
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLi-doped 2D aza-fused covalent organic framework: a promising avenue for hydrogen storage†
Preeti
Beniwal
 and
T. J.
Dhilip Kumar
*
and
T. J.
Dhilip Kumar
*
Department of Chemistry, Indian Institute of Technology Ropar, Rupnagar 140001, India. E-mail: dhilip@iitrpr.ac.in
First published on 20th January 2025
Abstract
Designing an efficient high-capacity hydrogen storage material is a critical challenge for advancing clean energy storage. Through detailed density functional theory calculations and ab initio molecular dynamics simulations, we found that the recently synthesized two-dimensional (2D) aza-fused covalent organic framework (aza-COF) doped with Li exhibits considerable promise for hydrogen storage applications. Despite a H2 storage capacity of 10.3 wt%, pristine aza-COF adsorbs H2 molecules via weak van der Waals interactions, limiting its viability under ambient conditions. The strategy relies on increasing more active sites for H2 adsorption, thereby improving the interactions between H2 and positively charged Li atoms. Li-doped aza-COF adsorbs H2 molecules with a combined effect of electrostatic and van der Waals interactions, resulting in enhanced H2 adsorption energy, ranging from −0.22 to −0.33 eV. The H2 storage capacity reaches 13.9 wt%, higher than that of the pristine aza-COF and the 5.5 wt% target of the U. S. Department of Energy. With appropriate structural stability, H2 adsorption energy, desorption temperature, hydrogen occupation number and high H2 storage ability, Li-doped 2D aza-COF exhibits great potential as a hydrogen storage material.
1 Introduction
The growing conflict between increasing energy demands and the need for environmental protection has intensified the search for clean energy alternatives.1,2 Hydrogen presents a compelling option as an energy carrier on account of its energy density, minimal ecological footprint, low emissions and natural abundance.3 However, a key challenge remains in the onboard storage of hydrogen, as its flammable and explosive properties require compression at high pressures and the development of advanced containment systems to prevent leaks and potential hazards. Current advancements in hydrogen storage research are primarily aimed at overcoming the constraints of high pressure and low temperatures, striving for solutions operable under ambient conditions.4,5 Porous materials including activated carbons, metal–organic frameworks (MOFs), porous aromatic frameworks (PAFs), zeolites, covalent organic frameworks (COFs), carbon nanotubes (CNTs) and carbon aerogels are ideal candidates due to their ability to store gases via weak van der Waals forces.6–9 Despite their potential, many adsorbents like MOFs are unstable in humid environments, and others, like activated carbon and zeolites, lack structural control and customizable functionalities. Taking these considerations, COFs are projected to be viable and efficient hosts for hydrogen storage.COFs, with their crystalline structure and composition of light elements (C, H, O, N), demonstrate low density and ease of processing, rendering them efficient for membrane use.10 By promoting extensive electron delocalization through their π-conjugated skeletons, these materials exhibit a substantial improvement in electrical conductivity.11 The tunability, porosity, and chemical stability of COFs make them highly suitable for critical applications in energy storage,12 catalysis,13 and hydrogen storage,14,15 alongside drug delivery,16 pseudocapacitors,17,18 rechargeable batteries,19,20 and electrochemical systems.21 This level of control over their structural features enhances their functionality in these domains, allowing for optimized performance in each specific application.
Tylianakis14 explored the hydrogen adsorption properties of COFs, highlighting that both experimental and theoretical analyses showed that COFs exhibit comparable or superior hydrogen storage capacities at 77 K when compared to other materials. Despite their promising performance at low temperatures, COFs struggle to meet the practical hydrogen storage standards set by the U. S. Department of Energy (DOE) at room temperature. Nonetheless, both experimental and computational studies consistently demonstrate that hydrogen storage in COFs is markedly improved through metal doping15,22–28 and organic group functionalization,29 providing a viable strategy for meeting these rigorous performance standards. Zhao et al. demonstrated that Li-decorated COF-1 achieves a hydrogen uptake of 5.26 wt% under ambient conditions, adsorbing 3H2 molecules per Li atom.30 Using density functional theory (DFT) and Grand Canonical Monte Carlo (GCMC) simulations, Xia et al. reported that Li-doped COF-320 leads to a 30.9% increase in gravimetric density.31 Cao et al. reported that Li-doped COF-105 and COF-108 showed hydrogen uptakes of 6.84 wt% and 6.73 wt%, respectively, at 298 K and 100 bar, making them competitive candidates for efficient hydrogen storage.24 Lan et al. reported that Li doping significantly improved the hydrogen uptake of COF-202, with the gravimetric storage capacity increasing from 1.52 wt% for the pristine aza-COF to 4.39 wt% for Li-COF-202, under conditions of 298 K and 100 bar.25 This marked enhancement underscores the crucial contribution of Li doping to the notable enhancement of hydrogen uptake in COF-202. Ke et al. showed that Li-doped COF-108 frameworks exhibited an enhanced hydrogen interaction energy, and the Li-doped C60-impregnated COF-108 achieved a notable hydrogen uptake of 4.56 wt% at 233 K and 100 bar.15
Aza-COF, a microporous 2D COF, has a periodic arrangement of well-dispersed pyridinic nitrogen sites.32 Its periodic microporosity contributes to its robust chemical stability and high conductivity, coupled with uniform chemical environments and minimal structural evolution.33,34 Recently, Yang et al.32 have synthesized the aza-COF via the polycondensation of 1,2,4,5-benzenetetramine tetrahydrochloride and triquinoyl octahydrate with a focus on its application in oxygen evolution reactions (OERs). In another study, the crystalline aza-COF was synthesized and used as a cathode material in sodium-ion batteries by Shehab et al.35 Meng et al. proposed a generalized strategy for facilitating efficient proton conduction within aza-COFs.10 Their comprehensive investigation revealed that chemical composition, porosity, and crystallinity are crucial factors that govern the modulation and optimization of proton transport in these materials. Very recently, Vasanthakannan et al.27 reported 8.43 wt% hydrogen uptake of a Sc-decorated aza-COF with −0.36 eV per H2 adsorption energy.
