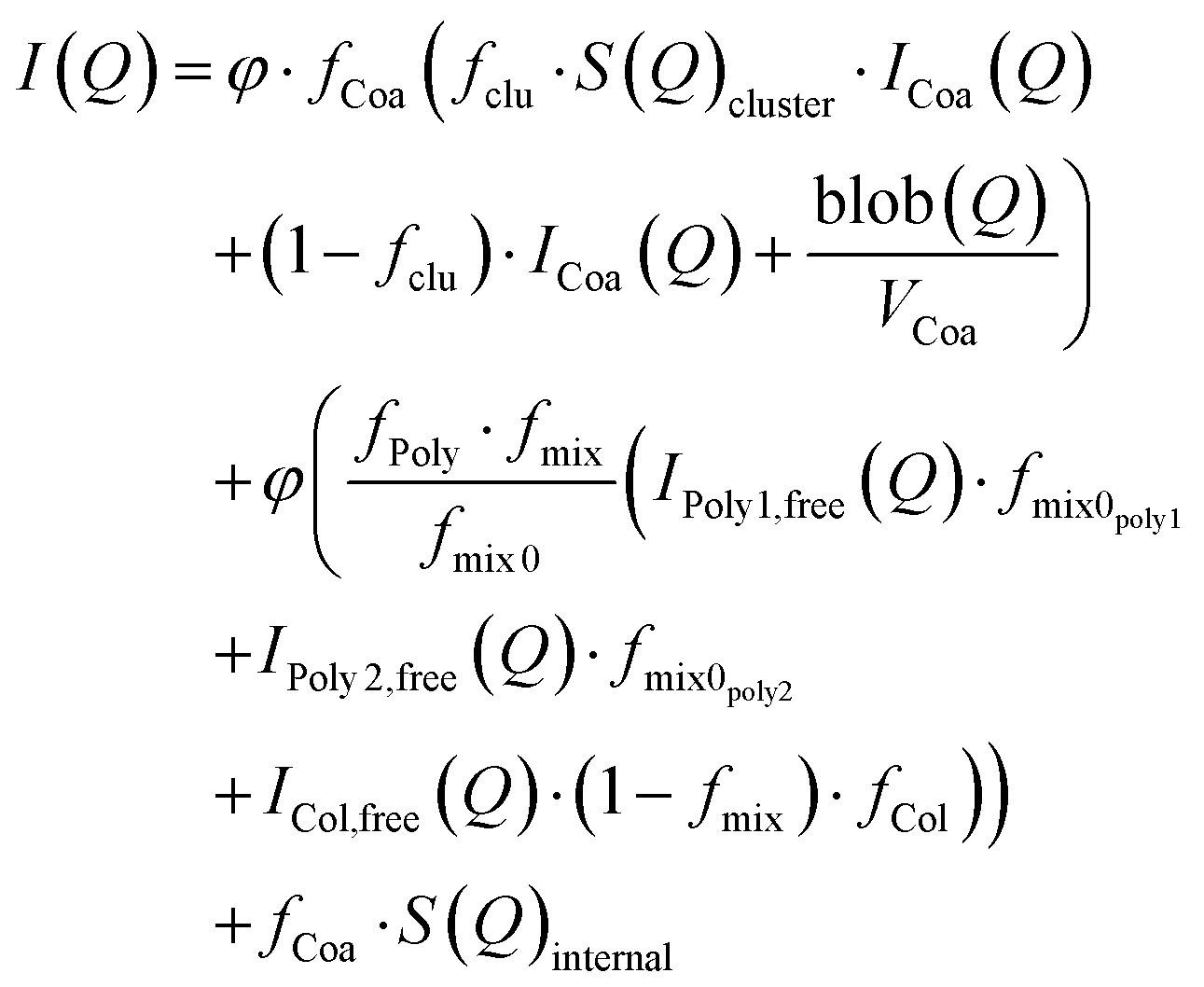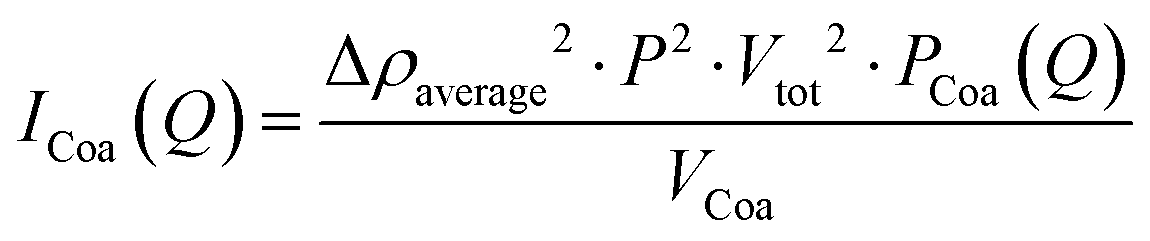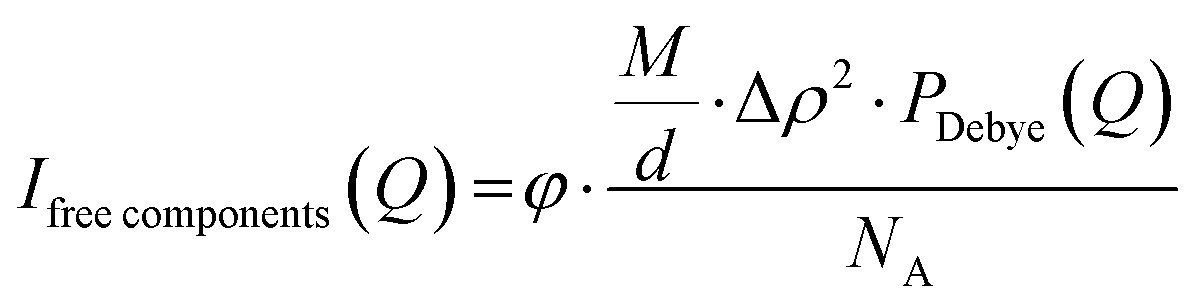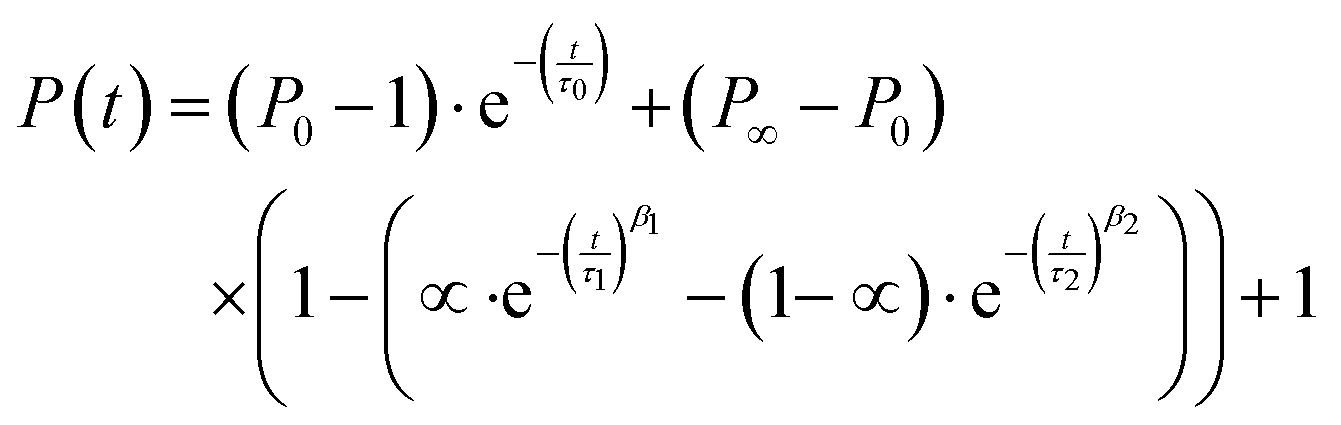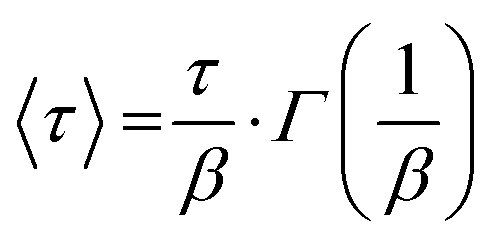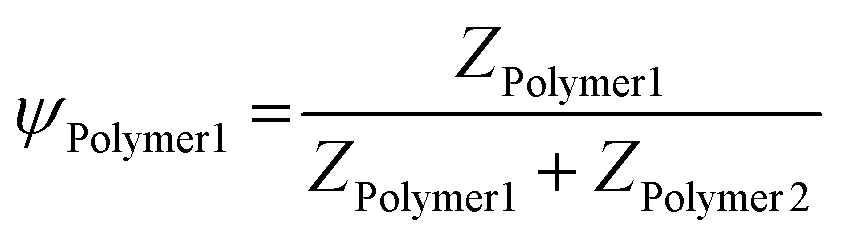DOI:
10.1039/D4SM01157K
(Paper)
Soft Matter, 2025,
21, 903-913
Size-controlled antimicrobial peptide drug delivery vehicles through complex coacervation†
Received
3rd October 2024
, Accepted 20th December 2024
First published on 23rd December 2024
Abstract
Due to the escalating threat of the pathogens’ capability of quick adaptation to antibiotics, finding new alternatives is crucial. Although antimicrobial peptides (AMPs) are highly potent and effective, their therapeutic use is limited‚ as they are prone to enzymatic degradation, are cytotoxic and have low retention. To overcome these challenges, we investigate the complexation of the cationic AMP colistin with diblock copolymers poly(ethylene oxide)-b-poly(methacrylic acid) (PEO-b-PMAA) forming colistin–complex coacervate core micelles (colistin–C3Ms). We present long-term stable kinetically controlled colistin–C3Ms that can be prepared from several block lengths of PEO-b-PMAA polymers, where the polymerisation degree governs the overall micellar size. To achieve precise control over size and polydispersity, which are crucial for drug delivery applications, we investigate the hybridisation of PEO-b-PMAA polymers with varying chain lengths or PMAA homopolymers in ternary complex coacervation systems with colistin. This results in size-tunable colistin–C3Ms, ranging, depending on the mixing ratios, from micellar sizes of 26 nm to 100 nm. With size tunability at rather narrow size distributions and high stability, ternary colistin–C3Ms offer potential advancements in C3M drug delivery, paving the way for more effective and targeted treatments for bacterial infections in precision medicine.
