
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer nanoparticle photocatalysts realized in non-aqueous solvents†
Max M. O'Connor a,
Steven C. Haydenb,
Melissa K. Gish‡
b,
Justin L. Ratkovec‡a,
Lily H. Harmona,
Yadong Zhangc,
Stephen Barlowc,
Seth R. Marderacd,
Obadiah G. Reid*bc and
Garry Rumbles*abc
a,
Steven C. Haydenb,
Melissa K. Gish‡
b,
Justin L. Ratkovec‡a,
Lily H. Harmona,
Yadong Zhangc,
Stephen Barlowc,
Seth R. Marderacd,
Obadiah G. Reid*bc and
Garry Rumbles*abc
aUniversity of Colorado Boulder, Department of Chemistry, Boulder, CO 80303, USA
bNational Renewable Energy Laboratory, Chemistry and Nanoscience Center, Golden, CO 80401, USA. E-mail: obadiah.reid@nrel.gov; garry.rumbles@nrel.gov
cUniversity of Colorado Boulder, Renewable and Sustainable Energy Institute, Boulder, CO 80303, USA
dUniversity of Colorado Boulder, Department of Chemical and Biological Engineering, Boulder, CO 80303, USA
First published on 22nd May 2025
Abstract
Colloidal organic nanoparticles (oNPs) have emerged as a promising category of photocatalyst, thanks to their long-lived surface-bound charges, electronic tunability, and strong absorption in the visible spectrum. Our previous research has established a direct correlation between charge generation in oNPs and their photocatalytic activity, highlighting their effectiveness as a framework for stable, long-lived free carriers. However, oNPs have been restricted to use only in aqueous environments as a result of being synthesized via either nano-emulsion or nano-precipitation procedures. Herein, we present a method for transferring oNP photocatalysts from water into polar non-aqueous solvents while retaining their long-term colloidal stability. We observed that the polymer chains in the solvent-transferred oNPs rearrange from a predominantly H-aggregate structure in water to a combination of H- and J-aggregate characteristics in N,N-dimethylformamide, suggesting a dynamic rearrangement in response to the new solvent environment. Importantly, transient absorption and time-resolved microwave conductivity measurements confirm that the solvent-transferred oNPs maintain their ability to generate free charges at an internal heterojunction. This development opens unique opportunities for eventually leveraging light-generated, long-lived electrons and holes in synthetic redox chemistry across diverse solvent environments, a direction that will be explored in future studies.
1 Introduction
Photocatalysis is a rapidly growing field as we strive to economically and effectively produce solar-fuels1–6 and value-added chemicals7 using sunlight, an abundant source of energy. Photocatalysis is promising for its ability to perform energetically demanding chemical transformations at ambient temperatures and pressures. However, traditional organometallic photocatalysts are typically made of extremely rare precious metals and can be unstable in the presence of air or water.7Recently, organic nanoparticles (oNPs) photocatalysts have been shown to perform the hydrogen evolution reaction (HER) in water when the oNPs are loaded with a platinum cocatalyst and combined with a sacrificial hole scavenger, typically ascorbic acid.8,9 These nanoparticles are typically composed of an electron-donating semiconducting polymer (‘donor’) and an electron accepting semiconducting small molecule (‘acceptor’), resulting in a bulk heterojunction-like morphology. When illuminated, excitons in the oNP migrate to a donor/acceptor heterojunction boundary where they undergo photoinduced electron transfer to generate free carriers. Our past work has shown that charge generation within a donor/acceptor blend oNP correlates linearly with their photocatalytic activity,10 and corroborates findings that oNPs are an effective framework for long-lived surface-available free carriers.10–12
oNP photocatalysts have several benefits over traditional molecular organometallic photocatalysts; they are composed of earth-abundant elements and require only small amounts of precious metal co-catalyst to perform HER.8 Moreover, their modular structure allows their overall redox potential to be tuned by swapping the donor and/or acceptor phases with different polymers or small molecules, allowing the electronic properties of these photocatalysts to be tuned toward different target reactions.9,13
Polymer oNPs are currently restricted to use in aqueous environments because they are typically synthesized through nano-precipitation or nano-emulsion methods, both of which rely on water as an ideal solvent.14–16 While oNPs synthesized via a nano-precipitation procedure can theoretically be prepared in non-aqueous environments, oNPs made via this procedure are typically quite dilute and often suffer from poor colloidal stability due to large particle size and lack of a surfactant coating.14,17,18 The conditions for this procedure need to be optimized for each solvent, and desired solvents must be amenable for direct ultra-sonication, which is not recommended for flammable or chlorinated solvents. As a result, oNPs are used widely for HER8,9,19 and even CO2 reduction in water,13 but are seldom, if ever, used to perform photoredox catalysis for chemical synthesis because few reactants for synthetic redox reactions are soluble in water.
Herein, we describe a water-in-oil emulsion method for transferring oNPs from water into polar non-aqueous solvents while retaining both their long-term colloidal stability and their ability to generate free charges at an internal heterojunction. Our approach can be used to create stable suspensions of oNPs in a diverse array of solvents with minimal tailoring to each system. Compared to traditional methods, a wider array of solvents can be used because the procedure relies on a gentle bath-sonication method rather than a high-intensity ultrasonication step. Our reported solvent transfer process is a unique addition to the organic nanoparticle synthesis literature and a significant step toward enabling the use of oNPs' long-lived surface charges to catalyze diverse chemistries beyond HER in synthetically relevant solvents, either directly or via a secondary surface-bound photocatalyst—an area of ongoing research that will be explored in future studies. Furthermore, solvent-transferred oNPs are a useful system for understanding how solvent dielectric properties impact the availability of free charges within a semiconducting organic polymer, an area of research that is broadly relevant for organic photovoltaic (OPV) research. Herein, we first summarize the solvent-transfer procedure, then demonstrate that single-phase PTB7-Th oNPs remain colloidally stable upon transfer into N,N-dimethylformamide (DMF), and finally demonstrate that solvent-transferred two-phase PTB7-Th (donor)/N2200 (acceptor) oNPs remain capable of generating free carriers in their new non-aqueous environment.
2 Results & discussion
2.1 Solvent transfer overview
Our reported solvent transfer process relies on an intermediate water-in-oil emulsion step, where oNPs are transferred first from water into oleic acid, then from oleic acid into the target solvent. A more detailed description of the oNP fabrication and transfer processes can be found in the methods section, but an overview is provided here and is summarized in Fig. 1. | ||
| Fig. 1 Schematic of the solvent-transfer process. | ||
oNPs are synthesized in water using a chloroform-in-water micro-emulsion method, as reported previously.10 This micro-emulsion is stabilized by the amphiphilic surfactant TEBS, and results in a aqueous stock solution of oNPs. The oNPs are then transferred into oleic acid using another emulsifying step: the aqueous oNP stock solution is bath-sonicated with oleic acid to form a water-in-oil emulsion. The water droplets are then boiled off, yielding a solution of oNPs suspended in oleic acid. Finally, the oleic acid/oNP solution is centrifuged until the oNPs crash out as a pellet; the oNPs are then washed with acetone before suspension in DMF. Chemical structures of each species are shown in Fig. 2.
 | ||
| Fig. 2 Chemical structures of (a) PTB7-Th, (b) N2200, (c) TEBS surfactant, and (d) oleic acid. | ||
Oleic acid is a commonly used ligand in nanomaterial synthesis protocols,20 and we hypothesize that residual oleic acid remains in the solvent-transferred oNP solutions, either as a surface coating on the oNPs or freely dissolved in the solvent. This is supported by the appearance of oleic acid absorption peaks at 270 nm in the spectrum of solvent-transferred samples, as shown in Fig. 3. We find that including an intermediate oleic acid stage helps to both precipitate the oNPs out of solution during centrifugation and also allows for reliable re-suspension of the oNPs in their destination solvent. Moreover, oleic acid's immiscibility with water and its high boiling point allow for the effective removal of water via a water-in-oil emulsion. The precise mechanism by which oleic acid stabilizes the particles—whether through chemical bonding or physical adsorption—has yet to be determined. Investigating all-organic systems, where the nanoparticle and the ligand inherently share chemical and vibronic properties, presents significant challenges. Advancing our understanding of such systems will require innovative approaches to adapt existing characterization techniques developed for probing inorganic nanomaterial–ligand interactions.
