metal nodes (i.e. linker-to-metal transfer) which is triggered by light irradiation.8 As such, they can act as semiconductor photocatalysts which absorb light, generate electron/hole pairs, and then initiate either the hydrogen evolution reaction (HER) or the oxygen evolution reaction (OER).
UiO-66s (Universite i Oslo) are amongst the most important MOFs and have gained intensive attention due to their thermal and chemical stabilities and facile design.9,10 Ce-UiO-NH2 is an analogue of the Zr-UiO, comprising hexanuclear clusters constituted by cerium-oxo [Ce6(O)4(OH)412+] and 2-aminoterephthalic acid.11,12 With Ce metal node substitution, it is possible to achieve better visible light absorption and catalytic properties due to the Ce(III)/(IV) redox properties (Ce(III)/(IV) couple at circa 1.61 V vs. NHE) and the presence of low energy 4f orbitals.13 However, UiO-66 in isolation only shows minimal photocatalytic activity. Significant efforts have been made to improve their photocatalytic HER efficiency through methods such as loading noble metal co-catalysts (Pt, Au),14,15 using amino-substituted organic linkers to increase the visible light photoresponse,16 or combining with conventional semiconductors in a heterojunction.17 Heterojunction design is considered as one of the most promising strategies due to its potential for improved charge separation and the advantages of combining different catalysts. Cadmium sulfide (CdS) nanoparticles are considered to be a good junction material for MOFs due to its strong visible light absorption (optical band gap 2.3 eV). However, its function can be seriously limited by self-corrosion and the rapid recombination of photogenerated electrons and holes.18,19 In 2015, Shen and co-workers reported CdS/UiO-66 composites decorated with MoS2 for enhanced HER. Jiang's group studied the dynamics of CdS/UiO-66 composites for enhanced HER in 2018, indicating the potential of UiO-66 working as an electron acceptor.17,20
In our previous work, UiO-66s were compared as supports for CdS nanoparticles (NPs) to enhance HER efficiency under visible light in sodium sulfide/sodium sulfite (Na2S/Na2SO3) aqueous solution.21 The maximum EQE was obtained using a Ce-substituted UiO-NH2. Spectral and electrochemical measurements indicated that Ce-UiO-NH2 is a more efficient partner with CdS due to its smaller band gap and more efficient charge transfer to CdS. However, the underlying charge dynamics of CdS/Ce-UiO-NH2 have not previously been reported, and there is no direct evidence indicating whether Ce-UiO-NH2 acts as electron donor or acceptor in this heterojunction. In the work herein, we improved the HER efficiency by optimizing the loading of CdS onto the Ce-UiO-NH2 framework and using methanol as the sacrificial regent. The charge separation dynamics in the CdS/Ce-UiO-NH2 heterojunction were then investigated by transient absorption spectroscopy. The optimized heterojunction was found to exhibit a much higher density of long-lived (ms) charges, consistent with its enhanced photocatalytic efficiency. Furthermore, the direction of charge transfer direction was studied using transient absorption spectroscopy and spectroelectrochemistry, determining the role of Ce-UiO-NH2 as the electron donor in this heterojunction.
Experimental
Materials preparation
Chemicals were in analytic grade and were used without further purification. Terephthalic acid (BDC, 99%), Ce(NH4)2(NO3)6, Cd(CH3COO)2, Na2S, acetonitrile (ACN) and N,N-dimethylformamide (DMF) were from Sigma-Aldrich. 2-Aminoterephthalic acid (ATA, 98%) were purchased from Thermo Scientific Chemicals. Catalysts were synthesized according to the previous work.21 Ce-UiO-NH2: Ce-UiO-NH2 was prepared by solvent-assistant-linker-exchange procedure.12 Ce-UiO66 (Ce-UiO) was firstly prepared as follows,11 terephthalic acid (143 mg, 0.86 mmol) was dissolved in 4.9 mL of DMF. After that, 1.6 mL of a 0.535 M aqueous solution of Ce(NH4)2(NO2)6 was added and the mixture was heated at 100 °C under stirring conditions for 15 min. Then, the precipitate was collected by centrifugation and purified three times with DMF and acetone respectively to remove the residue of organic ligands and DMF. The final product was dried overnight (60 °C). Second, for synthesis of Ce-UiO-NH2, Ce-UiO (81.6 mg, 0.25 mmol) and ATA (181.1 mg, 1 mmol) were dispersed in methanol (25 mL). The mixture was stirred for dissolving completely and then sonicated for 5 min, followed by purifying with DMF and ethanol, respectively. The final product was collected by centrifugation and dried under vacuum (60 °C, 12 h). xCdS/Ce-UiO-NH2: neat CdS NPs and 20–50 wt%CdS/Ce-UiO-NH2 (20–50%CdS/Ce-UiO-NH2 for short), were prepared by two-step-loading following the recipe in Table S1.† Typically, Ce-UiO-NH2 (60 mg) was activated under vacuum (40 °C, 12 h) and dispersed in 15 mL DI water. Afterwards, Cd(CH3COO)2 aqueous solution (2 mL, 103.8 M) was dropwise added into the solution with stirring for 2 h, and then with Na2S (2 mL, 103.8 M). After stirring for 5 h continuously, the product (40% CdS/Ce-UiO-NH2) was collected by centrifugation, purified with DI water and ethanol and dried under vacuum (60 °C, 12 h).Materials characterizations
Powder X-ray diffraction pattern (XRD) was obtained on a Rigaku diffractometer with Cu Kα as X-ray. The absorption spectra were recorded in an Agilent Cary 5000 UV-visible NIR spectrometer. The high-resolution transmission electron microscopic images (HRTEM) were taken by JEM-2100F at 150 kV accelerating voltage. Fourier transform infrared microscopy (FTIR) was collected using a Cary 630 FT-IR spectrometer in the wavenumber range 500–4000 cm−1. The steady-state photoluminescence (PL) spectra were recorded using an Edinburgh FLS1000 photoluminescence spectrometer.Hydrogen evolution
The photocatalytic H2 evolution was measured using methanol as a sacrificial electron donor. In a 100 mL sealed flat bottom flask, 25 mg of catalyst were dispersed in 50 mL aqueous solution (methanol, 10 vol%). Before irradiation, the reactor was purged for 30 min with argon to remove the oxygen and stirred at room temperature with irradiated by four 3 W LED lamps (420 nm), located at 1.0 cm away from the solution surface. H2 evolution was analyzed by gas chromatography (GC) equipped with a thermal conductivity detector (Agilent GC7820A). The stability test was performed with 40%CdS/Ce-UiO-NH2, with 120 min of irradiation. After one run was complete, the system was purged with argon for 30 min and irradiated again, without extra addition of new catalyst and sacrificial reagent.The EQE was measured under the same conditions as H2 evolution measurements. The illumination intensity was measured by using a UV-A radiometer (0.1 J cm−2, from Beijing Shida Technology Co., Ltd), and the EQE was calculated by the following equation:
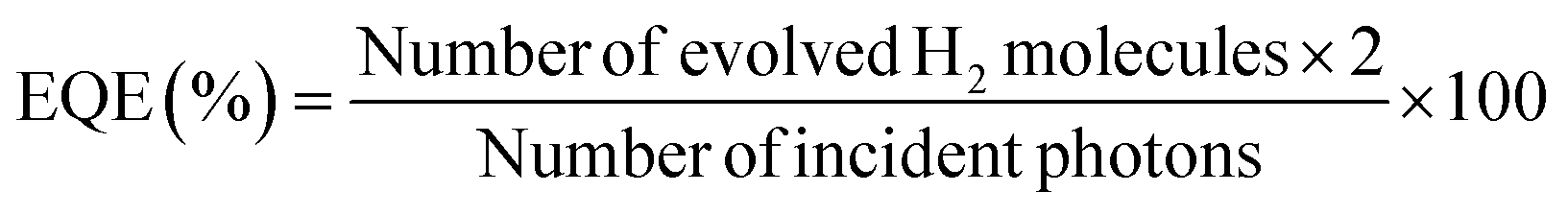
where Ep is the total energy of incident photons (J), h is the Planck constant (J s−1), ν is the frequency of light (Hz).