Introduction
The ever-increasing ability of pathogens to adapt and become resistant to antimicrobials poses a severe worldwide threat, leading to high morbidity and mortality rates.1,2 In 2019, it was estimated that antimicrobial resistance (AMR) was directly linked to 1.27 million deaths and that drug resistance played a role in the death of an additional 3.68 million people annually.2,3 As antibiotics struggle to maintain their efficiency, and considering the absence of antibiotics entering the market, new alternatives are of great importance to reduce the rise of impactful infectious bacterial diseases.4,5 As an alternative, antimicrobial peptides (AMPs) emerge as promising candidates.6 They are small molecules that play a vital role in the innate immune system of organisms among all classes of life. AMPs function by causing lysis of the bacterial cell by attacking intracellular targets when there is an infection.7,8 One class in the world of AMPs of substantial interest is the polymyxins.3,9,10 Polymyxin E (better known as colistin) is an AMP used as a last-resort drug in clinical settings when conventional antibiotics are contraindicated or ineffective.11–13 However, colistin faces several challenges associated with administration due to its high nephrotoxicity, poor stability and low bioavailability.9,10,13 To resolve these challenges, the encapsulation potential through complex coacervation of the cationic colistin with oppositely charged block copolymers has previously been investigated.14,15 Complex coacervation is a primarily entropically and electrostatically driven process between oppositely charged macromolecules in solution, leading to an associative liquid–liquid phase separation.16 From the complexation of the (partly) anionic poly(ethylene oxide)-b-poly(methacrylic acid) (PEO-b-PMAA) polymers with the cationic colistin (the charge is +5 under physiological conditions), we could form well-defined and colloidally stable micelles. Here, the oppositely charged PMAA and colistin formed a core while PEO provided a protective shell.15 These so-called complex core coacervate micelles (C3Ms) were found to improve the stability of colistin as well as protect the drug from enzymatic degradation while maintaining its antibacterial activity. This observed antimicrobial effect was found to be likely due to the subsequent release of colistin from the micelles, although the exact mechanism remains unclear.
In recent years, more attention has been given to the potential of C3Ms as drug delivery vehicles, as they have been shown to increase drug circulation times in the body significantly.17–19 Even though multiple drugs, from DNA to proteins and peptides, have been shown to be effectively encapsulated, C3Ms have not reached the progression from in vitro to (pre-) clinical testing.20–25 One of the major caveats of C3Ms as drug delivery vehicles is the shortcoming of control over structural reproducibility and optimisation of their loading capability.19 To obtain better control, the fundamental principles of complex coacervation must be better understood. By obtaining control over these systems, the morphology, structure, size, and stability can be steered, depending on the desired purpose.18,19 Vital steps in obtaining control over C3Ms are the comprehension of their dynamics and their structure–function relationships.18
Kinetic studies on C3Ms are scarce because of the requirements of challenging experimental setups.18,26–31 Important processes relevant to the properties of C3Ms as carriers include kinetics of micellisation that influence reproducibility and,26,28,32 chain exchange,33–35 and disassembly kinetics that govern the stability and drug release.27,29,36 In previous studies, we elucidated the formation kinetics of colistin-PEO-b-PMAA C3Ms in two different experimental setups: in slow motion through prodrug hydrolysis37 and through in situ high speed-mixing millisecond resolution TR-SAXS.32 We found that C3Ms are kinetically controlled, which mainly affects the final stability of the C3Ms under different formation conditions, like different concentrations, charge ratios, and ionic strengths. Only minor changes in size, structure, and morphology were reported under these different conditions. In line with the more widely studied structure–function relationships of C3Ms, we have observed previously that larger block-length polymers formed larger-sized C3Ms.15,16,18,19,32,38,39 In contrast to the well-reported direct correlation between stability and polymer block length,16,19,40 the preparation of colistin-C3Ms with larger polymers significantly reduced the stability reflecting in an increased tendency towards aggregation/clustering. Most likely, this is caused by the increasingly sluggish dynamics of larger polymers that lead to non-equilibrated and inhomogeneous micelles that are more likely to cluster into ill-defined structures.15,32 Understanding the fundamental connection between the polymer block length with size (at narrow size distributions) and its stability could be valuable to better unlock the potential of C3Ms as drug delivery vehicles. Additionally, the morphology and size (distribution) of C3Ms predominately determine their in vivo behaviour and drug delivery potential, particularly affecting circulation time, degradation by macrophages and phagocytes, filtration pathways, distribution, and permeability.41–44 Moreover, the optimal sizing is generally between 10 and 150 nm, as it minimises rapid renal filtration for particles below 10 nm and reduces macrophage clearance for particles above 150 nm.45,46
One of the potential ways to obtain a higher degree of control over the size, morphology, and, therefore, functionality of C3Ms is the hybridisation of complexation agents. Some studies have been conducted on so-called ternary complex coacervate systems where three complexing agents were complexed in different ratios.47–49 By adjusting the fractions of each component, a higher level of control and tunability over the system could be obtained.47,48 Priftis et al.47 reported the systematic study of highly tunable complex coacervates prepared from a mixture of PAH and branched polyethyleneimine (BPEI) with polyacrylic acid (PAA) and poly(N,N-dimethyl aminoethyl methacrylate) (PDMAEMA). Zhao et al.48 could prepare property and composition-tunable ternary complex coacervates by mixing branched polyethyleneimine (BPEI) with a binary mixture of polyacrylic acid (PAA) and poly(4-styrenesulfonic acid) (SPS).
Inspired by these works, we investigate the formation of ternary colistin C3Ms through mixed polymer–colistin complexation. Employing small-angle X-ray scattering (SAXS) with detailed modelling, we elucidate the micelle characteristics, such as the composition, size, and aggregation number. We demonstrate a high degree of size control by simple adjustments of the macroscopic composition. With ternary complex coacervation, we prepare stable colistin-C3Ms with sizes ranging from 26 to 100 nm that have a narrower size distribution than binary complex coacervate systems. We believe that this enhanced control will aid in the more tailored development of colistin drug delivery systems, optimising therapeutic efficacy for precision medicine.
Experimental
Materials
Four poly(ethylene oxide)-b-poly(methacrylic acid) (PEO-b-PMAA) polymers composed of different block lengths (PEO45-b-PMAA15, PDI = 1.10; PEO45-b-PMAA41, PDI = 1.20; PEO45-b-PMAA81, PDI = 1.15 and PEO114-b-PMAA81, PDI = 1.15) were purchased from Polymer source, Inc. (Canada). Colistin sulfate (PHR1605), poly(methacrylic acid) (PMAA), sodium salt (Mw ≈ 4000 g mol−1, obtained by SAXS analysis with the Debye model), Trizma© base, Trizma© hydrochloride, hydrochloric acid (HCl), and sodium chloride (NaCl) were purchased from Sigma-Aldrich/Merck (Germany). PMAA, sodium salt, was modified to obtain a salt-free polymer. Other chemicals were not purified further.
Methods
Complex coacervate preparation.
(PEO-b-)PMAA polymers and colistin sulfate were dissolved in 0.05 M tris buffer (pH = 7.4). The polymer solutions were mixed before (for ternary complex coacervate systems) and then mixed with the colistin solution. Mixing concentrations of polymer solutions and colistin solutions were based on a fixed final concentration of either 0.50 wt%, 0.25 wt% or 0.125 wt%, at equal volumes of polymer and colistin solutions and charge-matching conditions (total charges of colistin and polymer(s) are equal).
Purification of the PMAA sodium salt.
The PMAA sodium salt was dissolved in water to a 20–30 wt% solution. A 10% total volume of 12.1 M HCl was added without stirring to protonate the PMAA, which triggered precipitation. The supernatant was removed from the precipitate on a filter in a Büchner tract, and the precipitate was transferred to a filter paper and rinsed several times with a small amount of water to avoid dissolution.
Static small-angle X-ray scattering (SAXS) at BM29.