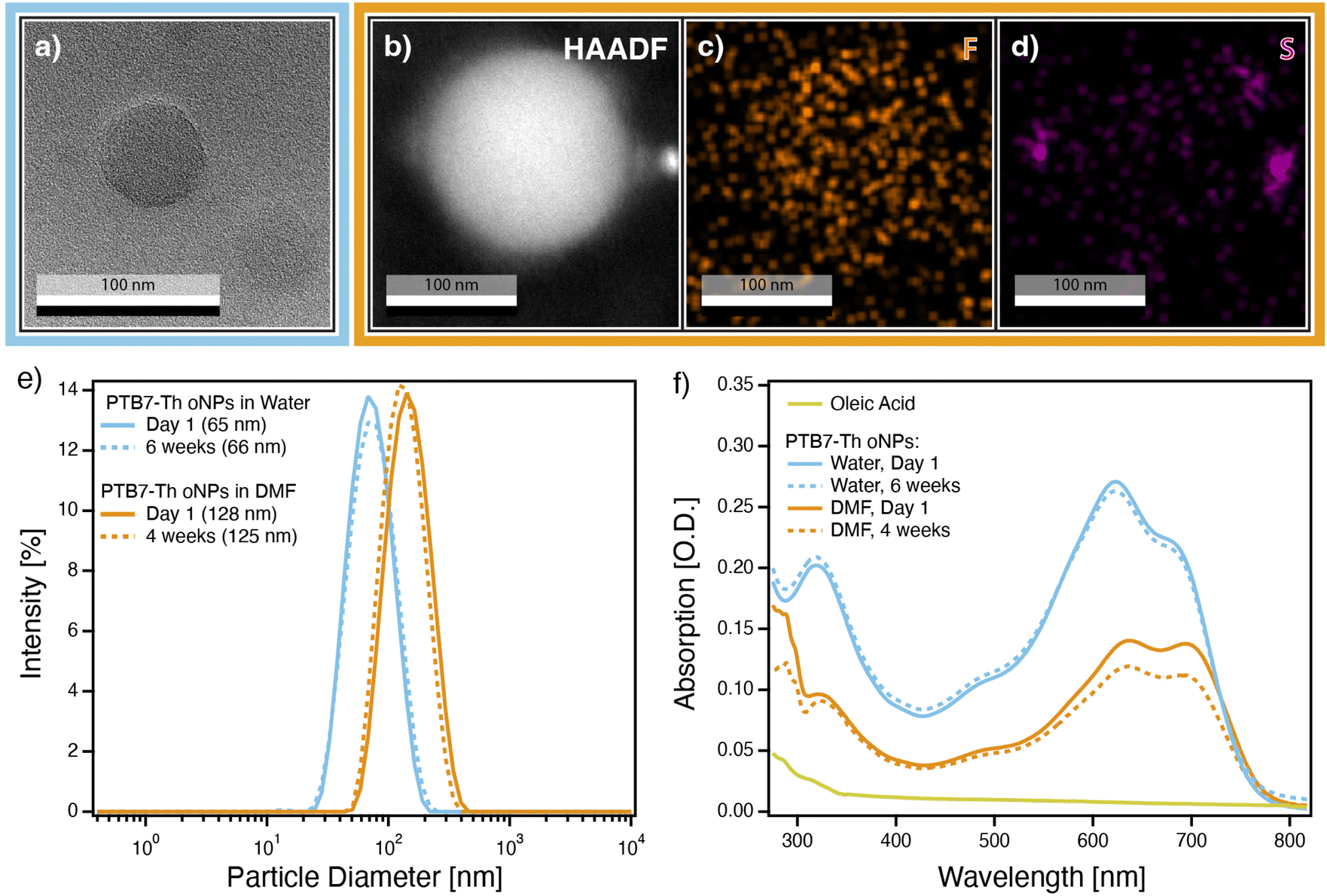 | ||
| Fig. 3 (a) Bright-field TEM image of PTB7-Th oNPs cast from water; (b) HAADF image of PTB7-Th oNP cast from DMF; EDS (c) fluorine and (d) sulfur elemental maps of image shown in (b); (e) DLS size distributions and (f) UV-Vis absorption spectra of PTB7-Th oNPs in water (light blue traces) and DMF (orange traces) on the day of sample preparation (solid traces) and after several weeks of aging (dashed traces). | ||
This procedure concentrates all contents of the water into the oleic acid, and therefore it is important to minimize contamination in the aqueous oNP stock solution. We observe via TEM imaging that trace ions like Si or Ca can condense into solid nanostructures during the water-in-oil emulsion process (ESI, Fig. S16 & S17†), as reported elsewhere in the literature.21 Therefore, all aqueous samples are prepared from MiliQ water and stored in Teflon in order to minimize silicon leeching into the aqueous stock.22 Steps towards reducing ionic contamination are described in full in the ESI, Section S6.†
It is important to highlight that this solvent-transfer process is effective only for moving polymer oNPs into ‘bad’ solvents, i.e., solvents in which the polymer is insoluble. Transferring the oNPs into a good solvent for the polymer results in their disassembly. We made significant efforts to adapt the solvent transfer process for good solvents by introducing a covalent cross-linking step, described by Dahlström et al.,23 to lock the oNPs in their spherical shape, preventing their disassembly. While these attempts showed promising results, it remains unclear whether the cross-linking was effective in preserving particle integrity. The work discussed in this report does not utilize the cross-linking step, but data related to these efforts are included in ESI, Section S8.†
2.2 Colloidal stability
Transmission electron microscopy (TEM), dynamic light scattering (DLS), and UV-Vis absorption spectroscopy show that single-phase PTB7-Th oNPs in water and in DMF are both chemically and colloidally stable for several weeks. TEM imaging confirms the presence of intact, spherical oNPs in the original aqueous stock solution (Fig. 3a), in DMF immediately after the solvent transfer (Fig. 3b), and after a week of aging in DMF (ESI, Fig. S24e†). TEM images indicate that oNPs cast from DMF appear slightly larger and less dense than those cast from water, and often form small multi-particle clusters (ESI, Fig. S24b†). We hypothesize this is due to the fact that PTB7-Th is slightly soluble in DMF but completely insoluble in water, causing the oNPs to swell more with DMF than with water. Indeed, semiconducting polymers are known to swell slightly in the presence of a bad solvent like water,2 and theoretically much more in the presence of a better solvent like DMF. We confirm that the nanoparticles seen in the TEM imaging are indeed composed of PTB7-Th polymer, as opposed to an ionic contaminant, using an energy dispersive X-ray spectroscopy (EDS) fluorine map (Fig. 3c), which shows that fluorine atoms, which are present only in PTB7-Th, map over the image of the oNP. EDS data also reveals clusters of sulfur around the outside of the oNPs/DMF (Fig. 3d), which we attribute to residual TEBS surfactant from the initial aqueous oNP synthesis.DLS data (Fig. 3e) confirm that the oNPs are colloidally stable over several weeks. The average diameter of oNPs in water remains approximately 65 nm over the course of 6 weeks, with minimal increase in polydispersity index (PDI) (ESI, Table S2†). Likewise, the oNPs in DMF are colloidally stable by DLS for at least 6 weeks, with an average diameter of approximately 127 nm. We hypothesize the two-fold increase in diameter as the oNPs move from water into DMF is largely a result of particles swelling with solvent, as seen from the increased particle diameter in the TEM data. Finally, absorption data reveal that the oNPs in water and in DMF are fairly chemically stable when stored in the dark under air, experiencing only 15% bleaching over the course of 4 weeks (Fig. 3f).
The transfer process and resulting oNP colloidal stability has proven to be generalizable to other solvents and to oNPs composed of other polymers beyond PTB7-Th. We've shown that this process can be applied to transfer oNPs composed of PM6, another high-performance semiconducting polymer, into DMF and methanol, and oNPs composed of an N2200/PTB7-Th blend into dimethoxyethane (DME). These data are shown ESI, Sections S8 & S5,† and the conclusions drawn from them closely mirror the conclusions from the PTB7-Th data set.