Inspired by such incredible applications of aza-COFs, we have studied the hydrogen storage in a Li-doped aza-COF by employing periodic DFT calculations in conjunction with ab initio molecular dynamics (AIMD) simulations. The optimal Li doping site is identified by evaluating and comparing binding energies at various locations of the aza-COF. A substantial improvement in hydrogen adsorption is observed for the Li-doped aza-COF when compared to the pristine framework. To determine the feasibility, we have assessed the stability of the Li-doped aza-COF by addressing the absence of Li clustering tendencies and thermal stability of the framework. The underlying interaction mechanisms during Li doping and H2 adsorption on the framework are thoroughly examined by computing partial density of states (PDOS) plots, charge density difference plots, and Bader charge calculations. The hydrogen desorption behavior is explored through calculations of the hydrogen occupation number, AIMD simulation and desorption temperature.
2 Computational methodology
DFT-based calculations were performed within the Vienna Ab initio Simulation Package (VASP),36,37 with a plane-wave basis set. Core-electron interactions were modeled using the projector augmented wave method.38 For the exchange–correlation energy contribution, we have employed the Perdew–Burke–Ernzerhof (PBE) functional within the framework of the generalized gradient approximation (GGA).39 To provide more accurate description, the band structure calculations were also done using a hybrid functional (HSE06).40 Self-consistent calculations were carried out using a 4 × 4 × 1 Monkhorst–Pack mesh,41 with a kinetic energy cutoff of 500 eV and a vacuum layer thickness of 20 Å, to reduce periodic image effects along the z-axis. The electronic self-consistency loop was considered to have converged, when the energy difference reached 1 × 10−5 eV, and geometry optimization continued until the Hellmann–Feynman force on each atom was reduced to below 0.01 eV Å−1. For an accurate description of van der Waals forces between the H2 adsorbate and aza-COF surface, Grimme's D2 dispersion correction (DFT-D2) was applied.42 To evaluate the system behavior at different temperatures, AIMD simulations were performed. The protocol involved heating the system from 0 K to the target temperature, followed by equilibration in the canonical (NVT) ensemble with the Nosé–Hoover thermostat43 method for 10 ps with a 1 fs time-step. To precisely ascertain the energy barrier associated with Li diffusion, the Climbing Image Nudged Elastic Band (CI-NEB)44 approach was adopted.3 Results and discussion
3.1 Structural stability and hydrogen storage potential of the aza-COF
Fig. 1a displays the optimized structure of the aza-COF supercell, where the red dashed box marks the unit cell. Our calculated lattice parameters of the aza-COF unit cell, a = b = 16.56 Å, match quite well with the experimental reports,10 confirming that the optimization method applied in this work is credible and efficient. AIMD simulations are conducted at 300 and 500 K in order to evaluate the system behavior at room and high temperature, respectively. As demonstrated in the ESI in Fig. S1a–d,† the simulations exhibit stable and uniform fluctuations in total energy and different bond lengths around their mean values. Insets of Fig. S1a and c† display the final snapshots of aza-COF after 10 ps at 300 and 500 K, respectively, showing intact geometry and confirming structural stability at both room and high temperatures, respectively. Moreover, the vibrational density of states (VDOS) vs. frequency plot in Fig. S1e† provides insights into the distribution and dynamics of vibrational modes within the framework at 300 K. The non-negative VDOS spectrum suggests that all vibrational modes are stable, indicating the dynamical stability of the framework. The pristine aza-COF shows a hydrogen storage capacity of 10.2 wt%, corresponding to the adsorption of 30H2 molecules at −0.17 eV, which is above the DOE's 5.5 wt% target. The optimized structure of aza-COF + 30H2 is shown in Fig. S2a in the ESI.