The small-angle X-ray scattering (SAXS) profiles were collected using the BioSAXS beamline BM29 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France.50–52 50 μL of each sample was injected using an autosampler into a quartz 1 mm glass capillary at 20 °C. Subsequently, ten scattering frames of 1.0 s each were detected using a Pilatus 3 × 2 M detector (E = 12.5 keV) that was positioned at 2.87 m from the sample. This resulted in a Q-range of roughly 0.007–0.50 Å−1. The background sample (0.05 M tris buffer at pH = 7.4) was measured before and after each sample, followed by an automatic cleaning process of the capillary. Water was used as a primary standard to scale the data to absolute intensities. All frames were manually checked for radiation damage, followed by averaging, buffer subtraction, and binning (from 1000 points to 280), resulting in the final static scattering curves.
Time-resolved (TR)-SAXS at ID02.
TR-SAXS measurements were carried out at the TRUSAXS beamline ID02 at the ESRF (Grenoble, France). The solutions were mixed using an SFM-4000 stopped-flow adapted for in situ TR-SAXS (Bio-Logic Science Instruments, France). Solutions of PEO-b-PMAA mixtures and colistin were mixed at 20 °C in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and equal charge contribution from each polymer (ΨPEO45-b-PMAA41 = 0.50) at a total flow rate of 6.0 mL s−1, resulting in a 2.9 ms dead time. Between the dead time and 2 minutes after mixing, frames were detected using an Eiger2-4 M detector with a detector distance of 2.0 from the stopped-flow capillary (diameter ≈ 1.25 mm), covering a Q-range of 0.003–0.40 Å−1. The total concentrations of polymer–colistin mixtures were fixed at 0.125 wt% under stoichiometric charge matching conditions. 31 frames were in total collected for each sample for a minimum of 8 reproducible shots. 30 successive frames with a 5 ms exposure time were measured at increasing time gaps evolving with a factor of 1.35, spanning a time scale from 2.9 ms to 86 s. A final frame was taken with an exposure time of 0.1 s when the formation of the micelles was completed (manually checked in advance), approximately 2 minutes after mixing (set to t = 120 s). The background sample (0.05 M tris buffer, pH = 7.4) was measured before each set of timed measurements. The data were again scaled to absolute intensities using water as a reference. Each frame was manually checked for radiation damage and bubbles, followed by averaging of the shots at all 31-time points, buffer subtraction, and binning (from ≈ 1600 points to ≈ 280 points), resulting in the final time-resolved scattering curves.
:1 and equal charge contribution from each polymer (ΨPEO45-b-PMAA41 = 0.50) at a total flow rate of 6.0 mL s−1, resulting in a 2.9 ms dead time. Between the dead time and 2 minutes after mixing, frames were detected using an Eiger2-4 M detector with a detector distance of 2.0 from the stopped-flow capillary (diameter ≈ 1.25 mm), covering a Q-range of 0.003–0.40 Å−1. The total concentrations of polymer–colistin mixtures were fixed at 0.125 wt% under stoichiometric charge matching conditions. 31 frames were in total collected for each sample for a minimum of 8 reproducible shots. 30 successive frames with a 5 ms exposure time were measured at increasing time gaps evolving with a factor of 1.35, spanning a time scale from 2.9 ms to 86 s. A final frame was taken with an exposure time of 0.1 s when the formation of the micelles was completed (manually checked in advance), approximately 2 minutes after mixing (set to t = 120 s). The background sample (0.05 M tris buffer, pH = 7.4) was measured before each set of timed measurements. The data were again scaled to absolute intensities using water as a reference. Each frame was manually checked for radiation damage and bubbles, followed by averaging of the shots at all 31-time points, buffer subtraction, and binning (from ≈ 1600 points to ≈ 280 points), resulting in the final time-resolved scattering curves.
SAXS data modelling.
To model the SAXS data, we used an extended version of a previously published fuzzy-interface complex coacervate core micelle model.15 In this work, we included the contributions of a second polymer into the mass balance calculations and scattering equations. The model is based on fixed partitioning, charge matching between colistin and polymers, and equal charge participation of polymers in the core of the micelles. Three separate contributions of scattering were considered: (1) complex coacervate scattering, (2) non-bound colistin and polymers, and (3) an additional structure factor describing positional polyelectrolyte charge correlations. The total scattering model (on absolute scale) is presented in eqn (1).| | 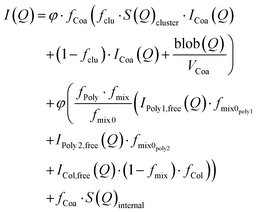 | (1) |
where φ is the volume fraction (based on the concentration), fCoa is the fraction of molecules forming coacervates, fclu is the fraction that is forming clusters, S(Q)cluster is the cluster structure factor (eqn (S1)–(S6), ESI†), ICoa(Q) is the intensity from complex coacervates, blob(Q) is the scattering contributions from the blobs of the polyelectrolytes in the core, VCoa is the dry volume of one complex coacervate, fPoly is the free fraction of the polymer, fmix is the molar fraction of the polymer in the aqueous phase surrounding the complex coacervates, fmix0 is the molar fraction of the polymer in the complex coacervate micelles and is fixed based on equal charge partitioning, IPoly,free(Q) is the Debye scattering from the two non-incorporated polymers, fmix0poly is the molar fraction of the two polymers (assuming equal participation in micellisation of each polymer specie), ICol,free(Q) is the Debye scattering from colistin that is not incorporated into the micelles, and S(Q)internal (eqn (S7), ESI†) is the additional polyelectrolyte charge correlation structure factor. The scattering contribution of the coacervates (ICoa(Q)) (eqn (2)) is based on a fuzzy-sphere form factor (PCoa(Q) (eqn (S8)–(S12), ESI†)).53,54| | 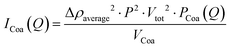 | (2) |
where Δρaverage is the average scattering length density of the complex coacervates, P is the aggregation number (number of molecules per micelle), and Vtot is the total dry volume of the coacervates. The scattering of unbound polymers and colistin is calculated and given in eqn (3). We use the Debye form factor (PDebye(Q)) for polymers and electrolytes (eqn (S13), ESI†).55| | 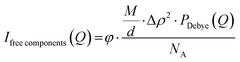 | (3) |
where M is the molecular weight of each free component, d is the solution density, Δρ is the scattering contrast, and NA is the Avogadro number. Least-squares fit routines were used with initially the following floating parameters: total aggregation number (P), the core radius including its density distribution (width of the interface) and polydispersity index (Rin, σin, and PDI) and the free fraction of colistin (fCol). After achieving an initial fit for the low and intermediate Q ranges, the high Q range data were fitted by considering the scattering from the blobs. To account for the polyelectrolyte charge correlations (internal structure) and the blob scattering, the relative width at high Q(W), fractal scattering (C(Q)), the internal scattering location (Qlocal), the blob fraction (fblob), and the blob correlation length (ξ) were fitted to the data. Lastly, all fitting parameters were simultaneously fitted to the data, resulting in the final fits. An overview of the parameters used in the model, including their roles and descriptions, is summarised in Table S1 (ESI†). The standard deviation of the fitting parameters was determined through manual parameters changing, maximally allowing a 10% increase in X2, the parameter that we used to ensure least-squares fitting. From the final fits and the mass balances, the molecular weight (Mw) (eqn (S14), ESI†) and the fraction of water in the micelles (fw) can be calculated (eqn (S15)–(S19), ESI†). In addition, the form factors and structure factors of the model, as well as a more detailed explanation of the mass balances, can be found in the ESI† and our previous publication.15
Dynamic light scattering (DLS).
Dynamic light scattering (DLS) measurements were conducted with a DLS instrument from LS Instruments (Switzerland), equipped with a Cobolt high-performance DPSS laser 100 mW (660 nm). The samples were filtered through 0.45 μm poly(vinylidene difluoride) (PVDF) filters (Millipore, Merck) directly into precleaned 2 mm NMR tubes to avoid dust contamination. The polymer–colistin mixtures were either measured at 0.25 wt% or 0.125 wt% at 20 °C and were regularly checked to avoid multiple scattering.
Formation kinetics: modelling.
To model the formation kinetics of the ternary complex coacervate systems, we used a previously published32 double-stretch exponential model for colistin complex coacervate kinetics where the kinetics are modelled based on three distinctive kinetic processes: nucleation (0), fusion (1) and insertion exchange (2). The model is specified in eqn (4).| | 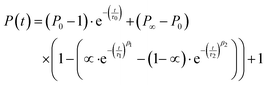 | (4) |
where P∞ represents the final aggregation number, P0 is the aggregation number from the first frame (at t = 2.9 ms), α is the amplitude weighting factor between the fusion (1) and insertion exchange kinetic (2) processes, quantifying the fraction contributed by the fusion. τ0, τ1, and τ2 correspond to the relaxation times associated with “nucleation” (0) and the two distinct growth processes (1 and 2), while β1 and β2 are the stretching exponents for each growth process. The mean relaxation times are then calculated using eqn (5).| | 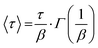 | (5) |
Results and discussion
As complex coacervate systems are dynamic, environmental conditions and intrinsic properties can substantially affect their structure/function properties.16,35,56 To gain control over the sizing and morphology and consequently achieve a structure–function understanding of C3M drug delivery vehicles, several strategies can be employed. These strategies include adjusting mixing properties, ionic strength, charge ratios, concentration effects, and intrinsic properties of the complexing agents, such as charge density and block lengths.16,19,57
In this work, we focus on the intrinsic properties of the components causing the complexation, as we have previously seen that changes in the external conditions have a negligible effect on the sizing and morphology of colistin-PEO-b-PMAA C3Ms without affecting their stability negatively.15,32,37 Through scaling laws, the link between the block length/charge densities and the resulting size and morphologies of the C3Ms has already been established in the literature.18,40,58,59 Above a threshold minimum block length/charge, the larger the charged block of the polyelectrolyte in the core, the bigger the micelle.59,60
Binary colistin complex coacervate systems
To visualise the effect of different block-length polymers on the sizing of colistin-C3Ms, the binary complexation of four different block-length PEO-b-PMAA di-block copolymers, as well as PMAA homopolymer with colistin, was investigated with SAXS and DLS (Fig. 1). The typical scattering pattern for C3Ms is shown for illustration purposes showing the large difference in intensity. The fits from the Debye model for the PEO-b-PMAA SAXS data (radius of gyration, Rg) and the CONTIN fits of the DLS correlation functions (hydrodynamic radius, Rh) are given in Table 1.