2.3 Morphological rearrangement
UV-Vis absorption spectroscopy reveals key electronic differences between oNPs in water vs. DMF, likely as a result of differences in polymer chain packing. Peak broadness, relative absorbance, and position are used to understand the nature of aggregation in each of the samples. Molecular aggregates are broadly understood as being either J-type or H-type, where J-aggregates have transition dipole moments that are aligned ‘head-to-tail’ and H-aggregates have transition dipole moments that are aligned parallel to one another.24 An isolated polymer chain can be roughly approximated as a J-aggregate due to the head-to-tail electronic coupling between sequential monomer units.25 However, as polymer chains aggregate, π–π stacking between adjacent chains can introduce significant H-aggregate character. Due to the diversity of possible polymer configurations and morphologies, polymer systems can ultimately have a dynamic mixture of H- and J-aggregate influence where intra-chain alignment and order between monomer units (J-aggregation) competes with inter-chain electronic coupling (H-aggregation), described as the ‘HJ-Aggregate Model’.25,26 It has been shown that absorption peaks of conjugated polymers broaden with increasing degrees of aggregation-induced disorder.27 With the exception of this broadening, absorption spectra for J-aggregates tend resemble the non-aggregated monomer; they have greater absorbances in their S00 peak than their S10 peak, but absorption peaks tend to be red-shifted relative to those of the monomer subunit. Meanwhile H-aggregates tend to have greater absorbance in their S10 peak than their S00 peak and these peaks are blue-shifted relative to the monomer subunit. J-aggregates also tend to be highly emissive since emission from their lowest energy excited state is allowed, while the lowest energy excited state of an H-aggregate has minimal oscillator strength, resulting in quenched emission.In order to better understand the oNP's electronic environment, absorption spectra for oNPs in water and oNPs in DMF are compared to absorption spectra of a solid-state bulk thin film of PTB7-Th and bulk PTB7-Th in chloroform in Fig. 4a. As expected, PTB7-Th in chloroform (pink trace) indeed appears to resemble a J-aggregate, evidenced by the greater absorbance of the S00 peak relative to the S10 peak. The absorption peaks for the parent aqueous oNPs (light blue trace) are broader and blue-shifted relative to the bulk polymer in CHCl3, an indication that the polymer chains are H-aggregated within the aqueous oNP. Moreover, the S10 peak is larger than the S00 peak in the aqueous oNP absorption spectrum, confirming a significant degree of H-aggregation within the aqueous oNP.
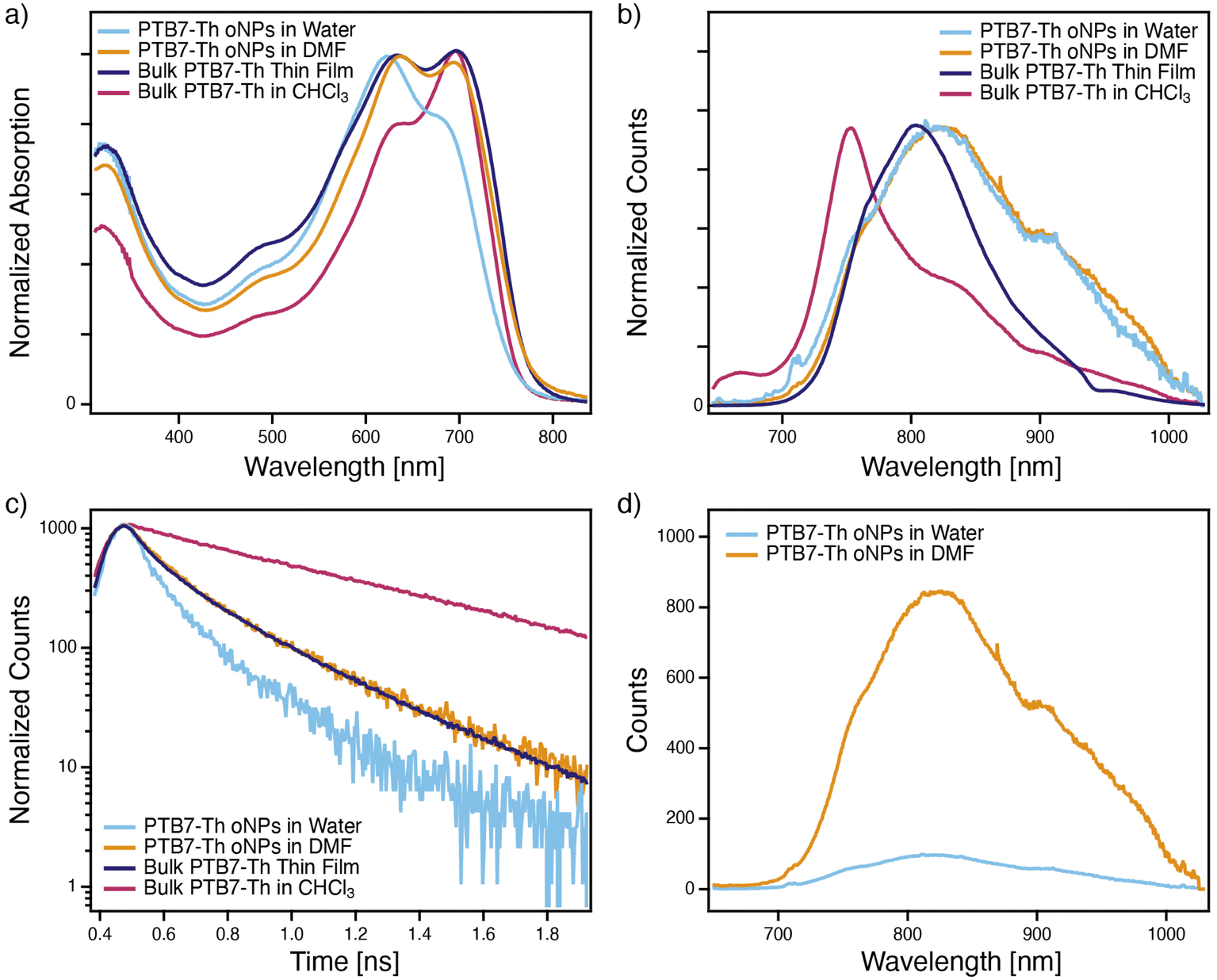 | ||
| Fig. 4 (a) Normalized UV-Vis absorption spectra, (b) normalized PL emission spectra, and (c) normalized TRPL kinetics of PTB7-Th oNPs in water (light blue trace), PTB7-Th oNPs in DMF (orange trace), a bulk PTB7-Th thin film (dark purple trace), and a bulk PTB7-Th solution in chloroform (pink trace); (d) non-normalized PL emission spectra of PTB7-Th oNPs in water vs. DMF. | ||
Absorption spectra shown in Fig. 4a of the PTB7-Th oNPs in DMF (orange trace) and bulk PTB7-Th thin films (purple trace) closely mirror one another, indicating the samples may share important morphological similarities. The primary difference between the spectra is their degree of broadening; the absorption peaks of the bulk PTB7-Th thin film are broadest, as expected for an inhomogeneously aggregated solid-state thin film,27 while the peaks of the oNPs in DMF, like the oNPs in water, are slightly narrower. Unlike in the aqueous oNP absorption spectrum, the S00 and S10 peaks in the absorption spectra for both the bulk thin film and the oNPs in DMF display equal absorbances, and their absorption onsets are equivalent to that of the bulk polymer in CHCl3. We interpret this to mean that both the bulk thin film and oNPs in DMF have a mixture of H- & J-aggregation.
Characterizing the oNPs part-way through the transfer process, when they are in oleic acid, shows that the oNP absorption spectrum does not come to mirror the bulk thin film until they're in DMF (ESI, Fig. S1a†). Rather, the absorption spectrum for the oNPs in oleic acid shows a greater contribution from J-aggregates than either the aqueous oNPs or the oNPs in DMF, evidenced by the greater relative absorbance of the S00 peak. This supports the conclusion that the oNPs are swollen with solvent and suggests that polymer chains experience dynamic morphological reorganization from primarily H-aggregation in water, to primarily J-aggregation in oleic acid, to finally a mixture of H & J-aggregation in DMF and the solid state. This type of stimulus-responsive change in packing structure has been observed in other polymer28–31 and small molecule32 systems and typically occurs in response to changes in temperature or solvent environment.
Steady-state photoluminescence (PL) data confirm similarities between the oNPs in DMF and the bulk thin film (Fig. 4b). As before, oNPs in water and DMF are compared to bulk PTB7-Th polymer in solution (CHCl3) and as a solid thin film. The bulk free polymer in CHCl3 is highly emissive, with a narrow emission peak around 750 nm and a small peak at 670 nm, which we hypothesize is due to low molecular weight monomer contamination within the polymer batch. By comparison, both oNPs in water and DMF have emission spectra that are quenched, broad, and have greater intensity in the S01 peak than the S00 peak. This peak intensity pattern and quenching is expected for H-aggregates and resembles the bulk thin film PL spectrum, which peaks around 800 nm but is slightly narrower than the oNP emission peaks. Interestingly, the oNPs in DMF are 12-fold more emissive than the oNPs in water (Fig. 4d), supporting the hypothesis that oNPs in DMF have a greater contribution from J-type aggregation.
