
Ethanol as a hydrogen carrier with a value-added co-product†
Andrew R.
Rander
 a,
Shayna R.
Kohl
a,
Valeriy
Cherepakhin
a,
Long
Zhang
a,
Van K.
Do
a,
Hanna
Breunig
b and
Travis J.
Williams
*a
a,
Shayna R.
Kohl
a,
Valeriy
Cherepakhin
a,
Long
Zhang
a,
Van K.
Do
a,
Hanna
Breunig
b and
Travis J.
Williams
*a
aDonald P. and Katherine B. Loker Hydrocarbon Research Institute, Wrigley Institute for Environment and Sustainability, Department of Chemistry, University of Southern California, Los Angeles, California 90089-1661, USA. E-mail: travisw@usc.edu
bSustainable Energy Systems Group, Energy Analysis and Environmental Impacts Division, Lawrence Berkeley National Lab, 1 Cyclotron Road, 90-2012, Berkeley, California 94720, USA
First published on 28th November 2024
Abstract
Traditional liquid organic hydrogen carriers (LOHCs) rely on return/re-charge of the carrier and financial subsidy. We show here that ethanol, available at scale from fermentation, can be a revenue-positive hydrogen carrier, owing to the value of its potassium acetate co-product, itself an emerging fertilizer.
High efficiency liquid organic hydrogen carriers (LOHCs) are an essential link in H2 storage, because they enable storage densities superior to the parent compressed gas and are easy to transport. Traditionally, the spent fuel that results from H2 release from a 2-way carrier is then transported to a source of low-cost hydrogen for re-charge. Commercialization of this concept has seen the use of functionalized toluenes; however, these only operate with financial subsides.1 While ethanol to ethyl acetate has been demonstrated as a 2-way hydrogen carrier that releases one equivalent of H2 per ethanol,2 we reexamine ethanol here, contemplating a 1-way carrier strategy that produces two equivalents of H2 and potassium acetate, an advantageous, emerging fertilizer.
Potassium acetate, a high value agrochemical, has emerged as a superior source of potassium for plants, with a market projected to reach $290.66 million by the end of 2030.3,4 Thus, we see ethanol as a 1-way H2 carrier that is “self-subsidizing” by the value created in its dehydrogenated co-product; in fact at present pricing the value increase is ca. 4×. Incumbent technologies for potassium acetate manufacturing involve multi-step syntheses, including the Monsanto or Cativa process, relying on a homogeneous rhodium or iridium catalysts respectively to affect the carbonylation of methanol derived from petrochemical sources.5
While a direct catalytic ethanol-to-acetate process is a straightforward extension of known alcohol-to-carboxylate dehydrogenation methods,6–13 such reactions typically require a high boiling co-solvent and pressurized systems have never been developed. Further, process commercialization would also require scalable, high turnover, solvent-free conditions and a pressure tolerant catalytic system; technology that has not been introduced. Selectivity also frustrates alcohol dehydrogenation catalysts: while several examples of catalytic ethanol dehydrogenation form the Tishchenko product, ethyl acetate, or the Guerbet product, 1-butanol, none focus on the dehydrogenation subsidizing the production of hydrogen with the carboxylate.2,9,13–21 We, therefore, see a need to introduce dehydrogenation of ethanol to potassium acetate with the ability to self-pressurize the produced hydrogen for on-demand hydrogen production.
Based on our group's work with iridium chelates for the dehydrogenation of formic acid,22–24 we screened complex 1 (Fig. 1) at 0.01 mol% in neat ethanol with an excess of potassium hydroxide. 1H NMR confirms acetate production after a 24 hours reaction. Complex 1 (CCDC 1415049) and its ruthenium congener (3; CCDC 1510912)25 both perform modestly with the acetate yields of 22% and 27%, respectively (Table 1, entries 1 and 2). NMR suggests that 3 has greater selectivity than 1, as the latter contained Guerbet products such as 2-ethyl-1-butanol while the former contains only product and trace butyrate (see ESI†).