Transient absorption spectroscopy (TAS)
Microsecond to second TAS data were acquired on previously-described home-built setups.22 420 nm or 560 nm laser excitation was generated from a Nd:YAG laser (OPOTEK Opolette 355 II, 6 ns pulse width). The probe beam was generated from a quartz halogen lamp. The light was directed through the sample to a monochromator, which selected the probe wavelength and was relayed to a Si photodiode detector (Hamamatsu S1722-01). Data on the sub-ms timescale were amplified with an electronic amplifier box (Costronics 2011 amplifier) and recorded on an oscilloscope (Tektronix DPO 3012). Data on the ms timescale were simultaneously recorded on a National Instruments DAQ card (National Instruments USB-6361). Pulse energies were measured by energy meter (OPHIR Photonics, VEGA P/N 7Z01560). Film samples were made by drop casting on FTO glass with acetonitrile (ACN) suspension (2 mg catalysts, 20 μL Nafion and 400 μL ACN). Films were measured in DI water or aqueous solution and purged with argon to remove oxygen. To ensure equal numbers of absorbed photons, all films were deposited such that they had the same ground state absorbance at the excitation wavelength (absorbance = 0.6 at 420 nm excitation and absorbance = 0.25 at 560 nm excitation).Electrochemical spectroscopy
Electrochemical measurements were conducted using a three-electrode setup with NaClO4 electrolyte (pH 7), sample as working electrode, Pt wire counter electrode, and an Ag/AgCl reference electrode (saturated KCl). A PGSTAT204 potentiostat (Metrohm-Autolab, The Netherlands) was used to apply linear sweep voltammograms with potential steps of 50 mV at rate of 50 mV s−1. Absorbance changes from 400–1000 nm were collected at each applied potential with a Maya2000Pro spectrometer (OceanOptics) using home-built LabView software (https://opensourcespectroscopy.com/sec_code/). All measurements were conducted on three separate batches of samples, to verify reproducibility and to calculate standard deviation. All potentials are presented against relative hydrogen electrode (RHE) through the following equation:| E (V vs. RHE) = E (V vs. AgCl/Ag) + 0.059 × pH + 0.197 |
Results and discussion
Heterojunction design and characterization
CdS/Ce-UiO-NH2 samples were synthesized in accordance with the protocol described in the previous work.21 Ce-UiO-NH2 was firstly synthesized by a solvothermal method followed by a solvent-assisted-linker-exchange procedure. Then CdS was loaded onto Ce-UiO-NH2 frameworks using a two-step loading to form heterojunctions (Fig. 1a). The structure and composition of all materials were investigated by XRD, HRTEM, FTIR and N2 sorption. According to powder XRD (Fig. 1d), Ce-UiO-NH2 has similar topological structure to other UiO-66 MOFs,10,11 showing the characteristic peaks at 7.20, 8.30, 11.70, 13.76, 16.52, 18.02, 21.28 and 24.86°, respectively. CdS had a XRD pattern in accord with that for cubic CdS. After CdS loading, the heterojunctions exhibited visible diffraction patterns of both components. The characteristic powder XRD pattern of CdS increased in intensity and the pattern of Ce-UiO-NH2 decreased in intensity as the CdS loading was increased (Fig. S1a†). In HRTEM images (Fig. 1b and c), small particles (5–10 nm) on large octahedrons (200–300 nm) were observed. The former had a distinct lattice spacing of 0.34 nm which matches well with the (111) facet of CdS, demonstrating that CdS NPs were evenly distributed on Ce-UiO-NH2.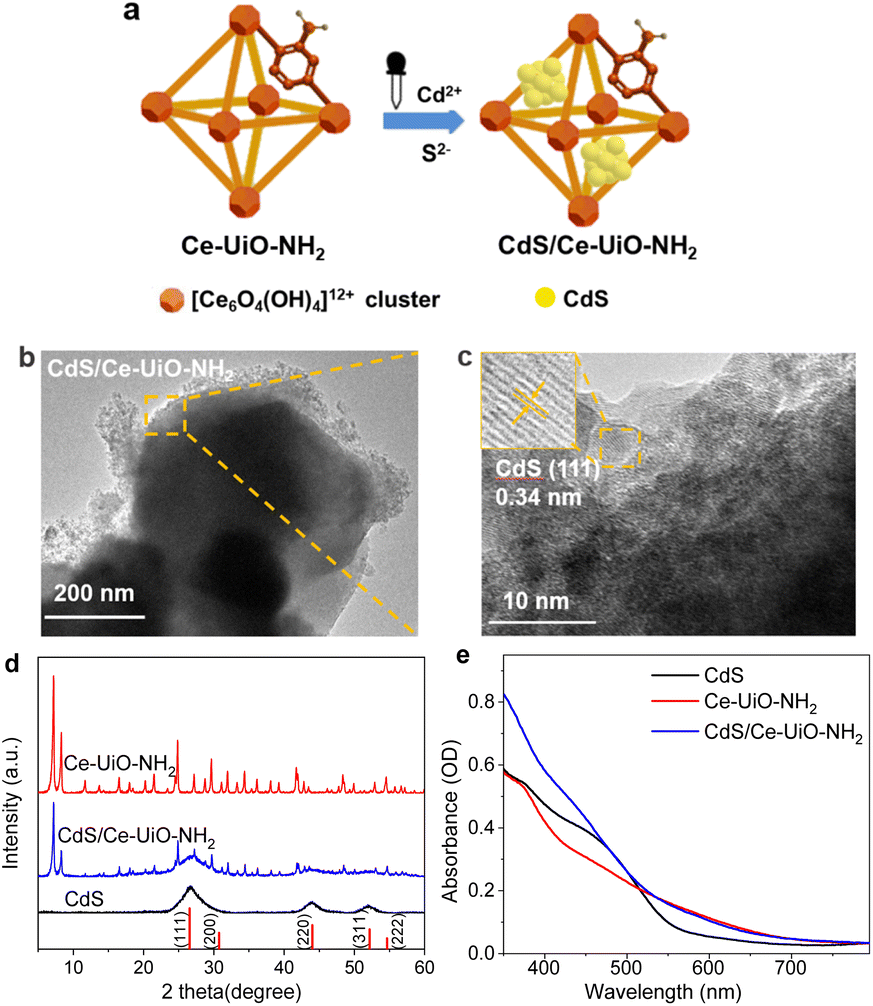 | ||
