
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmmonia synthesis from water and nitrogen using electricity with a hydrogen-permeable membrane electrochemical cell with Ru catalysts and molten hydroxide electrolyte: integration with ammonia separation and unreacted gas recirculation†
Raisei
Sagara
,
Eriko
Watanabe
and
Jun
Kubota
 *
*
Department of Chemical Engineering, Fukuoka University, 8-19-1, Nanakuma, Jonan-ku, Fukuoka 814-0180, Japan. E-mail: jkubota@fukuoka-u.ac.jp
First published on 15th April 2025
Abstract
There is considerable interest in synthesizing NH3 directly from abundant H2O and N2 using electricity from renewable energy sources, for applications such as synthetic fuels, artificial fertilizers, and raw materials for plastics. NH3 synthesis from N2 and H2O was investigated using an electrochemical setup featuring Ru/Cs+/C catalysts, Pd alloy membrane cathodes, NaOH–KOH molten electrolytes, and Ni anodes operated at 250 °C and 1.0 MPa (absolute). This electrochemical setup was integrated with a refrigerated gas/liquid separator at −75 °C to concentrate NH3 and a recirculation pump for unreacted H2 and N2. As a single-pass reactor, if NH3 separation and unreacted gas recirculation were not used, this electrochemical device produced NH3 at 1.0 MPa and 250 °C, with an apparent current efficiency of 32–20% at 10–100 mA cm−2. This efficiency was limited by the chemical equilibrium, which is calculated to be 36%. The study achieved a 90% apparent current efficiency, with a 320 nmol s−1 cm−2 production rate of NH3 at 100 mA cm−2, 250 °C, and 1.0 MPa with NH3 separation and unreacted gas recirculation. The remaining 10% of the apparent current efficiency was used for H2 production. The reaction kinetic properties and scalability of the present system were discussed.
1 Introduction
Ammonia (NH3) is a fundamental chemical substance produced at a rate of 180 million tons per year worldwide, with 85% of it being used as a raw material for chemical fertilizers or directly as a chemical fertilizer to sustain the food supply for humanity.1,2 If NH3 is obtained from renewable energy rather than fossil resources, it can be considered an ideal artificial carbon-free fuel.3 The majority of the NH3 currently produced is obtained from hydrogen (H2) derived from fossil resources and nitrogen (N2) from the atmosphere, typically through a high-temperature, high-pressure chemical process known as the Haber–Bosch process.4 It is often stated that NH3 synthesis uses ca. 2% of the human energy consumption.5 However, the characterization of NH3 synthesis as a process with high energy losses because of its high temperature and high pressure is a significant misconception. It is stated that NH3 has recently been produced at 28–29 GJ ton, which converts to 480 kJ mol−1.1,2 NH3 is an energy-rich chemical substance with a heat of combustion of 382 kJ mol−1,6 which is 80% of consumed energy. In the first place, the synthesis of NH3 requires 1.5H2, which corresponds to an energy as the heat of combustion of 429 kJ mol−1.6 Obtaining H2 from fossil resources requires additional energy for processes of endothermic reactions such as steam reforming. It is important to recognize that producing H2 from fossil fuels consumes a significant amount of energy and results in the emission of carbon dioxide (CO2). Considering this fact, the Haber–Bosch process is extremely efficient in synthesizing NH3 from H2 and N2, with the energy consumption almost equal to that of the raw material of H2.7 If high temperature and high pressure are not maintained, efficient heat recovery from the exothermic reaction with high exergy, the ease of liquefaction and separation of NH3, and the recycling of unreacted gases will not be possible, resulting in a decrease in process efficiency. It is important to recognize that the process operates at optimal pressure and temperature chosen according to the size of the operation, and while it may involve high temperature and pressure, these factors do not lead to excessive energy consumption. If NH3 is produced from H2 originated from fossil resources, the Haber–Bosch process is the most efficient method of NH3 synthesis that humanity has achieved.In moving towards establishing a carbon-neutral society without reliance on fossil resources, development of methods to derive the NH3 needed by humanity from renewable energy sources is essential.8 Furthermore, if NH3 is obtained from renewable energy, it possesses significant value not just as an artificial fertilizer but also as an artificially synthesized fuel.9 NH3 can be synthesized using H2 obtained from water electrolysis powered by hydropower and N2 from the atmosphere through the Haber–Bosch process, a method that has been still utilized in some countries worldwide for a long time.2 However, instead of the two separate stages of water electrolysis and the Haber–Bosch process, developing a method to directly obtain NH3 from water and nitrogen using electricity in a single reactor would offer a simpler approach that can better accommodate fluctuating operations. Particularly with the increasing reliance on renewable energies such as solar and wind power, which are variable energy sources rather than hydroelectric energy, establishing processes for fuel production from these power sources is crucially important.10,11 Among the various renewable energy sources, photovoltaic, solar thermal, and wind power have the potential to be deployed on a large scale in many regions worldwide, unlike biomass and other sources that are constrained by limited availability.
Various methods have been suggested for the electrochemical synthesis of NH3 from H2O and N2 in the past several years.12–18 The important point is to synthesize NH3 by evolving O2 from H2O and N2. If NH3 is synthesized from H2 and N2, it is a downhill exothermic reaction that does not require external energy of electricity; using a catalyst, as in the Haber–Bosch process, allows the reaction to proceed spontaneously without external energy. It must be noted that most of the electrochemical synthesis processes of NH3 are still in their early stages and far from being practically applicable.19,20 One of the most promising approaches seems to be Li-mediated electrochemical synthesis of NH3, although it has the drawback of demanding high overpotentials due to reduction to metallic Li.21–24 Another promising approach seems to be the application of proton-conducting solid oxide electrochemical cells operating at 400–500 °C.25 However, due to these high temperatures, high pressure like in the Haber–Bosch process is required to have a sufficient conversion ratio in the equilibrium. On the other hand, a huge number of studies have been underway to electrochemically synthesize NH3 using aqueous solutions and polymer electrolytes at around room temperature, but the reliability of NH3 production remains uncertain, making industrial application seem unlikely.19,20
The U.S. Department of Energy set numerical targets around 2017 for the development of technology to synthesize NH3 through an electrochemical pathway, with goals of 300 mA cm−2 current density, 90% current efficiency, and 60% energy conversion efficiency.10,26 While these values do represent a crucial initial step towards industrial demonstration tests, it should be noted that these benchmarks diverge significantly from the practical characteristics of electrochemical NH3 synthesis.
In this context, our research group has been conducting studies on an electrochemical device that combines water electrolysis and catalytic NH3 synthesis for the electrochemical production of NH3 from H2O and N2, operating within a temperature range of approximately 250 °C. This method aims to hydrogenate N2 catalytically, rather than reducing N2 electrochemically directly. For the electrolysis at 250 °C, phosphate-based electrolytes have been applied.27–30 Since phosphoric acid fuel cells (PAFCs) have been successfully commercialized some time ago, there is high potential for the realization of electrochemical devices utilizing phosphoric acid and phosphate electrolytes.31–34 The rate of NH3 production is essentially governed by the chemical equilibrium involving NH3, H2, and N2. Consequently, in synthesis conducted at 250 °C and 1.0 MPa in our previous paper, a maximum of approximately 28% of the current is used for NH3 production, while the remaining 72% is directed toward H2 production. The thermodynamic equilibrium limit for current efficiency under these conditions was calculated to be 36%, and the experimental results obtained were almost entirely determined by this equilibrium limitation. This equilibrium limitation is absolute even when using electrochemical methods and cannot be overcome unless a reactor or electrochemical cell capable of separating the produced NH3 from H2 and N2 is employed. While N2 activation and hydrogenation via adsorbed hydrogen atoms migrating on the surface from a hydrogen-permeable membrane can occur, surpassing chemical equilibrium might be possible if an ideal catalyst surface is designed that strongly suppresses H2 desorption. However, constructing such a surface is not realistic.