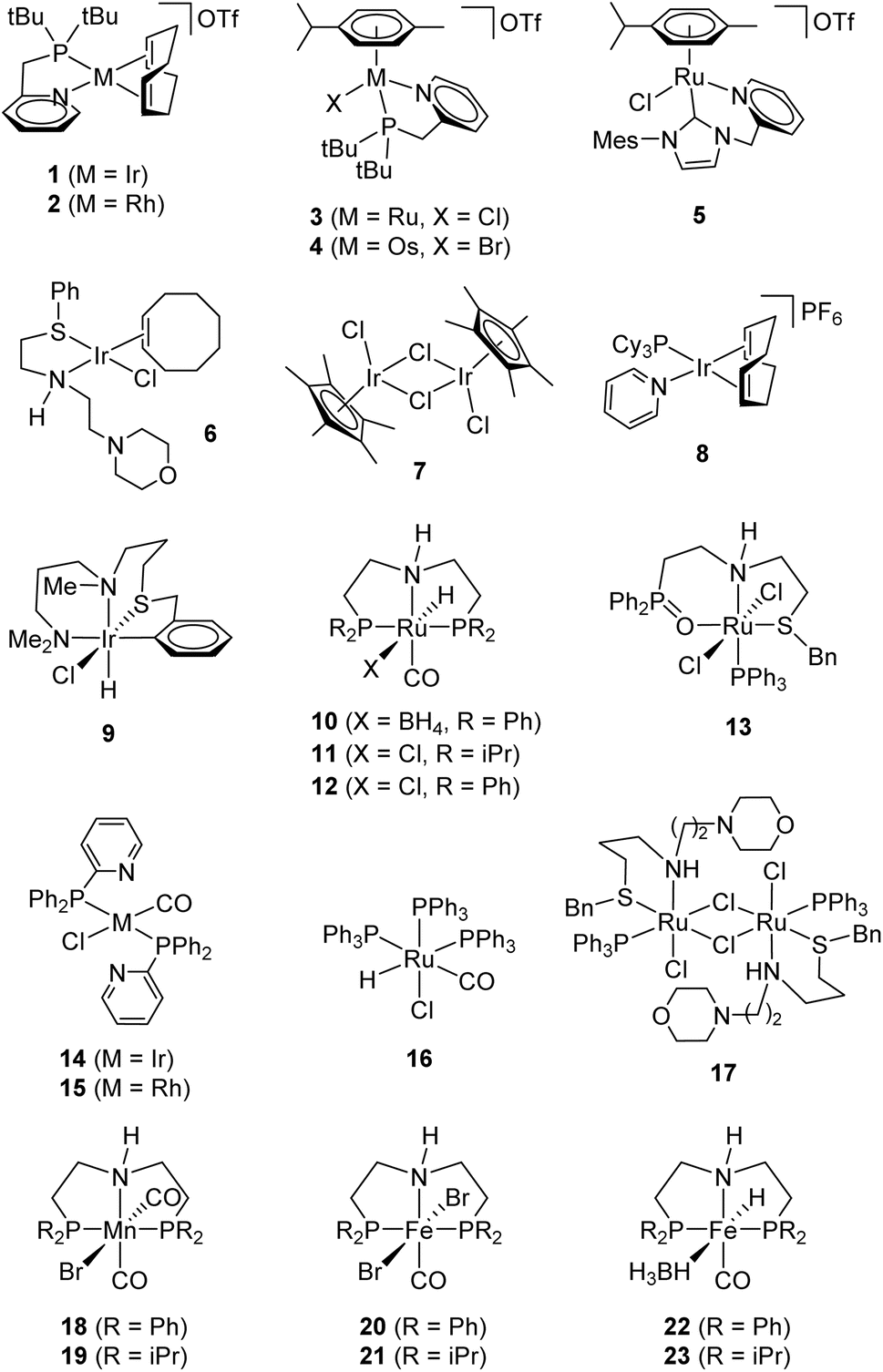 | ||
| Fig. 1 Compounds tested as ethanol dehydrogenation catalysts. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Acetateb (%) | Anionsc (mol%) | |||
| OH− | Formate | Acetate | Butyrate | |||
| a Conditions: catalyst, KOH·1/2H2O (0.56 g, 9 mmol), ethanol (0.92 g, 20 mmol), and toluene (10 mL) were heated at reflux (oil bath, 120 °C) under N2 for 42 h. b Determined by 1H NMR using DMF as an internal standard. c Determined by acid–base back titration and 1H NMR. | ||||||
| 1 | 1 | 10 | 76 | 2 | 22 | 0 |
| 2 | 2 | 29 | 63 | 22 | 15 | 0 |
| 3 | 3 | 26 | 60 | 12 | 27 | 1 |
| 4 | 4 | 37 | 71 | 0 | 28 | <1 |
| 5 | 5 | 87 | 18 | 3 | 79 | 0 |
| 6 | 6 | 18 | 58 | 1 | 40 | <1 |
| 7 | 7 | 7 | 80 | 1 | 19 | 0 |
| 8 | 8 | 19 | 71 | 1 | 28 | 0 |
| 9 | 9 | 1 | 63 | 2 | 35 | 0 |
| 10 | 10 | 80 | 5 | 3 | 90 | 1 |
| 11 | 11 | 75 | 21 | 7 | 71 | 1 |
| 12 | 12 | 68 | 14 | 3 | 83 | 0 |
| 13 | 13 | 14 | 66 | 0 | 32 | 2 |
| 14 | 14 | 17 | 73 | 1 | 26 | 0 |
| 15 | 15 | 12 | 87 | 1 | 11 | <1 |
| 16 | 16 | 7 | 64 | 32 | 4 | 0 |
| 17 | 17 | 24 | 56 | 25 | 19 | 0 |
| 18 | 18 | 26.5 | 51 | <1 | 49 | <1 |
| 19 | 19 | 7 | 61 | 4 | 39 | 2 |
| 20 | 20 | 66 | 52 | 2 | 47 | <1 |
| 21 | 21 | 8 | 70 | 4 | 30 | <1 |
| 22 | 22 | 15 | 83 | 3 | 20 | <1 |
| 23 | 23 | 9 | 69 | 3 | 31 | 5 |
We screened a group of compounds (Fig. 1) as catalysts in the ethanol dehydrogenation (Table 1). The reaction was conducted between ethanol and KOH·1/2 H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in boiling toluene for 42 h with 0.1 mol% catalyst. 1H NMR analysis of the products consistently shows acetate (1.90 ppm), formate (8.56 ppm), and butyrate ions (2.13 ppm). Quantitative 1H NMR (vs. internal DMF standard) and acid–base back titration provide the molar distribution of the carboxylates and the conversion of KOH. Several complexes (entries 1–4, 6–9, 13–17) performed poorly generating less than 40%.22,25–31 Complexes 2, 3, 16, and 17 produced large portions of formate, apparently generated by decarbonylation of an intermediate acetaldehyde; ruthenium(II) complex 16 prefers this pathway (8:1, formate to acetate).25–27 The complexes that afforded the highest yields of acetate are ruthenium-based (entries 5, 10–12, 70–90%).21,32,33 For example, we designed 5 based on the known efficiency of NHC-ligated iridium dehydrogenation catalysts, which maintain both good reactivity for H2 abstraction while remaining resistant to deactivation.34