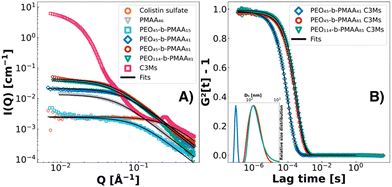 |
| | Fig. 1 SAXS patterns at 0.25 wt% of colistin (orange circles), homopolymer PMAA46 (grey triangles) and PEO-b-PMAA polymers PEO45-b-PMAA15 (light blue squares), PEO45-b-PMAA41 (dark blue rhombuses), PEO45-b-PMAA81 (red circles) and PEO114-b-PMAA81 (green triangles) including fits from the Debye model (A). The typical scattering pattern of C3Ms is also illustrated but not fitted (pink squares). Correlation curves of the corresponding complexes formed at 0.50 wt% are measured through DLS and fitted using CONTIN analysis (B), giving a size distribution of the diameter of hydration (Dh) (inset). | |
Table 1 Radii of the PEO-b-PMAA polymers through SAXS Debye model fitting and their complexation with colistin analysed with DLS CONTIN algorithm fitting
| Polymer |
Characteristic size of polymers from SAXS (Rg) (nm) |
Characteristic size of C3Ms (Rh) (DLS) (nm) |
| PMAA46 |
2.9 ± 0.2 |
Aggregation |
| PEO45-b-PMAA15 |
1.5 ± 0.1 |
No complexation |
| PEO45-b-PMAA41 |
2.3 ± 0.2 |
18 ± 2 |
| PEO45-b-PMAA81 |
3.3 ± 0.3 |
76 ± 6 |
| PEO114-b-PMAA81 |
3.4 ± 0.3 |
85 ± 8 |
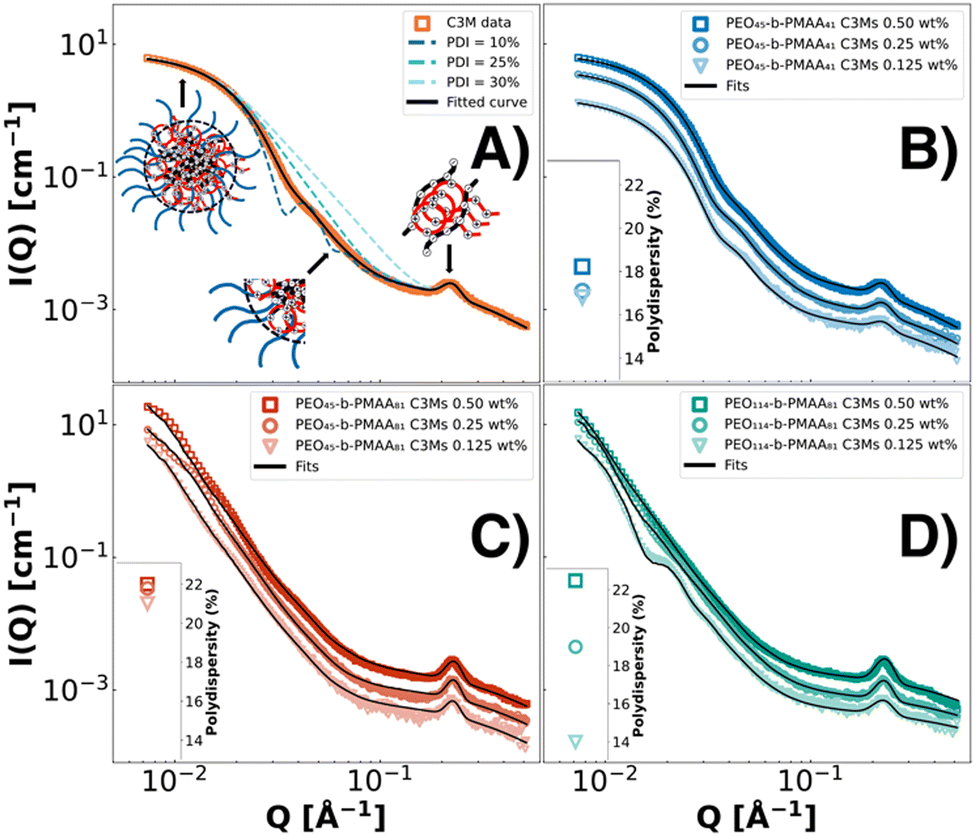
The colistin and polymer SAXS fits all display a Guinier region (flat at low Q) and weak Q−2 scattering at high Q, indicating a structure that resembles a random chain (Fig. 1). Three polymers could be complexed with colistin into three differently sized C3Ms, measured by DLS. The homopolymer (PMAA46) complexation resulted in precipitation, showing the importance of the neutral block in stabilising these systems through steric repulsion.16,61 PEO45-b-PMAA15 did not form complexes, most likely because the PMAA block was below the minimum block length requirements for micellar complexation.60,62 In line with the findings in the literature,16,18,19,59 the PMAA blocks with 81 units resulted in the formation of substantially larger complex coacervates than with 41 units, while the increase in the PEO block length from 45 to 114 resulted in much smaller increases in size. Since we established the formation of colistin complex coacervate systems from three different PEO-b-PMAA polymers, the next step is to elucidate its morphology and polydispersity with SAXS techniques. As these systems are kinetically controlled, the concentration at which complexes are prepared has a decisive effect.63–65 Therefore, in Fig. 2, we analysed three concentrations (0.50 wt%, 0.25 wt% and 0.125 wt%) of the three binary complexes at stoichiometric charge matching and consequently modelled them with the fuzzy-surface complex coacervate core model (eqn (1)) of which its features are highlighted in Fig. 2A. Polydispersity indexes from the fits are indicated in the insets.
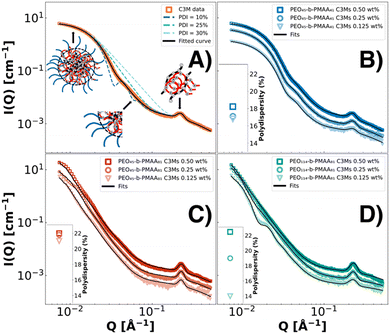 |
| | Fig. 2 SAXS patterns of colistin complex coacervate core micelles prepared, modelled with the fuzzy-interface C3M model, where the polydispersity and its general features are illustrated in (A). The micelles and polydispersity (insets) from three differently sized polymers PEO45-b-PMAA41 (B, blue symbols), PEO45-b-PMAA81 (C, red symbols) and PEO114-b-PMAA81 (D, green symbols) are shown at three concentrations (0.50 wt%: squares, 0.25 wt%: circles, and 0.125 wt% triangles), indicating kinetic control. | |
All scattering curves indicate the formation of “crew-cut”-like spherical micellar complexes (Fig. 2). In the low Q region, the size of the micelles is indicated where a flattening at low Q values, a so-called Guinier plateau, can be observed for PEO45-b-PMAA41–colistin–C3Ms. In Fig. 2C and D, no Guinier plateau is visible, indicating the presence of bigger and partly aggregated structures. The steep decay at intermediate Q values for all three systems resembles spherical structures. This region is also affected by polydispersity effects, where more monodisperse systems show more pronounced oscillations and steeper decays (Fig. 2A). At high Q values, we observe correlation peaks at around 0.22 Å−1, indicating an optimal packing distance between the colistin and PMAA chains within the core, which is referred to as electrostatic positional charge correlations of blobs.66 Based on the fit analysis, the total radii (Rtot) of the PEO45-b-PMAA41–colistin–C3M is 15.7 ± 0.3 nm, similar to the DLS results (Table 1 and Table S2, ESI†). Larger discrepancies between the DLS and SAXS fit analysis were found for the larger polymers, where PEO45-b-PMAA81-C3Ms and PEO114-b-PMAA81-C3Ms showed to have Rtot values of 33 ± 3 nm and 34 ± 4 nm, respectively (Tables S3 and S4, ESI†). This is likely related to the effect of clustering/slight aggregation, resulting in larger (weight-averaged) Rh values and a larger deviation with Rtot obtained from SAXS.37,67 Lastly, these structures are on the edge of the Q-range where precise diameters can be resolved for the instrument. From the SAXS patterns and the fits, we see that the concentration affects the structure progressively more for the larger polymers, indicating kinetic control. The lowest PDI values are found at the lowest concentration of 0.125 wt% for all systems. This was previously reported to be related to increased collisions at higher concentrations, resulting in kinetically trapped states that deviate further from the system's energy minimum.32 Therefore, the 0.125 wt% concentration is more suitable for further investigation.