Phosphate electrolytes were effectively used for NH3 synthesis, but increasing the current density led to corrosion issues at the anode. We have achieved stable operation at higher current densities in our electrochemical system using NaOH–KOH molten salt electrolyte at around 250 °C.35 As discussed later in this paper, the NaOH–KOH molten salt becomes a concentrated aqueous solution under humidified conditions and exhibits good ionic conductivity. The most efficient method currently employed by our group for synthesizing NH3 from N2 and H2O involves using Ru catalysts, Pd–Ag hydrogen-permeable membrane cathodes, NaOH–KOH molten salt electrolytes, and Ni anodes at temperatures of 230–250 °C.35 The attempt to electrochemically synthesize NH3 using molten NaOH–KOH is not the first of its kind. There is an example from a 2014 paper reporting NH3 production using molten NaOH–KOH at 200–500 °C with an Fe2O3 cathode, but the paper was retracted in 2020, likely due to the observation of impurities.36–38 The electrolysis using molten NaOH–KOH in this study is not aimed at generating NH3 by the reduction of N2 at the cathode/electrolyte interface. It is difficult for gaseous N2 to penetrate and adsorb at the interface between the cathode and the electrolyte. In the present electrochemical cell, the water electrolysis and NH3 synthesis sections are separated, and the Ru catalyst provides a clean surface for catalytic reactions at the solid–gas interface. Therefore, this study is completely different from previous research using molten NaOH–KOH, but it is similar in that it attempts to utilize a temperature of around 250 °C.
In situations where a reaction is restricted by equilibrium, a straightforward method to enhance conversion involves the separation of unreacted reactants from the product and their reintroduction into the reaction vessel. This method is employed commonly in various catalytic processes, NH3 synthesis, methanol synthesis, Fischer–Tropsch synthesis, and so on.7 This article aims to present our research findings regarding the rate and current efficiency of NH3 synthesis from H2O and N2 through our electrochemical system when integrated with gas–liquid separation and unreacted gas recirculation equipment. Our goal is not to discover a new electrochemical reduction reaction mechanism for N2, but rather to propose a technology for synthesizing NH3 efficiently and simply from N2 and H2O using electricity.
This paper summarizes the results of investigating the characteristics of NH3 synthesis using an electrochemical system with NaOH–KOH as the electrolyte and a Ru catalyst for NH3 synthesis from H2O and N2, in which NH3 separation and unreacted gas recirculation systems were employed. The study aims to demonstrate the potential of this method for practical applications. A schematic diagram of the experimental setup is shown in Fig. 1.
 | ||
| Fig. 1 Schematic diagram of the reaction system in this study. | ||
2 Experimental details
2.1. Catalyst preparation
A 30 wt%-Ru/Cs+/C catalyst (Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cs+ = 1:1 molar ratio) weighing 0.25 g was used in the electrochemical cell, placed on the back side of a Pd–Ag membrane cathode. The catalyst was synthesized via an impregnation method, where carbon black (Vulcan XC-72R, Cabot Co.) was impregnated with a tetrahydrofuran (THF) solution of Ru3(CO)12 (TANAKA Kikinzoku Kogyo K. K.) for 4 h with continuous stirring at room temperature. The THF solvent in the carbon black suspension was removed using a rotary evaporator below 40 °C under reduced pressure to prevent the decomposition of the carbonyl complex. The resulting Ru3(CO)12/C powder was then treated at 400 °C under vacuum to yield the Ru/C catalyst. The addition of the Cs+ promoter was carried out by impregnating the Ru catalyst with an aqueous solution of CsNO3. The Cs+-modified Ru/C catalyst was finally treated at 400 °C in a flow of H2. The state of Cs+ compounds was considered to be either CsOH, Cs2O, or Cs2CO3 due to exposure to the atmosphere during storage or impurities during the reaction. However, as this is not definitive, it is referred to as Cs+. The detailed procedure can be referenced in our previous paper.26–29,33
:Cs+ = 1:1 molar ratio) weighing 0.25 g was used in the electrochemical cell, placed on the back side of a Pd–Ag membrane cathode. The catalyst was synthesized via an impregnation method, where carbon black (Vulcan XC-72R, Cabot Co.) was impregnated with a tetrahydrofuran (THF) solution of Ru3(CO)12 (TANAKA Kikinzoku Kogyo K. K.) for 4 h with continuous stirring at room temperature. The THF solvent in the carbon black suspension was removed using a rotary evaporator below 40 °C under reduced pressure to prevent the decomposition of the carbonyl complex. The resulting Ru3(CO)12/C powder was then treated at 400 °C under vacuum to yield the Ru/C catalyst. The addition of the Cs+ promoter was carried out by impregnating the Ru catalyst with an aqueous solution of CsNO3. The Cs+-modified Ru/C catalyst was finally treated at 400 °C in a flow of H2. The state of Cs+ compounds was considered to be either CsOH, Cs2O, or Cs2CO3 due to exposure to the atmosphere during storage or impurities during the reaction. However, as this is not definitive, it is referred to as Cs+. The detailed procedure can be referenced in our previous paper.26–29,33
The prepared catalyst was observed using a transmission electron microscope (TEM; JEOL JEM-2100F). Additionally, using a home-built ultra-high vacuum system combined with a quadrupole mass spectrometer, the temperature-programmed desorption (TPD) properties of H2 from Ru catalysts were investigated. The catalysts were subjected to H2 reduction treatment at 400 °C and 50 kPa, followed by evacuation at 400 °C, and their H2 desorption behaviour was measured after adsorption of H2 at −183 °C and 5.0 kPa.
2.2. Assembly of electrochemical cells
The electrochemical cell used in this study is similar to the one detailed in previous publications,27–30,35 as briefly illustrated in Fig. 2A. The cathode was a Pd–Ag membrane with a thickness of 0.1 mm (Pd:Ag = 3:1 in atomic ratio), with a section of the circle with a diameter of 20 mm in contact with the electrolyte. The NaOH–KOH aqueous solution was a mixture of 0.20 mol NaOH, 0.20 mol KOH, and 20 mL H2O, and ca. 5.0 mL of the solution was introduced into the cell. H2O in the electrolyte was evaporated as the cell was gradually heated to the operating temperature. The anode was a Ni disk with a diameter of 10 mm and a thickness of 5 mm, which was smaller in diameter than the cathode to facilitate the release of generated O2. The surface of the anode disc was positioned directly facing the cathode, with a separation of about 3 mm between them. The current density in this study was calculated based on the area of the cathode (3.14 cm2). Since the sidewall of the anode was also in contact with the electrolyte, it was difficult to accurately define the effective area of the anode.