:1) in boiling toluene for 42 h with 0.1 mol% catalyst. 1H NMR analysis of the products consistently shows acetate (1.90 ppm), formate (8.56 ppm), and butyrate ions (2.13 ppm). Quantitative 1H NMR (vs. internal DMF standard) and acid–base back titration provide the molar distribution of the carboxylates and the conversion of KOH. Several complexes (entries 1–4, 6–9, 13–17) performed poorly generating less than 40%.22,25–31 Complexes 2, 3, 16, and 17 produced large portions of formate, apparently generated by decarbonylation of an intermediate acetaldehyde; ruthenium(II) complex 16 prefers this pathway (8:1, formate to acetate).25–27 The complexes that afforded the highest yields of acetate are ruthenium-based (entries 5, 10–12, 70–90%).21,32,33 For example, we designed 5 based on the known efficiency of NHC-ligated iridium dehydrogenation catalysts, which maintain both good reactivity for H2 abstraction while remaining resistant to deactivation.34
Pincer complexes 10–12 show promising reactivity (entries 10–12); we attribute the advantage of 10 over 12 to the known lability of the borohydride ligand to facilitate catalyst initiation.21,31,32 Finally, we measured the performance of 10 and a series of manganese(I) and iron(II) PNP-pincer complexes 18–23 (entries 18–23), which are known to catalyze alcohol dehydrogenation and hydrogen transfer reactions.35–40 Despite being derived from the earth-abundant metals, complexes 18–23 show inferior activity (TOF = (1–5) × 10−3 s−1) compared to their ruthenium congener 10 (TOF = 1.3 s−1).35–40 Unlike 10, these also suffer from photosensitivity (19) and hydrolysis (21) making them unfit for the practical use.20,40 Of all of the catalysts we screened, 23 returned the highest portion of butyrate, apparently through a Guerbet pathway. We had previously suggested that the ruthenium metal center is of upmost importance in catalytic dehydrogenation efficiency which is consistent with our findings.7,20
We selected catalyst 10 to proceed to optimize conditions (Table 2) due to its rate and selectivity. We found that reducing the KOH loading to 0.5 eq. or less suppresses the side processes and increases the acetate yield to 99% (entries 5 and 6). Furthermore, we showed that a high yield of acetate (96%) is attainable in neat ethanol at 80 °C (entry 7). By contrast, we observe (1H NMR) that operating the reaction in excess base lowers yields and compromises purity. These observations point to an operational scenario of slow addition of ethanol/hydroxide solution in a continuous process.