| Fig. 1 (a) Synthesis of CdS/Ce-UiO-NH2 heterojunctions, (b and c) HRTEM images for 40%CdS/Ce-UiO-NH2, (d) XRD patterns and (e) absorption spectra for CdS, Ce-UiO-NH2 and 40%CdS/Ce-UiO-NH2, measured on film samples. The red columns represent the patterns for cubic CdS (JCPDS 80-0019). | ||
FTIR spectroscopy was conducted to investigate the organic structures inside Ce-UiO-NH2. As shown in Fig. S1c,† some specific peaks of Ce-UiO-NH2 were observed: at 1260 cm−1 (stretching vibration of Car–N); at 3500–3200 cm−1 (two stretching vibration N–H peaks caused by the amino groups); at 1650–1450 cm−1 (stretching vibrations caused by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); and at 1655 cm−1 (stretching vibrations of CO). The lower frequency bonds at 800–450 cm−1 are attributed to the bending vibration of O–H and C–H, mixed with Ce–O bonds.23
C); and at 1655 cm−1 (stretching vibrations of CO). The lower frequency bonds at 800–450 cm−1 are attributed to the bending vibration of O–H and C–H, mixed with Ce–O bonds.23
N2 sorption measurements were performed to evaluate the porosity of the heterojunctions. The results are shown in Table S2 and Fig. S1b.† The sorption isotherm of Ce-UiO-NH2 shows the type I adsorption isotherm curve shape, whilst CdS exhibits type IV. The specific BET surface area (Asp) of Ce-UiO-NH2 was calculated to be 942 m2 g−1, with a large micropore volume (Vmp, 0.416 cm3 g−1) and total pore volume (Vtp, 0.521 cm3 g−1). With increasing content of CdS, the isotherm transitions from type I to type IV with increasing amounts of hysteresis. Asp, Vmp and Vtp of the composites decreased, respectively. These changes are ascribed to the load of CdS on the Ce-UiO-NH2. It is notable that the measured values were much smaller than those calculated from neat CdS and Ce-UiO-NH2, whilst the ratio of Vmp to Vtp dramatically decreased from 0.80 to 0.32 as the CdS content increased from 0 to 50% (Table S2†). All these decreases suggest that the CdS NPs are filling up the pores of Ce-UiO-NH2via the two-step loading procedure. Meanwhile, the reduced Vmp and increased pore size (dp) with CdS loaded were also in agreement with the fact that the pore space of Ce-UiO-NH2 was filled with CdS NPs.
Fig. 1e shows the steady state UV-vis absorption spectra. The spectrum of neat CdS NPs displays visible light absorption with an absorption edge at 550 nm (optical band gap: 2.25 eV), which can be attributed to the intrinsic band gap absorption of cubic CdS. For Ce-UiO-NH2, there is a broad absorption tail that extends across the visible up to 700 nm. This absorption is clearly redshifted compared to Zr-based UiO66-NH2 (450 nm) and is attributed to the narrower bandgap of Ce-UiO-NH2. This is because the orbital energy of Ce 4f, which contributes to the lowest unoccupied linker orbitals, is lower than the Zr 4d orbital energy and exhibits more favorable linker-to-metal charge transfer in UiO-NH2.13 Meanwhile, the broad visible light absorption starting from 700 nm is suggestive of a complex band-to-band structure in Ce-UiO-NH2. For CdS/Ce-UiO-NH2, the absorbance spectrum combines the features of neat CdS and Ce-UiO-NH2. These observations are in agreement with our previous work,21 and confirm the successful and consistent synthesis of the neat and heterojunction materials.