Time-resolved photoluminescence (TRPL) kinetics (Fig. 4c) of the bulk free polymer in CHCl3 are characterized by a long lifetime, single-exponential exciton decay kinetics, and a PL spectrum that is invariant over time (ESI, Fig. S6†). The smaller 670 nm peak has a slightly longer lifetime than the larger 750 nm peak (ESI, Fig. S7†), in agreement with its assignment as a trace monomer fragment. TRPL kinetics of oNPs in DMF and water resemble the bulk polymer thin film and are characterized by short lifetimes and multi-exponential exciton decay kinetics. Peaks within these PL spectra red-shift slightly at long time scales (ESI, Fig. S6†) and their shorter exciton lifetimes and multiexponential decays are characteristic for H-aggregated polymers. Normalized transients of the oNP/DMF and bulk thin film samples overlay closely with one another, while the oNPs in water have a faster decay. This further supports the conclusion that the PTB7-Th oNPs/water have a greater degree of H-aggregation than the PTB7-Th oNPs/DMF and the bulk PTB7-Th thin film, which appear to have more of a combination of H- and J-aggregation.
2.4 Retained charge-generation functionality
For photocatalysis applications, solvent-transferred oNPs need to generate long-lived free electrons and holes, similar to how they perform in water. In this section, we use transient absorption (TA) spectroscopy and time resolved microwave conductivity (TRMC) to examine two-phase “bulk heterojunction-like” PTB7-Th (donor)/N2200 (acceptor) oNPs and compare their functionality in water and non-aqueous solvents. Steady-State Microwave Conductivity (SSMC)33 was used to identify the optimal donor:acceptor sample ratio to use within each of these experiments (ESI, Section S2†). SSMC data reveals that charge generation within both blend oNPs and bulk blend thin films remains fairly consistent across intermediate blend ratios but is ultimately optimized at different ratios. Based on these results, samples with a composition of approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PTB7-Th:N2200 were used for transient absorption measurements, while samples with a composition of approximately 1:1 PTB7-Th:N2200 were used in TRMC experiments. Charge generation within bulk and oNP thin films is expected to be comparable to one another at each of these compositions.
:1 PTB7-Th:N2200 were used for transient absorption measurements, while samples with a composition of approximately 1:1 PTB7-Th:N2200 were used in TRMC experiments. Charge generation within bulk and oNP thin films is expected to be comparable to one another at each of these compositions.
TA spectra (Fig. 5) reveal that, like a bulk blend (PTB7-Th/N2200) thin film (purple traces), 2-phase oNPs generate free charges in both water (light blue traces) and DMF (orange traces). As in the case for the single-phase oNPs, absorption spectra of the solvent-transferred 2-phase PTB7-Th/N2200 oNPs indicate a rearrangement from primarily H-aggregate-like in water to a combination of H- and J-aggregate-like in DMF (ESI, Fig. S8†). Successful charge generation is evidenced by a peak at 1124 nm, corresponding to the PTB7-Th cation, and at 838 nm, corresponding to both the N2200 anion34,35 and the PTB7-Th cation (PTB7-Th cation reference spectrum shown in ESI, Fig. S10†). Interestingly, charges in the oNPs/DMF appear slightly longer lived than those in the oNPs/water, based on the persistence of peaks at 838 nm and 1124 nm and a slower recovery of both the N2200 and PTB7-Th bleach peaks. This can be seen more clearly from kinetic traces at each of these features (Fig. 6), though kinetics at 1124 nm are inconclusive due to overlap between the PTB7-Th cation peak at 1124 nm and the broad PTB7-Th exciton peak around 1300 nm.36 TA spectra for the oNPs/water, oNPs/DMF, and bulk blend thin film show a gradual red-shifting of the ground state bleach signals and the PTB7-Th cation peak, which we attribute to the relaxation of polarons into a lower energy conformation.
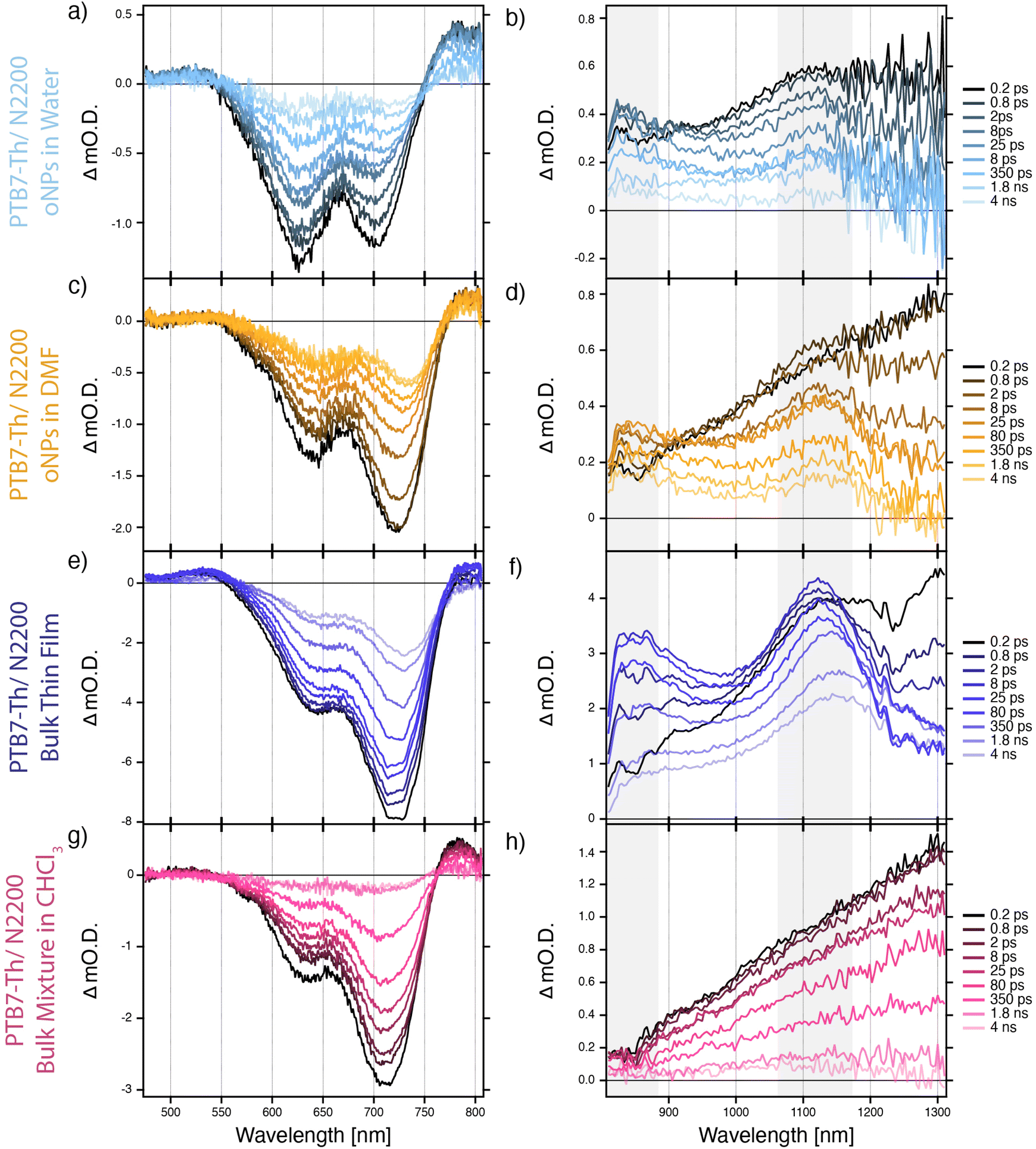 | ||
| Fig. 5 Visible (left column) and NIR (right column) transient absorption spectra of 2:1 PTB7-Th:N2200 oNPs in water (a & b), 2:1 PTB7-Th:N2200 oNPs in DMF (c & d), 2:1 PTB7-Th:N2200 bulk thin film (e & f), and a 2:1 mixture of bulk PTB7-Th + bulk N2200 in chloroform (g & h). Wavelengths of interest, highlighted in gray, correspond to kinetics presented in Fig. 6. | ||
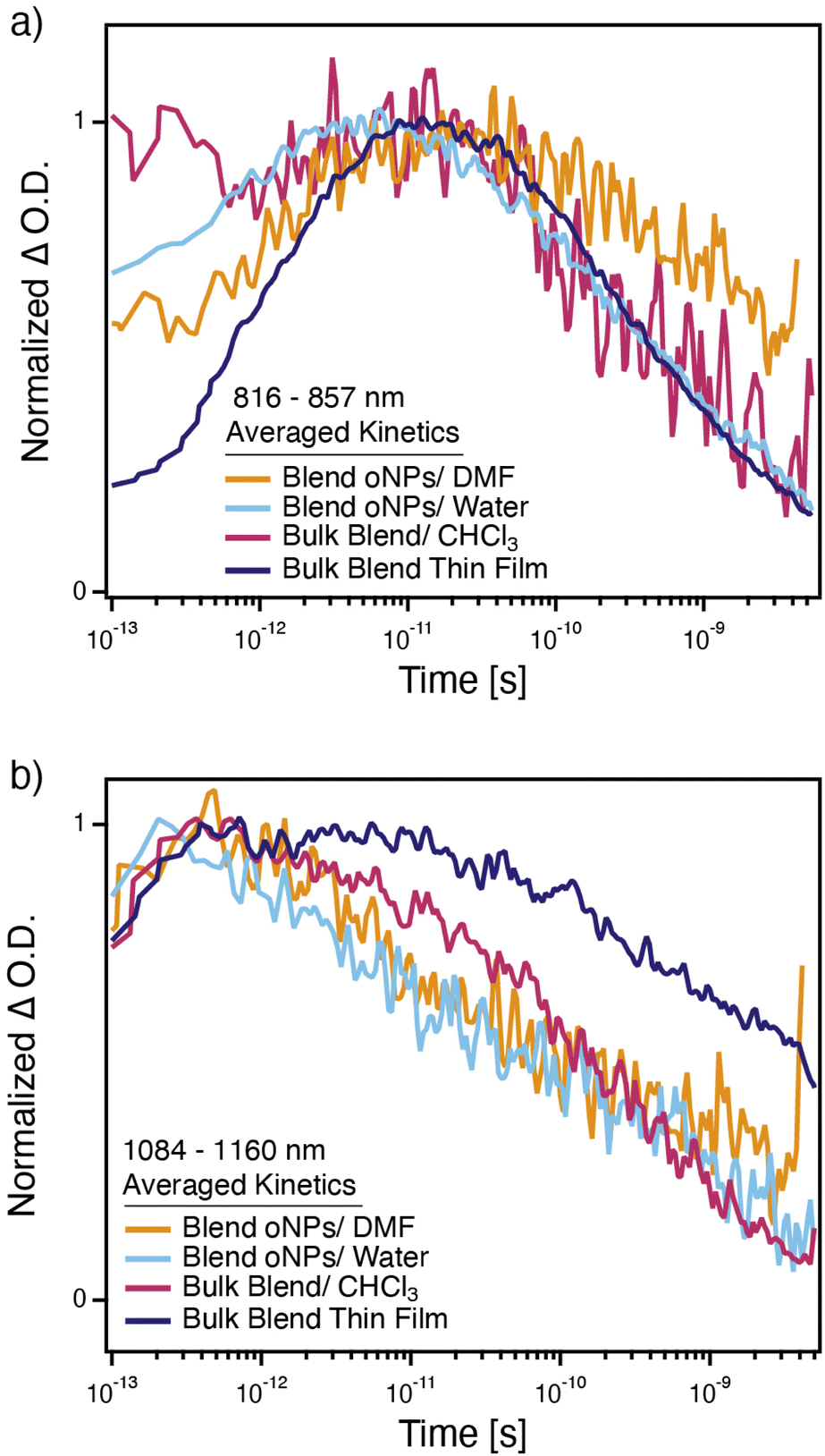 | ||
| Fig. 6 Averaged and normalized transient absorption kinetics from (a) 816–857 nm; and (b) 1084–1160 nm for PTB7-Th/N2200 blend oNPs in water (light blue trace), PTB7-Th/N2200 blend oNPs in DMF (orange trace), a bulk PTB7-Th/N2200 thin film (dark purple trace), and a solution of bulk PTB7-Th and N2200 in chloroform (pink trace). All samples are photo-excited at 600 nm. | ||
Negative control samples, including a chloroform solution of bulk PTB7-Th mixed with bulk N2200 (Fig. 5, pink traces) and an aqueous solution of single-phase PTB7-Th oNPs mixed with single-phase N2200 oNPs (ESI, Fig. S9†), show only the loss of the PTB7-Th exciton absorption at 1300 nm and rapid recovery of the ground state bleaches. These samples show no evidence of long-lived charge generation, as we would expect for an organic semiconducting mixture lacking an efficient heterojunction structure.