| Entry | KOH (eq.) | 10 (mol% vs. EtOH) | Carboxylatesb (mol%) | Acetatec (%) |
|---|---|---|---|---|
| a Conditions: 10, KOH·1/2H2O, ethanol (0.92 g, 20 mmol), and toluene (10 mL) were heated at reflux (oil bath, 120 °C) under N2 for 42 h. b Determined by acid–base back titration. c Determined by 1H NMR. d Conditions: 10, KOH·1/2H2O, and ethanol (5.53 g, 120 mmol) were heated at reflux (80 °C) under N2 for 42 h. | ||||
| 1 | 1.97 | 0.13 | 48 | 27 |
| 2 | 1.32 | 0.09 | 47 | 18 |
| 3 | 0.95 | 0.10 | 95 | 77 |
| 4 | 0.74 | 0.10 | 98 | 73 |
| 5 | 0.49 | 0.10 | 99 | 94 |
| 6d | 0.14 | 0.02 | 96 | 87 |
Gas compression costs can contribute up to 80% of hydrogen delivery costs,41 depending on the application, and the entropy of dehydrogenation can supply this pressure cost-free.23 We thus investigated self-pressurizing conditions for our reaction. While complex 10 delivers a modestly lower yield of (74%) when run under self-pressurizing conditions (Table 3, entry 4), complex 1 demonstrates improved performance under self-pressurizing conditions, achieving 67% yield (entry 1) compared to 22% yield under ambient conditions.
| Entry | Catalyst | EtOH (eq.) | Time (h) | Final pressure (bar) | Carboxylatesb (mol%) | Acetatec (%) |
|---|---|---|---|---|---|---|
| a Conditions: catalyst (0.1 mol% vs. ethanol), KOH·1/2H2O (1.92 g, 29.5 mmol), ethanol, and toluene (10 mL) were heated in a 125 mL Parr reactor at 120 °C. b Determined by acid–base back titration. c Determined by 1H NMR. d Conditions: 10 (13 mg, 0.04 mol% vs. KOH), KOH·1/2H2O (3.7 g), and ethanol (16 g) were heated in the Parr reactor at 120 °C. | ||||||
| 1 | 1 | 1 | 45 | 8 | 67 | 64 |
| 2 | 1 | 2 | 25 | 6 | 50 | 28 |
| 3 | 5 | 1 | 47 | 9 | 66 | 70 |
| 4 | 10 | 1 | 45 | 9 | 74 | 82 |
| 5 | 10 | 2 | 45 | 14 | 98 | 95 |
| 6d | 10 | 6 | 68 | 26 | 98 | >99 |
We've previously shown that 1 activates to a multi-metal cluster via carbonylation, which is facilitated by pressurization.23 Similar trends appear when varying the ratio of reactants at pressure as seen under ambient conditions (entries 4 and 5): decreasing the concentration of hydroxide resulted in 98% yield of acetate in a reaction of 10. Further, conditions with six equivalents of ethanol to hydroxide resulted in 98% yield, demonstrating that co-solvent is unnecessary. With high yielding conditions, we turned to determine the efficiency of selected catalysts at increased scale (see ESI†). Over the course of several months, 10 and 1 were each allowed to react on a 10 gram (KOAc) scale under our optimized ambient pressure, batch reaction conditions.
Through three reactions using the same catalytic mixture, 10 showed 55000 turnovers without loss of reactivity (See ESI Table S4†). Catalyst 1 delivered 168000 turnover numbers over one three-month reaction, similarly without deactivation (See ESI Table S5†). Catalyst 10 and 1 can be recovered and reused through several directed cycles presaging the use of a continuous operation reactor.