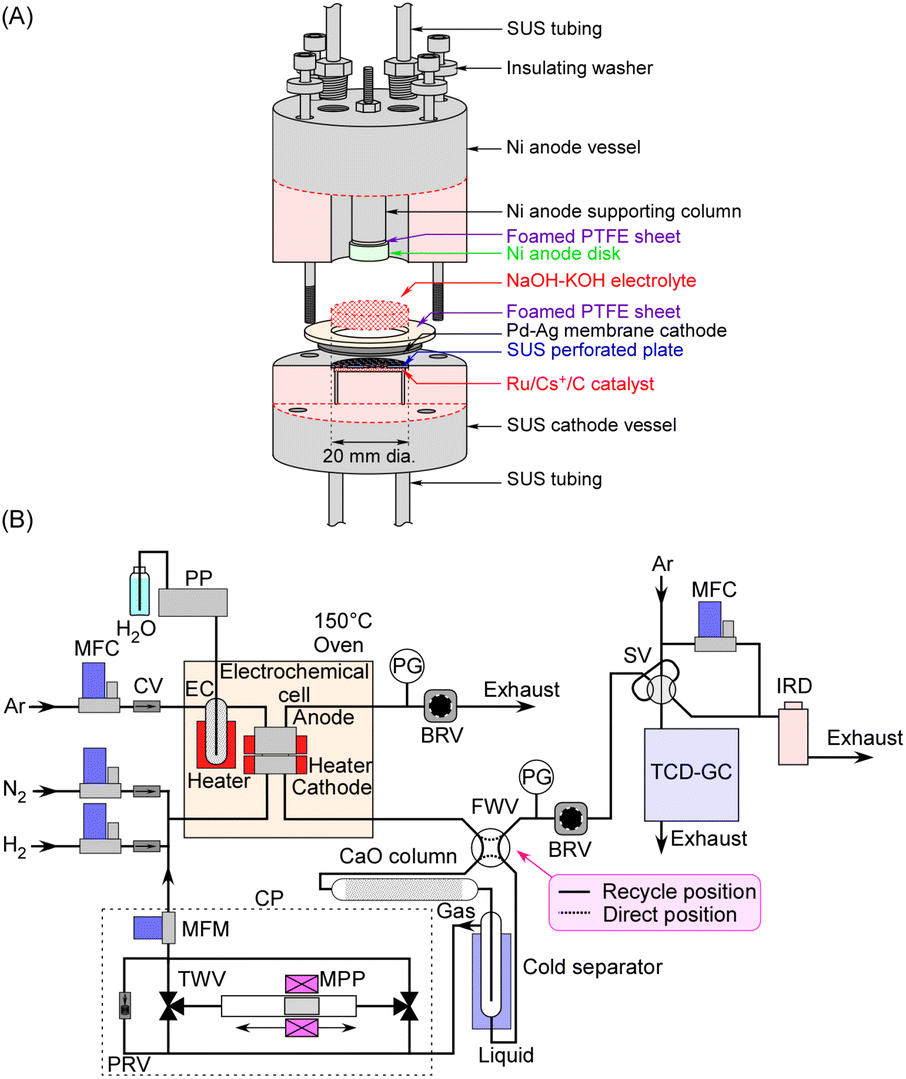 | ||
| Fig. 2 Detailed cross-sectional view of the electrochemical cell (A) and diagram of the gas lines (B). PP: plunger pump, MFC: mass flow controller, CV: check valve, EC: evaporation chamber, PG: pressure gauge, BRV: back-pressure regulating valve, FWV: four-way valve, CP: circulation pump, MFM: mass flow meter, PRV: pressure relief valve, TWV: electric three-way valve, MPP: magnetic piston pump, SV: sampling valve, TCD-GC: gas chromatograph with a thermal-conductivity detector, and IRD: infrared absorption NH3 detector. | ||
Our previous paper reported that a polytetrafluoroethylene (PTFE) vessel was used for the anode side while a steel use stainless (SUS) vessel was used for the cathode side,35 to prevent corrosion by the NaOH–KOH molten salt. However, a Ni vessel was employed for the anode side in this work, which exhibited greater mechanical strength than PTFE during operation at 250 °C and adequately withstood pressurization at 1.0 MPa. The Ni parts of the vessel, except for the anode disc, were electrically insulated, so they did not function as electrodes even when in contact with the electrolyte.
2.3. NH3 separation and unreacted gas recirculation
The gas lines are depicted in Fig. 2B. Notably, the setup includes a gas–liquid separator and a circulation pump. The gas–liquid separator was mounted on the cold head of a Stirling refrigerator which can be cooled to −100 °C. For NH3 separation, the gas–liquid separator was controlled to be at −75 °C to prevent the freezing of NH3 (melting point at −77.7 °C). The vapor pressure of NH3 at −75 °C is approximately 7 kPa, resulting in an NH3 concentration of 0.7 vol% in the recirculated gas during operation at 1.0 MPa, after passing through the gas–liquid separator. For reference, the vapor pressure curve of NH3 is shown in Fig. S1 in the ESI.†Inside the gas–liquid separator, liquefied NH3 accumulates at the bottom of the trap tube, and when the liquid blocks the tube, it is pushed out together with excess N2 relative to the stoichiometric amount, discharged from the gas–liquid separator, and vaporizes when returning to room temperature. Since NH3 is not discharged until the liquid blocks the tube is expelled and liquid accumulates again, the NH3 concentration fluctuates. The average NH3 concentration over several hours is used to calculate the NH3 production rate.
The gas–liquid separator has an inner diameter of 4.35 mm, while the liquid discharge tube has an inner diameter of 2.17 mm. The introduction tube to the gas–liquid separator has an outer diameter of 1.58 mm and an inner diameter of 1.0 mm.
To prevent the freezing of water derived from impurities in N2 from blocking the tube in the gas–liquid separator, the exhaust from the electrochemical cell was passed through a column of quick lime (CaO) to remove moisture.
The circulation pump was custom-built and featured a magnetic piston with an outer diameter of 10 mm. The piston was driven by a reciprocating motion generated by a moving magnet positioned outside the piston cylinder, which was constructed from a nonmagnetic SUS tube. The moving magnet was fixed to a linear stage that reciprocated with a 150 mm stroke, powered by a stepper motor, and provided a displacement capacity of 11.8 cm3 per stroke. Two electric solenoid-operated 3-port valves were connected to both ends of the piston cylinder, alternating to enable one-way flow. The system was capable of circulating gas at flow rates ranging from 0 to 200 cmSTP3 min−1 at a pressure of 1.0 MPa. The circulating flow rate was monitored using a mass flow meter (Model 3810DSII, KOFLOC Corp.). In case of a tubing blockage, a pressure relief valve was installed between the inlet and outlet of the pump to release excess pressure. In this paper, all pressures are expressed as absolute pressures, and gas flow rates are expressed under standard temperature and pressure conditions (STP: 0 °C and 101.3 kPa).
For unreacted gas recirculation experiments, the exhaust gas from the liquid side of the gas/liquid separator and NH3 and H2 productions were quantified using an infrared absorption NH3 detector with full scales of 1.0 or 15% (Model E12-15, Analytical Technology, Inc. (ATI)) and a gas chromatograph (Model 4200, GL Sciences Inc.) with a thermal conductivity detector, respectively. When the NH3 concentration exceeded the full scale of the NH3 detector, the sample gas was diluted with a specified flow rate of Ar before being sent to the NH3 detector. The exhaust gas from the gas side of the gas/liquid separator was introduced to the cathode side of the electrochemical cell via the circulation pump. The gas/liquid separator cannot operate at the exact stoichiometric H2/N2 ratio (=3) because the liquid NH3 is pushed out along with the excess N2 discharged beyond the stoichiometric ratio. Furthermore, along with the discharge of excess N2, H2 is also released with a few % in an apparent current efficiency from the liquid phase side of the separator.
A gas line for H2 was connected to the cathode side of the cell. However, H2 was not used for NH3 synthesis but solely for the pretreatment of the Ru catalyst when installing a fresh catalyst, as well as for conducting a leak test of the system using a portable H2 detector (Testo 316-2, Testo SE & Co. KGaA).