Photocatalytic activity for hydrogen evolution
After determining the structure of CdS and Ce-UiO-NH2 photocatalysts, their performance for H2 evolution was measured under 420 nm LED excitation, using 10 vol% methanol aqueous solution as a hole scavenger. Fig. 2a shows the result of H2 evolution on xCdS/Ce-UiO-NH2 samples. The amount of H2 evolved approximately linearly with time. We note that the H2 evolved in the first 30 min was slightly lower than expected for linear H2 evolution, likely due to the initial system heating. To assess the relative activity of those catalysts, the amount of H2 evolved at 2 h and EQE were used as measure. Neat Ce-UiO-NH2 showed negligible H2 evolution within 2 h, whilst neat CdS also performed poorly. After loading with 20, 30, 40 and 50% CdS, however, the amount of H2 evolution in 2 h increased to 60.2, 84.6, 103.2 and 89.9 μmol, respectively. The maximum EQE at 420 nm was 2.2%, observed at x = 40%, which was 11.0 times higher than the EQE of individual CdS (0.2%). It is worth noting that the 40%CdS/Ce-UiO-NH2 heterojunction evolves hydrogen approximately 10 times more efficiently than Pt-loaded Ce-UiO-NH2 (Table S3†). Further, the HER performance of the CdS/Ce-UiO-NH2 was generally of similar order of magnitude to other reported CdS/MOFs. However, the vastly different experimental conditions used in the literature (sacrificial reagents, light intensity, etc.) prevent meaningful comparison.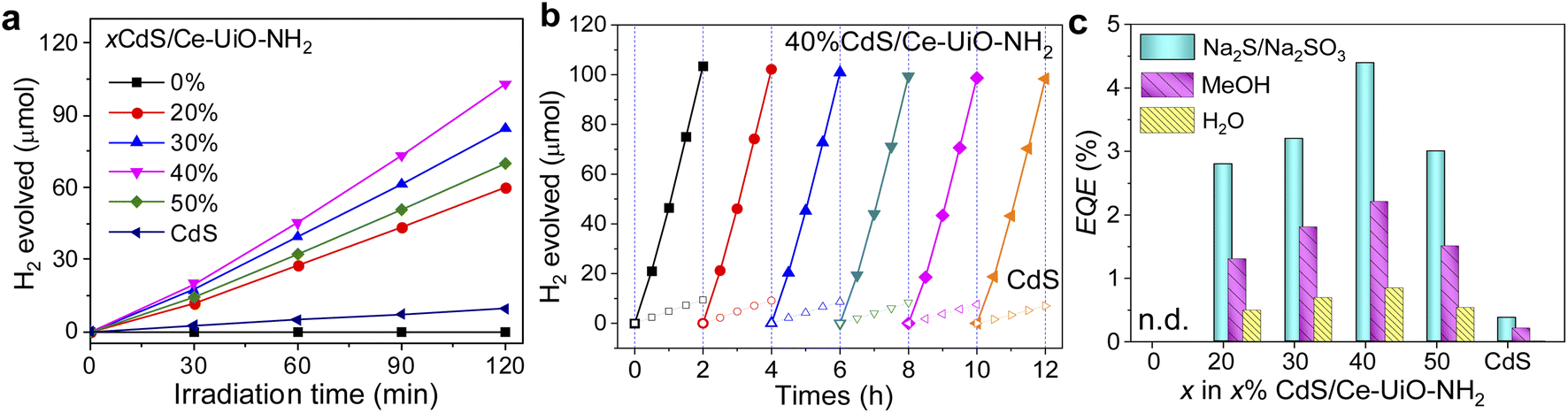 | ||
| Fig. 2 (a) Photocatalytic evolution of H2 for 25 mg xCdS/Ce-UiO-NH2 in 10 vol% methanol solutions, where x was 0, 20, 30, 40, and 50 wt%, measured for 2 h. (b) 6 sequential 2 h H2 evolution tests for CdS (dotted line) and 40%CdS/Ce-UiO-NH2 (solid line), excited with 420 nm LED lamps in a 10 vol% methanol solution. (c) EQE at 420 nm, measured in 0.35 M Na2S/0.25 M Na2SO3 (unstable), 10 vol% methanol solutions and without sacrificial agents, respectively. | ||
Fig. 2b shows six successive 2 h tests for H2 evolution. From the first run to the sixth, the amount of H2 evolved decreased by only 3.75% for 40%CdS/Ce-UiO-NH2 (from 102.17 μmol to 98.34 μmol) while the neat CdS decreased by 25.3% (from 9.43 μmol to 7.04 μmol, Fig. S2a†), demonstrating the improved HER stability of the heterojunction (marked as HJ for short). The stability of samples was further confirmed by XRD and XPS (Fig. S2†). For neat CdS, the diffraction peaks (such as those at 2θ ≈ 26.5°, 44.0°, and 52.2° for CdS) decreased in intensity after hydrogen evolution test, indicating a lowered crystallinity in CdS (Fig. S2b†). XPS of CdS after hydrogen evolution tests revealed that the S0 peak at 163.78 eV and the S6+ peak at 168.28 eV had formed, indicating the presence of higher-valence sulfur resulting from the oxidation of divalent sulfur (Fig. S2f†). These observations confirm that neat CdS undergoes photocorrosion which likely contributes to its decreasing HER performance over time. In contrast, the HJ exhibits no obvious changes in XRD and XPS patterns after 12 h of HER test, thereby demonstrating its improved stability.
These results suggest there is a cooperative effect between CdS and Ce-UiO-NH2 which contributes to the enhanced photocatalytic activity and stability of the CdS/Ce-UiO-NH2 heterojunctions. The Asp of a catalyst is often correlated to its photocatalytic activity. As MOFs are usually considered as a good catalyst support, we normalized the EQEs to Asp to isolate the influence of Asp on the CdS/Ce-UiO-NH2 heterojunctions' performance. As shown in Table S2,† the corresponding EQE/Asp values for CdS/Ce-UiO-NH2 are larger than those of CdS (0.041 g m−2) when the CdS content is between 30% (0.047 g m−2) and 50% (0.058 g m−2). This indicates that the EQE change of CdS/Ce-UiO-NH2 heterojunction is not only due to the increase of Asp introduced by Ce-UiO-NH2 but also to cooperation between CdS and Ce-UiO-NH2.
In Fig. 2c, the CdS/Ce-UiO-NH2 heterojunction also shows enhanced HER performance vs. neat CdS both in pure water and in the presence of Na2S/Na2SO3. The EQEs follow the same trend with CdS content in all three cases, thereby showing that interaction with the hole scavenger does not explain the EQE trend with CdS content. We note that our previous work showed higher EQE in Na2S/Na2SO3 than in methanol and pure H2O.21 However, this system was not stable enough for in-depth spectroscopic analysis.
Photophysical characterization
Now, we turn to the photophysical analysis of the origin of the enhanced efficiency of CdS/Ce-UiO-NH2 heterojunctions. 40%CdS/Ce-UiO-NH2, which showed the maximum EQE in H2 evolution, was chosen as the HJ sample for comparison with neat CdS and Ce-UiO-NH2. The PL spectrum was used to gain more insight into electron–hole separation processes in the catalysts (Fig. S1d†). Upon 330 nm excitation, the PL spectrum of CdS showed an intense peak at 550 nm (2.25 eV), which well matches to the band-to-band transition (Eg = 2.25 eV), obtained from the optical absorption edge. The PL spectrum of Ce-UiO-NH2 showed a peak centered at 430 nm. The PL spectrum of HJ exhibited two visible peaks centered at 430 and 515 nm. The 515 nm emission peak is attributed to CdS in the HJ, with the blue-shift relative to pure CdS indicating CdS in the HJ forms smaller particles than pure CdS. Both PL peaks were strongly quenched relative to the neat materials, suggesting that the HJ enables charge separation between CdS and Ce-UiO-NH2. This is in agreement with the enhanced EQE of the HJ for H2 evolution.We have previously undertaken analyses of the energetics and electrochemistry of neat CdS and UiO-66s, and based on these analyses made preliminarily conclusions on the relative band alignment and direction of charge separation.21 However, these studies did not include direct measurement of the photogenerated charges. Moreover, the crystallite size reduction of CdS induced by the MOF in the HJ is likely to significantly impact the CdS due to quantum size effects (Fig. 1b and Table S2†). As such, we employ herein both optical transient spectroscopy and spectroelectrochemistry to investigate the underlying charge dynamics and energetics of CdS/Ce-UiO-NH2 HJ photocatalysts.