We use flash-photolysis TRMC to confirm that solvent-transferred 2-phase blend oNPs are capable of generating free charges from excitons. TRMC is a perturbative technique capable of measuring the time-dependent evolution of a material's electron density distribution, either in molecules in solution37 (where it is known as time-resolved dielectric loss, TRDL) or in semiconducting solids.38,39 In this technique, our samples are placed in a cavity containing a standing wave of the amplified microwave probe and photo-excited at their absorption maximum (λ = 600 nm). The sample's complex photoconductivity (Δσ = Δσ′ − iΔσ′′) is measured by monitoring the shift in resonance characteristics of the microwave cavity after photo-excitation.37,38,40 As shown in Fig. 7a, changes in the depth of cavity resonance curve correspond approximately to photo-induced changes in the real component of the sample's complex conductivity (Δσ′), which is associated with diffusion of free charges in a solid or change in dipole for a molecule tumbling in solution. Changes in the frequency of the cavity resonance curve correspond approximately to photo-induced changes in the imaginary component of the sample's complex conductivity (Δσ′′), which is associated with changes in polarizability.
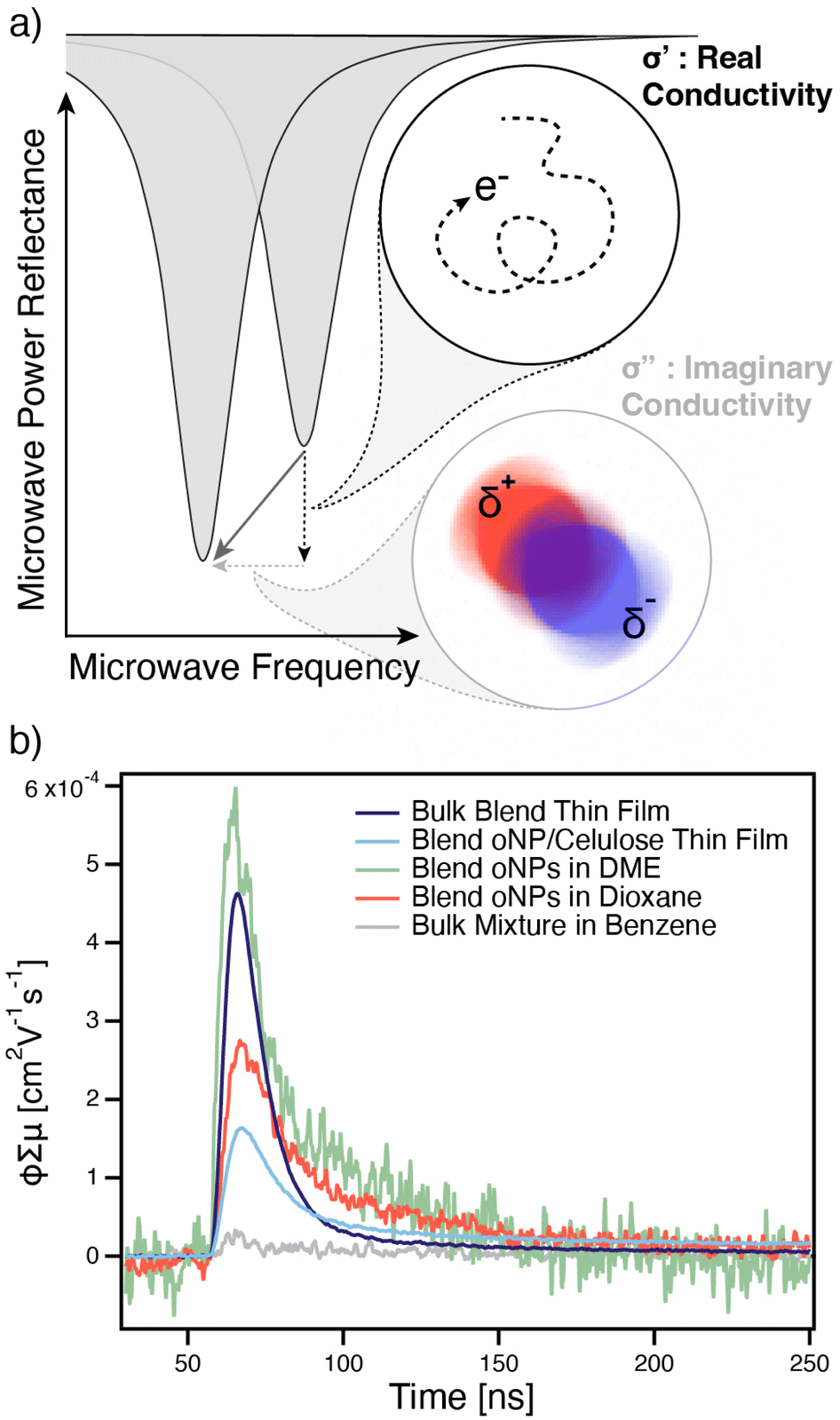 | ||
| Fig. 7 (a) Schematic of concepts describing time-resolved microwave conductivity (TRMC) spectroscopy. Changes in depth (y) of the cavity's microwave resonance describe the diffusion of free charges (real conductivity, Δσ′), while changes in frequency (x) of the cavity's microwave resonance describe changes in polarizability (imaginary conductivity, Δσ′′); (b) TRMC transients corresponding to changes in real conductivity (Δσ′) for PTB7-Th:N2200 blend oNPs cast from water as a cellulose thin film (light blue trace), a PTB7-Th:N2200 bulk blend thin film (dark purple trace), PTB7-Th:N2200 blend oNPs in 1,4-dioxane (red trace), PTB7-Th:N2200 blend oNPs in DME (green trace), and a 1:1 mixture of bulk PTB7-Th + bulk N2200 in benzene (gray trace). | ||
TRMC/TRDL is a powerful technique owing to its ability to directly quantify the yield-mobility product (ϕΣμ) of photo-generated free charges in solid-state samples,38,39 or transient changes in a molecule's dipole moment and polarizability in solution samples.37,41 However, solution-phase samples must be in solvents with a dielectric within the approximate range of 2–3 to minimize the contributions from thermal artifacts.37 These thermal artifacts dominate the TRMC or TRDL signal in solvents with relatively large dielectric constants, thus prohibiting the study of oNPs in bulk water or DMF. Therefore, we use TRMC to study 2-phase PTB7-Th/N2200 oNPs in dimethoxyethane (DME) and 1,4-dioxane in addition to a solid thin film of 2-phase PTB7-Th/N2200 oNPs in hydroxypropyl cellulose (cast from water) and a bulk PTB7-Th/N2200 blend thin film. Transient changes in real conductivity are shown in Fig. 7b, while changes in imaginary conductivity are shown in ESI, Fig. S15a.† All samples demonstrate successful charge generation, evidenced by a positive real conductivity signal, whilst the negative control sample of bulk PTB7-Th polymer mixed with bulk N2200 polymer free in benzene solution shows negligible photoconductivity. All of the samples exhibit nearly identical photoconductivity kinetics, independent of the environment and whether the sample is formed into nanoparticles or a bulk heterojunction thin film. This is similar but not identical to past results on a different donor:acceptor system.10,12 However, we do observe variations in the total photoconductivity, expressed as yield-mobility product in Fig. 7b. The oNPs in DME exhibit a ϕΣμ amplitude nearly identical to that of the bulk heterojunction thin-film, whilst nanoparticles in either dioxane or in thin-films of cellulose binder show reduced photoconductivity. This may be connected with variations in the polymer microstructure we observe via shifts in the absorbance spectra in Fig. 4a, ESI, S15c & d,† and is consistent with the greater residual ground-state bleach we observe for the oNPs in DMF and the bulk heterojunction blend at 5 ns delay in the TA results (ESI, Fig. S13a†). Notably, the relatively low yield-mobility product that all samples exhibit is attributed to the fact that it was necessary to measure these transients at relatively high (∼2.4 × 1015 cm−2) photon fluence, which results in extensive exciton-charge annihilation limiting the effective carrier yield (ϕ).10,42–44 (see ESI, Fig. S34†).
Imaginary conductivity signals (ESI, Fig. S15a†) for all solid-state samples are very short-lived (instrument-response-limited) and are attributed to a combination of the polarizability of the initial exciton and contributions from the polarizability of free charges and charge-transfer states. However, the contributions from the intrachain polarizability of excitons41 must be small, given that the mixture of free polymers in benzene exhibits substantially weaker signals than the donor/acceptor blends.
3 Conclusion