† Despite this, the weak van der Waals interactions, as evidenced by the low adsorption energy below the ideal threshold of −0.2 eV, limit its practicality for efficient hydrogen storage. The van der Waals interactions are a type of non-covalent interactions (NCIs).45,46 To investigate these types of interactions, the NCI isosurface is mapped for the aza-COF + 30H2 complex (Fig. S2b†), using the Multifin and Visual Molecular Dynamics (VMD) software. The green disc between the H2 molecule and aza-COF further confirms the van der Waals interactions. Consequently, pristine aza-COF is unsuitable for storage under ambient conditions. The desorption temperature is determined via the van't Hoff equation: | (1) |
 | ||
| Fig. 1 (a) Optimized structure of the 2 × 2 × 1 supercell of aza-COF with the red dashed box denoting the unit cell. (b) Potential sites of aza-COF explored for Li doping. | ||
3.2 Structure and stability of 6Li@aza-COF
To strengthen the hydrogen interaction with the aza-COF, we doped it with the Li atoms, probing various potential binding sites, as depicted in Fig. 1b. During structural optimization, the Li atom is displaced across multiple sites, giving four unique configurations, provided in Fig. S4,† considered as energetically stable. The corresponding Li binding energies for these configurations are calculated using the following equation:| Eb = (EmLi@aza-COF − Eaza-COF − mELi)/m | (2) |
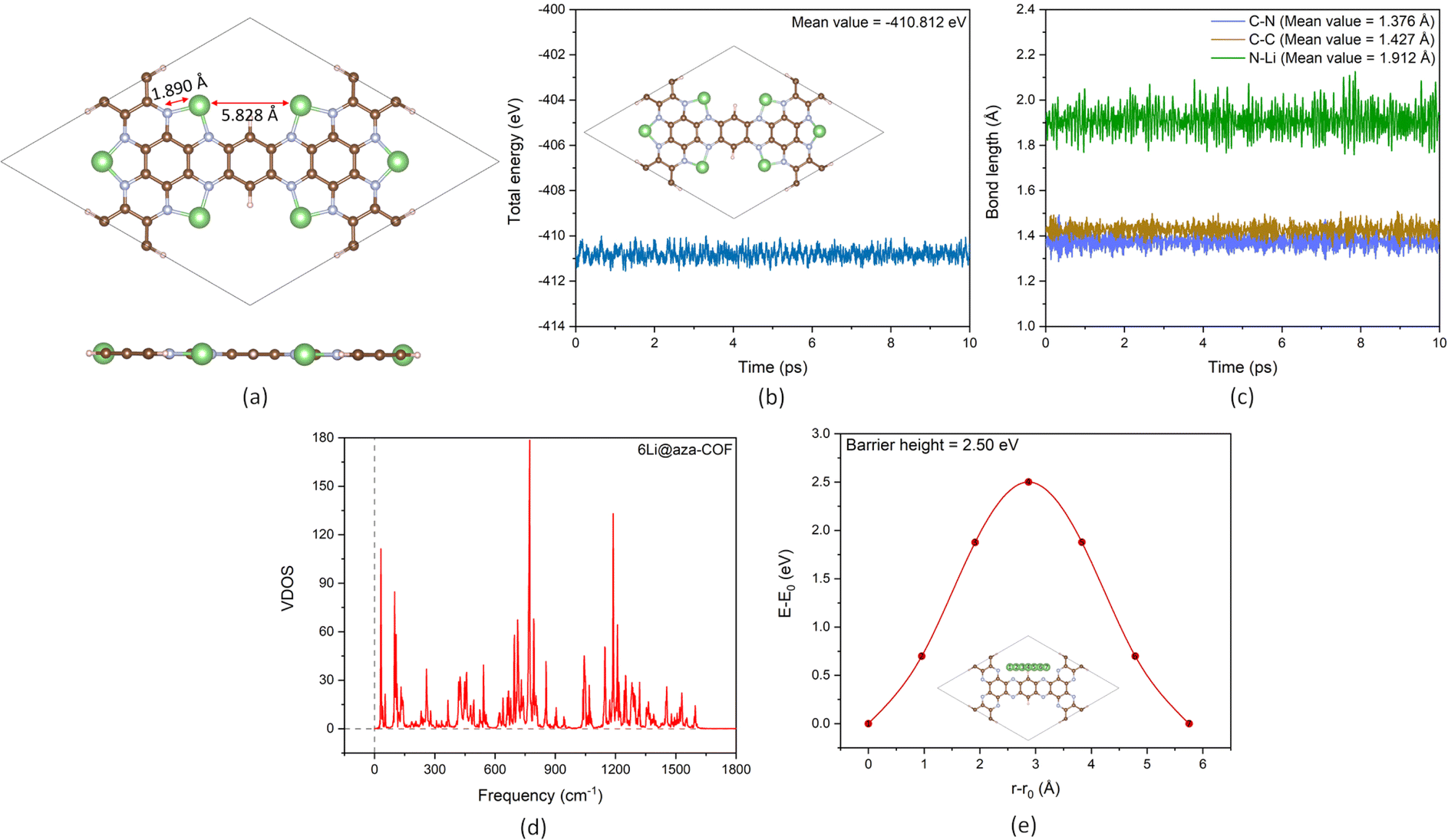 | ||
| Fig. 2 (a) Optimized structure of 6Li@aza-COF. Atom color representation: C-brown; N-silver; H-peach; and Li-green. (b) Total energy and (c) bond length fluctuations during the AIMD simulation at 300 K of 6Li@aza-COF. (d) VDOS vs. frequency plot at 300 K of 6Li@aza-COF. (e) Energy barrier for Li diffusion computed using the Cl-NEB method. | ||
The thermal stability of 6Li@aza-COF is evaluated using the same AIMD protocol employed for pristine aza-COF. Simulation results, in Fig. 2b and c, reveal stable, uniform fluctuations in total energy and different bond lengths, respectively. Furthermore, the absence of imaginary frequencies in the VDOS spectrum in Fig. 2d further affirms the dynamical stability of the framework. Minimal fluctuations in total energy and bond lengths in Fig. S6a and b† indicate the stability of 6Li@aza-COF at 500 K, while the final snapshots (insets of Fig. 2b and S6a†) show that Li atoms remain firmly intact at 300 and 500 K, confirming that there is no clustering tendency. These findings underscore the exceptional structural stability of 6Li@aza-COF, emphasizing its potential for hydrogen storage applications.
As previously outlined, in 6Li@aza-COF, Li atoms bind with a stronger Eb of −3.47 eV per Li, which is 2.1 times the Li cohesive energy. Although AIMD simulations confirm that no metal clustering occurs in 6Li@aza-COF, with Li atoms remaining intact to the framework even at a high temperature of 500 K, the possibility is further checked by computing the Li diffusion energy barrier. The CI-NEB calculation, with five intermediate images between two equivalent P sites, provided the energy profile shown in Fig. 2e. The calculated Li diffusion energy barrier of 2.50 eV, higher than Li cohesive energy, is sufficiently large to prevent Li diffusion across the framework, and thereby metal clustering. With the stability demonstrated through AIMD simulations, dynamic analysis, and the calculated diffusion barrier, we conclude that 6Li@aza-COF is an excellent candidate for reversible hydrogen storage.