Ternary colistin complex coacervate systems
As we observe a clear relationship between the block length of the anionic block and the resulting properties, we can exploit this property to control the size of these systems. One example of this is polymer hybridisation, resulting in ternary complex coacervation systems. By mixing polymers with different PMAA block lengths, the interactions are modified, potentially resulting in an enlarged palette of size control and morphology. Therefore, eight ternary systems comprising different sets of two previously analysed (PEO-b-)PMAA polymers with colistin were investigated (Table 2). In these ternary systems, the polymer charge contributions (Ψ) (eqn (6)) were varied, while stoichiometric charge matching conditions were maintained at a total concentration of 0.125 wt% (eqn (7)).| | 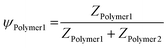 | (6) |
| | | ZColistin = ZPolymer1 + ZPolymer2 | (7) |
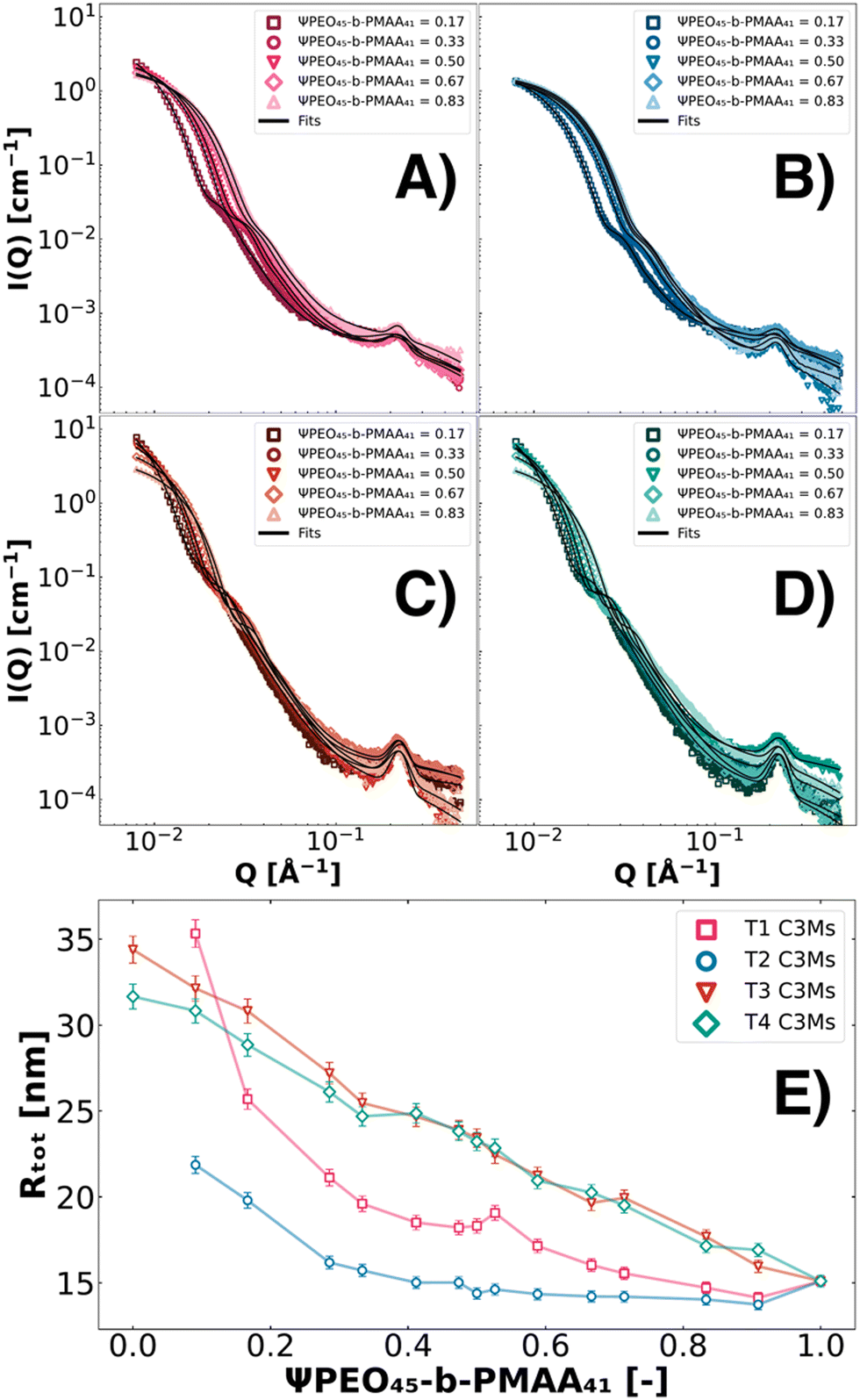
where Z is the total number of charges of PMAA from the polymers or charges from colistin at physiological pH (pH = 7.4). The ternary systems were analysed along a complete set of polymer molar charge fraction Ψ values (0 ≤ Ψ ≤ 1) with static SAXS and DLS, which served as an internal validation. As DLS data at low Ψ-values were skewed by clustering effects, the SAXS data and fits proved to be the more precise sizing technique.67 To quantify the findings, the SAXS data were fitted with the fuzzy-surface C3M model. It was found that systems containing PEO45-b-PMAA41 (T1–T4) showed the most promising characteristics for size-tunability with low polydispersity and high colloidal stability. The ternary systems T5–T8 suffer from a relatively high PDI and offer smaller size ranges (Fig. S1, ESI†). For all ternary systems, the tendency to observe clustering in SAXS was found to be significantly lower than for binary systems. This decrease in clustering results in improved colloidal stability for ternary systems. Moreover, based on visual inspection and DLS measurements over long time scales, the ternary systems showed long-lasting stability (several months). In Fig. 3, a selection of SAXS patterns of T1, T2, T3, and T4 C3Ms are depicted with fits (Fig. 3A–D). From the fits, the Rtot parameter (the sum of Rin and σin) is reported in this work, as the σin parameter itself did not show trends throughout different ternary systems and polymer molar charge fractions. Moreover, the total radius (Rtot) is shown along the complete polymer molar charge fraction set 0 ≤ Ψ ≤ 1 (Fig. 3E).
Table 2 Ternary complex coacervate systems investigated consisting of two different (partly) anionic polymers mixed with the cationic colistin to form complex coacervate structures
| #Ternary system |
Anionic component 1 |
Anionic component 2 |
Cationic component |
| T1 |
PEO45-b-PMAA41 |
PMAA46 |
Colistin |
| T2 |
PEO45-b-PMAA15 |
| T3 |
PEO45-b-PMAA81 |
| T4 |
PEO114-b-PMAA81 |
| T5 |
PMAA46 |
PEO45-b-PMAA81 |
| T6 |
PEO114-b-PMAA81 |
| T7 |
PEO45-b-PMAA15 |
PEO45-b-PMAA81 |
| T8 |
PEO114-b-PMAA81 |
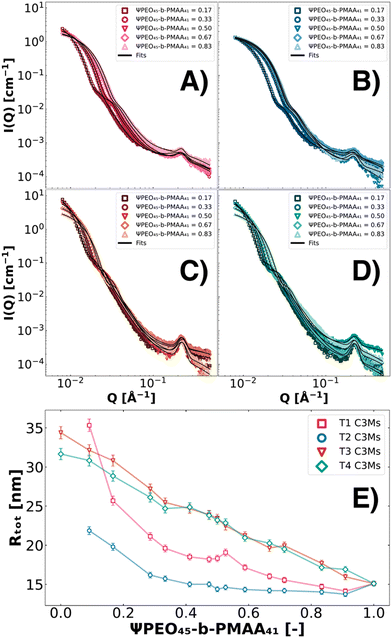 |
| | Fig. 3 SAXS patterns of colistin–C3Ms formed from four different ternary coacervate formulations at 0.125 wt%. C3Ms were prepared from a combination of colistin, PEO45-b-PMAA41 and second polymers PMAA46 (T1, pink symbols, A), PEO45-b-PMAA15 (T2, blue symbols, B), PEO45-b-PMAA81 (T3, red symbols, C), and PEO114-b-PMAA81 (T4, green symbols, D). The complete polymer molar charge fraction set 0 ≤ Ψ ≤ 1 was investigated and fitted with the fuzzy-surface C3M model, while for visibility reasons, only a couple of SAXS patterns are shown. The fit parameter Rtot is shown for T1, T2, T3, and T4 along the complete polymer molar charge fraction set (E). | |
We show the dependence of the fraction of other molecular weight polymers on the sizes of T1–T4 systems (Fig. 3). Inspecting the SAXS data, we observe two different types of size behaviours. In T1 and T2 systems, the maximum intensity is only subtly impacted by the addition of relatively more PEO45-b-PMAA41 whereas T3 and T4 systems show a significant gradual decrease of intensity at low Q values with increasing ΨPEO45-b-PMAA41, indicating a significant decrease in aggregation numbers. All systems show however substantial changes at intermediate Q values, indicating a change in size, which is summarised in Fig. 3E showing the total radius (Rtot) as a function of composition. With the relative increase in PEO45-b-PMAA41, the size of the C3Ms decreases for all systems. For T1 and T2, this size decrease follows an exponential drop, whereas for T3 and T4, the drop in size appears linear.