In this study, we have introduced a solvent-transfer procedure for transferring organic polymer oNPs from water into non-aqueous organic environments. Our findings demonstrate that single-phase PTB7-Th oNPs transferred into DMF are both chemically and colloidally stable, as confirmed by UV-Vis, DLS, and TEM analysis. PTB7-Th oNPs in water appear to have a high degree of H-aggregate character, indicated by blue-shifted absorption peaks, a reduced absorbance of the S00 peak, and pronounced emission quenching. Conversely, PTB7-Th oNPs in DMF more closely resemble the bulk solid PTB7-Th, and both appear to be more of a combination of H- and J-aggregates, with balanced absorbances in the S00 and S10 peaks and no shift relative to free PTB7-Th in solution. Notably, the emission of the oNPs in DMF is less quenched than the oNPs in water, suggesting dynamic rearrangement of the polymer chains within the oNPs upon transfer from water into a new solvent environment. We also demonstrate that these solvent-transferred oNPs retain their ability to generate free charges from excitons. Specifically, 2-phase PTB7-Th/N2200 oNPs in DMF can generate free charges as evidenced by transient absorption measurements, while those in DME and dioxane produce a long-lived free charge signal detected by TRMC which is generally greater than that observed for the native oNPs cast from water in a cellulose thin-film. Our reported methods represents an important new tool for transferring organic NP photocatalysts from water to more synthetically relevant solvents like DMF and DME, paving the way for broader applications in diverse chemical reactions. Future efforts will focus on investigating how solvent conditions affect the availability of free charges on the surface of these organic NP photocatalysts and exploring their potential in catalyzing a wider range of synthetic reactions beyond hydrogen evolution.4 Methods
4.1 Materials
The semiconducting polymer poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b;4,5-b′]dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-)-2-carboxylate-2-6-diyl] (PTB7-Th, also known as PCE-10, Batch # MKCT7241, MW = 180000) and the Oleic Acid (Technical Grade, 90%, Batch #MKCT2808) were purchased from Sigma Aldrich. The surfactant 3-(ethyloxy-4-butyl sodium sulfonate)thiophene (TEBS, Lot # 23B0310) was purchased from Solaris Chem. The semiconducting polymer poly[N,N′-bis(2-octyldodecyl)naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl]-alt-5,5′-(2,2′-bithiophene) (N2200, also known as P(NDI2OD-T2), Lot # YY23108DC, MW = 130000) was purchased from 1-Material. All solvents were purchased from Sigma-Aldrich. Oleic acid was purified before use by ultra-centrifugation at 22000 rpm for 3 hours (Beckman Coulter Optima XE-90 Ultracentrifuge, SW 32 Ti swinging bucket rotor) to precipitate any trace colloidal contaminants, after which the supernatant was collected. All other materials were used as received.
4.2 Aqueous nanoparticle fabrication
Stock solutions of each bulk semiconducting polymer (PTB7-Th and/or N2200) in chloroform (0.5 mg mL−1) are stirred at 80 °C for 8 hours, then stirred at room temperature for another 7 hours. A stock solution of TEBS surfactant in MiliQ water (0.5 wt%) is prepared in a Teflon vial and used fresh on the day of preparation. To make single-phase PTB7-Th oNPs, PTB7-Th/CHCl3 stock solution (2.5 mL) is combined with aqueous TEBS surfactant solution (5 mL) in a 30 mL Teflon vial, and tip-sonicated at 30% amplitude for 1.5 min (Cole-Parmer CP 750 Ultrasonic Processor, 1/4 inch tapered tip). To make 2-phase PTB7-Th/N2200 oNPs, PTB7-Th/CHCl3 stock solution is combined with N2200/CHCl3 stock solution in the desired blend ratio (total volume = 2.5 mL). The organic mixture is combined with aqueous TEBS surfactant solution (5 mL) in a 30 mL Teflon vial, and tip-sonicated using the same conditions noted above. In either case, the resulting emulsion is stirred for 30 min at 55 °C in an aluminum heating block while being bubbled with N2 to evaporate the organic phase, yielding a surfactant-stabilized suspension of oNPs in water. The aqueous oNP stock is transferred into a clean, lidded Teflon vial for storage. Aqueous oNPs are approximately 65 nm in diameter, as determined by dynamic light scattering.4.3 oNP solvent transfer
Oleic acid (9 mL) and aqueous oNP stock solution (300 μL) are combined in a 30 mL Teflon beaker and bath sonicated for 45 min, during which time a water-in-oil emulsion gradually forms. During the sonication process, the mixture is vigorously stirred every 15 min to ensure that all water is incorporated into the emulsion. After 45 min, the emulsion appears homogeneously opaque white/light blue. The resulting emulsion is stirred for 5 min at 130 °C in an aluminum heating block while being bubbled with N2 to evaporate off most of the aqueous phase. After 5 min, the oleic acid phase should be clear blue and there may be small aqueous droplets remaining at the bottom of the vial. The sample is removed from heat and allowed to cool for 10 min, then bath sonicated for another 45 min and stirred at 15-min intervals to emulsify any remaining water into the oleic acid fraction. The emulsion is again stirred at 130 °C in an aluminum heating block while being bubbled with N2 until all water is evaporated off and the oleic acid fraction is clear blue, between 2–5 min. The oleic acid solution is removed from heat and allowed to cool for 10 min, then bath sonicated for one more hour to ensure stable colloidal dispersion of the oNPs. The resulting oleic acid solution is clear blue and contains a monodisperse population of oNPs by DLS (ESI, Fig. S1b†).