3.3 Electronic properties of aza-COF and 6Li@aza-COF
Electron localization function (ELF) provides a detailed visualization of electron localization patterns and bonding nature within the frameworks, aza-COF and 6Li@aza-COF, depicted in Fig. 3a and b, respectively. Fig. 3a and b reveal pronounced electron localization around N and H atoms, with uniform electron distribution along C–N and C–C bonds clearly highlighting the conjugated π-bonding network within the frameworks. In Fig. 3b, the localization of electrons around the Li atoms and no electron localization between N and Li atoms (blue color) indicates the formation of partially ionic bonds between Li and the aza-COF framework. Moreover, ELF maps delineate a well-ordered covalent network with a pervasive π-electron delocalization, ensuring both structural integrity and electronic stability. | ||
| Fig. 3 ELF maps of (a) aza-COF and (b) 6Li@aza-COF. (c) Electronic band structure and (d) PDOS plot of aza-COF. (e) Electronic band structure and (f) PDOS plot of 6Li@aza-COF. | ||
The electronic band structure of the aza-COF is calculated with the PBE functional, shown by dashed bands in Fig. 3c, which indicates the semiconductor nature of aza-COF corresponding to a direct band gap of 1.04 eV (at the Γ point). Furthermore, the PDOS plot, in Fig. 3d, reveals that spin has no influence on the electronic structure of this COF. Considering the limitations of the PBE functional in band gap prediction, to improve accuracy, the band structure of aza-COF is recalculated using the screened hybrid functional HSE06, shown by solid bands in Fig. 3c. Using the HSE06 functional, the band gap of aza-COF is determined to be 1.64 eV. It is evident from the band structures that for aza-COF, the PBE functional underestimates the band gap by 0.6 eV. The calculated band gap of aza-COF aligns well with prior theoretical results.33,48 Li doping reduces the band gap, resulting in an indirect band gap of 0.38 eV (PBE/GGA) and 0.73 eV (HSE06), as shown in Fig. 3e. Li doping lowers the Fermi level from −5.22 eV to −1.72 eV in 6Li@aza-COF, evidencing significant charge transfer by Li, further confirmed by PDOS and charge analysis. This Fermi level shift results from an increase in the valence band maximum upon Li doping, with a smaller rise observed in the conduction band minimum. The electron density influx from the Li-2s orbitals contributes to a more densely populated conduction band than in the pristine aza-COF, resulting in a decrease in the band gap. In analogy to aza-COF, the electronic structure of 6Li@aza-COF is spin-independent, as shown by the symmetric spin-up and spin-down states in the PDOS plot (Fig. 3f). In the range of −3 to −6 eV, the appearance of new electronic states of the N-2p orbital and the shift of electronic states of C-2p and N-2p orbitals toward the valence region display the charge gain from the Li-2s orbitals.
Bader charge analysis reveals that Li atoms lose 0.890e charge to nearby N and C atoms and acquire positive charge. These positively charged Li atoms now act as an effective adsorption site for H2 adsorption and result in intensified interaction compared to pristine aza-COF. The increase in the average negative charge on N from −1.113 to −1.322e and the decrease in the positive charge on the nearby C atoms (Table S1†) further evidenced the charge gain. The differential charge density distribution, resulting from Li doping, illustrates the charge transfer process. The yellow color on N and C atoms in the charge density difference plot, Fig. 4a, reflects the charge accumulation region while the cyan color over Li atoms denotes the charge depletion. For a deeper insight into the mechanism involved in Li doping, we computed the work function for both aza-COF and 6Li@aza-COF. The work function of a system is a key parameter that provides valuable insight into the charge flow interactions. Mathematically, the work function, ϕ, of a system can be expressed as:
| ϕ = V − Ef | (3) |
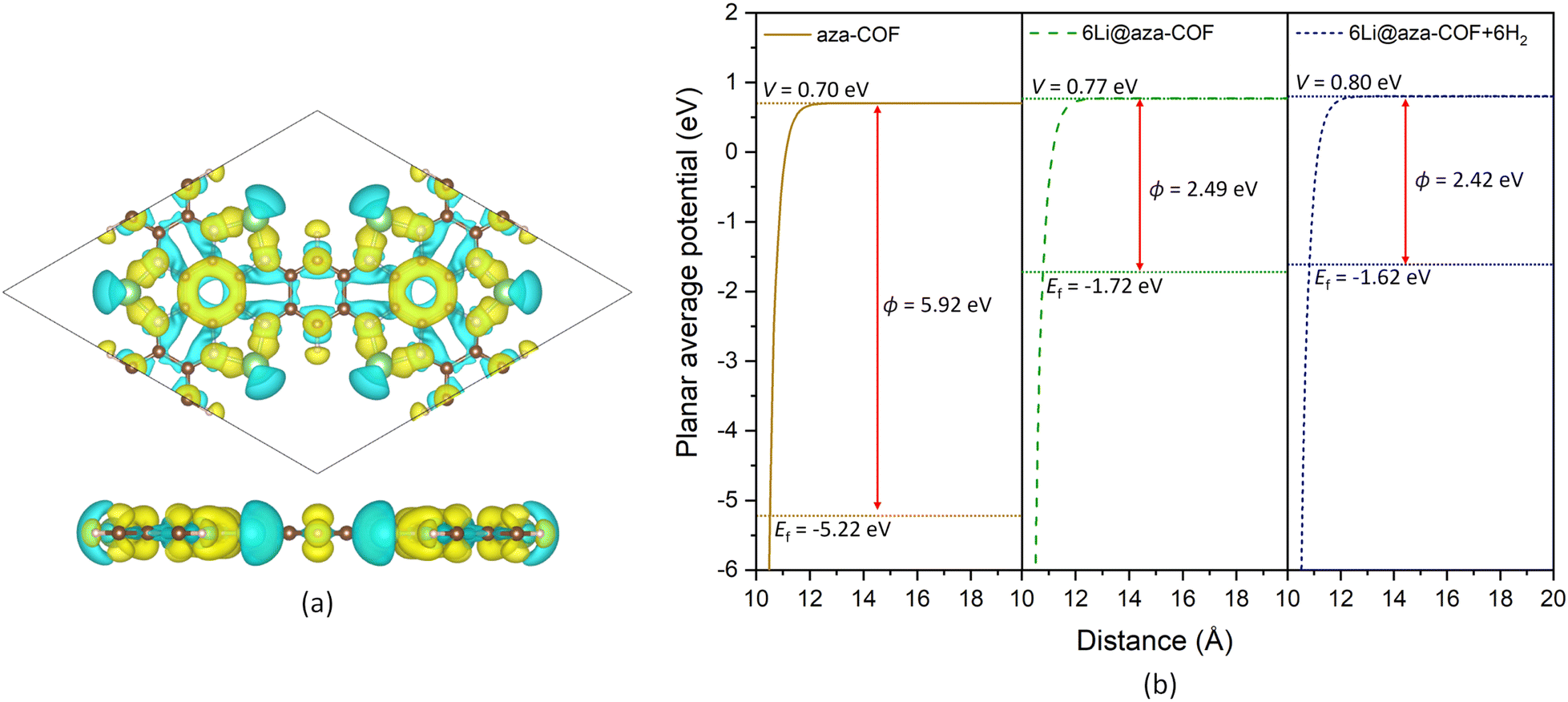 | ||