For single pass experiments, NH3 was sometimes quantified by absorption of the exhaust gas from the cathode side into 1 mM H2SO4 aqueous solutions with monitoring of the electroconductivity, which was used in our previous studies.27–30,35
3 Results and discussion
3.1. Condition of the electrolyte
In this study, since a NaOH–KOH eutectic molten salt was used as the electrolyte, its state will first be explained. An NaOH–KOH (1:1 molar ratio) electrolyte was used as a eutectic molten salt with a melting point at 171 °C, which is lower than the operating temperature of the electrochemical cell. The melting points of NaOH and KOH are 318 and 360 °C, respectively, both of which are higher than the operating temperature. Even when the steam supply to the anode side is stopped, the electrolyte remains as a melted eutectic molten salt. However, under the humidified anode gas conditions, the molten salt is considered to become deliquescent by the steam. The anode gas consisted of a mixture of Ar at 10 cmSTP3 min−1 and liquid H2O at 5 μL min−1, resulting in a steam concentration of 38 vol%. According to the literature, Henry's law constant for water in NaOH–KOH (63.1:36.9 mol%) eutectic molten salt was reported as 0.0095 atm mol−1 kg at 250 °C. Based on this constant, the amount of H2O at a total pressure of 0.10 MPa is 40 mol (720 g) per 1 kg of NaOH–KOH melt, and at 1.0 MPa, it is 400 mol (7200 g) per 1 kg of NaOH–KOH melt. While NaOH–KOH is a eutectic molten salt under dry conditions, under the humidified conditions of this experiment, it can be considered a concentrated aqueous solution. The detailed water concentration in the eutectic molten salt was calculated as shown in Fig. S2 in the ESI.†
3.2. One pass reaction without unreacted gas recirculation
First of all, properties of NH3 synthesis from N2 and H2O using the electrochemical cell without NH3 separation and unreacted gas recirculation as a function of current density are shown in Fig. 3. NH3 and H2 formation rates increased with increasing current density. The apparent current efficiencies (CE) for NH3 (CENH3) and H2 (CEH2) formations are defined as follows:27–30,35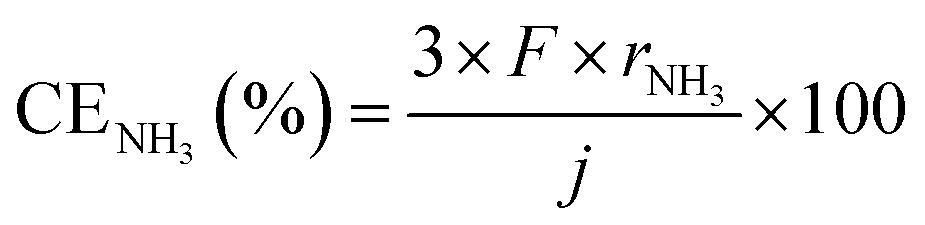
where rNH3 and rH2 are electrode area-normalized formation rates of NH3 and H2, respectively, in mol s−1 cm−2. The variable j expresses the current density in A cm−2, and F expresses the Faraday constant of 96
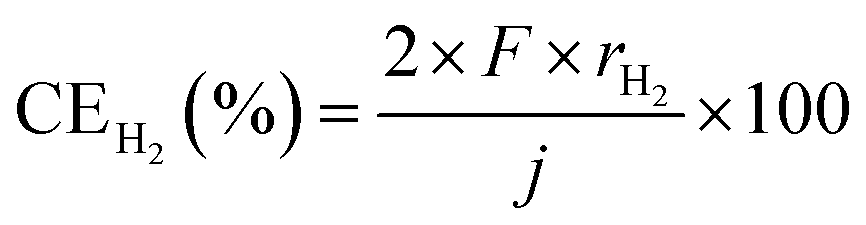 485 C mol−1. The term “apparent current efficiency” is used because, in this study, the NH3 synthesis reaction does not involve direct charge transfer to N2, but rather occurs through a hydrogenation reaction by H2 generated via electrolysis. In particular, during the experiment with unreacted gas recirculation, some NH3 is synthesized from the unreacted H2 and the current does not directly participate in the recirculation process. Therefore, we refer to it as the “apparent” current efficiency. It represents the efficiency of the current during the overall operation of the system in relation to the rate of NH3 produced.
485 C mol−1. The term “apparent current efficiency” is used because, in this study, the NH3 synthesis reaction does not involve direct charge transfer to N2, but rather occurs through a hydrogenation reaction by H2 generated via electrolysis. In particular, during the experiment with unreacted gas recirculation, some NH3 is synthesized from the unreacted H2 and the current does not directly participate in the recirculation process. Therefore, we refer to it as the “apparent” current efficiency. It represents the efficiency of the current during the overall operation of the system in relation to the rate of NH3 produced.
 | ||
| Fig. 3 Formation rates of NH3 and H2, and apparent current efficiency for NH3 and H2 formation from H2O and N2 at 250 °C and 1.0 MPa as a function of current density for one-pass experiments without NH3 separation and unreacted gas recirculation. The anode gas was a mixture of Ar at 10 cmSTP3 min−1 with 5 μL min−1 of liquid H2O. The feeding rate of N2 was set at 0.10 cmSTP3 min−1 per 1 mA cm−2 and was adjusted in proportion to the current density. | ||
The apparent current efficiency for NH3 formation was 32% at 10 mA cm−2 and gradually decreased with increasing current density. On the other hand, the apparent current efficiency for H2 formation was 66% at 10 mA cm−2 and gradually increased with increasing current density. The change in current efficiency for NH3 formation and H2 formation as a function of current density is complementary, and their sum is approximately 100%.
As mentioned earlier, the equilibrium limit under these conditions is a current efficiency of 36%, and the NH3 formation rate and its current efficiency were found to be largely constrained by thermodynamic equilibrium. Apparent current efficiency for NH3 formation decreased from 32 to 20% on increasing current density from 10 to 100 mA cm−2, and this was due to the insufficient catalyst activity of the Ru catalyst. This is confirmed by the fact that as the current efficiency for NH3 decreases, the current efficiency for H2 formation increases. Our previous paper indicated that both the current efficiencies for NH3 and H2 formation decreased with increasing current density over 30 mA cm−2. In this study, due to the improved mechanical precision of the cell, up to a current density of 100 mA cm−2, the changes in current efficiency for NH3 formation and H2 formation were complementary, and the overall current efficiency was maintained at 100%.
However, when attempting NH3 synthesis at a current density of 300 mA cm−2, the overall current efficiency deviated significantly from 100%, indicating the occurrence of product cross-leakage. The current efficiency recovers when the current is reduced, so this is not due to irreversible degradation of electrodes and electrolytes, such as electrode corrosion. This could be due to O2 bubbles generated at the anode interface coming into contact with the Pd–Ag cathode interface, or hydrogen produced at the cathode being unable to be absorbed as H atoms by Pd–Ag and instead generating H2 gas on the electrolyte side. It is speculated that improvements could be made through the use of separator materials or surface treatment of Pd–Ag, but this will be addressed in future research and development. Although the overall current efficiency was operated at nearly 100% at a current density of 100 mA cm−2, which is one-third of the DOE target in 2017 of 300 mA cm−2, this is considered a promising value for achieving the DOE target.10,26
Next, the dependency on the N2 flow rate at 100 mA cm−2, 1.0 MPa, and 250 °C was examined, as shown in Fig. 4. The current efficiency for NH3 formation reached a maximum at an N2 flow rate of around 5–10 cmSTP3 min−1. In contrast to the behavior observed for NH3 formation, the apparent current efficiency for H2 formation decreased at an N2 flow rate of around 5–10 cmSTP3 min−1, thus maintaining the total current efficiency at 100%. The stoichiometric N2 flow rate for 100 mA cm−2 is 0.729 cmSTP3 min−1, meaning that the optimum N2 flow rate was roughly estimated to correspond to an H2/N2 = 0.3. In our previous paper, the optimum N2 flow rate was found to be H2/N2 = 0.07 under the conditions of 3.2 mA cm−2, 0.1 MPa, and 250 °C.29 In the present experiment, the optimal N2 flow rate conditions also deviated significantly from the stoichiometric H2/N2 = 3, with the highest current efficiency for NH3 formation being achieved where N2 was in great excess. The reason why these N2-excess conditions are optimal for the NH3 synthesis rate is that, in NH3 synthesis reactions using Ru-based catalysts, the reaction order with respect to the partial pressure of H2 is generally negative.39,40 In other words, in NH3 synthesis from the stoichiometric ratio of 3H2 + N2, even though H2 is a reactant, the reaction rate decreases as the partial pressure of H2 increases. Therefore, the NH3 formation rate reaches its maximum under conditions where N2 is in excess.