The second round of bath sonication is crucial for enhancing the yield of the solvent-transfer process. After the initial heating, any remaining water separates from the oleic acid phase, and this water appears dark blue, indicating the presence of oNPs. If at this point, heating of the oil–water mixture is continued until the water evaporates, the oNPs within the water droplet will settle onto the wall of the vial instead of being included in the final oleic acid solution. To ensure the oNPs are instead incorporated into the oleic acid, it is necessary to perform a second round of bath sonication followed by heating.
oNP/oleic acid solution (∼14 mL) is ultracentrifuged at 16000 rpm for 2 hours, then at 18000 rpm for 1 more hour (Beckman Coulter Optima XE-90 Ultracentrifuge, SW 32 Ti swinging bucket rotor) such that the oNPs precipitate out as a pellet. The oleic acid supernatant is decanted and the oNP pellet is re-dispersed in 6 mL of acetone then bath sonicated for 1 min. The oNP/acetone solution is ultracentrifuged at 12000 rpm for 5 min to precipitate the oNPs as a pellet. The acetone supernatant is decanted, and the oNP pellet is re-dispersed in the target solvent. oNP/solvent samples were bath sonicated for 1 hour, then filtered with a 0.45 μm glass fiber syringe filter.
Note: Data for this report were collected using the procedure described above. Subsequently, it was found that the yield of the solvent-transfer procedure can be maximized by adding acetone (1 mL) to the oNP/oleic acid solution (14 mL) and centrifuging the mixture at a higher speed (22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000 rpm) for 3 hours prior to the acetone wash. All other steps in the process remain the same.
000 rpm) for 3 hours prior to the acetone wash. All other steps in the process remain the same.
4.4 Thin film preparation
4.5 Dynamic light scattering (DLS) measurements
The hydrodynamic diameter of oNPs in solution were measured on a Zetasizer Nano-ZS ZEN3600 from Malvern Instruments Limited. oNP diameter values are averaged from 3 sequential measurements.4.6 Steady state absorption and photoluminescence (PL) measurements
Steady-state UV-Visible absorption spectra were collected on a Cary 6000i Spectrophotometer from Agilent Technologies. Absorption spectra are baseline subtracted and baselines are collected on a 1 cm quartz cuvette containing neat solvent. Calibrated steady-state photoluminescence spectra were collected on oNP/solvent samples on the day following their transfer into solvent using a custom-built Princeton Instruments spectrometer. Samples were diluted such that their absorption at 600 nm, the excitation wavelength, was normalized to ca. 0.08 O.D. Vis-NIR PL spectra were intensity calibrated using an IntelliCal USB-LSVN (9000–410) calibration lamp and collected on a liquid nitrogen-cooled, front-illuminated Si CCD (PyLoN). Dual monochromators (HRS 500) were used to achieve pseudo-monochromatic excitation from an Energetiq EQ99x laser driven light source, with typical FWHM bandwidths ≈16 nm using a 1200 g mm−1, 750 nm blaze grating. A single monochromator was used for detection (Princeton HRS-300) with a 150 g mm−1 (800 nm blaze) grating.4.7 Time-resolved photoluminescence (TRPL) measurements
Time-resolved photoluminescence data were collected using a Hamamatsu Streak Camera (300–900 nm, C10910-04) with an NKT supercontinuum fiber laser (SuperK EXU-6-PP) routed to an acousto-optic tunable filter (SuperK Select). The samples were photoexcited at 600 nm with a repetition rate of approximately 6.5 MHz. Samples were diluted such that their absorption at the excitation wavelength was normalized to ca. 0.08 O.D. Solution phase samples were excited within a 1 cm quartz cuvette using front-face excitation and a 605 nm long-pass filter was inserted in front of the detector to remove excitation light. Transient data were collected within a 2 ns time window. Gain (25), number of exposures (75000), and exposure time (35.212 ms) were kept the same for all samples.
4.8 Scanning transmission electron microscopy (STEM) & energy-dispersive X-ray spectroscopy (EDS)
Scanning transmission electron microscopy (STEM) was performed using a high angle annular dark field (HAADF) detector on a Spectra200 S/TEM (ThermoFisher Sciences). The microscope was equipped with an X-CFEG source and was operated at an acceleration voltage of 200 kV. Samples were prepared as described above. The resulting oNP solutions (5 μL) were drop-cast onto 300 mesh Cu grids coated with lacey carbon (Ted Pella) and allowed to dry before being loaded into the microscope. Energy dispersive X-ray spectroscopy (EDS) was used to analyze the spatial distribution of elemental composition in the particles. EDS maps were acquired using a dwell time of 20–50 μs.4.9 Transient absorption spectroscopy
Transient absorption measurements were performed using a Ti:Sapphire laser (Coherent Astrella) with an 800 nm fundamental (1 kHz rep rate, 90 fs pulse width). The beam was split at the outset to create a pump and a probe pulse where the pump is generated in an optical parametric amplifier (Light Conversion, TOPAS-C) and the probe travels through a mechanical delay stage and focused into a sapphire crystal to generate a supercontinuum white light probe. Depending on the thickness of the sapphire, we accessed probe wavelengths from 440–800 nm (thin sapphire) and 850–1600 nm (thick sapphire). The probe was focused into the sample and spatially overlapped with the pump. The time-delay between the pump and probe is controlled by the position of the delay stage. Changes in the absorption spectrum are monitored by a fiber-coupled multi-channel CMOS spectrometer (Ultrafast Systems) while the pump is chopped at 500 Hz. A portion of the probe beam is picked off before the sample and guided to a reference detector to maximize the signal-to-noise ratio. Data were collected by Helios software (Ultrafast Systems) and analyzed in Surface Xplorer (Ultrafast Systems).4.10 Time-resolved microwave conductivity (TRMC)
TRMC utilizes a pump-continuous-probe design to selectively measure mobile charges, electric dipole moments, and polarizability volumes of photoexcited molecular systems.37–39 A Nd:YAG laser (Spectra-Physics) coupled to an optical parametric oscillator produces a 600 nm pump pulse (30 Hz rep rate, 7 ns pulse width) to excite the sample, which is held within a microwave resonant cavity. The X-band microwave field (8.2–12.4 GHz) within the cavity produced via an analog signal generator (Rhode&Schwarz SMA 100B) probes changes to the sample's complex conductivity following the pump pulse.37,38 Solutions of suspended oNPs are held within a quartz tube,37,45 whereas solid-state oNPs samples are cast onto a quartz substrate and measured as a thin film.38,39 Each sample is placed within a microwave resonant cavity at a position where the electric field generated by the probing microwaves are maximized, therefore, any changes to the complex conductivity of the sample affect the amount of microwave power reflected out of the cavity observed via an oscilloscope. Transient changes in the reflected power of the microwave field after photoexcitation of the samples are collected at 5 different frequencies centered around the microwave frequency where the maximum of microwaves are absorbed by the sample in the ground-state. Analysis of these transient changes to the resonant microwave field within the cavity containing the photoexcited sample allows for the separation of the contribution from each part of the complex photoconductivity,37 thus providing selective information on the production of free charge carriers and their lifetimes within oNPs.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. M. O., O. G. R., and G. R. collaborated on project conceptualization and administration. O. G. R. and G. R. collaborated on funding acquisition and supervision. O. G. R. also designed and built the TRMC technique, wrote the TRMC analysis software, and contributed to TRMC data analysis. M. M. O. designed and performed experiments (including developing the solvent-transfer procedure, sample preparation, and sample characterization via DLS, UV-Vis, TRPL, TRMC, and transient absorption), analyzed and visualized such data, created all figures, and wrote the original manuscript draft. S. C. H. performed S/TEM and EDS experiments, processed image data, wrote relevant methods section, and advised on experimental technique. M. K. G. collaborated on transient absorption spectroscopy data collection and analysis and wrote relevant methods section. J. L. R. collaborated on TRMC data collection and analysis, and wrote relevant methods section. L. H. H. identified the optimal donor/acceptor composition of oNP and bulk thin films using SSMC. Y. Z., S. B, and S. R. M. provided the covalent cross-linker. Efforts to use this covalent cross-linker to stabilize the oNPs throughout the transfer process are detailed in the ESI, Section S8.† All parties participated in manuscript editing and review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored in part by the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding was provided by the US Department of Energy, Office of Science, as part of BioLEC EFRC under grant DE-SC0019370. M. K. G. acknowledges funding for time-resolved spectroscopy and assistance with data analysis provided by the Solar Photochemistry Program, Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy. The cross-linker used in this study (see ESI, Section S8†) was synthesized with support from Office of Naval Research through awards no. N00014-21-1-2087 and N00014-20-1-2587. The authors acknowledge Dr Ross Kerner and Dr David Moore for performing inductively coupled plasma mass spectrometry (ICP-MS) measurements and analyses to identify contaminants within commercially acquired polymers (See ESI, Section S6†). The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004, 57, 39–44 CrossRef CAS.