Neither the homopolymer (PMAA46) nor PEO15-b-PMAA45 can form micelles in binary colistin complex coacervate systems (Table 1). This leads us to hypothesise that their non-linear size behaviour (T1–T2) and their maximum intensity may be attributed to swelling effects, potentially driven by increased instability of the micellar core at low ΨPEO45-b-PMAA41. To investigate size dependency more closely, we calculated the average PMAA block length across the polymer molar charge fraction set for T1–T4 and compared it to existing scaling laws for anionic block sizes with size (Rtot) and aggregation numbers (P) obtained from the SAXS fit analysis (Fig. 4).
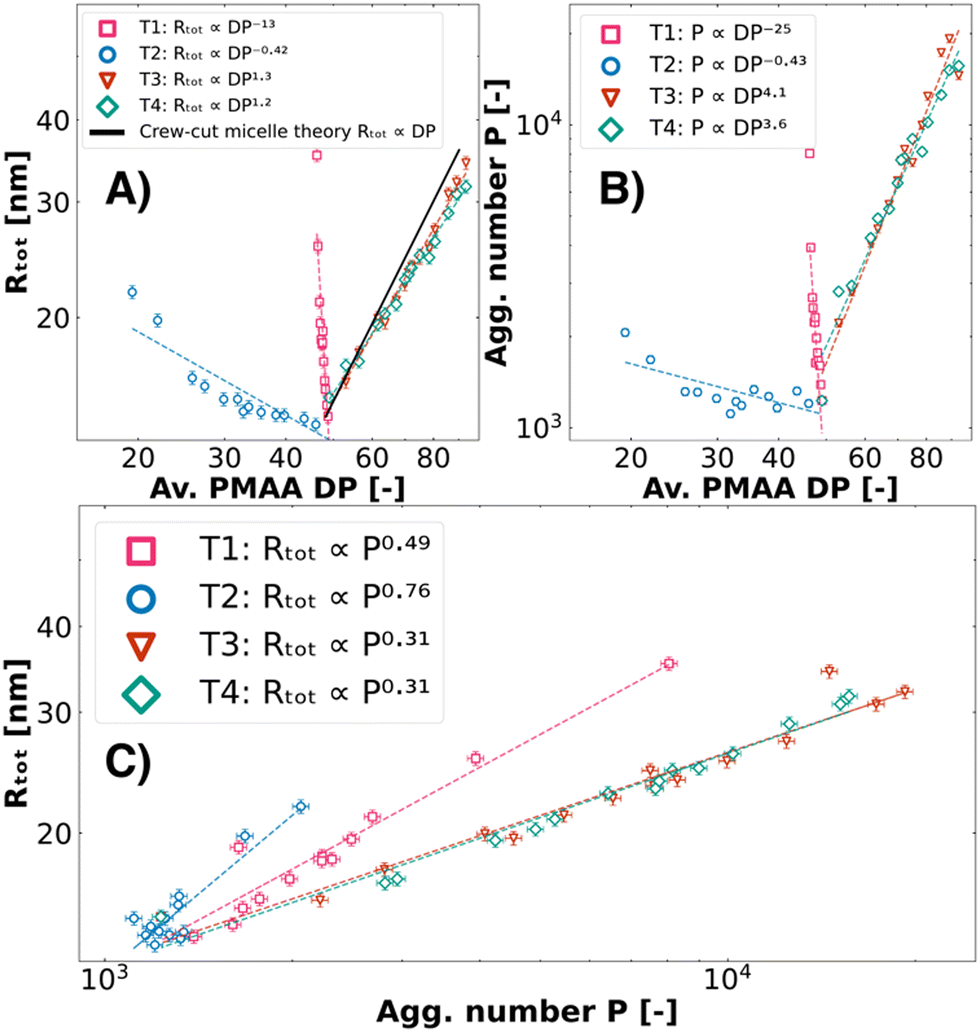
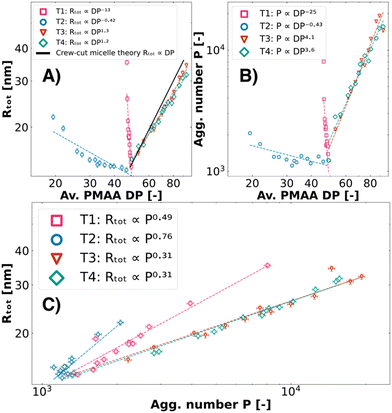 |
| | Fig. 4 Scaling law behaviour of T1, T2, T3, and T4 ternary colistin complex coacervate systems. From the fuzzy-surface C3M model fits, the total radius (Rtot) (A) and aggregation number P (B) were plotted against the average degree of polymerisation DP of the PMAA blocks. The theoretical scaling law of Rtot ∝ Px, x = 1.0 for crew-cut micelles was added for illustration purposes. In (C), the Rtot was plotted against P. C3Ms were prepared from a combination of colistin, PEO45-b-PMAA41 and second polymers PMAA46 (T1, pink squares), PEO45-b-PMAA15 (T2, blue circles), PEO45-b-PMAA81 (T3, red triangles), and PEO114-b-PMAA81 (T4, rhombuses). | |
T1 and T2 show negative scaling laws between the total radius (Rtot) and the aggregation number (P) with the average block length (DP) of PMAA. These negative scaling laws can be attributed to being artefacts, as the polymer combinations do not behave as an average effective block length but rather cause swelling, confirmed by the atypical Rtot ∝ Px scaling (Fig. 4C). In contrast, the Rtot of T3 and T4 scales with the block length to the power x = 1.3 and 1.2, respectively (Fig. 4A). This is similar to previously published work on so-called “crew-cut” micelles which are dominated by the free energy contributions of the core, where Rg ∝ DP1.0 from different binary coacervate micellar systems was found.18,59,68,69 Unlike the charged block, the neutral block length is known to not significantly impact the size of crew-cut micelles.59,69 This is in line with our findings, as we observe no significant differences between T3 and T4 in all scaling laws (Fig. 4). We therefore conclude that these two ternary complex coacervate systems form micelles with a size that corresponds to their average PMAA block length.
Swelling of C3M systems caused by mismatches in the polymer size
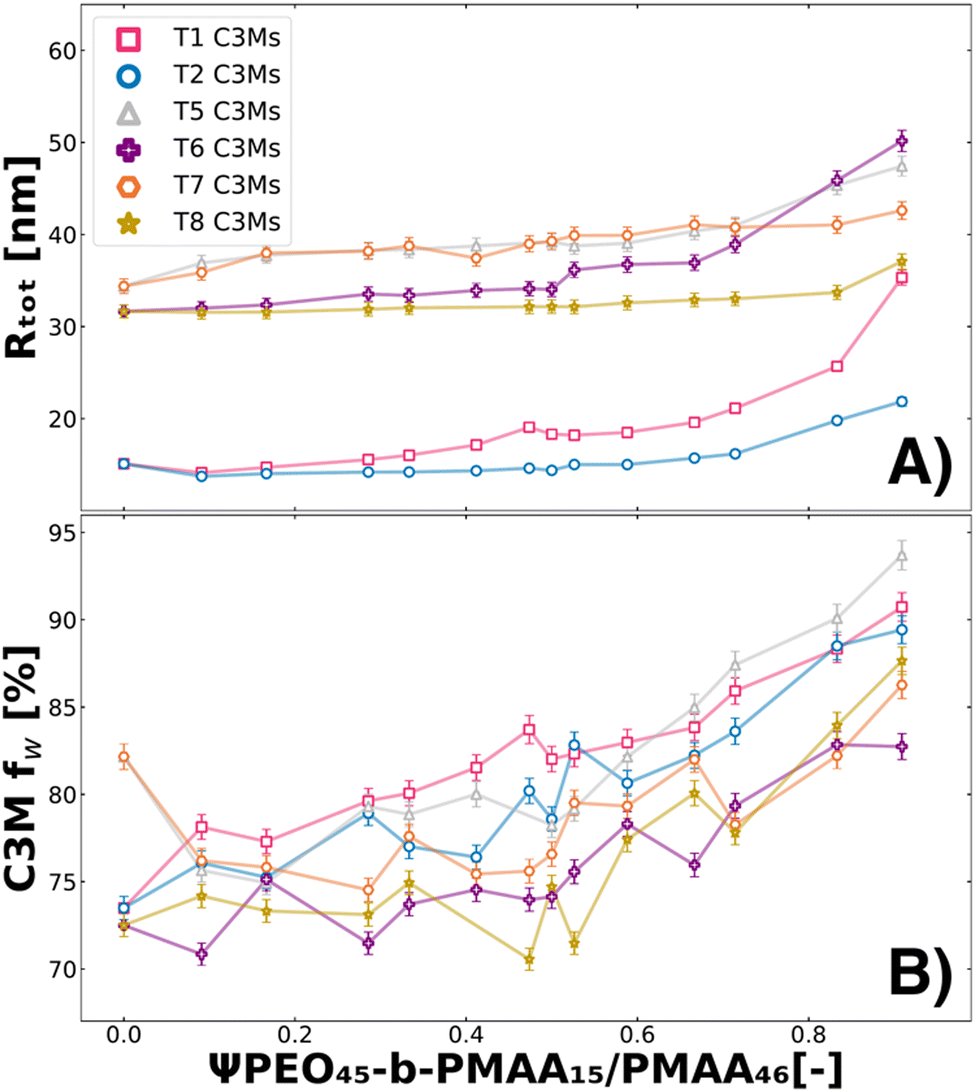
Since the dynamics causing the swelling in T1 and T2 remain unclear, we also investigated T5, T6, T7, and T8 systems. All these systems have in common that they contain components that do not form micelles by themselves (PMAA46 or PEO15-b-PMAA45) (Table 1). We analysed these systems with SAXS and fitted the data with the fuzzy-surface complex coacervate model (Fig. S1, ESI†). From the fits, we extracted the water content in the micelles (fw) to observe the swelling behaviour next to other fit parameters such as Mw, P, σin, PDI and Rtot. In Fig. 5, we show the Rtot (A) and fw (B) behaviour over the polymer molar charge fraction of ΨPEO45-b-PMAA15/PMAA46 for T1, T2, T5, T6, T7 and T8.