We first employ transient absorption (TA) spectroscopy to explore the charge transfer mechanism in our CdS/Ce-UiO-NH2 heterojunctions. To achieve efficient HER, the charge carrier lifetimes must be long enough to enable the efficient extraction of holes by methanol and the reduction of protons to molecular H2. Fig. 3 shows the TA spectrum of CdS, Ce-UiO-NH2 and HJ films in water on the μs–s timescale under 420 nm excitation. It is notable that the HJ exhibited a broad photoinduced absorption spectrum from 500–1000 nm, with two absorption peaks at 700 nm and 1000 nm. This absorbance has a half-life of 1 ms, which is substantially longer than the half-life of CdS (2 μs) and Ce-UiO-NH2 (sub-μs) alone. The amplitude of the TA kinetic probed at 700 nm was up to 12 times higher for the HJ than for neat CdS and Ce-UiO-NH2, indicating that there is a much higher photogenerated charge density in the HJ on these timescales (Fig. 3d). Interestingly, the trend in photogenerated charge density on the μs timescale (HJ > CdS > Ce-UiO-NH2) matches the trend in HER efficiency of the three materials.
 | ||
| Fig. 3 (a–c) Transient absorption spectra at different time delays, excited at 420 nm (840 μJ cm−2) and (d) decay dynamics of CdS (black), Ce-UiO-NH2 (red) and the HJ (blue) films in water, probed at 700 nm. | ||
Scavenging tests were further conducted to identify the charge species observed in our TAS data; methanol was used as the hole scavenger, and O2 was used as the electron scavenger. As shown in Fig. S3,† for neat CdS, the bleach probed at 500 nm was partly quenched by O2 and increased by methanol, which indicates this is an electron-dominated signal. The positive absorption signal at 700 nm is quite small and near the resolution limit of our instrument. However, we observed that adding methanol partly quenched this signal, which might suggest it is a hole-dominated signal. The absence of a strong signal from holes in CdS on this microsecond timescale is consistent with reports that trapped holes in CdS have short (ps) lifetimes when measured in ACN.24–26 For the neat Ce-UiO-NH2, there was negligible TA signal even with the addition of oxygen or methanol, demonstrating that charge recombination in the MOF occurs on the sub-microsecond timescale and is faster than charge transfer to methanol or oxygen. For the HJ, the TA spectrum from 500–1000 nm was completely quenched on the sub-μs timescale by 10 vol% methanol (Fig. S3c†), indicating that the long-lived TA spectrum of the HJ is a hole-dominated spectra and 10 vol% methanol is able to extract holes from the HJ on sub-μs timescales.
So far, the samples were excited at 420 nm, which excites both CdS and Ce-UiO-NH2. Now we excite at 560 nm, which preferentially excites Ce-UiO-NH2, to investigate whether Ce-UiO-NH2 acts as a light sensitiser or just as a charge separator. The results are shown in Fig. 4. For neat CdS, no TAS signal was observed (Fig. 4a), as expected, because it has no ground state absorption at 560 nm (Fig. 1e). Neat Ce-UiO-NH2 also exhibited no signal (Fig. 4a), which was attributed to rapid charge recombination on the sub-microsecond timescale and is consistent with its TA spectrum under 420 nm excitation (Fig. 3b). However, the TA spectrum of the HJ showed the same broad positive absorbance at 600–1000 nm alongside a negative signal at 500–600 nm (Fig. 4b). These data prove that Ce-UiO-NH2 can act as a light absorber and photogenerate long-lived holes. The 500 nm bleach in the HJ (Fig. 4c) is not observed when exciting at 420 nm (Fig. 3c), possibly because the overlapping positive MOF signal dominates over the negative CdS blench. The bleaching of the CdS ground state in Fig. 4c and the increase in the MOF hole signal in Fig. 4d indicate that in the HJ there is electron transfer from the photoexcited MOF to the unexcited CdS. Overall, these observations based on 560 nm excitation suggest that Ce-UiO-NH2 acts as light absorber and electron donor in our HJ.