| Fig. 4 (a) Charge density difference plot for 6Li@aza-COF. The isosurface level is 0.0026 e bohr−3. The cyan region indicates the charge depletion and the yellow region represents the charge accumulation. (b) Planar average potential of aza-COF, 6Li@aza-COF, and 6Li@aza-COF + 6H2 with increasing distance in the z direction representing the computed vacuum potential (V), Fermi energy (Ef), and work function (ϕ). | ||
3.4 Hydrogen storage potential of 6Li@aza-COF
The structural modification of aza-COF through Li doping is undertaken to improve its adsorption site availability. The hydrogen storage potential of 6Li@aza-COF is analyzed, where H2 molecules are introduced nearby Li atoms. H2 introduction is performed in a sequential manner, as each time one H2 per Li is added and resulting structures are fully optimized. The resulting optimized structures of H2-adsorbed 6Li@aza-COF configurations are displayed in Fig. 5. From this analysis, it is concluded that each Li atom adsorbs 6H2 molecules and the adsorption energies corresponding to each H2 adsorption are detailed in Table 1 with the respective gravimetric densities. The adsorption energy lies in the range of −0.2 to −0.7 eV, which is regarded as an optimal range for the effective performance of hydrogen storage materials under ambient conditions and indicative of physisorption of H2 molecules. Also, the gravimetric density for 36H2 adsorption is 11.2 wt%, significantly passing the DOE targets. After each H2 adsorption geometrical changes in terms of H–H bond length and Li–H2 distances are evaluated. H–H bond lengths of adsorbed H2 are elongated compared to 0.750 Å (in the free H2 molecule), evidencing the interaction between H2 and Li atoms. For adsorption distances exceeding 4 Å, the hydrogen bond length remains unchanged, indicating no interaction with the substrate.49 The maximum measured H2 distance from the Li atoms is 3.128 Å in 6Li@aza-COF + 36H2 (Table 1), consistently below 4 Å and aligning well with prior research findings.50 The H–H bond length and the average values of Li–H2 distances are plotted in Fig. 6a. This adsorption energy analysis evidenced the intensified H2 interaction on Li-doped aza-COF compared to the pristine aza-COF. | ||
| Fig. 5 Optimized structures of H2-adsorbed 6Li@aza-COF configurations: (a) 6Li@aza-COF + 6H2, (b) 6Li@aza-COF + 12H2, (c) 6Li@aza-COF + 18H2, (d) 6Li@aza-COF + 24H2, (e) 6Li@aza-COF + 30H2 and (f) 6Li@aza-COF + 36H2. Atom color representation: C-brown; N-silver; H-peach, blue; and Li-green. | ||
| H2-adsorbed configurations | Adsorption energy (eV) | Li–H2 distance (Å) | Gravimetric density (wt%) |
|---|---|---|---|
| 6Li@aza-COF + 6H2 | −0.30 | 1.952–1.954 | 2.06 |
| 6Li@aza-COF + 12H2 | −0.33 | 1.937–1.941 | 4.03 |
| 6Li@aza-COF + 18H2 | −0.29 | 1.987–2.008 | 5.93 |
| 6Li@aza-COF + 24H2 | −0.26 | 1.972–2.652 | 7.75 |
| 6Li@aza-COF + 30H2 | −0.25 | 1.948–2.964 | 9.51 |
| 6Li@aza-COF + 36H2 | −0.24 | 1.953–3.128 | 11.2 |
| 6Li@aza-COF + 46H2 | −0.22 | 1.945–3.366 | 13.9 |
 | ||
| Fig. 6 (a) H–H bond length and average Li–H2 distance plots for H2-adsorbed 6Li@aza-COF configurations: 6Li@aza-COF + nH2, n = 6, 12, 18, 24, 30, and 36. (b) Optimized structure of 6Li@aza-COF + 46H2. | ||
With additional adsorption sites present on 6Li@aza-COF, we further checked the H2 adsorption on H1 and H3 sites. The optimized structures of H2 adsorption on the H1 site (6Li@aza-COF + 40H2) and H3 site (6Li@aza-COF + 42H2) are provided in Fig. S7a and b,† respectively. Considering both H1 and H3 sites, 10 more H2 molecules are effectively stored by 6Li@aza-COF + 36H2 with the adsorption energy of −0.22 eV. These 10H2 molecules relaxed in the range of 2.009 to 2.737 Å (within 4 Å) with the average H–H bond length of 0.752 Å. The optimized structure of 6Li@aza-COF + 46H2 is given in Fig. 6b. The decision to limit the adsorption of 10 additional H2 molecules is based on the optimal range of −0.2 to −0.7 eV and adsorption distance up to 4 Å, as further addition results in adsorption energy below the −0.2 eV threshold and H2 distance from the surface beyond 4 Å. Li doping results in charge redistribution on C and N atoms (as observed from Bader charges on C and N atoms in Table S1†) and thus increased adsorption sites, which could be attributed to the greater interaction of H2 molecule compared to pristine aza-COF. With the adsorption of 46H2 molecule, 6Li@aza-COF shows a maximum gravimetric density of 13.9 wt%.