The mechanism of alcohol dehydrogenation with ruthenium Noyori type catalysts is well known.21,42–48 System 10 appears to conform: the deprotonation of NH and dissociation of BH4− give the active catalyst 10a (Scheme 1).42,44–46 Analysis of the post-catalytic mixture suggests the presence of two complexes 10b and 10c (89% yield combined).49 These are fac- and mer-[(PNP)RuH2(CO)] featuring hydride signals at δH = −6.62 ppm (10b), and −5.77, −5.99 ppm (10c). The 31P{1H} NMR contains signals at δP = 64.72 ppm (10c) and 55.39 ppm (10b) with no shared correlations in the 1H–31P HMBC.
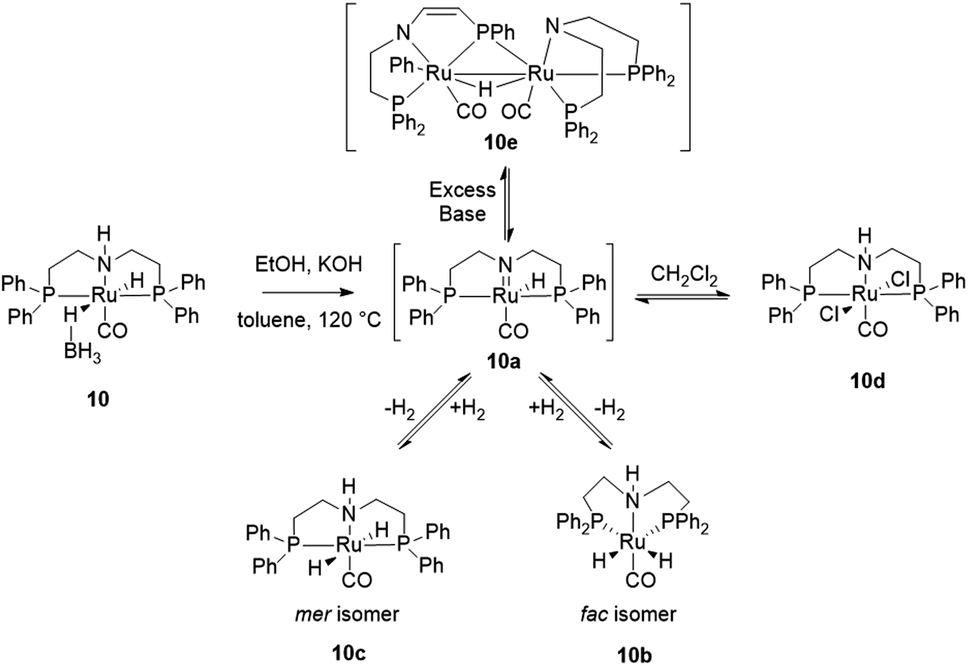 | ||
| Scheme 1 Derivatization of 10 under the ethanol dehydrogenation conditions. | ||
Both compounds have the identical mass (m/z = 572 Da, MALDI)50 and elemental composition. A 1H{31P} experiment (see ESI†) shows interconversion between 10c and 10b, appropriate for respective mer- and fac-isomers. Complexes 10b and 10c are both active in the ethanol dehydrogenation. Interestingly, 10b and 10c are not stable to chlorinated solvents: a known dichloro derivative 10d (δP = 48 ppm)49 is formed within a few days in dichloromethane. Heating the reaction mixture past ethanol consumption results in further derivatization of 10b and 10c. MALDI suggests formation of a previously characterized diruthenium complex 10e (m/z = 1141.63 Da).45 Its formation can be seen as a base-induced tautomerization of 10a, followed by insertion of Ru(0) to one of the phenyl–phosphorus bonds.