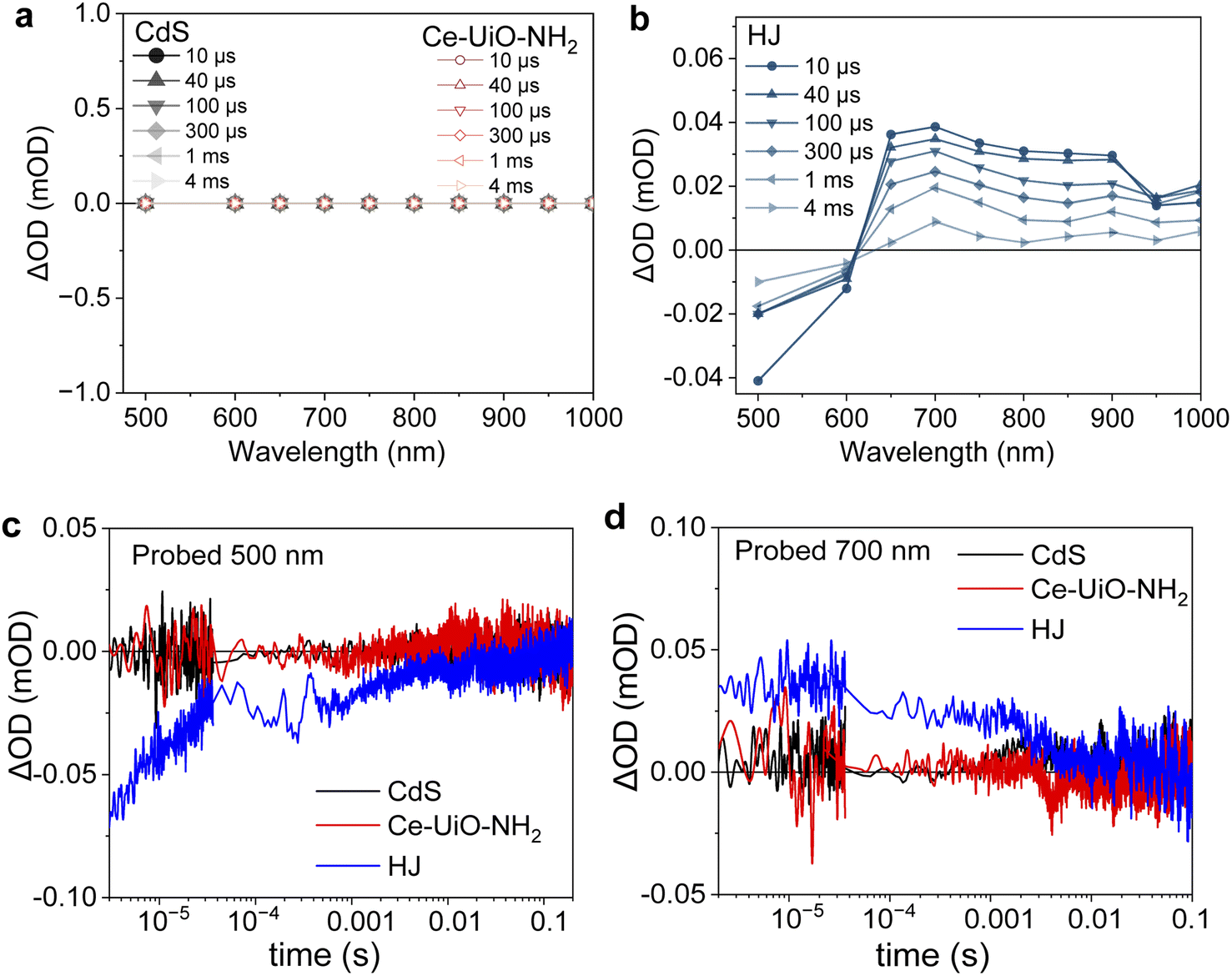 | ||
| Fig. 4 Transient absorption spectra at different time delays of (a) CdS and Ce-UiO-NH2 and (c) HJ, excited at 560 nm (840 μJ cm−2) and comparison of the decay dynamics of CdS (black), Ce-UiO-NH2 (red) and HJ (blue) films in water, probed at (b) 500 nm and (c) 700 nm. | ||
Our TAS data suggest that the long-lived photoinduced absorption observed for our HJ samples results from Ce-UiO-NH2 holes. To provide further support to this conclusion, spectroelectrochemical measurements (SEC) were conducted to investigate the electron and hole spectra of CdS and Ce-UiO-NH2, respectively. As shown in Fig. 5a, the application of a positive bias to CdS films results in broad bleaching of the absorbance change spectrum, which increases towards the blue visible light region. This bleaching is tentatively assigned to hole formation associated with oxidation of S2− species in CdS, which is one of the origins of CdS self-corrosion and often limits its photocatalytic performance.24,27 The application of a comparable positive potential to Ce-UiO-NH2 resulted in a broad absorption spectrum across the entire visible spectrum and is assigned to Ce-UiO-NH2 holes. A very similar broad absorption spectrum is observed when positive bias is applied to the HJ (Fig. 5c), suggesting that the Ce-UiO-NH2 is more readily oxidized than the CdS and that oxidation of the HJ happens on the Ce-UiO-NH2. This SEC hole spectrum for Ce-UiO-NH2 (Fig. 5a) is also similar to the hole-dominated signal observed in our HJ TAS measurements (Fig. 3c), confirming our assignment of this TAS HJ signal to the photogeneration of long-lived holes on the Ce-UiO-NH2. For SEC reduction spectra (Fig. 5b and d), the similarity of the SEC spectra measured on CdS and on the HJ suggests that in the HJ, reduction happens more readily on the CdS. The behaviour of the HJ under applied bias, in which oxidation happens on the Ce-UiO-NH2 and reduction happens on the CdS, is consistent with our conclusion that charge separation in our photoexcited HJ results in hole accumulation on the Ce-UiO-NH2 and electron accumulation primarily on the CdS.
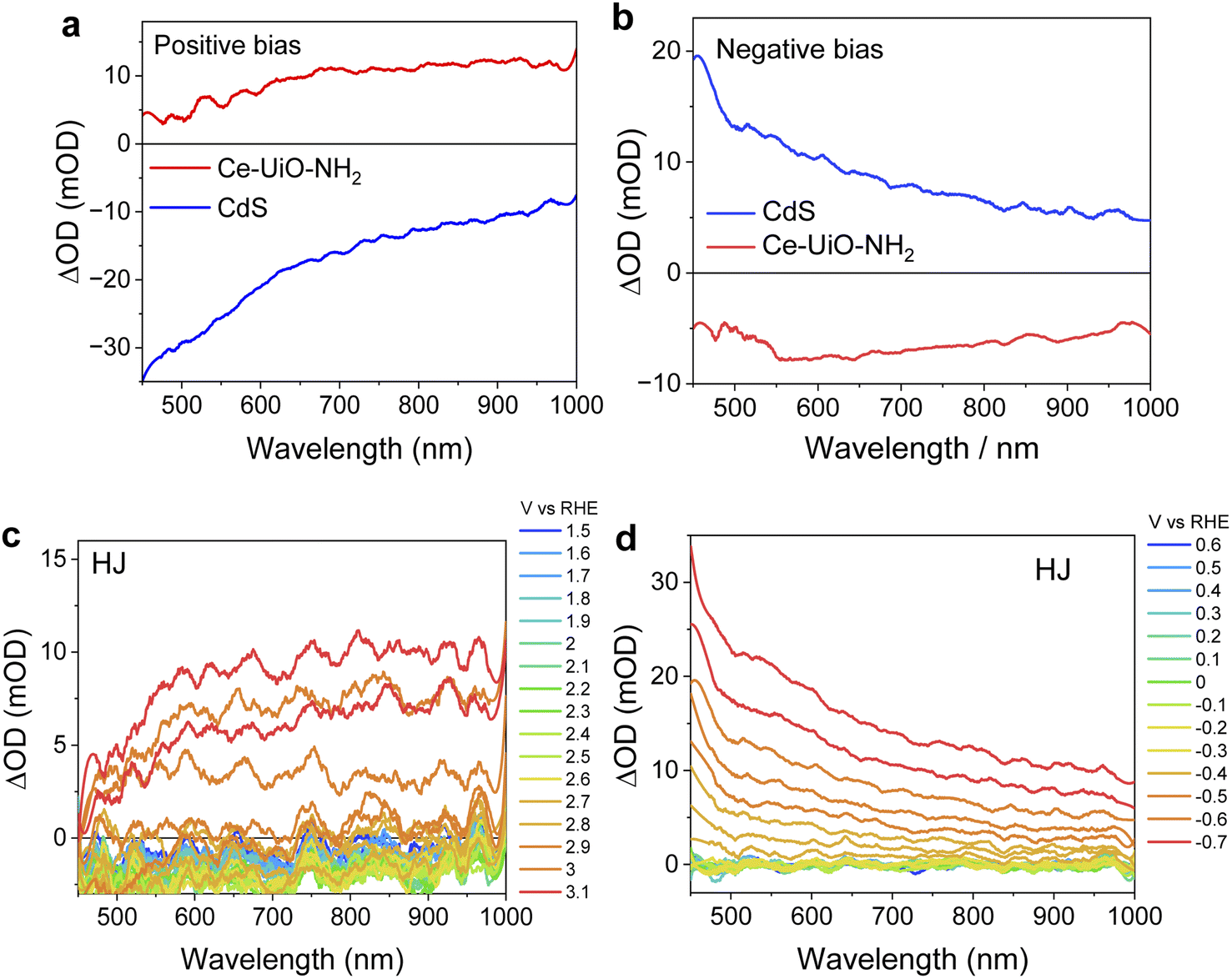 | ||
| Fig. 5 Absorbance difference spectra at positive bias for (a) CdS and Ce-UiO-NH2, and (c) HJ, respectively and at negative bias for (b) CdS and Ce-UiO-NH2 and (d) HJ, respectively. All films were measured in 0.1 M NaClO4 solution, pH = 7. | ||