3.5 Mechanism of H2 adsorption on 6Li@aza-COF
To investigate the mechanism of H2 interaction with 6Li@aza-COF, the PDOS plot is analyzed for the 6Li@aza-COF + 6H2 configuration. A negligible redistribution in the electronic states of C-2p, N-2p and Li-2s is observed on H2 adsorption. In Fig. 7a, no apparent change in the electronic states of Li-2s orbital can be observed, indicating that there is no orbital overlapping between H-1s and Li-2s orbitals. Thus, the electrostatic interaction may be the reason why H2 molecules are attracted toward positively charged Li atoms. H2 adsorption on 6Li@aza-COF induces a minimal shift in the Fermi energy (to −1.62 eV) and work function (to 2.42 eV), plotted in Fig. 4b. Work function analysis reveals that H2 interacts weakly with 6Li@aza-COF compared to the interaction between Li and the aza-COF framework.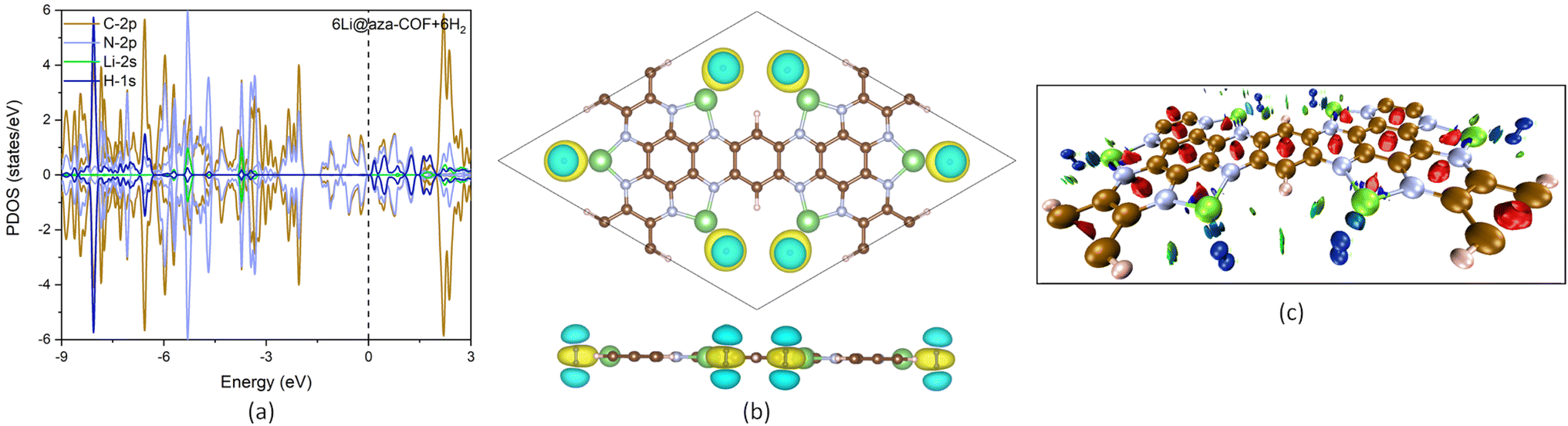 | ||
| Fig. 7 (a) PDOS plot of 6Li@aza-COF + 6H2. (b) Charge density difference plot for 6Li@aza-COF + 6H2. The isosurface level is 0.0039 e bohr−3. (c) NCI isosurface mapped for 6Li@aza-COF + 6H2. | ||
From the charge density difference plots (Fig. 7b), an enhanced yellow region evidences the electrostatic interaction between H2 molecules and Li atoms, as the electronically rich bonds of H2 molecule experience attraction toward positively charged Li atoms, which induce polarization in H2. Furthermore, the Bader charge analysis validates this polarization, as on the adsorbed H2 molecule, one H atom acquires positive charge and another one acquires negative charge (Table S2†). This charge distribution confirms the polarization in H2 molecules, which subsequently elongates the H–H bond length. Thus, the electrostatic interaction that exists between Li and H2 molecules facilitates the polarized H2 adsorption.
To investigate the NCIs, the isosurface is mapped for the 6Li@aza-COF + 6H2 complex, as shown in Fig. 7c. The blue disc observed between the H2 molecules and Li atoms confirms the electrostatic interaction, while the greenish disc reflects the van der Waals interaction with other atoms. Based on these findings, it can be concluded that H2 adsorption over 6Li@aza-COF occurs via a combined effect of electrostatic and van der Waals interactions.