While some estimate negative carbon impact of ethanol as a gasoline additive, we calculate positive carbon impact in its use as a hydrogen carrier. Steam methane reforming (SMR) for H2 production generates 11.9 kgCO2-eq. per kgH2. As corn or soybean production for ethanol or glycerol (via biodiesel), respectively, requires fertilizer and energy for conversion, the hydrogen generated from these carriers will have a carbon intensity. If 2 equivalents of H2 are liberated, and the carbon intensity of manufacturing ethanol is 51.4 gCO2-eq MJ−1, the minimum carbon intensity of H2 would be 8.03 kgCO2 per kgH2 without the production of co-products. The process emits biogenic CO2 along with H2, which in our case would eventually be emitted upon the use of the acetate co-product. Allocation of emissions between H2 and coproducts can be done based on mass, energy content, or economic value: with a 1 to 41 weight ratio of H2 to sodium acetate, mass allocation would result in a carbon intensity of 0.09 kgCO2 per kgH2. For perspective, wind-powered electrolysis has a carbon intensity of 0.6 kgCO2 per kgH2 assuming an electricity emission intensity of 11 g kW−1 h−1.51
The power of our reaction is the ability to produce pure H2 with a coproduct whose value can negate production costs. The vast majority of hydrogen sold today is produced from non-renewable, hydrocarbon streams with only 4% of global hydrogen being considered “green hydrogen” typically produced by electrolysing water, which still faces challenges of gas separation, selectivity, and metal cost.52 Furthermore, the largest electrolysis units are owned and operated by the agrochemical industry,52 a sector with a need for pure hydrogen and an efficient fertilizer production. While our method addresses both, its H2 product is not “green” according to the electrolysis-based definition. Hydrogen derived from biomass is not classified in the current colour definition, so we refer to ours as “clear” hydrogen, submitting this as a suitable designation for hydrogen derived from biomass.
In sum, we have demonstrated the first direct one-pot synthesis of potassium acetate from neat ethanol, providing two equivalents of H2, potentially at pressure, and a biologically advantageous fertilizer. The strategy obviates co-products, CO and CO2, associated with formic acid and methanol dehydrogenation,53–57 and eliminates nearly 99% of the carbon intensity of ethanol-based H2 available for other methods. Several catalysts are effective for the reaction, with each offering useful features for use cases requiring pressure, longevity, or both. Popular Ru-MACHO systems are particularly attractive, and we show their speciation under ethanol dehydrogenation conditions. Overall, favorable carbon impact, yield, and selectivity of the system makes ethanol a very attractive 1-way liquid hydrogen carrier.
Data availability
All data supporting this communication has been included and is available as part of the ESI† free of charge. Crystallographic, spectral, and experimental information, X-ray data for 2, X-ray data for 5.Author contributions
Conceptualization, data curation, formal analysis, investigation, methodology, project administration, validation, visualization: A. R. R, S. R. K., V. C. Original draft: A. R. R. Synthesis of 5: L. Z. Synthesis and crystal structure of 2: V. D. Life-cycle analysis: H. B. Funding acquisition, resources, supervision, writing review & editing: T. J. W.Conflicts of interest
T. J. W. is a founder of the Catapower Inc., which sells some complexes discussed herein.Acknowledgements
This work is sponsored by the US Department of Energy, DE-EE0011096 (USC) and DE-AC02-05CH11231 (LBNL). A. R. R. thanks the Wrigley Institute for Environment and Sustainability for fellowship assistance. We thank the NSF (CHE-2018740, DBI-0821671, CHE-0840366), the NIH (S10 RR25432), and USC Research and Innovation Instrumentation Awards for analytical tools. We thank Mr Justin Lim and Mr Anushan Alagaratnam for insightful discussions.Notes and references
- A Final Link in the Global Hydrogen Supply Chain.
- B. L. Tran, S. I. Johnson, K. P. Brooks and S. T. Autrey, ACS Sustainable Chem. Eng., 2021, 9(20), 7130 CrossRef CAS.
- W. E. Shafer and D. W. Reed, J. Plant Nutr., 1986, 9(2), 143–157 CrossRef CAS.
- Potassium Acetate Market Size 2023-2030 Global Industrial Analysis, Market Share, Top Key Players, https://www.linkedin.com/pulse/potassium-acetate-market-size-2023-2030-global-analysis-aiwale-ixipf/?trk=article-ssr-frontend-pulse_more-articles_related-content-card, (accessed March 2024).
- A. W. Budiman, J. S. Nam, J. H. Park, R. I. Mukti, T. S. Chang and J. W. Bae, et al. , Catal. Surv. Asia., 2016, 20(3), 173–193 CrossRef CAS.
- V. Cherepakhin and T. J. Williams, ACS Catal., 2018, 8(5), 3754–3763 CrossRef CAS PubMed.
- V. Cherepakhin and T. J. Williams, Synthesis, 2021, 53(6), 1023–1034 CrossRef CAS.