Mechanism and discussion
Scheme 1 summarizes the charge separation generated in the optimized HJs determined herein. Our TA studies showed that a much higher yield of long-lived charges survives to the μs–s timescale in the HJ when compared to its individual components. This is strong evidence that the HJ helps generate charges which survive long enough to contribute to HER.28 TAS measurements were also undertaken using selective Ce-UiO-NH2 excitation, indicating that Ce-UiO-NH2 acts as a light absorber and electron donor. The direction of charge transfer in HJ was further confirmed by SEC. The reduced HJ and reduced CdS SEC spectra have similar shapes, indicating electron accumulation in the HJ primarily occurs on the CdS. On the other hand, the oxidised HJ SEC and oxidised Ce-UiO-NH2 SEC spectra have similar shapes, indicating that hole accumulation in the HJ primarily occurs on the Ce-UiO-NH2. These hole spectra matched the photoexcited TA spectrum of Ce-UiO-NH2 in the presence of methanol, indicating that sub-μs hole extraction by methanol enables hole transfer to, and accumulation on, the MOF.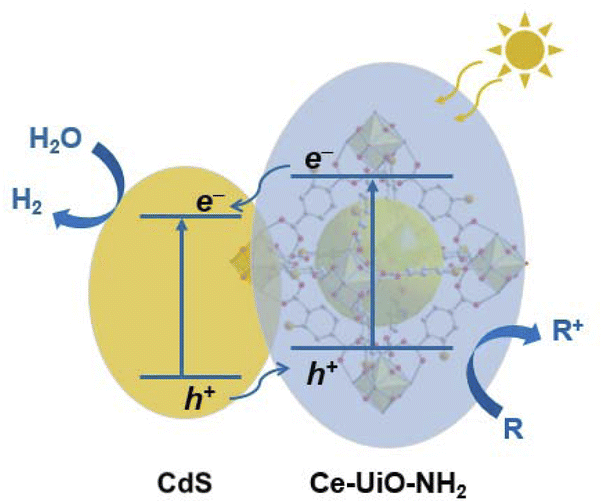 | ||
| Scheme 1 Mechanism of the CdS/Ce-UiO-NH2 heterojunction for enhanced HER in the presence of a sacrificial electron donor ‘R’. | ||
We note that the direction of charge transfer reported herein is the inverse of that concluded in previous studies of CdS/Zr-UiO-66 and CdS/Zr-UiO-66-NH2 heterojunctions.17,29,30 This may be the result of Ce-doping facilitating the oxidation of our MOF.13,31 It may also result from the different timescales employed for TAS studies. On ultrafast (picosecond) timescales, charge transfer may proceed prior to charge trapping and the direction of transfer is therefore dominated by band edge alignment. On longer (microsecond) timescales, which are likely more relevant to catalysis (e.g.: HER), charges will localize on the most energetically favorable trapping sites within the heterojunction, which may result in a different charge distribution between the two components of the HJ. For the HJ herein, our observation of hole localization on the MOF is particularly advantageous in minimizing CdS corrosion driven by hole accumulation on this material – as evidenced by the high stability of our HJ.