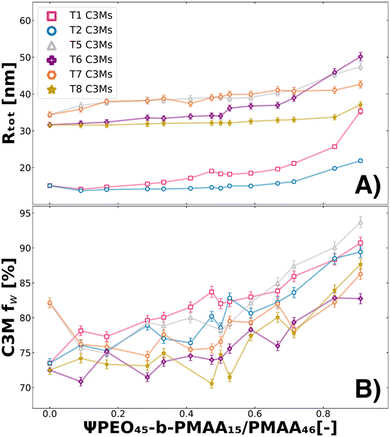 |
| | Fig. 5 Swelling of ternary colistin complex coacervate systems caused by the addition of PEO45-b-PMAA15 and PMAA46 for the complete polymer molar charge fraction set (0 ≤ Ψ ≤ 1). From the fuzzy-surface C3M model fits, the total radius (Rtot) (A) and the C3M water content (fw) (B) were plotted against the polymer molar charge fraction of either PMAA46 (T1, pink squares; T5, grey triangles pointing up; T6, purple plus signs) or PEO45-b-PMAA15 (for T2, blue circles; T7, orange hexagons; T8, yellow stars) polymers. | |
From the Rtot, we see similar behaviours for T5–T8 as observed for T1–T2, as the size increases with the addition of PEO45-b-PMAA15/PMAA46 (Fig. 5A). However, it must be noted that for T5–T8 systems, we are close to the boundaries of the experimental resolution of SAXS analysis, and therefore the fit parameter values should be interpreted with care. The fw fit parameter shows the swelling to be causing this size increase, as increases of 10–20% pt% of water can be observed (fw of T3 and T4 in Fig. S2, ESI†) when ΨPEO45-b-PMAA15/PMAA46 is increased. The addition of PMAA46 (T1, T5, and T6) causes the largest swelling, likely caused by the increased mismatch in the packing of the coacervate core from the absence of a PEO block. The addition of PEO45-b-PMAA15 (T2, T7, T8) leads to relatively little swelling behaviour until higher Ψ values. This is most likely due to the better compatibility of the small block copolymer with the other block copolymers than the homopolymer. While the swelling indicates reduced stability, and therefore could potentially lead to quicker in vivo release of colistin, as electrostatic interactions are weakened, it does not necessarily lead to micelle destabilisation.70
Polydispersity effects of ternary coacervate systems
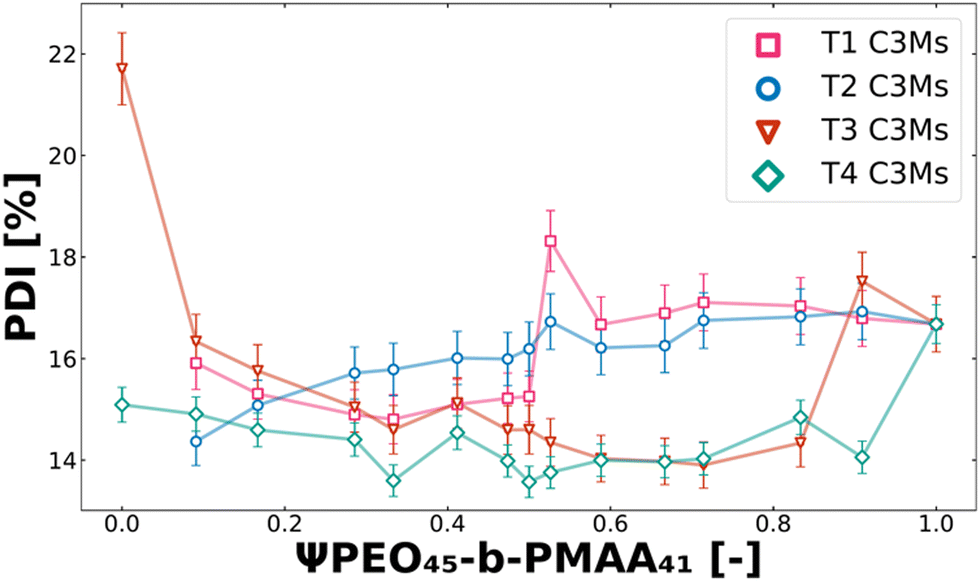
Another crucial factor in the design of drug delivery vehicles, in addition to size control, is their size distribution (polydispersity).67,71,72 It is known that drug formulations with good size properties, particularly those exhibiting a narrow size distribution (as indicated by a low polydispersity index, PDI), are less prone to in vivo blood clearance and degradation. This allows a larger fraction of the formulation to remain effective in therapeutic applications.73–75 The polydispersity also plays a crucial role in solution for the colloidal stability, where systems with higher PDI are more prone to Ostwald ripening and other slow equilibration processes.62,76 Therefore, when optimising these systems, it is desired to assess the effect of polymer mixtures on the polydispersity (PDI). In Fig. 6, the PDI is shown for T1, T2, T3 and T4 systems along the set of polymer molar charge fractions of PEO45-b-PMAA41. The PDI behaviour of T5–T8 can be found in the ESI,† in Fig. S3.
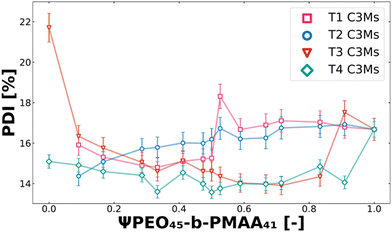 |
| | Fig. 6 Polydispersity index (PDI) behaviour of T1 (pink squares), T2 (blue circles), T3 (red triangles), and T4 (green rhombuses) ternary colistin complex coacervate systems for the complete polymer molar charge fraction set (0 ≤ Ψ ≤ 1). From the fuzzy-surface C3M model fits, the PDI values were plotted against the polymer molar charge fraction of PEO45-b-PMAA41. | |
Different minima and maxima could be observed for each ternary complex coacervate system (Fig. 6 and Fig. S3, ESI†). This means that for optimisation, differently sized complex coacervates can be obtained with a narrow size distribution. Apart from increasing polydispersity caused by swelling for T1 and T2, there are no clear trends in PDI for T3 and T4 systems (Fig. 6). For T5–T8 systems, the swelling does not directly increase or decrease the PDI; thus, only minor changes in PDI could be observed (Fig. S3, ESI†). However, the addition of PEO45-b-PMAA41 to PEO45-b-PMAA81, even at low fractions, results in a significant decrease in the polydispersity (T3). The polydispersities of T3 and T4 are relatively low compared to their binary counterparts, demonstrating not only the ability of ternary systems to achieve precise size control but also to result in a narrower size distribution.