- E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nature Chem., 2013, 5(2), 122–125 CrossRef CAS PubMed.
- A. Sarbajna, I. Dutta, P. Daw, S. Dinda, S. M. W. Rahaman and A. Sarkar, et al. , ACS Catal., 2017, 7(4), 2786–2790 CrossRef CAS.
- D. R. Pradhan, S. Pattanaik, J. Kishore and C. Gunanathan, Org. Lett., 2020, 22(5), 1852–1857 CrossRef CAS PubMed.
- L. Zhang, D. H. Nguyen, G. Raffa, X. Trivelli, F. Capet and S. Desset, et al. , ChemSusChem, 2016, 9(12), 1413–1423 CrossRef CAS PubMed.
- H. Li and M. B. Hall, J. Am. Chem. Soc., 2014, 136(1), 383–395 CrossRef CAS PubMed.
- P. Sponholz, D. Mellmann, C. Cordes, P. G. Alsabeh, B. Li and Y. Li, et al. , ChemSusChem, 2014, 7(9), 2419–2422 CrossRef CAS PubMed.
- M. Nielsen, H. Junge, A. Kammer and M. Beller, Angew. Chem., Int. Ed., 2012, 51(23), 5711–5713 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127(31), 10840–10841 CrossRef CAS PubMed.
- D. G. Gusev, ACS Catal., 2016, 6(10), 6967–6981 CrossRef CAS.
- H. Aitchison, R. L. Wingad and D. F. Wass, ACS Catal., 2016, 6(10), 7125–7132 CrossRef CAS.
- N. V. Kulkarni, W. W. Brennessel and W. D. Jones, ACS Catal., 2018, 8(2), 997–1002 CrossRef CAS.
- S. Chakraborty, P. E. Piszel, C. E. Hayes, R. T. Baker and W. D. Jones, J. Am. Chem. Soc., 2015, 137(45), 14264–14267 CrossRef CAS PubMed.
- D. H. Nguyen, Y. Morin, L. Zhang, X. Trivelli, F. Capet and S. Paul, et al. , ChemCatChem, 2017, 9(14), 2652–2660 CrossRef CAS.
- M. Bertoli, A. Choualeb, A. J. Lough, B. Moore, D. Spasyuk and D. G. Gusev, Organometallics, 2011, 30(13), 3479–3482 CrossRef CAS.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, N. J. Lo and T. J. Williams, Nat. Commun., 2016, 7(1), 11308 CrossRef CAS PubMed.
- V. K. Do, N. A. Vargas, A. J. Chavez, L. Zhang, V. Cherepakhin and Z. Lu, et al. , Catal. Sci. Technol., 2022, 12(23), 7182–7189 RSC.
- P. J. Lauridsen, Z. Lu, J. J. A. Celaje, E. A. Kedzie and T. J. Williams, Dalton Trans., 2018, 47(38), 13559–13564 RSC.
- J. J. A. Celaje, X. Zhang, F. Zhang, L. Kam, J. R. Herron and T. J. Williams, ACS Catal., 2017, 7(2), 1136–1142 CrossRef CAS.
- P. A. Dub, B. L. Scott and J. C. Gordon, Organometallics, 2015, 34(18), 4464–4479 CrossRef CAS.
- P. A. Dub and J. C. Gordon, Polydentate Ligands and Their Complexes for Molecular Catalysis, WO pat. 2015191505A1, 2015 Search PubMed.
- R. H. Crabtree, H. Felkin and G. E. Morris, J. Organomet. Chem., 1977, 141(2), 205–215 CrossRef CAS.
- P. A. Dub, R. J. Batrice, J. C. Gordon, B. L. Scott, Y. Minko, J. G. Schmidt and R. F. Williams, Org. Process Res. Dev., 2020, 24(3), 415–442 CrossRef CAS.
- G. Franciò, R. Scopelliti, C. G. Arena, G. Bruno, D. Drommi and F. Faraone, Organometallics, 1998, 17(3), 338–347 CrossRef.
- P. Farr, M. M. Olmstead, C. H. Hunt and A. L. Balch, Inorg. Chem., 1981, 20(4), 1182–1187 CrossRef.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki and S. Tanaka, et al. , Org. Process Res. Dev., 2012, 16(1), 166–171 CrossRef CAS.
- W. Kuriyama, T. Matsumoto, Y. Ino and O. Ogata, Novel Ruthenium Carbonyl Complex Having a Tridentate Ligand and Manufacturing Method and Usage Therefor, WO pat. 2011048727A1, 2011.