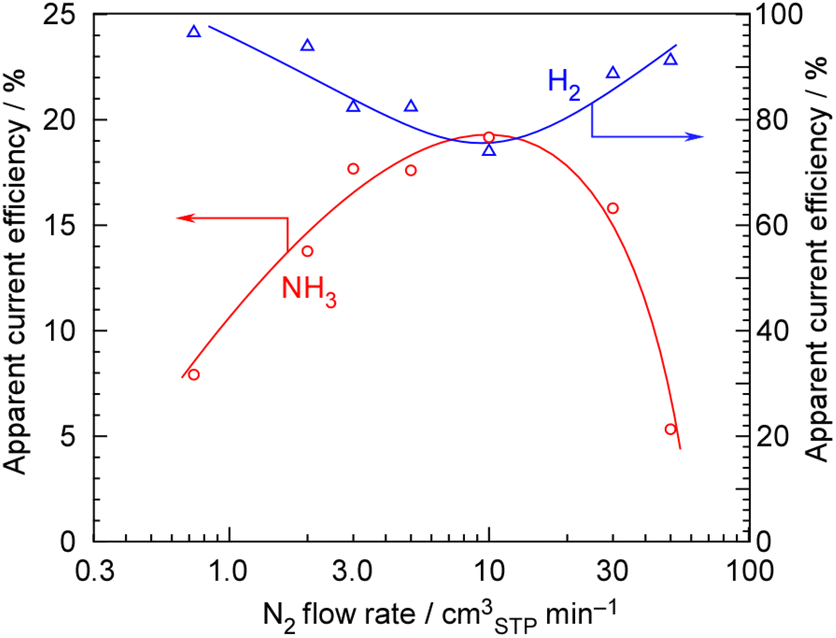 | ||
| Fig. 4 Apparent current efficiency for NH3 and H2 formation from H2O and N2 at 250 °C and 1.0 MPa as a function of the N2 flow rate for one-pass experiments without NH3 separation and unreacted gas recirculation. The anode gas was the mixture of Ar at 10 cmSTP3 min−1 with 5 μL min−1 of liquid H2O. | ||
3.3. Reaction with NH3 separation and unreacted gas recirculation
The results of NH3 synthesis from H2O and N2 at 30 and 100 mA cm−2 with NH3 separation at −75 °C and unreacted gas recirculation are shown in Fig. 5 and 6, respectively. Formation rates of NH3 and H2, the NH3 concentration at the outlet of the liquid side of the separator, and apparent current efficiency for NH3 and H2 formation are plotted against the feeding flow rate of N2 for each graph. The operation temperature and pressure were 250 °C and 1.0 MPa. | ||
| Fig. 5 NH3 and H2 formation rates, NH3 concentration in the exhaust gas, and apparent current efficiency for NH3 synthesis and H2 formation from H2O and N2 at 250 °C and 1.0 MPa as a function of the N2 flow rate, with NH3 separation at −75 °C and unreacted gas recirculation. The current density was 30 mA cm−2 and recirculation flow rate was 150 cmSTP3 min−1. The stoichiometric flow rate of N2 is 0.219 cmSTP3 min−1 for 30 mA cm−2. The anode gas was provided from the mixture of Ar at 10 cmSTP3 min−1 with 5 μL min−1 of liquid H2O. | ||
 | ||
| Fig. 6 NH3 and H2 formation rates, NH3 concentration in the exhaust gas, and apparent current efficiency for NH3 synthesis and H2 formation from H2O and N2 at 250 °C and 1.0 MPa as a function of the N2 flow rate, with NH3 separation at −75 °C and unreacted gas recirculation. The current density was 100 mA cm−2 and recirculation flow rate was 150 cmSTP3 min−1. The stoichiometric flow rate of N2 is 0.729 cmSTP3 min−1 for 100 mA cm−2. The anode gas was provided from the mixture of Ar at 10 cmSTP3 min−1 with 5 μL min−1 of liquid H2O. | ||
Fig. 5 (30 mA cm−2) shows that the formation rate of NH3 gradually increased with the decreasing flow rate of N2. In contrast, the formation rate of H2 gradually increased with the decreasing flow rate of N2. The stoichiometric flow rate of N2 is 0.219 cmSTP3 min−1 for 30 mA cm−2, so the minimum N2 flow of 0.50 cmSTP3 min−1 was 2.3 times. Below a N2 flow of 1.0 cmSTP3 min−1, the formation rate of NH3 became maximum and constant, while the formation rate of H2 became minimum and constant. The absolute accuracy of the formation rate of H2 was higher than that of NH3; the apparent current efficiency for the NH3 formation was estimated to be 94% after subtracting the apparent current efficiency for the H2 formation about 6% from 100%.
On the other hand, at a current density of 100 mA cm−2 as shown in Fig. 6, the apparent current efficiency for H2 formation was evaluated to be approximately 10% when the N2 flow rate was below 3.0 cm3 min−1, so the apparent current efficiency for NH3 formation was evaluated to be 90%. At any N2 flow rate and current density, the current efficiencies for NH3 and H2 formations add up to approximately 100%, indicating that there is almost no current loss and that either NH3 or H2 is being generated.
The total current efficiency may deviate by a few percent from 100%, but this is considered experimental error. NH3 and H2 are analyzed using an infrared detector and gas chromatography, and these measurements are quantified relative to the N2 flow rate. We frequently calibrate the N2 flow rate as much as possible and conduct experiments carefully to avoid gas leakage, but experimental errors of a few percent to around 10% relative to 100% are unavoidable.
One important point here is that the optimal N2 flow rate differs significantly from 5–10 cmSTP3 min−1 shown in Fig. 4 for a single-pass system without recirculation. Fig. 4 shows the optimal flow rate for achieving the maximum NH3 formation rate from a kinetic standpoint in a single-pass system. However, the optimal flow rate in a system that combines NH3 separation and unreacted gas recirculation is not determined by kinetics. In a system with NH3 separation and unreacted gas recirculation, an excess of N2, stoichiometrically relative to the H2 produced by electrolysis, is discharged. In this case, since the system relies on the displacement of liquid NH3, the presence of excess N2 is essential. However, if there is too much N2, the system will discharge the unreacted H2 in nearly 80% current efficiency, along with the excess N2. While it is crucial to minimize excess N2 flow as much as possible in a system with NH3 separation and unreacted gas recirculation, this suggests that operating in this way is kinetically disadvantageous, as indicated in Fig. 4.
Fig. 7 shows apparent current efficiency for NH3 and H2 formation as a function of the recirculation flow rate. Without recirculation (flow rate of 0), the apparent current efficiency for NH3 formation was 20.0%, and for H2 formation, it was 74.7%. However, since the horizontal axis of the figure is shown on a logarithmic scale, zero could not be plotted. With the increasing recirculation flow rate, the apparent current efficiency for NH3 formation increased in contrast to the decreasing apparent current efficiency for H2 formation. At any recirculation rate, the current efficiencies for NH3 and H2 formations add up to approximately 100%, indicating that there is almost no current loss and that either NH3 or H2 is being generated.
 | ||
| Fig. 7 Apparent current efficiency for NH3 synthesis and H2 formation from H2O and N2 at 250 °C and 1.0 MPa as a function of recirculation flow rate, with NH3 separation at −75 °C and unreacted gas recirculation. The current density was 100 mA cm−2 and N2 flow rate was 3.0 cmSTP3 min−1. The anode gas was the mixture of Ar at 10 cmSTP3 min−1 with 5 μL min−1 of liquid H2O. | ||
Summarizing the results in Fig. 5–7, it was found that keeping the N2 flow rate below five times the stoichiometric amount and keeping the recirculation rate above 50 times the N2 flow rate are crucial for achieving an apparent current efficiency of over 90% for NH3 production.
3.4. Electric properties of electrochemical cells
Time courses of current density and cell voltage are depicted in Fig. 8. In this experiment, electrolysis was conducted by connecting a constant current power supply to the cell to maintain a steady production rate. Therefore, the current remained constant, and the cell voltage fluctuated depending on the conditions of the electrolysis. Fig. 8A shows the cell voltage at current densities ranging from 10 to 300 mA cm−2. Between 10 and 100 mA cm−2, the cell voltage was between 1.5 and 1.8 V. At 300 mA cm−2, the cell voltage was significantly higher at 3 V compared to the cell voltage at 100 mA cm−2 or below. Furthermore, the cell voltage was unstable and exhibited significant fluctuations. As seen in the results of Fig. 3, the cell did not function properly at 300 mA cm−2, and the total current efficiency was not 100%. | ||
| Fig. 8 Time courses of cell voltage at various current densities (A) and cell voltage changes over several weeks of operation at 100 mA cm−2 (B) for NH3 synthesis and H2 formation from H2O and N2 at 250 °C. The inset of (A) shows the relationship between current density and cell voltage. The error bar at 300 mA cm−2 in (A) represents three times the standard deviation of the data fluctuation. | ||
The inset of Fig. 8A shows the relationship between current density and cell voltage. Despite the instability at 300 mA cm−2, the current density and cell voltage showed an approximately linear relationship. If the activation overpotential of an electrochemical reaction determines the cell voltage, the relationship between current density and cell voltage should follow an exponential curve based on the Tafel equation. However, since the experimental results were linear, it can be concluded that the cell voltage does not depend on the activation overpotential. It is suggested that factors, such as resistance due to the ionic conductivity of the electrolyte, bubble generation, or mass transport, determine the magnitude of the overpotential.
An important factor to consider when evaluating cell voltage is the reduction in energy efficiency caused by overpotential. Table 1 shows thermodynamic parameters for NH3 synthesis from H2O and N2. For reference, the values for the electrolysis of H2O to H2 and O2 are also included in Table 1. The energy is presented in kJ mol−1 with respect to H2 and converted to volts based on the corresponding number of electrons. The values are calculated for standard conditions, for liquid water at 25 °C, and for steam at temperatures of 25 °C and 250 °C. The value at 25 °C for steam is hypothetical. In the NH3 synthesis from H2O and N2, the equilibrium potential at 250 °C is 1.15 V, and the experimental results were between 1.5 and 1.8 V, which are higher than the equilibrium potential due to the overpotential.
| Reaction | Temp. (°C) | H2O | ΔGr equilibrium potential | ΔHr thermoneutral potential | ||
|---|---|---|---|---|---|---|
| kJ molH2−1 | V | kJ molH2−1 | V | |||
| NH3 synthesis | 25 | Liq. | 226 | 1.17 | 255 | 1.32 |
| 25 | Steam | 218 | 1.13 | 211 | 1.09 | |
| 250 | Steam | 223 | 1.15 | 211 | 1.09 | |
| H2O electrolysis | 25 | Liq. | 237 | 1.23 | 286 | 1.48 |
| 25 | Steam | 229 | 1.19 | 242 | 1.25 | |
| 250 | Steam | 218 | 1.13 | 244 | 1.26 | |
In terms of energy efficiency, the voltage efficiency is calculated based on the thermoneutral potential rather than the equilibrium potential, and when multiplied by the current efficiency, it gives the energy efficiency of the electrochemical cell itself. When calculated from liquid water under standard conditions, NH3 is an energy material with a potential of 1.32 V. This means that a voltage range of 1.5 to 1.8 V corresponds to a voltage efficiency of 88% to 73%. At a current density of 30 mA cm−2, the apparent current efficiency was 94%, and since the electrolytic voltage was 1.7 V, the voltage efficiency was 78%, resulting in an energy efficiency of 73%. Naturally, this energy efficiency does not account for losses due to the evaporation of H2O, heat loss from NH3 separation and unreacted gas recirculation, or the heat required for NH3 liquefaction, and it only represents a very simplified efficiency.
Fig. 8B shows the fluctuation of cell voltage for 100 mA cm−2 operation. During the several weeks of experimentation, the cell voltage fluctuated between 1.75 and 1.85 V, but no signs of degradation, such as an increase in cell voltage, were observed.
To investigate whether the cause of increased overvoltage or instability at high current densities lies in the anode or the cathode, it is useful to use a three-electrode cell equipped with a reference electrode to measure the overvoltage of each electrode. In our previous work with phosphate electrolytes, we constructed a three-electrode cell and attempted to measure the overvoltage of each electrode, ultimately identifying the anode as the cause of the increased and unstable cell voltage. Similar experiments should be considered for the current alkali molten salt electrolyte. However, since the electrolyte is in liquid form and conducting three-electrode cell experiments is difficult with our current equipment, we have left this as a subject for future investigation. Additionally, AC impedance measurements were conducted in our previous work with phosphate electrolytes, which proved useful in determining the overvoltage due to the ion conductivity of the electrolyte. For the alkali molten salt electrolyte, however, this remains a future task.
3.5. Catalyst characterization
A brief explanation of the properties of the catalyst used is provided below. A transmission electron microscope (TEM) image of the 30 wt%-Ru/Cs+/C (Cs+/Ru = 1 mol mol−1) catalyst is shown in Fig. 9. In the TEM image, it can be seen that small particles, around a few nanometers in size, are dispersed on the surface of carbon black particles which are approximately 40 nm in size. These small particles are identified as Ru particles. The Cs+ promoter has been added due to its electron-donating effect on the N2 adsorption sites of Ru, and this electron-donating effect has been clarified through nitrogen adsorption studies using infrared absorption spectroscopy. However, the TEM image does not provide information about the state of Cs+. | ||
| Fig. 9 Transmission electron microscopy image of the 30 wt%-Ru/Cs+/C (Cs+/Ru = 1 mol mol−1) catalyst. | ||
The effect of addition of alkali-metal compound promoters to Ru catalysts has been extensively discussed using infrared spectroscopic studies on molecularly adsorbed N2 on Ru sites.40–42 Although electronic changes in Ru particles on alkali-metal compound-promoted support materials are difficult to observe using photoelectron spectroscopy, they significantly impact the infrared spectra of molecularly adsorbed N2 on metallic Ru surfaces.40–44 The alkali-metal compound promoters cause a large low wavenumber shift of approximately 300 cm−1 in the infrared absorption near 2100 cm−1, which is attributed to the stretching vibration of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond in the adsorbed N2 molecule. When placing a highly electronegative substance, such as an alkali-metal compound, near metal catalyst particles such as Ru, Fe, or Fe, electrons are donated to the metal catalyst particles, making them electron-rich. This electron donation to the anti-bonding orbital, which is the lowest unoccupied molecular orbital (LUMO) of the NN triple bond of the adsorbed N2 molecule, promotes the dissociation of the adsorbed N2.45 This explanation has also been commonly cited in describing the function of industrial alkali-metal compound promoters and structure-directing promoters in doubly-promoted Fe catalysts. However, in this study, the effect of adding the Cs+ compound promoter was examined through H2 adsorption and desorption studies.
N triple bond in the adsorbed N2 molecule. When placing a highly electronegative substance, such as an alkali-metal compound, near metal catalyst particles such as Ru, Fe, or Fe, electrons are donated to the metal catalyst particles, making them electron-rich. This electron donation to the anti-bonding orbital, which is the lowest unoccupied molecular orbital (LUMO) of the NN triple bond of the adsorbed N2 molecule, promotes the dissociation of the adsorbed N2.45 This explanation has also been commonly cited in describing the function of industrial alkali-metal compound promoters and structure-directing promoters in doubly-promoted Fe catalysts. However, in this study, the effect of adding the Cs+ compound promoter was examined through H2 adsorption and desorption studies.
Temperature-programmed desorption (TPD) spectra of H2 from 30 wt%-Ru/Cs+/C (Cs+/Ru = 1 mol mol−1) and 30 wt%-Ru/C catalysts were measured from −183 °C as shown in Fig. 10. The TPD measurement of powder catalysts at low temperatures can be referenced in our previous papers.46 For Ru/C, desorption peaks of H2 were observed at −147, −55, and 113 °C. From single crystal Ru(0001) surfaces, desorption peaks were reported to appear at 52 °C (β1) and 127–167 °C (β2).47 On the other hand, from Ru(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surfaces, a peak was observed at −53 °C (α1).48 These peaks are attributed to the desorption through the recombination of dissociatively adsorbed hydrogen atoms. However, this difference is not due to a difference in surface orientation, as the α1 peak is observed even on Ru(0001) when hydrogen is adsorbed at low temperatures.49 In any case, the TPD results obtained under ultra-high vacuum using single-crystal metals show two main peaks: an α peak below room temperature and a β peak at around 100 °C. The two low-temperature peaks observed for Ru/C in this experiment are thought to correspond to the α peak, while the high-temperature peak is classified as the β peak. Naturally, since the catalyst used in this experiment is a realistic powder catalyst, there are various effects such as the presence of sites strongly influenced by the support, diverse surface orientations, and crystal edge effects. Therefore, the results are not exactly the same as those obtained with single crystals, but the observed peaks generally correspond to the phenomena observed with single-crystal metals.
0) surfaces, a peak was observed at −53 °C (α1).48 These peaks are attributed to the desorption through the recombination of dissociatively adsorbed hydrogen atoms. However, this difference is not due to a difference in surface orientation, as the α1 peak is observed even on Ru(0001) when hydrogen is adsorbed at low temperatures.49 In any case, the TPD results obtained under ultra-high vacuum using single-crystal metals show two main peaks: an α peak below room temperature and a β peak at around 100 °C. The two low-temperature peaks observed for Ru/C in this experiment are thought to correspond to the α peak, while the high-temperature peak is classified as the β peak. Naturally, since the catalyst used in this experiment is a realistic powder catalyst, there are various effects such as the presence of sites strongly influenced by the support, diverse surface orientations, and crystal edge effects. Therefore, the results are not exactly the same as those obtained with single crystals, but the observed peaks generally correspond to the phenomena observed with single-crystal metals.
 | ||
| Fig. 10 TPD spectra of H2 from 30 wt%-Ru/C and 30 wt%-Ru/Cs+ (Cs:Ru = 1:1)/C. The ramping rate of temperature was 10 °C min−1. The samples were treated by reduction treatment at 400 °C, followed by evacuation at 400 °C, prior to TPD measurements. The adsorption of H2 was performed at −183 °C and 5.0 kPa. | ||
A very interesting result is the TPD spectrum from the Ru/Cs+/C sample, in which the Cs+ promoter was added. The low-temperature desorption peak classified as α completely disappeared for Ru/Cs+/C. Furthermore, the peak at around 100 °C clearly split into two peaks at 90 °C and 180 °C. In studies conducted on the Ru(0001) surface under ultra-high vacuum, the TPD spectra of H2 desorption from a surface modified with K metal on Ru(0001) have also been investigated,48 but the disappearance of the α peak due to the addition of K has not been reported. Of course, the modification with K in surface science studies involves partially covering the Ru surface with metallic K, which is entirely different from the addition of Cs+ compounds as promoters in practical catalysts.
The effect of alkali metal compound promoters has primarily been highlighted in terms of the dissociation process of molecularly adsorbed N2, which is the rate-determining step in the NH3 synthesis reaction. This effect involves the donation of electrons to the adsorbed N2 through Ru, weakening the bond between nitrogen atoms and promoting dissociation. However, as explained in the previous section, adsorbed hydrogen atoms have an exclusive inhibitory effect on N2 adsorption. Since the addition of Cs+ compound is related to the presence of weakly adsorbed hydrogen atoms, it has emerged that this could indirectly affect the rate of the NH3 synthesis reaction. The effect of additives on the strength of hydrogen adsorption is a new perspective. It has been reported that with certain supports and additives, the hydrogen poisoning effect, the negative reaction order for partial pressure of H2 in NH3 synthesis, changes from negative to positive.50,51 This may be linked to this effect on hydrogen adsorption by the additives. However, in the case of Ru/Cs+/C in this study, as discussed in Fig. 4, although weakly adsorbed hydrogen atoms were no longer observed in the TPD with Cs+ addition, hydrogen poisoning still occurred in the NH3 synthesis reaction and nitrogen-excess conditions were found to favor the reaction.
3.6. Considerations for practical applications
Assuming the operation at 100 mA cm−2 and 1.8 V, as achieved in this study, the power density is estimated to be 0.18 W cm−2, which is approximately 0.2 W cm−2. For example, to fully utilize the power generated by typical 2 MW-class wind turbines, which are commonly used as a standard in floating offshore wind farms,52,53 the scale of power is 10 million times that of our electrochemical cell. Therefore, it would be necessary to construct an electrochemical system with an electrode area of ca. 10 million cm2, or 1000 m2. This corresponds to 250 stacks of cells with square electrodes measuring 2 m per side. If the thickness of the single cell can be constructed to be within 2 cm, the total thickness of the stack would be less than 5 m. Since the diameter of the tower of a 2 MW-class wind turbine is approximately 5 m, it can be understood that the system can be housed within the tower. Converting the production rate of 320 nmol s−1 cm−2 obtained in this study to an electrode area of 10 million cm2, the production rate becomes 3.2 mol s−1, yielding 54 g s−1 of NH3, as being produced with a molecular weight of NH3 of 17 g mol−1. This equates to 190 kg per hour, or 4600 kg per day. It will be possible to produce the amount of NH3 equivalent to one tank truck with a capacity of 5 tons in one day using a 2 MW-class wind turbine. Although wind turbines in the future are expected to be larger, such as in the 12 MW-class,53 the cells will also become larger accordingly. Of course, this calculation is a simplified one that ignores the power required for N2 separation from the air, NH3 separation, refrigeration for unreacted gas recirculation, and pump power. However, these energy requirements are smaller compared to the energy needed to produce H2 by water splitting, so the NH3 production rate will not be reduced by half.If we calculate the price of NH3 produced at the current electricity rates, it turns out to be significantly more expensive than NH3 produced from natural gas, making this calculation meaningless. However, if surplus renewable energy, which cannot be connected to the power grid, becomes available in the future, it might be important to convert it into NH3 in this way and use it as fuel for power generation or transportation equipment.
The electrochemical system in this study is not in its final form. Since it uses a large amount of expensive and rare Pd, there is a need to reduce its usage. Even for the Ru catalyst, which is also rare, alternative catalysts are necessary. The efficiency of NH3 liquefaction and separation also needs to be improved to reduce energy losses. The current density of 100 mA cm−2 needs to be increased by at least several times, and if this is achieved, the system could be made smaller. Additionally, although there was no degradation after about 1 month of operation, longer term stability must also be investigated, and issues such as electrolyte evaporation need to be examined. Moreover, when scaling up, it is essential to ensure that the same performance is maintained. There are many challenges that must be addressed, but we would like to propose this method as the simplest approach for synthesizing NH3 from H2O and N2 using electricity.
4 Conclusions
NH3 synthesis from N2 and H2O was investigated using an electrochemical setup featuring Ru/Cs+/C catalysts, Pd alloy membrane cathodes, NaOH–KOH molten electrolytes, and Ni anodes operated at 250 °C and 1.0 MPa. This electrochemical setup was integrated with a refrigerated gas/liquid separator at −75 °C to concentrate NH3 and a recirculation pump for unreacted H2 and N2. Without NH3 separation and unreacted gas recirculation, apparent current efficiency for NH3 formation was 32–20% for 10–100 mA cm−2 at 1.0 MPa and 250 °C, which was limited by the thermodynamic equilibrium limitation estimated to be 36%. To obtain products beyond chemical equilibrium, the most effective method is to use NH3 separation and unreacted gas recirculation. Using this apparatus setup, an apparent current efficiency of 90% and a production rate of 320 nmol s−1 cm−2 for NH3 production were achieved for 100 mA cm−2 at 1.0 MPa and 250 °C.The H2-TPD results showed a unique effect on hydrogen adsorption due to the addition of the Cs+ compound. This result may provide a new perspective on NH3 synthesis catalysts.