The CdS/Ce-UiO-NH2 heterojunctions employed in this work show a modest EQE of 2.2% for HER. This EQE is limited, at least in part, by the relatively modest light absorption of our photocatalyst suspension. Secondly, the two-step loading most likely results in CdS NPs filling into the pores. Such CdS may be less able to drive HER. It may also decrease the particle size of some of the CdS, shifting their bandgap to >3 eV.27 Thirdly, in the study herein, no co-catalyst was added for the H2 evolution experiments. For obtaining a higher EQE, adding an established co-catalyst like Pt is an obvious strategy.32–34 Moreover, alternate sacrificial regents should be explored. Our previous work showed higher EQE in Na2S/Na2SO3 than in methanol, but with a lower stability. Most related photocatalytic HER measurements in the literature have been conducted in either ACN with lactic acid solution; with Na2S/Na2SO3 solution for CdS; or with TEOA for MOFs. However, recent studies indicate that some of these sacrificial regents also cause instability and photocorrosion.35,36
Conclusions
In summary, we have reported CdS-loaded Ce-UiO-NH2 heterojunctions for enhanced H2 evolution under visible light in the presence of a methanol hole scavenger. Under visible light, Ce-UiO-NH2 has negligible H2 evolution. The EQE increased from 0 to 2.2% after 40% CdS loading. This HJ EQE was also 11 times higher than neat CdS. To understand the mechanism of this CdS/Ce-UiO-NH2 heterojunction, μs–s TA spectroscopy and spectroelectrochemistry measurements were further conducted. Firstly, long-lived charges on the millisecond timescale were observed in CdS/Ce-UiO-NH2 heterojunctions, indicating that the heterojunction can achieve effective charge separation. The photogenerated charge density on μs–s timescale follows the order of HJ > CdS > Ce-UiO-NH2, which correlates with their H2 evolution efficiency. Secondly, the specific role of Ce-UiO-NH2 as electron donor in heterojunction was confirmed by preferentially exciting Ce-UiO-NH2. Thirdly, the charge transfer direction was further confirmed by spectroelectrochemical measurements, comprising electron transfer from Ce-UiO-NH2 to CdS, and hole transfer from CdS to Ce-UiO-NH2. The direction of charge transfer is the opposite of recent studies of Zr-UiOs, with hole accumulation on the MOF preventing the corrosion of CdS in HJ. Thus, this work shows the potential of Ce-MOF in photocatalytic HER not only as a driver of charge separation but also as an electron donor, demonstrating that metal node substitution is a promising design strategy for enhanced photoactive MOFs and efficient heterojunctions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
W. H.: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, and writing – original draft. S. A. J. H.: formal analysis, methodology, software, validation, visualization, and writing – review & editing. S. G.-C.: formal analysis, investigation, validation, and visualization. S. Y.: data curation, investigation, and validation. H. Z.: data curation, formal analysis, and resources. J. R. D.: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from the China Scholarships Council (No. 202206320209) and the METHASOL project which receives funding from the European Union Horizon 2020 Research and Innovation Programme under Grant Agreement N101022649.References
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981–987 CrossRef CAS PubMed.
- M. Ball and M. Weeda, Int. J. Hydrogen Energy, 2015, 40, 7903–7919 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- Y.-C. Wang, X.-Y. Liu, X.-X. Wang and M.-S. Cao, Chem. Eng. J., 2021, 419, 129459 CrossRef CAS.
- M. A. Syzgantseva, C. P. Ireland, F. M. Ebrahim, B. Smit and O. A. Syzgantseva, J. Am. Chem. Soc., 2019, 141, 6271–6278 CrossRef CAS PubMed.
- Y. Li, H. Xu, S. Ouyang and J. Ye, Phys. Chem. Chem. Phys., 2016, 18, 7563–7572 RSC.
- Y. Zhao, J. Wang, W. Zhu, L. Liu and R. Pei, Nanoscale, 2021, 13, 4505–4511 RSC.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- F. A. Son, A. Atilgan, K. B. Idrees, T. Islamoglu and O. K. Farha, Inorg. Chem. Front., 2020, 7, 984–990 RSC.
- X. P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- J. D. Xiao, Q. C. Shang, Y. J. Xiong, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 9389–9393 CrossRef CAS PubMed.
- J. Qiu, X. Zhang, K. Xie, X.-F. Zhang, Y. Feng, M. Jia and J. Yao, J. Colloid Interface Sci., 2019, 538, 569–577 CrossRef CAS PubMed.
- X. Hou, L. Wu, L. Gu, G. Xu, H. Du and Y. Yuan, J. Mater. Sci.:Mater. Electron., 2019, 30, 5203–5211 CrossRef CAS.
- H.-Q. Xu, S. Yang, X. Ma, J. Huang and H.-L. Jiang, ACS Catal., 2018, 8, 11615–11621 CrossRef CAS.
- L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362–1391 RSC.
- D. Ayodhya and G. Veerabhadram, J. Sci.:Adv. Mater. Devices, 2019, 4, 381–391 Search PubMed.
- L. J. Shen, M. B. Luo, Y. H. Liu, R. W. Liang, F. F. Jing and L. Wu, Appl. Catal., B, 2015, 166, 445–453 CrossRef.
- W. Hou, C. Chen, Y. Wang and Y. Xu, Catal. Sci. Technol., 2022, 12, 4012–4019 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- I. Strauss, K. Chakarova, A. Mundstock, M. Mihaylov, K. Hadjiivanov, N. Guschanski and J. Caro, Microporous Mesoporous Mater., 2020, 302, 110227 CrossRef CAS.
- W. Wang, Y. Tao, J. Fan, Z. Yan, H. Shang, D. L. Phillips, M. Chen and G. Li, Adv. Funct. Mater., 2022, 32, 2201357 CrossRef CAS.
- Z. Yan, L. Du and D. Lee Phillips, RSC Adv., 2017, 7, 55993–55999 RSC.
- X. P. Wu, L. Gagliardi and D. G. Truhlar, J. Chem. Phys., 2019, 150, 041701 CrossRef PubMed.
- S. K. Haram, B. M. Quinn and A. J. Bard, J. Am. Chem. Soc., 2001, 123, 8860 CrossRef CAS PubMed.
- R. Godin and J. R. Durrant, Chem. Soc. Rev., 2021, 50, 13372–13409 RSC.
- Z. Lian, Z. Li, F. Wu, Y. Zhong, Y. Liu, W. Wang, J. Zi and W. Yang, Commun. Chem., 2022, 5, 93 CrossRef CAS PubMed.
- Z. Li, J. Zi, X. Luan, Y. Zhong, M. Qu, Y. Wang and Z. Lian, Adv. Funct. Mater., 2023, 33, 2303069 CrossRef CAS.
- P. Parnicka, W. Lisowski, T. Klimczuk, A. Mikolajczyk and A. Zaleska-Medynska, Appl. Catal., B, 2022, 310, 121349 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H.-L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- I. Vamvasakis, B. Liu and G. S. Armatas, Adv. Funct. Mater., 2016, 26, 8062–8071 CrossRef CAS.
- Y. Chen, W. Zhong, F. Chen, P. Wang, J. Fan and H. Yu, J. Mater. Sci. Technol., 2022, 121, 19–27 CrossRef CAS.
- J. Wang, A. S. Cherevan, C. Hannecart, S. Naghdi, S. P. Nandan, T. Gupta and D. Eder, Appl. Catal., B, 2021, 283, 119626 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01473a |
| ‡ We dedicated this paper to Prof. Yiming Xu, who was one of supervisors for Dr Wenqing Hou. He suddenly passed away in 2023 as we were studying this work. He was an incredible supervisor, co-worker and friend who is dearly missed. |
| This journal is © The Royal Society of Chemistry 2025 |