- L. S. Sharninghausen and R. H. Crabtree, Isr. J. Chem., 2017, 57(10–11), 937–944 CrossRef CAS.
- J. C. Borghs, Y. Lebedev, M. Rueping and O. El-Sepelgy, Org. Lett., 2019, 21(1), 70–74 CrossRef CAS PubMed.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138(28), 8809–8814 CrossRef CAS PubMed.
- W. Ma, S. Cui, H. Sun, W. Tang, D. Xue and C. Li, et al. , Chem. –Eur. J., 2018, 24(50), 13118–13123 CrossRef CAS PubMed.
- S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson and J. A. Krause, et al. , J. Am. Chem. Soc., 2014, 136(22), 7869–7872 CrossRef CAS PubMed.
- F. Schneck, M. Assmann, M. Balmer, K. Harms and R. Langer, Organometallics, 2016, 35(11), 1931–1943 CrossRef CAS.
- M. Andérez-Fernández, L. K. Vogt, S. Fischer, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig and M. Beller, Angew. Chem., Int. Ed., 2017, 56(2), 559–562 CrossRef PubMed.
- Office of Fossil Energy, Hydrogen Strategy Enabling A Low-Carbon Economy, chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/https://www.energy.gov/sites/prod/files/2020/07/f76/USDOE_FE_Hydrogen_Strategy_July2020.pdf, (accessed March 2023).
- P. A. Dub and J. C. Gordon, Nat. Rev. Chem, 2018, 2(12), 396–408 CrossRef CAS.
- E. Alberico, A. J. J. Lennox, L. K. Vogt, H. Jiao, W. Baumann and H. J. Drexler, et al. , J. Am. Chem. Soc., 2016, 138(45), 14890–14904 CrossRef CAS PubMed.
- P. A. Dub, B. L. Scott and J. C. Gordon, J. Am. Chem. Soc., 2017, 139(3), 1245–1260 CrossRef CAS PubMed.
- A. Anaby, M. Schelwies, J. Schwaben, F. Rominger, A. S. K. Hashmi and T. Schaub, Organometallics, 2018, 37(13), 2193–2201 CrossRef CAS.
- D. J. Tindall, M. Menche, M. Schelwies, R. A. Paciello, A. Schäfer and P. Comba, et al. , Inorg. Chem., 2020, 59(7), 5099–5115 CrossRef CAS PubMed.
- D. H. Nguyen, X. Trivelli, F. Capet, Y. Swesi, A. Favre-Réguillon and L. Vanoye, et al. , ACS Catal., 2018, 8(5), 4719–4734 CrossRef CAS.
- N. J. Oldenhuis, V. M. Dong and Z. Guan, Tetrahedron, 2014, 70(27), 4213–4218 CrossRef CAS PubMed.
- T. Otsuka, A. Ishii, P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135(26), 9600–9603 CrossRef CAS PubMed.
- N. Bampos, L. D. Field and B. A. Messerle, Organometallics, 1993, 12(7), 2529–2535 CrossRef CAS.
- Life Cycle Assessment Harmonization, https://www.nrel.gov/analysis/life-cycle-assessment.html, (accessed March 2024).
- M. Nayal, A. Kr. Sharma, S. Jain and V. P. Singh, Highly Efficient Thermal Renewable Energy Systems: Design, Optimization and Applications, ed. V. Verma, M. Chitt, S. Thangavel and A. Kumar, Taylor & Francis Group, Boca Raton, 2024, ch. 11, vol. 1, p. 168–193 Search PubMed.
- H. Park and S. Yoon, ACS Sustainable Chem. Eng., 2023, 11(32), 12036–12044 CrossRef CAS.
- A. Monney, E. Barsch, P. Sponholz, H. Junge, R. Ludwig and M. Beller, Chem. Commun., 2013, 50(6), 707–709 RSC.
- M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge and S. Gladiali, et al. , Nature, 2013, 495(7439), 85–89 CAS.
- R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nature Chem., 2013, 5(4), 342–347 Search PubMed.
- N. Onishi, G. Laurenczy, M. Beller and Y. Himeda, Coord. Chem. Rev., 2018, 373, 317–332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 2206834 and 2354122. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4se01524j |
| This journal is © The Royal Society of Chemistry 2025 |