3.6 H2 desorption from 6Li@aza-COF
We conducted a detailed study of the desorption behavior of hydrogen molecules by hydrogen occupation number, AIMD simulation at 300 K and desorption temperature. Hydrogen occupation number over a range of temperatures and pressures captures the adsorption trends under variable conditions.51,52 The ñPT plot for the 6Li@aza-COF + 36H2 configuration is shown in Fig. 8a, where ñ denotes the adsorbed H2 molecules under different P and T conditions. At the DOE's minimum temperature of 230 K and under moderate pressures of 10–20 bar, ∼25–29 of the initial 36H2 molecules remain adsorbed on the framework. With increasing temperature and decreasing pressure, the occupation number decreases, signifying the onset of desorption. At a maximum temperature of 360 K and 0–5 bar pressure, only 1 molecule remains adsorbed, marking near-complete desorption of H2 molecules. | ||
| Fig. 8 (a) ñPT plot at the pressure range of 1–60 atm and temperature range of 100–500 K. (b) Snapshot of 6Li@aza-COF + 36H2 after 10 ps of AIMD simulation at 300 K. H2 molecule color representation: purple – d < 2.5 Å; cyan – 2.5 < d < 4.0 Å; pink – d > 4.0 Å. d denotes the distance of H2 molecules from the surface of the framework. | ||
AIMD simulation at 300 K is performed in the NVT ensemble for the 6Li@aza-COF + 36H2 configuration, to check the hydrogen reversibility within the framework, providing a real-time view of molecular behavior under standard conditions. H2 molecules located within 2.5 Å are categorized as strongly attached to the host material, while those within the range of 2.5 Å to 4.0 Å are deemed weakly adsorbed. Beyond 4.0 Å, H2 molecules exhibit no interaction with the framework and are regarded as free H2 molecules, as established in previous research studies.49,50,53 Considering this, from the AIMD simulation results, provided in Fig. 8b, it is apparent that at 300 K, out of the initial 36H2 molecules, 12H2 molecules are present below 2.5 Å, 12H2 molecules are within 2.5–4.0 Å and 12H2 are released from the surface, showing the reversible hydrogen storage ability of 6Li@aza-COF.
For the maximum hydrogen adsorption of 6Li@aza-COF, i.e., for 6Li@aza-COF + 46H2, we computed the desorption temperature using the van't Hoff equation. The desorption temperature values are determined to be 281 K at 1 atm and 342 K at 5 atm, higher than the aza-COF. These analyses provide an insight into the temperature and pressure-responsive adsorption and desorption mechanisms, providing a comprehensive picture of the 6Li@aza-COF potential for efficient reversible hydrogen storage.
4 Conclusions
In conclusion, using DFT-coupled AIMD methodology, we have rationally designed a Li-doped 2D aza-COF that provides active sites for H2 adsorption toward hydrogen storage application. The findings indicate that Li can be successfully doped on the surface of the aza-COF without any concern of metal clustering, as evidenced by a stronger binding energy of −3.47 eV per Li for 6Li@aza-COF. AIMD simulations and VDOS confirmed the structural and dynamic stability of Li-doped aza-COF. Furthermore, the diffusion energy barrier calculation affirms the absence of any metal clustering tendency. Each Li atom has the capability to adsorb up to 6H2 molecules and with additional 10H2 adsorption, 6Li@aza-COF can effectively adsorb 46H2 molecules. The adsorption energy with respect to each H2 adsorption configuration ranges from −0.22 to −0.33 eV, indicative of the physisorption of H2 molecules. H2 molecule adsorption occurs with a combined effect of electrostatic and van der Waals interactions, confirmed by charge density difference and NCI calculations. The electrostatic interactions result in polarization in H2 molecules. The storage of 46H2 molecules in Li-doped aza-COF results in a maximum capacity of 13.9 wt%, fulfilling the DOE requirements. The reversible behavior of stored H2 molecules is confirmed by the AIMD simulations. The hydrogen occupation number calculation provides the number of stored H2 molecules under different P and T conditions. Therefore, our findings provide a comprehensive idea for the design of Li-doped aza-COF as a potential material toward hydrogen storage application.Data availability
All supporting data are provided in the ESI.†Author contributions
P. Beniwal: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft. T. J. Dhilip Kumar: conceptualization, funding acquisition, resources, methodology, formal analysis, supervision, writing – review & editing.Conflicts of interest
There are no financial disclosures or conflicts of interest to report by the authors.Acknowledgements
P. B. thanks the University Grants Commission (UGC) for awarding fellowship support. The authors sincerely thank IIT Ropar for computational resources and acknowledge the PARAM Smriti supercomputing facility at NABI, Mohali. The authors sincerely acknowledge the financial support provided by the CSIR, India [Sanction No. 01/3060/21/EMR-II].References
- X. Li, Mater. Chem. Front., 2021, 5, 2931–2949 RSC.
- W. Lv, Z. Shen, J. Liu, X. Li, F. Ding, D. Zhang, L. Miao, X. Lyu, R. Li, M. Wang, Y. Li, J. Meng and C. Xu, Sci. Bull., 2025, 70, 203–211 CrossRef PubMed.
- S. A. Sherif, F. Barbir and T. N. Veziroglu, Sol. Energy, 2005, 78, 647–660 CrossRef CAS.
- A. Hashmi, M. U. Farooq, I. Khan, J. Son and J. Hong, J. Mater. Chem. A, 2017, 5, 2821–2828 RSC.
- L. Luo, H. Han, D. Feng, W. Lv, L. Chen, L. Li, T. Zhai, S. Liu, S. Sun, Y. Li, W. Pei, J. Cui and Y. Li, Renewables, 2024, 2, 138–149 CrossRef.
- J. Lan, D. Cao, W. Wang, T. Ben and G. Zhu, J. Phys. Chem. Lett., 2010, 1, 978–981 CrossRef CAS.
- I. Rossetti, G. Ramis, A. Gallo and A. Di Michele, Int. J. Hydrogen Energy, 2015, 40, 7609–7616 CrossRef CAS.
- O. Czakkel, B. Nagy, G. Dobos, P. Fouquet, E. Bahn and K. László, Int. J. Hydrogen Energy, 2019, 44, 18169–18178 CrossRef CAS.
- L. Wang, S. Feng, C. Zhang, X. Zhang, X. Liu, H. Gao, Z. Liu, R. Li, J. Wang and X. Jin, ACS Appl. Mater. Interfaces, 2024, 16, 36444–36452 CrossRef CAS PubMed.
- Z. Meng, A. Aykanat and K. A. Mirica, Chem. Mater., 2019, 31, 819–825 CrossRef CAS.
- P. T. Parvatkar, S. Kandambeth, A. C. Shaikh, I. Nadinov, J. Yin, V. S. Kale, G. Healing, A.-H. Emwas, O. Shekhah, H. N. Alshareef, O. F. Mohammed and M. Eddaoudi, J. Am. Chem. Soc., 2023, 145, 5074–5082 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- E. Tylianakis, E. Klontzas and G. E. Froudakis, Nanoscale, 2011, 3, 856–869 RSC.