- E. L. Ratcliff, Z. Chen, C. M. Davis, E. H. Suh, M. F. Toney, N. R. Armstrong, O. G. Reid and A. L. Greenaway, ACS Energy Lett., 2023, 8, 5116–5127 CrossRef CAS PubMed.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- O. C. Anika, S. G. Nnabuife, A. Bello, E. R. Okoroafor, B. Kuang and R. Villa, Carbon Capture Sci. Technol., 2022, 5, 100072 CrossRef CAS.
- R. Schäppi, D. Rutz, F. Dähler, A. Muroyama, P. Haueter, J. Lilliestam, A. Patt, P. Furler and A. Steinfeld, Nature, 2022, 601, 63–68 CrossRef PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- H. Yang, X. Li, R. S. Sprick and A. I. Cooper, Chem. Commun., 2020, 56, 6790–6793 RSC.
- M. M. O'Connor, T. J. Aubry, O. G. Reid and G. Rumbles, Adv. Mater., 2024, 36, 2210481 CrossRef PubMed.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- S. Gonzalez-carrero, J. Kosco, T. Fei, I. Mcculloch and J. R. Durrant, Chem. Sci., 2024, 15, 19044–19056 RSC.
- M. Ferree, J. Kosco, N. Alshehri, L. Zhao, C. S. De Castro, C. E. Petoukhoff, I. McCulloch, M. Heeney and F. Laquai, Sustainable Energy Fuels, 2024, 8, 2423–2430 RSC.
- M. V. Pavliuk, S. Wrede, A. Liu, A. Brnovic, S. Wang, M. Axelsson and H. Tian, Chem. Soc. Rev., 2022, 51, 6909–6935 RSC.
- A. Holmes, E. Deniau, C. Lartigau-Dagron, A. Bousquet, S. Chambon and N. P. Holmes, ACS Nano, 2021, 15, 3927–3959 CrossRef CAS PubMed.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- P. Fortin, S. Rajasekar, P. Chowdhury and S. Holdcroft, Can. J. Chem., 2018, 96, 148–157 CrossRef CAS.
- F. Wang, M. Y. Han, K. Y. Mya, Y. Wang and Y. H. Lai, J. Am. Chem. Soc., 2005, 127, 10350–10355 CrossRef CAS PubMed.
- A. Liu, L. Gedda, M. Axelsson, M. Pavliuk, K. Edwards, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2021, 143, 2875–2885 CrossRef CAS PubMed.
- S. Mourdikoudis, M. Menelaou, N. Fiuza-Maneiro, G. Zheng, S. Wei, J. Pérez-Juste, L. Polavarapu and Z. Sofer, Nanoscale Horiz., 2022, 7, 941–1015 RSC.
- M. Sanchez-Dominguez, K. Pemartin and M. Boutonnet, Curr. Opin. Colloid Interface Sci., 2012, 17, 297–305 CrossRef CAS.
- S. Gin, J. M. Delaye, F. Angeli and S. Schuller, npj Mater. Degrad., 2021, 5, 42 CrossRef.
- S. Dahlström, S. Wilken, Y. Zhang, C. Ahläng, S. Barlow, M. Nyman, S. R. Marder and R. Österbacka, ACS Appl. Energy Mater., 2021, 4, 14458–14466 CrossRef PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- H. Yamagata and F. C. Spano, J. Chem. Phys., 2012, 136, 184901 CrossRef CAS PubMed.
- F. Panzer, M. J. Dyson, H. Bakr, S. Wedler, K. Schötz, M. Chauhan, P. N. Stavrinou, A. Köhler and N. Stingelin, Adv. Funct. Mater., 2024, 34, 2314729 CrossRef CAS.
- M. Li, H. Bin, X. Jiao, M. M. Wienk, H. Yan and R. A. Janssen, Angew. Chem., Int. Ed., 2020, 59, 846–852 CrossRef CAS PubMed.
- F. Panzer, H. Bässler and A. Köhler, J. Phys. Chem. Lett., 2017, 8, 114–125 CrossRef CAS PubMed.
- A. Köhler, S. T. Hoffmann and H. Bässler, J. Am. Chem. Soc., 2012, 134, 11594–11601 CrossRef PubMed.
- B. M. Langeveld-Voss, M. P. Christiaans, R. A. Janssen and E. W. Meijer, Macromolecules, 1998, 31, 6702–6704 CrossRef CAS.
- A. Sarbu, L. Biniek, J. M. Guenet, P. J. Mésini and M. Brinkmann, J. Mater. Chem. C, 2015, 3, 1235–1242 RSC.
- J. L. Blackburn, H. Zhang, A. R. Myers, J. R. Dunklin, D. C. Coffey, R. N. Hirsch, D. Vigil-Fowler, S. J. Yun, B. W. Cho, Y. H. Lee, E. M. Miller, G. Rumbles and O. G. Reid, J. Phys. Chem. Lett., 2020, 11, 99–107 CrossRef CAS PubMed.
- M. K. Gish, C. D. Karunasena, J. M. Carr, W. P. Kopcha, A. L. Greenaway, A. A. Mohapatra, J. Zhang, A. Basu, V. Brosius, S. M. Pratik, J. L. Bredas, V. Coropceanu, S. Barlow, S. R. Marder, A. J. Ferguson and O. G. Reid, J. Phys. Chem. C, 2024, 128, 6392–6400 CrossRef CAS PubMed.
- G. Wen, X. Zou, R. Hu, J. Peng, Z. Chen, X. He, G. Dong and W. Zhang, RSC Adv., 2021, 11, 20191–20199 RSC.
- Y. Tamai, Y. Fan, V. O. Kim, K. Ziabrev, A. Rao, S. Barlow, S. R. Marder, R. H. Friend and S. M. Menke, ACS Nano, 2017, 11, 12473–12481 CrossRef CAS PubMed.
- J. D. Earley, A. Zieleniewska, H. H. Ripberger, N. Y. Shin, M. S. Lazorski, Z. J. Mast, H. J. Sayre, J. K. McCusker, G. D. Scholes, R. R. Knowles, O. G. Reid and G. Rumbles, Nat. Chem., 2022, 14, 746–753 CrossRef CAS PubMed.
- O. G. Reid, D. T. Moore, Z. Li, D. Zhao, Y. Yan, K. Zhu and G. Rumbles, J. Phys. D: Appl. Phys., 2017, 50, 493002 CrossRef.
- T. J. Savenije, A. J. Ferguson, N. Kopidakis and G. Rumbles, J. Phys. Chem. C, 2013, 117, 24085–24103 CrossRef CAS.
- J. M. Schins and E. Talgorn, Rev. Sci. Instrum., 2011, 82, 064703 CrossRef PubMed.
- F. C. Grozema, R. Telesca, H. T. Jonkman, L. D. Siebbeles and J. G. Snijders, J. Chem. Phys., 2001, 115, 10014–10021 CrossRef CAS.
- A. J. Ferguson, N. Kopidakis, S. E. Shaheen and G. Rumbles, J. Phys. Chem. C, 2008, 112, 9865–9871 CrossRef CAS.
- O. G. Reid and G. Rumbles, J. Phys. Chem. Lett., 2013, 4, 2348–2355 CrossRef CAS.
- J. M. Hodgkiss, S. Albert-Seifried, A. Rao, A. J. Barker, A. R. Campbell, R. A. Marsh and R. H. Friend, Adv. Funct. Mater., 2012, 22, 1567–1577 CrossRef CAS.
- Z. Yuan, D. Dahliah, M. R. Hasan, G. Kassa, A. Pike, S. Quadir, R. Claes, C. Chandler, Y. Xiong, V. Kyveryga, P. Yox, G. M. Rignanese, I. Dabo, A. Zakutayev, D. P. Fenning, O. G. Reid, S. Bauers, J. Liu, K. Kovnir and G. Hautier, Joule, 2024, 8, 1412–1429 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00263j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |