
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGradients in cell density and shape transitions drive collective cell migration into confining environments†
Wan-Jung
Lin
 ,
Hongsheng
Yu
and
Amit
Pathak
*
,
Hongsheng
Yu
and
Amit
Pathak
*
Department of Mechanical Engineering & Materials Science, Washington University, St. Louis, USA. E-mail: pathaka@wustl.edu
First published on 30th December 2024
Abstract
Epithelial cell collectives migrate through tissue interfaces and crevices to orchestrate development processes, tumor invasion, and wound healing. Naturally, the traversal of cell collective through confining environments involves crowding due to narrowing spaces, which seems tenuous given the conventional inverse relationship between cell density and migration. However, the physical transitions required to overcome such epithelial densification for migration across confinements remain unclear. Here, in a system of contiguous microchannels of varying confinements, we show that epithelial (MCF10A) monolayers accumulate higher cell density and undergo fluid-like shape transitions before entering narrower channels. However, overexpression of breast cancer oncogene ErbB2 did not require such accumulation of cell density to migrate across matrix confinement. While wild-type MCF10A cells migrated faster in narrow channels, this confinement sensitivity was reduced after +ErbB2 mutation or with constitutively active RhoA. This physical interpretation of collective cell migration as density and shape transitions in granular matter could advance our understanding of complex living systems.
Introduction
As epithelia grow and migrate to sculpt organs, heal wounds, or escape tumours, they must traverse physically heterogeneous environments of varying stiffness, topography, and confinement. Collectively migrating epithelial cells migrate faster on stiffer substrates due to higher levels of traction forces associated with elevated actin–myosin activity.1,2 When cells encounter an interface between soft and stiff regions, they undergo ‘durotaxis’ and quickly migrate into the stiffer side due to higher forces.3 On surfaces with a gradient of stiffness, epithelial monolayers develop emergent durotaxis through force propagation across cell–cell junctions and seamlessly migrate from soft to stiff regions.4 Apart from stiffness, physical heterogeneities in the extracellular matrix (ECM) also arise from topography and confinement.5 According to previous work, both single and collective cells migrate faster in confined matrices, largely due to their alignment of forces and protrusion along a single-axis.6–8 However, unlike the known durotaxis phenomenon for cell migration from soft to stiff regions,3,4 it remains unclear how epithelial collectives migrate from open (unconfined) substrates into confined regions. Prior work has shown that collective cell migration is enhanced by cellular transformations of shape elongation, unjamming transition, and epithelial–mesenchymal transition (EMT),9–11 all of which are promoted in confinement.12–14 Thus, upon encountering confined topographies, it is possible that epithelial collectives get arrested while undergoing such cellular transformation or they could seamlessly enter confinement analogous to durotaxis from soft to stiff environment; however, these possibilities remain unexplored.Among the numerous physiological and pathological events related to collective cell behaviour, epithelial–mesenchymal transition (EMT) is notable in its role in critical cellular processes such as embryogenesis and cancer invasion.15–18 Within epithelial clusters, cells are usually cuboidal, sedentary, have definite top-bottom polarity, and are in close contact with neighbouring cells. During EMT, epithelial cells undergo a series of physiological changes, decreasing cell–cell junctions, elongating, and switching to front-rear polarity before migration. Mechanical properties of the environment, such as stiffness and topography, can also induce EMT. For example, stiff matrices trigger EMT in lung and breast epithelial cells without any growth factor addition.17,18 On nano-grooved surfaces, switch-like enhancement of EMT is mediated by the yes-associated protein (YAP) pathway.19 In breast cancer, the EMT-related oncogene ErbB2 (Her2/neu epidermal growth receptor family) was found to be overexpressed in advanced-stage patients,20 and its overexpression enhances cell migration in confined matrices.21 Furthermore, confinement and stiffness could induce EMT in epithelial clusters independently.13,14,22,23 Since aligned matrix topographies trigger mechano-active responses, including EMT, in epithelial cells,13,14,19,22,23 collectively migrating epithelia could exploit such topography-sensitivities to enter confined matrices.
Moving beyond EMT, flocking and migratory behaviours of epithelial cells have also been described biophysically as analogues of solid–liquid phase transitions.11,24–27 Within cell clusters, where cells are usually constrained by neighbouring cells, individual cell motions are coupled with their neighbours. At large distances and time scales, collective migration is analogous to particulate systems approaching a glass transition,24 wherein monolayer aging is analogous to solidifying glass particulates. Previous work has characterized shape and migratory variations of healthy and asthmatic human airway epithelial (HBEC; primary human bronchial epithelial cell) monolayers as “unjamming transition” (UJT). While baring some phenotypic resemblance with partial EMT (pEMT), the UJT triggered by compression is found to follow a distinct mode of cell migration.9–11,28 Existing theoretical and empirical models attribute these collective cellular transformations, via EMT, pEMT, or UJT, to dynamic competition and cooperation between cell–cell junctions and intracellular forces.9,10,29 In this balancing act of forces and cohesion, RhoA (Ras homolog family member A), a member of small Rho GTPases, affects actomyosin contractility as well as cell–cell junctions.30,31 By the differential levels of actomyosin contractility, leader-follower hierarchy within the migrating epithelial cluster is established and forward movement is ensured.16,32,33 Given RhoA itself could induce divergent downstream effects and thus opposing morphological changes,34 its activation in cell collectives could alter how they change shape and negotiate varying matrix topographies.
Given that cell migration is influenced by various properties of their microenvironment including confinement, stiffness, surface geometry, and even external shear, most reports of mechanosensitive cell migration are cell-type specific or assay-specific.35,36 Microchannels have been one of the popular models for confined cell migration37,38 since they allow easy manipulation and independent control of surface chemistry, material stiffness, and physical constraints.5,6,13,39 Emerging migratory modes inside microchannels versus channel conditions have been characterized either biochemically through signal pathways or physically by diffusivity and oscillation.7,8,13,14,40,41 Whereas researchers have previously studied collective cell migration in defined confinement, how cells collectively negotiate and move from unconfined to confined matrices remains unclear. We adopt a biophysical approach combining microchannels and mutant epithelial cells to study monolayer migration into confinement.
In this study, we use MCF10A (WT 10A) human mammary epithelial cells and their derivative cell lines with constitutively active RhoA (10A + RhoA) and overexpressed ErbB2 (10A + ErbB2) to form epithelial monolayers. We fabricate substrates with unconfined (open) regions adjacent to confined (narrow or wide) channels to study monolayer migration into confinement. By analyzing migration phenotypes, individual cell shapes, and cell density, we found that monolayers develop differentials of density and shape transitions between confined and unconfined regions. These migratory and shape transitions into confined regions reduce with EMT-like +ErbB2 mutant cells. Our findings indicate the collective cell migration into confinement is driven by cell-level physical changes of density and velocity.
Materials and methods
Fabrication of PDMS substrates with microchannels
Microchannels of polydimethylsiloxane (PDMS, Sylgard 184; Dow Corning, Midland, MI) were fabricated following the standard procedures of soft lithography, as depicted in Fig. 1. Spin-coated SU-8 2050 (Kayaku Advanced Materials, Westborough, MA) photoresist was exposed and developed to yield grooves in sets of microchannels, each of widths 50 μm (narrow) and 200 μm (wide), spaced 700 μm apart, and 60–70 μm height. For easy release of cured PDMS, the master SU-8 2050 photoresist was treated with trichloro (1H,1H,2H,2H-perfluorooctyl) silane (448![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 931, Sigma-Aldrich, Saint Louis, MO) in a desiccator for overnight. The monomers and crosslinkers of PDMS were mixed in a 10:1 ratio and degassed until clear. The mixed PDMS was spin-coated on the SU-8 master and incubated at 70 °C for at least 2 hours and let sit at room temperature overnight; after fully cured, the PDMS was peeled from the master. The cured PDMS microchannels were cut into pieces and attached to glass-bottom 12-well plates (P12-1.5H-N, Cellvis, Mountain View, CA). For the channel tops, thin film of PDMS were plasma-bonded to the bottom layer and treated for one additional minute with air plasma to ensure the hydrophilicity of microchannel surfaces. The microchannels were then immersed in phosphate buffered saline (PBS; Thermo Fisher, Waltham, MA) and UV-sterilized for 1 h and coated with 0.05 mg mL−1 rat tail collagen I (sc-136157, Santa Cruz Biotechnology, Dallas, TX) at 4 °C overnight.
931, Sigma-Aldrich, Saint Louis, MO) in a desiccator for overnight. The monomers and crosslinkers of PDMS were mixed in a 10:1 ratio and degassed until clear. The mixed PDMS was spin-coated on the SU-8 master and incubated at 70 °C for at least 2 hours and let sit at room temperature overnight; after fully cured, the PDMS was peeled from the master. The cured PDMS microchannels were cut into pieces and attached to glass-bottom 12-well plates (P12-1.5H-N, Cellvis, Mountain View, CA). For the channel tops, thin film of PDMS were plasma-bonded to the bottom layer and treated for one additional minute with air plasma to ensure the hydrophilicity of microchannel surfaces. The microchannels were then immersed in phosphate buffered saline (PBS; Thermo Fisher, Waltham, MA) and UV-sterilized for 1 h and coated with 0.05 mg mL−1 rat tail collagen I (sc-136157, Santa Cruz Biotechnology, Dallas, TX) at 4 °C overnight.
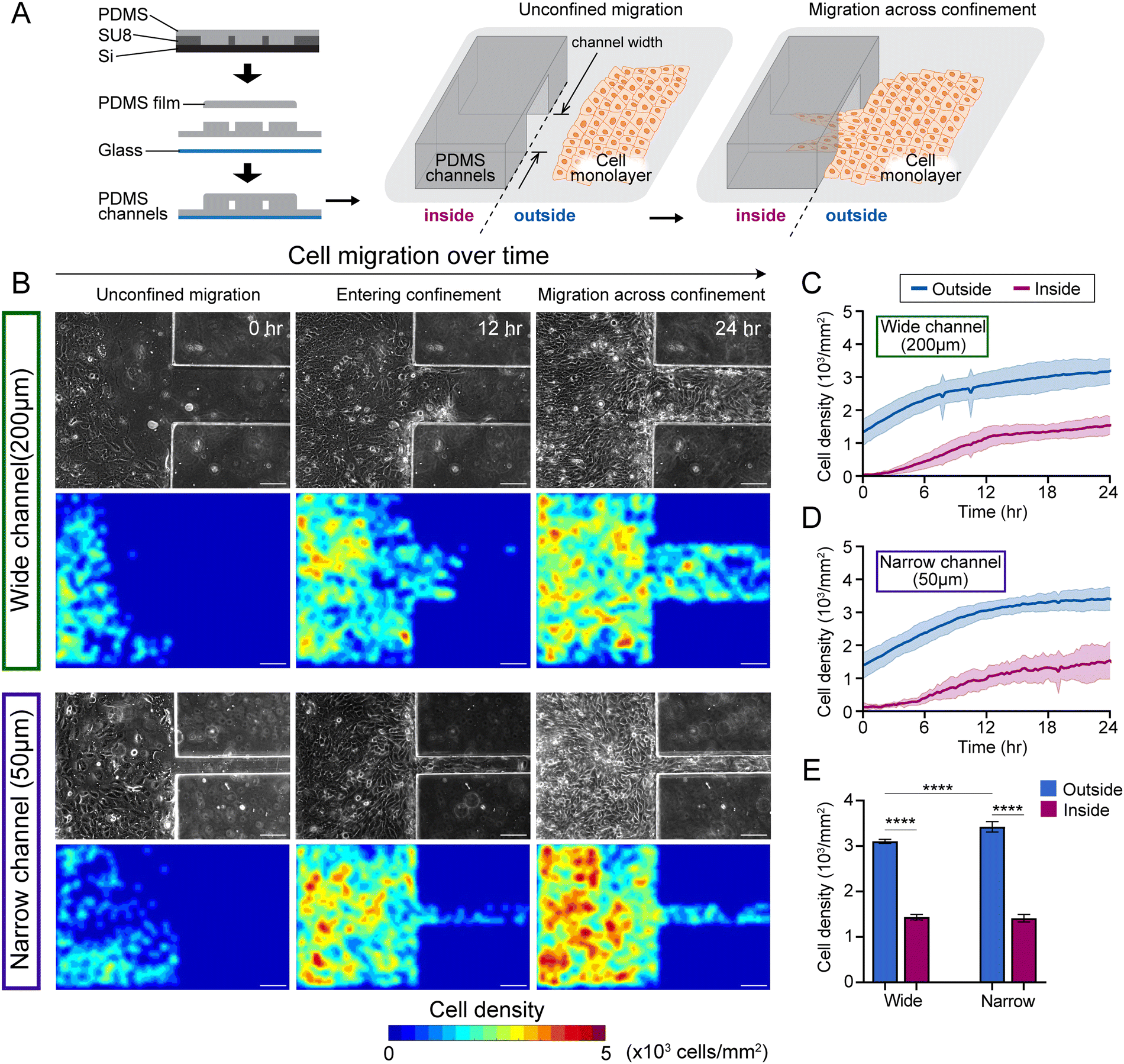 | ||
| Fig. 1 Gradients in cell density in collective migration across confining channels. (A) Experiment schematic. (B) Representative images of MCF10A epithelial monolayer entering 200 μm (wide) or 50 μm (narrow) microchannels from 0 h (monolayer reaches the channel wall) to 24 h (monolayer already establishes steady flow into the channel). The first row: phase contrast images; the second row: cell density heatmaps. Scale bar, 100 μm. (C) and (D) Cell density curves of wild-type (WT) monolayers entering wide and narrow microchannels over 24 hours, where blue and magenta curves represent average cell density outside and inside the microchannels, respectively. (E) Average cell density within the WT monolayers from 18–24 h outside (blue) and inside (magenta) microchannels in both wide and narrow groups (****, p < 0.0001, by two-way ANOVA). Data expressed as mean ± SD and n ≥ 8. | ||
Cell culture
Human mammary gland (MCF10A) epithelial cells with green fluorescence protein-labelled nuclei (WT 10A) and 10A + RhoA (courtesy of Gregory Longmore, Washington University in St. Louis) and 10A + ErbB2 (courtesy of Dihua Yu, The University of Texas MD Anderson Cancer Center) mutant cell lines derived from MCF10A were cultured using Dulbecco's modified Eagle's medium/nutrient mixture F-12 (DMEM/F12; Invitrogen, Waltham, MA), supplemented with 5% horse serum (Hyclone heat-inactivated, Cytiva, Marlborough, MA), 20 ng mL−1 epidermal growth factor (EGF; Miltenyi Biotec, Gaithersburg, MD), 0.5 mg mL−1 hydrocortisone (H0888, MilliporeSigma), 100 ng mL−1 cholera toxin (C8052, Sigma-Aldrich), 10 μg mL−1 insulin (I6634, Sigma-Aldrich) and 100 μg mL−1 proprietary antimicrobial (Normocin; InvivoGen, San Diego, CA). Cells were cultured at 90–95% confluency, every three to four days. To see epithelial monolayers, cell suspensions were prepared at 107 cells per mL and 10 μL volume (105 cells) droplets were used to grow monolayers in circular stencils of 3.7 mm diameter. These stencils were removed 1 h after seeding, and the wells were refilled with media. Epithelial monolayers formed within a few hours after seeding and migrating edges were 1–2 mm away from the channel entrance. A subset of 10A + ErbB2 monolayers were treated with 2 mM metformin (317240, Sigma-Aldrich) 2 h before live imaging, which was used to suppress mesenchymal phenotype triggered by over-expressing ErbB2.
Live imaging and analysis of collective cell migration
After epithelial cells formed monolayers on PDMS substrates and approached microchannel entrances, live time-lapse imaging was performed. 10A + RhoA and 10A + ErbB2 cell lines were live stained with NucBlue (3.3 μg mL−1; Thermo Fisher, Waltham, MA) for 8 min and verapamil (0.2 nM; Thermo Fisher) was used for preventing dye efflux from the nuclei. Images were acquired every 15 min for 36–72 h. Monolayer dynamics were analysed using the open-source image processing platform Fiji42 plugin TrackMate,43 which allowed us to track individual cell migration within the monolayers using the fluorescent nuclei as focal points. The “outside” region was defined as the 450 to 550 μm square region immediately in front of the microchannel entrance, and the “inside” region as the 450 to 550 μm square region immediately inside of the channel. The initial time point (t0) was defined as the instant the monolayers reached the channel walls (spanning length ≥ 25% in the field of view). TrackMate generated color-coded tracks according to the mean speeds and persistence throughout the experiment. The order parameter was defined as the projection of nucleus velocity along the channel direction. The persistence was defined as the net displacement a nucleus migrated divided by the total distance in 2 hours. The cell density heatmaps were obtained using nucleus positions given by TrackMate and custom MATLAB codes.Immunostaining and analysing cell morphology
Cells were fixed for immunostaining after live imaging. Hoechst 33258 (1:50; Thermo Fisher), the primary antibody of P120 (1:600, (D7S2M) XP; Cell Signaling Tech., Danvers, MA) and phalloidin (1:200, R415, Invitrogen) was used for labeling nuclei, P120 (δ-catenin) and F-actin. Phosphorylated myosin light chain (pMLC) staining was applied on 10A + RhoA cells on PDMS for characterization (Fig. S5, ESI†). Images of P120 immunostaining (or those overlaid with F-actin images as necessary) were first used to determine cell outlines. The Python program SeedWaterSegmenter (v0.5.7.1, SWS)44 was used for segmentation. Cell areas, aspect ratios, and shape indices were calculated accordingly. The heat maps of shape indices and aspect ratios were generated using a custom MATLAB code.
Results
Epithelial monolayers undergo crowding and higher cell density, before entering confinement
To image and analyse how epithelial monolayers enter matrix regions of narrowing confinement, we fabricated PDMS substrates with unconfined regions adjacent to microchannels of widths 200 μm and 50 μm to capture wide and narrow confinements, respectively (Fig. 1A). By tracking nuclei within monolayers entering microchannels, we calculated cell densities separately outside and inside the microchannels (Fig. 1C and D). Through live imaging over time (the frame that monolayer reaches the wall of PDMS channel is defined as 0 h), we observed that cell density is relatively higher in outside regions (unconfined) compared to inside microchannels, as shown in representative phase contrast and nuclei images (Fig. 1B), and cells aggregate near the entrance of the microchannels (Fig. S1A, ESI†). Cell monolayer shows higher density outside the narrow channels, compared to wide (middle column in Fig. 1B), which continues to hold even after cells migrate into the channels. We plotted average cell density over time and found that cell density rises over the first 12 hours and stays higher outside the microchannels, compared to inside (Fig. 1C, D and Fig. S2A, B, ESI†). Once the monolayers established stable flows inside microchannels, the cell density heatmaps revealed greater cell densities outside narrow microchannels, compared to those in the wide group (Fig. 1B and E).Thus, entry of collectively migrating epithelial cells into narrower confinement requires a rise in cell density. We wondered whether such accumulation of cell density near narrower confinement would also depend on cellular properties, specifically those regulating the cohesion of the epithelial population. Since cell monolayers maintained higher density outside (unconfined) compared to inside microchannels in the wild-type 10A case (WT 10A, Fig. 1), we calculated the cell density difference (Δρ) between outside and inside microchannels over time and found it to be slightly higher in case of entry into narrow confinement (Fig. 2B and Fig. S2C, ESI†). While WT monolayers entered the wide microchannels, the density difference Δρ peaked at 6 h and dropped at 12 h, while the density difference of monolayers entering the narrow microchannel plateaued after 6 h (Fig. 2B).
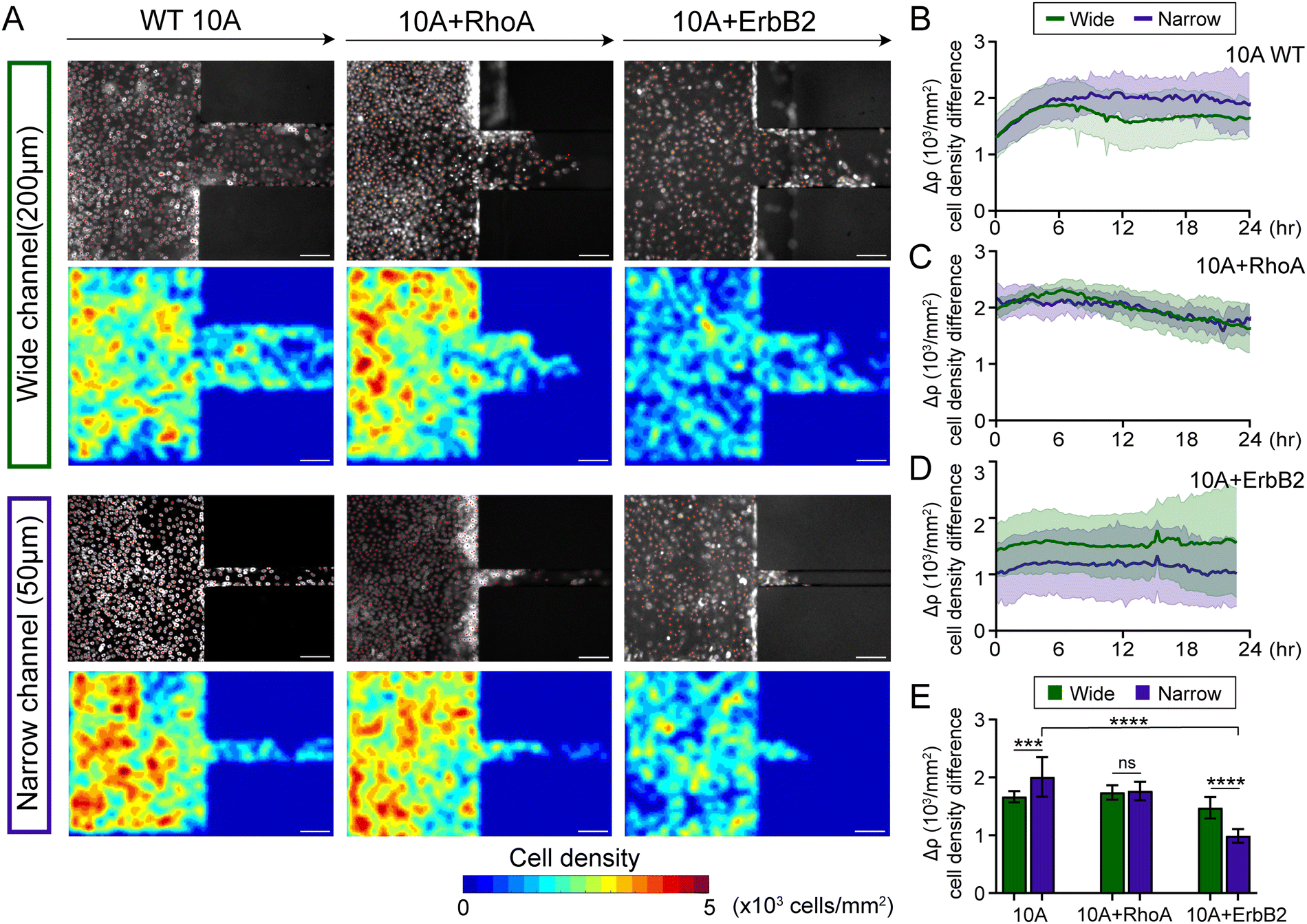 | ||
| Fig. 2 Cell density gradients across confinement vary with cellular changes. (A) Cell density distribution in wide and narrow channels for wild-type MCF10A (WT 10A), constitutively active RhoA (10A + RhoA), and ErbB2 overexpressing (10A + ErbB2) cell variants, with nuclear intensity and cell density heatmaps. (B)–(D) Temporal variation of difference in cell density (Δρ) between inside and outside the microchannels for WT 10A, 10A + RhoA and 10A + ErbB2 cells. (E) Cell density difference averaged from 18–24 hour. ***p < 0.001, ****p< 0.0001, by two-way ANOVA. Data expressed as mean ± SD and n ≥ 6. | ||
Next, we used MCF10A cells with constitutively active RhoA, which is known to enhance intracellular contractility and reinforce cell–cell adhesions.36,45 We found that for the first 24 hours after monolayers reached channel walls, 10A + RhoA cell monolayers maintained similar cell density differences between outside and inside both narrow and wide channels (Fig. 2A, C and Fig. S3A, C, ESI†). Although cells overexpressing RhoA also exhibited cell density accumulation outside of microchannels, they did not differentiate between narrow and wide channels. Furthermore, given cancer cells navigate confinement environments in tumor invasion and cancer metastasis, we used MCF10A cells with overexpressing oncogene ErbB2, which features in advanced-stage breast cancer and relates to EMT.20,46 We found that 10A + ErbB2 cell monolayers maintained higher cell density difference Δρ across confinement when entering wide channels, compared to narrow, over 24 h duration of migration (Fig. 2A, D and Fig. S3B, D, ESI†). For 10A + RhoA monolayers, the cell density difference while entering the wide and the narrow channels were almost identical (Fig. 2C), whereas in 10A + ErbB2 monolayers, the cell density difference necessary to enter the narrow channels was smaller than that necessary to enter the wide channel (Fig. 2D). Thus, compared to WT 10A and 10A + RhoA cells, this lower density accumulation of 10A + ErbB2 cells (Fig. 2E), particularly while moving to narrow channels, indicates that cancer-like mutations might make it easier for cells to enter confined regions, without requiring further density accumulation.21
Faster migration in confined regions and reduced confinement sensitivity by mutant cells
To assess the spatiotemporal evolution of migration speed into confinement, we plotted the average speed of individual cell migration tracks as monolayers of all three cell groups entered narrow or wide microchannels (Fig. 3A). In these representative images, wild-type (WT 10A) cells migrate faster than both mutant cells, especially when entering the channels. Upon plotting the root mean square cell velocity ( ) over time for WT 10A cells, we found higher cell speed inside microchannels compared to the outside unconfined regions (Fig. 3B and C). For both +RhoA and +ErbB2 cells, migration velocity of cells outside and inside channels were similar (Fig. 3D–G). We calculated the difference in
) over time for WT 10A cells, we found higher cell speed inside microchannels compared to the outside unconfined regions (Fig. 3B and C). For both +RhoA and +ErbB2 cells, migration velocity of cells outside and inside channels were similar (Fig. 3D–G). We calculated the difference in  between outside and inside channels (
between outside and inside channels (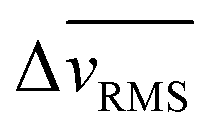 averaged over the last 6 hours of imaging), which indicates the degree of sensitivity for confinement by a given cell type (Fig. 3H). We found that wild-type cells displayed the highest difference between confined and unconfined cell migration speed, which further increased when monolayers attempt to enter narrow channels. This velocity difference was reduced for both mutants (+RhoA and +ErbB2), while cancer-like +ErbB2 cells moving into narrow channels exhibited the smallest velocity difference (Fig. 3H), indicating their lack of sensitivity for confinement.
averaged over the last 6 hours of imaging), which indicates the degree of sensitivity for confinement by a given cell type (Fig. 3H). We found that wild-type cells displayed the highest difference between confined and unconfined cell migration speed, which further increased when monolayers attempt to enter narrow channels. This velocity difference was reduced for both mutants (+RhoA and +ErbB2), while cancer-like +ErbB2 cells moving into narrow channels exhibited the smallest velocity difference (Fig. 3H), indicating their lack of sensitivity for confinement.
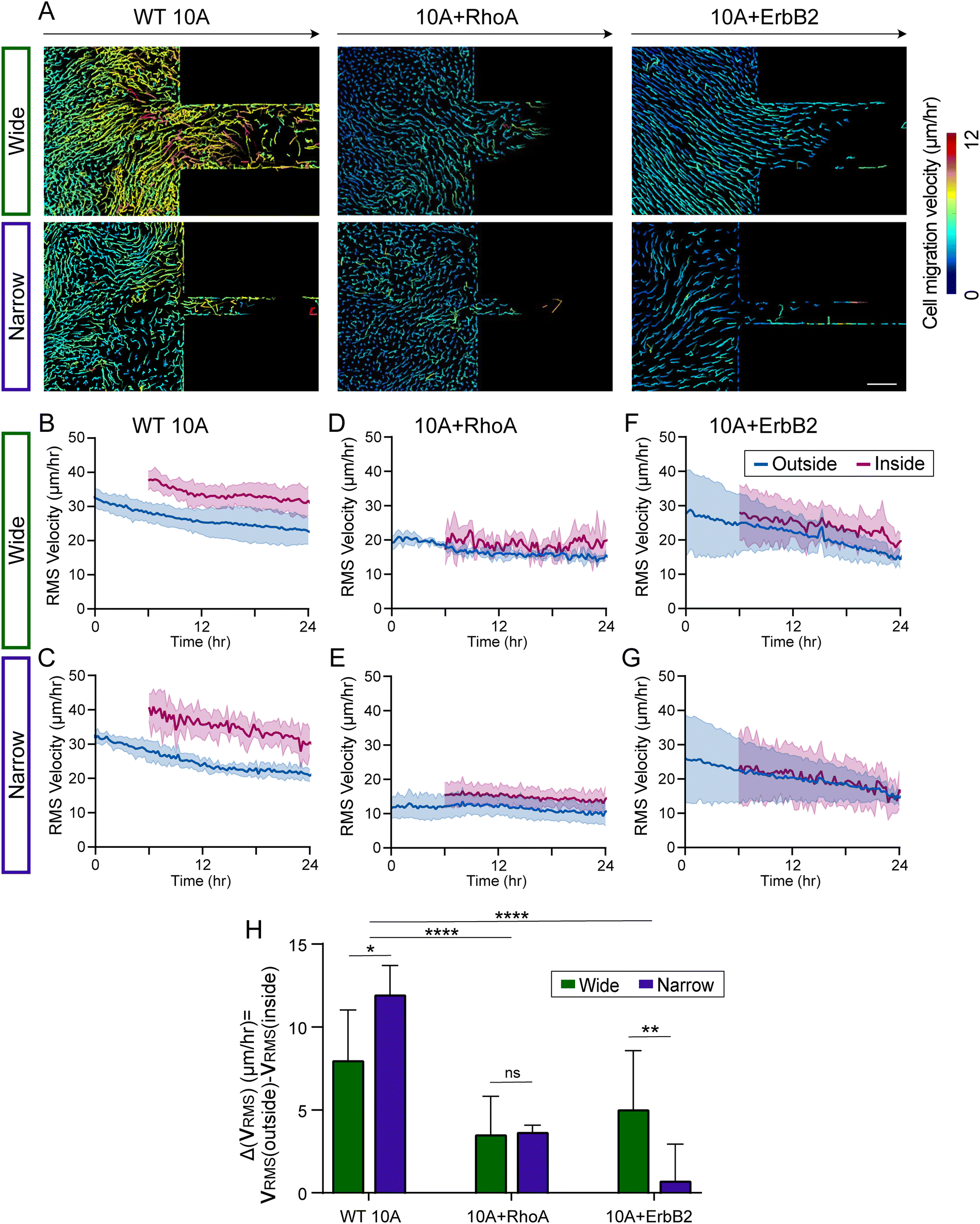 | ||
| Fig. 3 Velocity of cell migration across confinement varies with cell type. (A) Migration tracks of cells within WT 10A, 10A + RhoA, and 10A + ErbB2 monolayers entering wide and narrow channels, with tracks color-coded according to the track mean speed. (B)–(G) Root-mean-squared (RMS) velocities of WT 10A (B)–(C), 10A + RhoA (D)–(E), and 10A + ErbB2 (F)–(G) cells outside (blue) and inside (magenta) channels. (H) Average difference in RMS velocity of cells outside and inside the channels. *p < 0.05, **p < 0.01, ****p < 0.0001, by two-way ANOVA. Data expressed as mean ± SD and n ≥ 6. | ||
When epithelial cells collectively migrate from unconfined environments into confined regions, some cell groups enter the microchannel while others are left behind. This spatial heterogeneity could cause migratory disorder at the leading edge, which could also vary with healthy and cancer-like cells. By tracking cells within monolayers, we observed that WT 10A cells outside of microchannels persistently maintained their direction of migration over time (Fig. 4A) regardless of narrow or wide channels. In comparison, directional persistence of 10A + RhoA cells reduced during 12–24 h time-window (Fig. 4B–D). With +ErbB2 mutation, migrating monolayers showed higher and temporally stable directional persistence than both WT and +RhoA variants. We also calculated order parameter as cosine of angle between individual velocity vectors and direction of the channel, which largely resembled directional persistence trends (Fig. 4E–G). For +ErbB2 cells, while persistence was consistently higher in entering wide channels, order parameter for narrow channels increased over time, indicating a tendency of 10A + ErbB2 cells to expand regardless of confinement dimension.
 | ||
| Fig. 4 Persistence and order parameter of collective cell migration across confinement vary with cell types. (A) Persistence and order parameter within WT 10A, 10A + RhoA, and 10A + ErbB2 monolayers. The snapshots of cell tracks within the monolayers color-coded according to track-wise persistence. (B)–(G) Curves of persistence and order parameter within the monolayers across cell lines. Data expressed as mean ± SD and n ≥ 6. | ||
Cells undergo mixed morphological changes of jamming, unjamming, and elongation to enter confinement
Since epithelial monolayers develop gradients of density (Fig. 2) across confined and unconfined regions, we next wondered whether individual cell shapes change during these transitions of encountering and entering confinement. In previous studies, cell elongation has been calculated as aspect ratio (AR), and values of shape index (SI; perimeter divided by square root of area) ≥3.81 indicate unjamming.11 Furthermore, SI and AR of cells have been used to describe different states of UJT and pEMT9 such that low values of both SI and AR indicate jamming, high values of both SI and AR indicate UJT, and low AR combined with high SI indicate pEMT. To segment individual cell outlines with epithelial monolayers, we performed immunostaining for p120, which localizes on adherens junctions (Fig. 5A). Through custom cell segmentation tools, we visualized individual cells within migrating monolayers by color-coding for their AR (Fig. 5B), SI (Fig. 5C), and combined AR and SI traits (Fig. 5D). From spatial shape distributions, we observed that majority of cells had 1 ≤ AR ≤ 3 and 4 ≤ SI ≤ 5 (Fig. 5B–G), which suggests that most cells underwent moderate elongation yet developed UJT-like high shape index. In WT 10A and +RhoA monolayers (Fig. 5B and C), cells with very high AR (≥4) or highly unjammed SI (≥6) shape index, both in red hues, were found at the entry of microchannels, indicating their need to undergo larger shape transitions to enter confinement. In comparison, 10A + ErbB2 cells had more homogeneous distribution of AR and SI, with very few cells showing large shape changes, indicating their less requirement for dramatic shape transitions to enter confinement. We plotted average metrics for cell morphology across samples and found that cell area for WT and +RhoA cells was lower than +ErbB2 (Fig. 5E), further indicating the need for non-cancer cells to get squeezed to enter confinement. While the average AR of WT and +RhoA cells was higher than +ErbB2, differences in SI across cell lines were statistically insignificant when averaged over the whole population (Fig. 5F and G).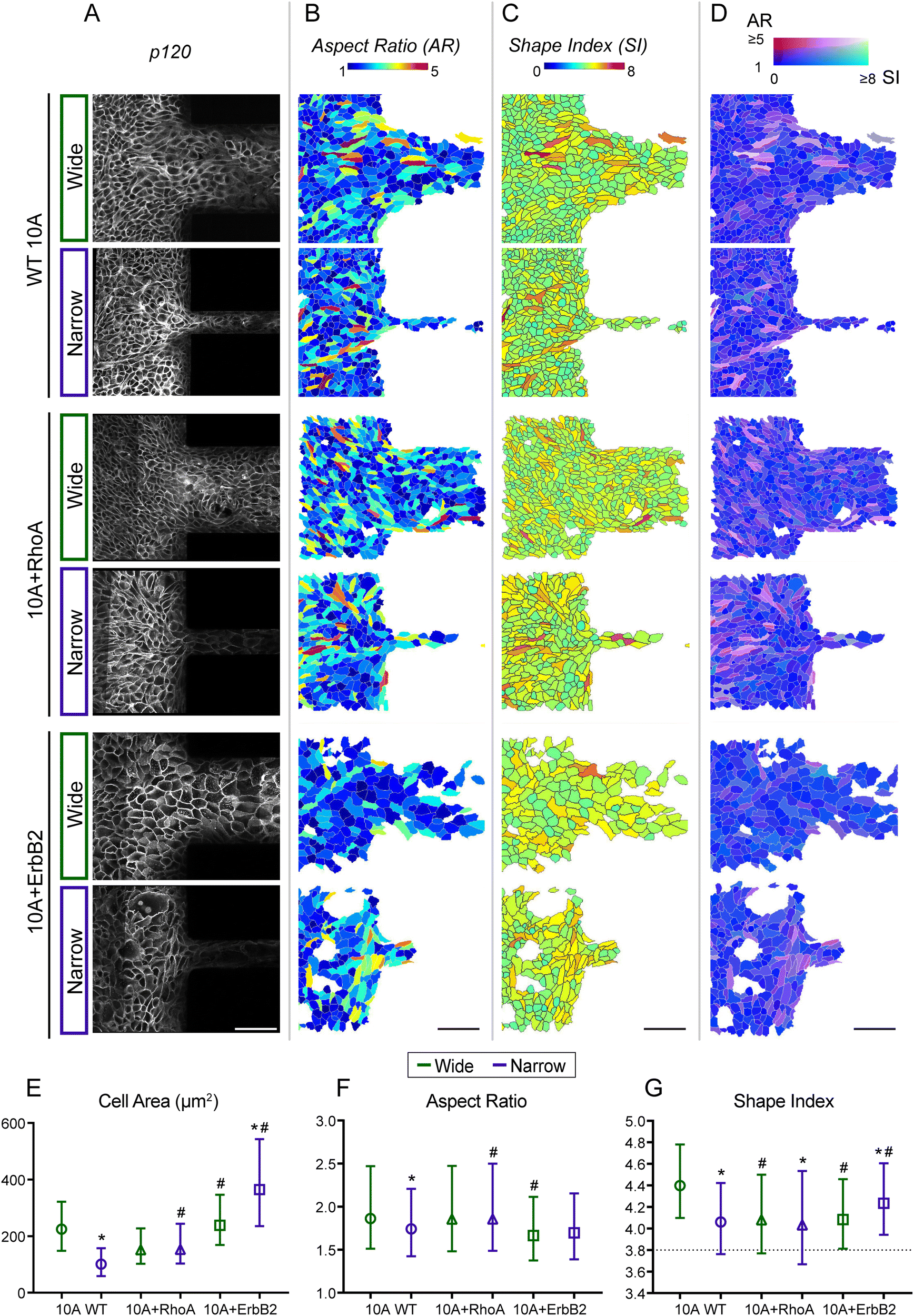 | ||
| Fig. 5 Cell shape transitions in migration across confinement. (A) Immunostaining of cell–cell junctions (P120), (B) aspect ratio (AR), (C) shape index (SI), and (D) combined representation AR and SI according to the indicated colormap. Scale bar, 100 μm. Comparison of averaged (E) cell area, (F) aspect ratio, and (G) shape index across cell type and confinement variations, where the markers and bars represent median and interquartile range, respectively. Kruskal–Wallis test is used here for pairwise comparison between groups, *p < 0.05 compared to wide channels. #p < 0.05 compared to WT. n ≥ 1700 cells from at least 4 ROIs. | ||
We also visualized the morphological state of each cell as a combined effect of AR and SI (Fig. 5D) to spot cells with relatively lower unjamming (SI) yet higher elongation (AR). This visualization allows assessment of whether cells get jammed despite getting elongated while entering confinement. Indeed, in WT 10A and 10A + RhoA monolayers, many cells showed higher AR and lower SI (magenta hues in Fig. 5D), which indicates that cells undergo some jamming despite shape elongation required to enter confinement. These jammed-elongated cells (dark magenta) seemed to be squeezed ahead of channel entry while unjammed-elongated cells (lighter magenta) migrated into channels. For +ErBb2 cells, the jammed-elongated (dark magenta) phenotype was minimal, and most cells showed unjamming (blue), particularly in wide channels. When migrating into narrow channels, +ErbB2 showed some jamming behaviour (magenta cells), but this phenotype was much less prevalent compared to WT or +RhoA cells.
Discussion and conclusion
Over the past decade, cell collectives have been physically described as jamming of deformable bodies at high density, while unjamming requires a rise in internal forces and shape transitions.11,24 Since the process of epithelial collectives approaching confinement would naturally incur crowding due to the narrowing space, cell density is expected to rise quickly within hours, without proliferation that needs days. Thus, our system of open-to-confined transmigration of collective cells allowed a unique opportunity to correlate cell shape transitions with density across cell lines.In our system of epithelia attempting to enter microchannels, some cells hit the channel walls and change their direction while others enter the microchannels (Fig. 3). This mixture of tissue collisions and free expansion is similar to a recent tri-tissue collision experiment,47 except with the complication of forced and rapid changes in cell crowding and density. Indeed, after part of the monolayer hits the channel wall, the cells that do not enter the microchannel cause crowding outside, which creates a density gradient between the inside and outside of the microchannel. When +RhoA cells approach confinement, their rise in density is similar in wide and narrow channels (Fig. 2). Since RhoA is associated with both cell contractility and cell–cell adhesions, even the confinement presented by wide channels may be rapidly sensed by this cell population due to their higher cohesivity. In wide type 10A cells, narrow channels are associated with slightly higher crowding than wide channels. By contrast, +ErbB2 cells show lower crowding ahead of narrower channels (Fig. 2B–E), which could be attributed to their reduced contact inhibition, relation with EMT, and the cancer-like ability to crawl on top of each other.21 As a result, +ErbB2 cells do not require standard crowding and higher density accumulation ahead of narrower channels. This differences between +RhoA and +ErbB2 cells could be attributed to stronger cell–cell contractility and cell-ECM adhesions downstream of RhoA32,36 and contact inhibition phenotype of +ErbB2. Since Wnt pathway is associated with intercellular separation and it involves the ErbB2 oncogene, we also inhibited Wnt signalling using metformin as an attempt to restore contact inhibition in +ErbB2 cells,48,49 which eliminate the cell density difference between wide and narrow channel in +ErbB2 cells (Fig. S4, ESI†).
According to our physical interpretations, despite dissimilar cell types, collective cell migration seems to follow some basic relationships between density and shape transition through migration into confinement. Our system of confinement gradient could also enable further studies of densification-driven migration, which is otherwise not feasible in situations with free boundary and growth-dependent densification of cell populations. Overall, these insights and physical analyses of cell migration could enable better understanding of how cell populations aggregate yet migrate in tumour invasion, wound healing, and embryogenesis.
Author contributions
AP and WL conceived the project. WL, HY performed experiments and analysed data. WL, HY, and AP designed experiments, interpreted findings, made figures, and wrote the manuscript. AP edited the manuscript, supervised the project, and acquired funding.Data availability
The data supporting this article have been included as part of the ESI.† Source code for analysis of collective cell migration (MATLAB license required) are freely available at https://github.com/sahuyu/SWS-Post-Processing.gitConflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the NIH/NIGMS MIRA (R35GM128764) grant to Amit Pathak. Microscopy with live nuclear tracking was carried out at the Washington University Centre for Cellular Imaging (WUCCI).References
- X. Trepat, M. R. Wasserman, T. E. Angelini, E. Millet, D. A. Weitz, J. P. Butler and J. J. Fredberg, Nat. Phys., 2009, 5, 426–430 Search PubMed.
- M. R. Ng, A. Besser, G. Danuser and J. S. Brugge, J. Cell Biol., 2012, 199, 545–563 CrossRef CAS PubMed.
- C.-M. Lo, H.-B. Wang, M. Dembo and Y. Wang, Biophys. J., 2000, 79, 144–152 CrossRef CAS PubMed.
- R. Sunyer, V. Conte, J. Escribano, A. Elosegui-Artola, A. Labernadie, L. Valon, D. Navajas, J. M. García-Aznar, J. J. Muñoz, P. Roca-Cusachs and X. Trepat, Science, 2016, 353, 1157–1161 CrossRef CAS PubMed.
- A. Pathak and S. Kumar, Integr. Biol., 2011, 3, 267 CrossRef CAS PubMed.
- A. Pathak and S. Kumar, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10334–10339 CrossRef CAS PubMed.
- S. R. K. Vedula, M. C. Leong, T. L. Lai, P. Hersen, A. J. Kabla, C. T. Lim and B. Ladoux, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12974–12979 CrossRef CAS PubMed.
- W. Xi, S. Sonam, T. Beng Saw, B. Ladoux and C. Teck Lim, Nat. Commun., 2017, 8, 1517 CrossRef PubMed.
- J. A. Mitchel, A. Das, M. J. O’Sullivan, I. T. Stancil, S. J. DeCamp, S. Koehler, O. H. Ocaña, J. P. Butler, J. J. Fredberg, M. A. Nieto, D. Bi and J.-A. Park, Nat. Commun., 2020, 11, 5053 CrossRef CAS PubMed.
- L. Atia, D. Bi, Y. Sharma, J. A. Mitchel, B. Gweon, S. A. Koehler, S. J. DeCamp, B. Lan, J. H. Kim, R. Hirsch, A. F. Pegoraro, K. H. Lee, J. R. Starr, D. A. Weitz, A. C. Martin, J.-A. Park, J. P. Butler and J. J. Fredberg, Nat. Phys., 2018, 14, 613–620 Search PubMed.
- J.-A. Park, J. H. Kim, D. Bi, J. A. Mitchel, N. T. Qazvini, K. Tantisira, C. Y. Park, M. McGill, S.-H. Kim, B. Gweon, J. Notbohm, R. Steward Jr, S. Burger, S. H. Randell, A. T. Kho, D. T. Tambe, C. Hardin, S. A. Shore, E. Israel, D. A. Weitz, D. J. Tschumperlin, E. P. Henske, S. T. Weiss, M. L. Manning, J. P. Butler, J. M. Drazen and J. J. Fredberg, Nat. Mater., 2015, 14, 1040–1048 CrossRef CAS PubMed.
- A. Pathak, Biophys. J., 2016, 111, 1496–1506 CrossRef CAS PubMed.
- S. Nasrollahi and A. Pathak, Sci. Rep., 2016, 6, 18831 CrossRef CAS PubMed.
- S. Nasrollahi and A. Pathak, MRS Commun., 2017, 7, 450–457 CrossRef CAS.
- P. Friedl and D. Gilmour, Nat. Rev. Mol. Cell Biol., 2009, 10, 445–457 CrossRef CAS PubMed.
- A. Haeger, K. Wolf, M. M. Zegers and P. Friedl, Trends Cell Biol., 2015, 25, 556–566 CrossRef PubMed.
- A. C. Brown, V. F. Fiore, T. A. Sulchek and T. H. Barker, J. Pathol., 2013, 229, 25–35 CrossRef CAS PubMed.
- S. C. Wei, L. Fattet, J. H. Tsai, Y. Guo, V. H. Pai, H. E. Majeski, A. C. Chen, R. L. Sah, S. S. Taylor, A. J. Engler and J. Yang, Nat. Cell Biol., 2015, 17, 678–688 CrossRef CAS PubMed.
- J. Park, D.-H. Kim, S. R. Shah, H.-N. Kim Kshitiz, P. Kim, A. Quiñones-Hinojosa and A. Levchenko, Nat. Commun., 2019, 10, 2797 CrossRef PubMed.
- J. Lu, H. Guo, W. Treekitkarnmongkol, P. Li, J. Zhang, B. Shi, C. Ling, X. Zhou, T. Chen, P. J. Chiao, X. Feng, V. L. Seewaldt, W. J. Muller, A. Sahin, M.-C. Hung and D. Yu, Cancer Cell, 2009, 16, 195–207 CrossRef CAS PubMed.
- A. Pathak and S. Kumar, Integr. Biol., 2013, 5, 1067 CrossRef CAS PubMed.
- B. Zhao, Q.-Y. Lei and K.-L. Guan, Curr. Opin. Cell Biol., 2008, 20, 638–646 CrossRef CAS PubMed.
- M. Aragona, T. Panciera, A. Manfrin, S. Giulitti, F. Michielin, N. Elvassore, S. Dupont and S. Piccolo, Cell, 2013, 154, 1047–1059 CrossRef CAS PubMed.
- T. E. Angelini, E. Hannezo, X. Trepat, M. Marquez, J. J. Fredberg and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4714–4719 CrossRef CAS PubMed.
- D. Bi, X. Yang, M. C. Marchetti and M. L. Manning, Phys. Rev. X, 2016, 6, 021011 Search PubMed.
- M. Gamboa Castro, S. E. Leggett and I. Y. Wong, Soft Matter, 2016, 12, 8327–8337 RSC.
- S. Garcia, E. Hannezo, J. Elgeti, J.-F. Joanny, P. Silberzan and N. S. Gov, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15314–15319 CrossRef CAS PubMed.
- M. De Marzio, A. Kılıç, E. Maiorino, J. A. Mitchel, C. Mwase, M. J. O’Sullivan, M. McGill, R. Chase, J. J. Fredberg, J.-A. Park, K. Glass and S. T. Weiss, Sci. Adv., 2021, 7, eabf1088 CrossRef CAS PubMed.
- D. Bi, J. H. Lopez, J. M. Schwarz and M. L. Manning, Nat. Phys., 2015, 11, 1074–1079 Search PubMed.
- M. M. Zegers and P. Friedl, Small GTPases, 2014, 5, e983869 Search PubMed.
- S. J. Terry, C. Zihni, A. Elbediwy, E. Vitiello, I. V. Leefa Chong San, M. S. Balda and K. Matter, Nat. Cell Biol., 2011, 13, 159–166 CrossRef CAS PubMed.
- M. Reffay, M. C. Parrini, O. Cochet-Escartin, B. Ladoux, A. Buguin, S. Coscoy, F. Amblard, J. Camonis and P. Silberzan, Nat. Cell Biol., 2014, 16, 217–223 CrossRef CAS PubMed.
- P. Friedl, K. Wolf and M. M. Zegers, Nat. Cell Biol., 2014, 16, 208–210 CrossRef CAS PubMed.
- J. Ju, H. N. Lee, L. Ning, H. Ryu, X. X. Zhou, H. Chun, Y. W. Lee, A. I. Lee-Richerson, C. Jeong, M. Z. Lin and J. Seong, Cell Rep., 2022, 40, 111080 CrossRef CAS PubMed.
- G. Charras and E. Sahai, Nat. Rev. Mol. Cell Biol., 2014, 15, 813–824 CrossRef CAS PubMed.
- P. Pandya, J. L. Orgaz and V. Sanz-Moreno, Curr. Opin. Cell Biol., 2017, 48, 87–96 CrossRef CAS PubMed.
- C. D. Paul, P. Mistriotis and K. Konstantopoulos, Nat. Rev. Cancer, 2017, 17, 131–140 CrossRef CAS PubMed.
- E. M. Balzer, Z. Tong, C. D. Paul, W. Hung, K. M. Stroka, A. E. Boggs, S. S. Martin and K. Konstantopoulos, FASEB J., 2012, 26, 4045–4056 CrossRef CAS PubMed.
- B. Burkel, B. A. Morris, S. M. Ponik, K. M. Riching, K. W. Eliceiri and P. J. Keely, J. Vis. Exp., 2016,(111), e53989 Search PubMed.
- A.-K. Marel, M. Zorn, C. Klingner, R. Wedlich-Söldner, E. Frey and J. O. Rädler, Biophys. J., 2014, 107, 1054–1064 CrossRef CAS PubMed.
- V. Petrolli, M. Le Goff, M. Tadrous, K. Martens, C. Allier, O. Mandula, L. Hervé, S. Henkes, R. Sknepnek, T. Boudou, G. Cappello and M. Balland, Phys. Rev. Lett., 2019, 122, 168101 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- D. Ershov, M.-S. Phan, J. W. Pylvänäinen, S. U. Rigaud, L. Le Blanc, A. Charles-Orszag, J. R. W. Conway, R. F. Laine, N. H. Roy, D. Bonazzi, G. Duménil, G. Jacquemet and J.-Y. Tinevez, Nat. Methods, 2022, 19, 829–832 CrossRef CAS PubMed.
- D. N. Mashburn, H. E. Lynch, X. Ma and M. S. Hutson, Cytometry, Part A, 2012, 81A, 409–418 CrossRef PubMed.
- S. Varadarajan, S. A. Chumki, R. E. Stephenson, E. R. Misterovich, J. L. Wu, C. E. Dudley, I. S. Erofeev, A. B. Goryachev and A. L. Miller, J. Cell Biol., 2022, 221, e202105107 CrossRef CAS PubMed.
- B. D'souza and J. Taylor-Papadimitriou, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 7202–7206 CrossRef PubMed.
- M. A. Heinrich, R. Alert, A. E. Wolf, A. Košmrlj and D. J. Cohen, Nat. Commun., 2022, 13, 4026 CrossRef CAS PubMed.
- G. Amable, E. Martínez-León, M. E. Picco, S. I. Nemirovsky, E. Rozengurt and O. Rey, J. Cell. Physiol., 2020, 235, 8334–8344 CrossRef CAS PubMed.
- E. Anear and R. W. Parish, FEBS Lett., 2012, 586, 1330–1335 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01240a |
| This journal is © The Royal Society of Chemistry 2025 |