- Z. Ke, Y. Cheng, S. Yang, F. Li and L. Ding, Int. J. Hydrogen Energy, 2017, 42, 11461–11468 CrossRef CAS.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- C. R. Mulzer, L. Shen, R. P. Bisbey, J. R. McKone, N. Zhang, H. D. Abruña and W. R. Dichtel, ACS Cent. Sci., 2016, 2, 667–673 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- K. S. Weeraratne, A. A. Alzharani and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2019, 11, 23520–23526 CrossRef CAS PubMed.
- B. C. Patra, S. K. Das, A. Ghosh, A. Raj K, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6, 16655–16663 RSC.
- K. Zhang, K. O. Kirlikovali, R. S. Varma, Z. Jin, H. W. Jang, O. K. Farha and M. Shokouhimehr, ACS Appl. Mater. Interfaces, 2020, 12, 27821–27852 CrossRef CAS PubMed.
- X. Zou, G. Zhou, W. Duan, K. Choi and J. Ihm, J. Phys. Chem. C, 2010, 114, 13402–13407 CrossRef CAS.
- P. Srepusharawoot, E. Swatsitang, V. Amornkitbamrung, U. Pinsook and R. Ahuja, Int. J. Hydrogen Energy, 2013, 38, 14276–14280 CrossRef CAS.
- D. Cao, J. Lan, W. Wang and B. Smit, Angew. Chem., Int. Ed., 2009, 48, 4730–4733 CrossRef CAS PubMed.
- J. Lan, D. Cao and W. Wang, J. Phys. Chem. C, 2010, 114, 3108–3114 CrossRef CAS.
- F. Li, J. Zhao, B. Johansson and L. Sun, Int. J. Hydrogen Energy, 2010, 35, 266–271 CrossRef CAS.
- V. M. Vasanthakannan and K. Senthilkumar, J. Power Sources, 2024, 604, 234505 CrossRef CAS.
- R. Y. Sathe, T. J. Dhilip Kumar and R. Ahuja, Int. J. Hydrogen Energy, 2023, 48, 12767–12795 CrossRef CAS.
- L. Xia, F. Wang and Q. Liu, Mater. Lett., 2016, 162, 9–12 CrossRef CAS.
- H. Zhao, Y. Guan, H. Guo, R. Du and C. Yan, Mater. Res. Express, 2020, 7, 035506 CrossRef CAS.
- L. Xia and Q. Liu, J. Solid State Chem., 2016, 244, 1–5 CrossRef CAS.
- H. Yang, F. Li, S. Zhan, Y. Liu, W. Li, Q. Meng, A. Kravchenko, T. Liu, Y. Yang, Y. Fang, L. Wang, J. Guan, I. Furó, M. S. G. Ahlquist and L. Sun, Nat. Catal., 2022, 5, 414–429 CrossRef CAS.
- Z.-D. Yang, W. Wu and X. C. Zeng, J. Mater. Chem. C, 2014, 2, 2902–2907 RSC.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS PubMed.
- M. K. Shehab, K. S. Weeraratne, T. Huang, K. U. Lao and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2021, 13, 15083–15091 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Ángyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- V. Ponnuchamy, A. Sandak and J. Sandak, Phys. Chem. Chem. Phys., 2020, 22, 28448–28458 RSC.
- P. Beniwal, P. Gautam, N. Duhan and T. J. Dhilip Kumar, ACS Appl. Energy Mater., 2024, 7, 3263–3273 CrossRef CAS.
- Z. Zhou, J. Zhao, X. Gao, Z. Chen, J. Yan, P. von Ragué Schleyer and M. Morinaga, Chem. Mater., 2005, 17, 992–1000 CrossRef CAS.
- C. Zhao, Y. Tian, L. Yan and Z. Su, J. Phys. Chem. C, 2021, 125, 23133–23141 CrossRef CAS.
- S. Dong, E. Lv, J. Wang, C. Li, K. Ma, Z. Gao, W. Yang, Z. Ding, C. Wu and I. D. Gates, Fuel, 2021, 304, 121351 CrossRef CAS.
- L.-C. Wang, Z.-C. Zhang, L.-C. Ma, L. Ma and J.-M. Zhang, Eur. Phys. J. B, 2022, 95, 1–14 CrossRef.
- P. Beniwal and T. J. Dhilip Kumar, ACS Appl. Energy Mater., 2023, 6, 6251–6261 CrossRef CAS.
- P. Beniwal, B. Chakraborty and T. J. Dhilip Kumar, Int. J. Hydrogen Energy, 2024, 53, 29–39 CrossRef CAS.
- V. Mahamiya, J. Dewangan and B. Chakraborty, Int. J. Hydrogen Energy, 2024, 50, 1302–1316 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: AIMD simulation results at 300 and 500 K for aza-COF; optimized structure and NCI isosurface for aza-COF + 30H2. AIMD simulation results at 300 and 500 K for aza-COF + 30H2; optimized structures of Li-doped aza-COF at different binding sites; different bond lengths of aza-COF and 6Li@aza-COF; AIMD simulation results at 500 K for 6Li@aza-COF; planar average potential of aza-COF, 6Li@aza-COF, and 6Li@aza-COF + 6H2; optimized structures of 6Li@aza-COF + 40H2 and 6Li@aza-COF + 42H2; Bader charges on C, N, H and Li atoms in aza-COF and 6Li@aza-COF; Bader charges on Li and H atoms of adsorbed H2 molecules in 6Li@aza-COF and 6Li@aza-COF + 6H2. See DOI: https://doi.org/10.1039/d4se01808g |
| This journal is © The Royal Society of Chemistry 2025 |