The current density and current efficiency of the electrochemical cell being studied have been confirmed to be suitable for scaling up to a system that can be integrated with wind and solar power generation. With the anticipated increase in surplus renewable energy, we hope that the development of such systems will be necessary in the future.
Data availability
Raw data were generated at Fukuoka University. Derived data supporting the findings of this study are available from the corresponding author Jun Kubota on request. The data that support the findings of this study are available from the corresponding author, Jun Kubota, upon reasonable request.Author contributions
Sagara was primarily involved in practical experimental studies and data analysis. Watanabe, along with Sagara, conducted TPD measurements and processed the data. Kubota played a multifaceted role including supervision in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of this article is based on results obtained from a project, JPNP14004, “Highly-efficient NH3 electrochemical synthesis technology from renewable energy electricity (FY2022-2023)”, commissioned by the New Energy and Industrial Technology Development Organization (NEDO), Japan.References
- I. Rafiqul, C. Weber, B. Lehmann and A. Voss, Energy, 2005, 30, 2487–2504 CrossRef CAS.
- A. Valera-Medina, F. Amer-Hatem, A. K. Azad, I. C. Dedoussi, M. de Joannon, R. X. Fernandes, P. Glarborg, H. Hashemi, X. He, S. Mashruk, J. McGowan, C. Mounaim-Rouselle, A. Ortiz-Prado, A. Ortiz-Valera, I. Rossetti, B. Shu, M. Yehia, H. Xiao and M. Costa, Energy Fuels, 2021, 35, 6964–7029 CrossRef CAS.
- R. F. Service, Science, 2018, 361, 120–123 Search PubMed.
- Ammonia: Catalysis and Manufacture, ed. A. Nielsen, Springer-Verlag, Berlin, 1995 Search PubMed.
- International Energy Agency, Ammonia Technology Roadmap, 2021 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. J. Rumble, 99th edn, CRC Press, 2018 Search PubMed.
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, Wiley-VCH, 2007 Search PubMed.
- H. Ishaq and C. Crawford, Energy Convers. Manage., 2024, 300, 117869 CrossRef CAS.
- U. Jafar, U. Nuhu, W. U. Khan and M. M. Hossain, Int. J. Hydrogen Energy, 2024, 71, 857–876 CrossRef CAS.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. F. Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustainable Chem. Eng., 2017, 6, 10231–10239 CrossRef.
- C. R. Santhosh and R. Sankannavar, Appl. Energy, 2023, 352, 121960 CrossRef CAS.
- B. Wang, T. Li, F. Gong, M. H. D. Othman and R. Xiao, Fuel Process. Technol., 2022, 235, 107380 CrossRef CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 Search PubMed.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- X. Guo, Y. Zhu and T. Ma, J. Energy Chem., 2017, 26, 1107–1116 CrossRef.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- I. A. Amar, R. Lan, C. T. G. Petit and S. Tao, J. Solid State Electrochem., 2011, 15, 1845–1860 CrossRef CAS.
- L. F. Greenlee, J. N. Renner and S. L. Foster, ACS Catal., 2018, 8, 7820–7827 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and Ib Chorkendorff, Nature, 2019, 570, 504–508 Search PubMed.
- A. Mangini, L. Fagiolari, A. Sacchetti, A. Garbujo, P. Biasi and F. Bella, Adv. Energy Mater., 2024, 14, 2400076 CrossRef CAS.
- N. Lazouski, Z. J. Schiffer, K. Williams and K. Manthiram, Joule, 2019, 3, 1127–1139 CrossRef CAS.
- A. Tsuneto, A. Kudo and T. Sakata, J. Electroanal. Chem., 1994, 367, 183–188 CrossRef CAS.
- Y. Ito, T. Nishikiori and H. Tsujimura, Faraday Discuss., 2016, 190, 307–326 RSC.
- C. A. Fernandez, N. M. Hortance, Y.-H. Liu, J. Lim, K. B. Hatzell and M. C. Hatzell, J. Mater. Chem. A, 2020, 8, 15591–15606 RSC.
- C. A. Fernandez and M. C. Hatzell, J. Electrochem. Soc., 2020, 167, 143504 CrossRef CAS.
- K. Imamura, M. Matsuyama and J. Kubota, Chem. Select, 2017, 2, 11100–11103 CAS.
- K. Imamura and J. Kubota, Sustain. Ener. Fuels, 2018, 2, 1278–1286 RSC.
- K. Imamura and J. Kubota, Sustain. Ener. Fuels, 2019, 3, 1406–1417 RSC.
- S. Nagaishi, R. Hayashi, A. Hirata, R. Sagara and J. Kubota, Sustain. Ener. Fuels, 2024, 8, 914–926 RSC.
- Y. Yuan, S. Tada and R. Kikuchi, Sustain. Ener. Fuels, 2022, 6, 458–465 RSC.
- Y. Yuan, S. Tada and R. Kikuchi, Mater. Adv., 2021, 2, 793–803 RSC.
- N. Mohammad, A. B. Mohamad, A. A. H. Kadhum and K. S. Loh, J. Power Sources, 2016, 322, 77–92 CrossRef CAS.
- T. Matsui, T. Kukino, R. Kikuchi and K. Eguchi, J. Electrochem. Soc., 2006, 153, A339–A342 CrossRef CAS.
- R. Sagara, R. Hayashi, A. Hirata, S. Nagaishi and J. Kubota, Energy Adv., 2024, 3, 1265–1270 RSC.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS.
- S. Licht, B. Cui, B. Wang, F.-F. Li, J. Lau and S. Liu, Retraction, 14 Aug. 2020, Science, 2014, 345, 637–640 CrossRef CAS.
- Y. Chen, H. Liu, N. Ha, S. Licht, S. Gu and W. Li, Nature Catal., 2020, 3, 1055–1061 CrossRef CAS.
- A. Ozaki and K. Aika, Catalysis-Science and Technology, ed. J. R. Anderson and M. Boudart, Springer-Verlag KG, Berlin, 1981, vol. 1, pp. 87–158 Search PubMed.
- K. Aika, Catal. Today, 2017, 286, 14–20 CrossRef CAS.
- J. Kubota and K. Aika, J. Phys. Chem., 1994, 98, 11293–11300 CrossRef CAS.
- K. Aika, J. Kubota, Y. Kadowaki, Y. Niwa and Y. Izumi, Appl. Surf. Sci., 1997, 121, 488–491 CrossRef.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nature Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- Y. Ogura, K. Tsujimaru, K. Sato, S. Miyahara, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustainable Chem. Eng., 2018, 6, 17258–17266 CrossRef CAS.
- C. N. R. Rao and G. R. Rao, Surf. Sci. Rep., 1991, 13, 223–263 CrossRef.
- K. Higuchi, H. Sugiyama and J. Kubota, J. Phy. Chem. C, 2017, 121, 14581–14588 CrossRef CAS.
- H. Shinizu, K. Christmann and G. Ertl, J. Catal., 1980, 61, 412–429 CrossRef.
- K. Christmann, Surf. Sci. Rep., 1988, 9, 1–163 CrossRef.
- G. H. Rocker, C. L. Cobb, C.-S. Jun, H. Metiu and R. M. Martin, Surf. Sci., 1989, 208, 205–220 CrossRef CAS.
- Y. Kadowaki and K. Aika, J. Catal., 1996, 161, 178–185 CrossRef CAS.
- S. Miyahara, K. Sato, Y. Kawano, K. Imamura, Y. Ogura, K. Tsujimaru and K. Nagaoka, Catal. Today, 2021, 376, 36–40 CrossRef CAS.
- Global Wind Energy Council (GWEC), Global Offshore Wind Report, 2024, https://www.gwec.net/.
- International Energy Agency (IEA), Offshore Wind Outlook, 2019, https://www.iea.org.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5se00348b |
| This journal is © The Royal Society of Chemistry 2025 |