Kinetics of formation of ternary C3M mixtures
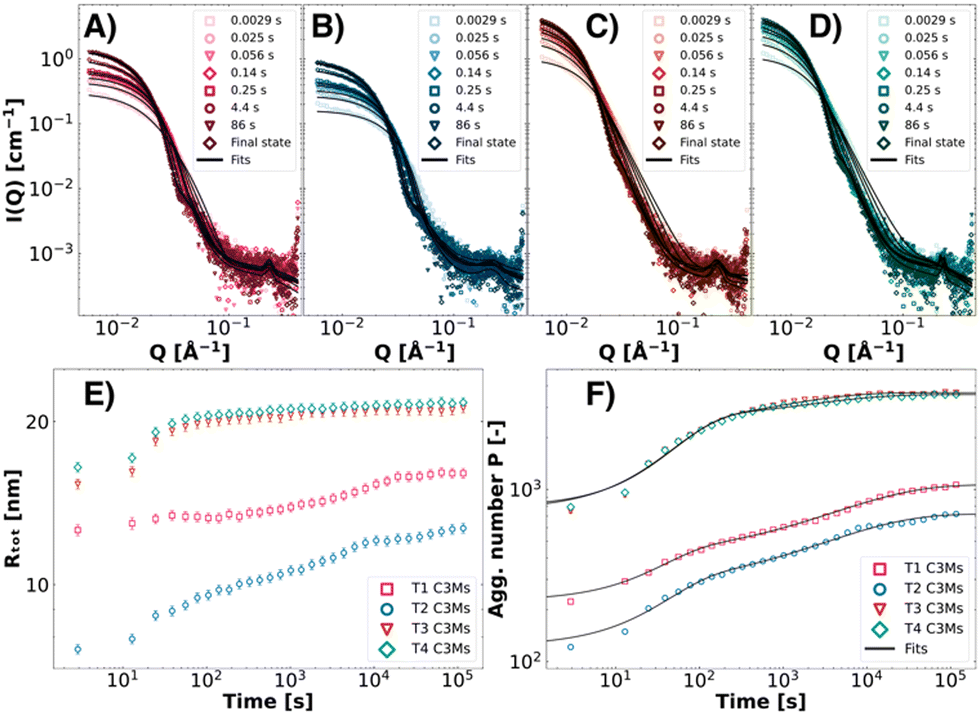
As colistin–C3Ms are kinetically controlled systems,32,65 the preparation method dictates the final size distribution. Therefore, careful consideration must be given to the formation kinetics of these systems. In a previous publication, we investigated the binary PEO-b-PMAA–colistin–C3M systems and found that the formation kinetics consisted of three stages: nucleation, fusion and exchange.32 As micelles with the same morphology are formed in ternary C3M systems, we expect that the same kinetic pathway is followed. However, since in these systems, two different polymers with different block lengths are now combined, the kinetics are expected to proceed differently. Therefore, we investigated the formation kinetics of T1–T4 with time-resolved SAXS (TR-SAXS) with millisecond resolution at equal polymer charges between PEO45-b-PMAA41 and the other polymer (ΨPEO45-b-PMAA41 = 0.50). The SAXS data, spanning from 2.9 ms (the dead time of mixing) to approximately 2 minutes (Fig. 7A–D), were analysed with the fuzzy-surface complex coacervate model, from which the Rtot and P over time were extracted (Fig. 7E and F, power law behaviour in Fig. S4, ESI†). The aggregation number P was then fitted with a double-stretched exponential kinetic model (Fig. 7F), comprising nucleation (0), fusion (1) and a chain insertion/reinsertion exchange stage (2) (eqn (4) and (5)). The fit parameters, including the amplitude weight factor α of the fusion and chain insertion processes and the relaxation times τ1 and τ2, are presented in Table 3. As the nucleation process is not resolved within the experimental time-window of TR-SAXS, >2.9 ms, τ0 was fixed to the arbitrary value of τ0 = 2.0 μs. The stretched exponentials were found to be β1 = 1.0 for the fusion process and β2 = 0.5 for the chain insertion exchange process.
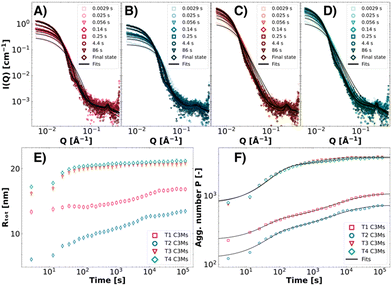 |
| | Fig. 7 Time-resolved SAXS data of ΨPEO45-b-PMAA41 = 0.50 T1 (pink symbols, A), T2 (blue symbols, B), T3 (red symbols, C), and T4 (green symbols, D) ternary complex coacervate systems. From the fuzzy-surface C3M model fits, the Rtot (E) and P (F) values were plotted over time to observe the growth of these ternary C3M systems. The P was modelled with the double-stretched kinetic model to obtain kinetic modelling parameters (eqn (4) and (5)). | |
Table 3 Kinetic fit parameters from modelling of the formation kinetics (eqn (4) and (5)) for T1, T2, T3 and T4 ternary complex coacervate systems
| Ternary system |
α (—) |
〈τ1〉 (ms) |
〈τ2〉 (s) |
P
0 × 102 (—) |
P
∞ × 102 (—) |
| T1 |
0.22 ± 0.01 |
47 ± 6 |
16 ± 1 |
2.2 ± 0.1 |
10.6 ± 0.3 |
| T2 |
0.24 ± 0.01 |
59 ± 7 |
13 ± 1 |
1.2 ± 0.1 |
7.2 ± 0.2 |
| T3 |
0.51 ± 0.02 |
75 ± 9 |
2.3 ± 0.4 |
7.5 ± 0.2 |
36 ± 1 |
| T4 |
0.62 ± 0.02 |
79 ± 9 |
5.2 ± 0.7 |
7.9 ± 0.2 |
35 ± 1 |
Similar to the previously published kinetic analysis of colistin–PEO-b-PMAA–C3Ms, the data show that nucleation could not be experimentally resolved.32 As shown by the SAXS patterns, micellar structures already emerge within the dead time of mixing (t = 2.9 ms). Moreover, features like a Guinier-like region can be observed for all investigated systems at low Q, as well as a steep decay at intermediate Q and an internal structure factor at high Q (Fig. 7A–D). The complex coacervates then grow within 2 minutes to their final states. The smallest complexes are formed from the T2 mixture, where C3Ms of Rtot,final = 12.7 ± 0.3 nm are formed (Fig. 7E). All C3Ms are slightly smaller but show the same trends in terms of size as the statically mixed T1–T4 C3Ms at Ψ = 0.50, an effect induced by rapid mixing in the stopped-flow setup in kinetic measurements.32,64 From the modelling of the aggregation number over time (Fig. 7F and Table 3), we see considerable differences in the kinetics depending on the system. For T1 and T2 systems, the micellar growth is majorly dominated by slower chain exchange processes, whereas for T3 and T4 ternary coacervate systems, the quicker fusion step plays a more dominant role. This distinction can likely be attributed to the higher molecular weights of the polymers in T3 and T4, facilitating less possibility for chain exchange (higher α), thereby contributing to the overall faster micellisation kinetics observed in these systems. The effect of the length of the PEO block can be directly observed by comparing T3 (DPPEO = 45) and T4 (DPPEO = 114). Even though the kinetics are even more fusion-dominated for T4, the chain insertion exchange kinetics are more than twice as slow, potentially due to the increase in steric repulsion from the PEO block, slowing down chain insertion. Since the polymer size has been shown to directly influence kinetic pathways, these findings not only reinforce the previously reported kinetic mechanisms of PEO-b-PMAA–colistin complex coacervate micelles but also improve the ability to understand and control the formation kinetics.
Conclusions
In this work, we demonstrate that ternary colistin complex coacervate micelle systems offer good control over size by mixing PEO-b-PMAA polymers of varying sizes with colistin. Using SAXS analysis with a customised fuzzy-surface complex coacervate model, we could determine the micelle composition and extract their sizes and size distribution. Stable ternary colistin-C3Ms could be formed with sizes ranging from 26 to 100 nm, depending on the composition. Next to size control, these ternary systems were found to have a narrower size distribution compared to their binary equivalents. We attributed the observed size changes to two distinct mechanisms: swelling and effective polymer miscibility, both resulting in highly stable C3M formulations. Swelling occurs due to mismatches between the anionic polymer blocks, reducing packing efficiency in the micelle core. In contrast, the mixing of larger polymers leads to an increase in micelle sizes by extending the effective length of the PMAA block. We found that ternary complex coacervate systems exhibit formation kinetics similar to those we have previously identified in binary PEO-b-PMAA-colistin systems, involving the kinetic processes of nucleation, fusion, and chain insertion. However, in ternary systems, the addition of larger polymers not only forms bigger micelles but also alters the kinetics toward being more fusion-driven, as the exchange of chains is more constrained by larger activation barriers for release and overall slower dynamics. At large, ternary complex coacervation offers good control of size, with a narrower distribution. This offers a handle to tune the size at narrower size distributions, which could be advantageous for the development of C3M-based drug delivery systems for therapeutic purposes.
Author contributions
T. D. Vogelaar: conceptualisation, methodology, investigation, software, analysis, and writing – original draft. H. Torjusen: conceptualisation, methodology, investigation, analysis, and writing – review and editing. R. Lund: conceptualisation, methodology, supervision, software, writing – review and editing, and funding acquisition.
Data availability
SAXS data for this article are available at ESRF at https://data.esrf.fr/doi/10.15151/ESRF-ES-1580560228.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the funding from the Norwegian Research Council (Project No. 315666). The static SAXS data collected at the BioSAXS beamline at BM29 at the European Synchrotron Radiation Facility (ESRF) (Grenoble, France) were collected with the assistance of M. Tully, P. Pernot, A. Calio, and S. Hutin. The authors thank the complete crew of the TRUSAXS beamline ID02 at the ESRF where the TR-SAXS data were obtained with the supervision of Theyencheri Nararyanan (proposal SC-5459). We are in general indebted to the ESRF for beam time through the Norwegian BAG proposal and providing the sample preparation infrastructure through the PSCM lab. Lastly, we highly acknowledge the use of the Norwegian Centre for X-ray Diffraction, Scattering and Imaging (RECX) for access to SAXS, which is supported by the Norwegian Research Council (NRC).
Notes and references
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. van der Meer, N. Van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef
 .
.
- I. N. Okeke, M. E. A. de Kraker, T. P. V. Boeckel, C. K. Kumar, H. Schmitt, A. C. Gales, S. Bertagnolio, M. Sharland and R. Laxminarayan, The Lancet, 2024, 403, 2426–2438 CrossRef CAS .
- C. J. Slingerland and N. I. Martin, ACS Infect. Dis., 2024, 10, 1056–1079 CrossRef CAS .
- J. Xuan, W. Feng, J. Wang, R. Wang, B. Zhang, L. Bo, Z.-S. Chen, H. Yang and L. Sun, Drug Resist. Updates, 2023, 68, 100954 CrossRef CAS .
- T. M. Uddin, A. J. Chakraborty, A. Khusro, B. R. M. Zidan, S. Mitra, T. B. Emran, K. Dhama, Md. K. H. Ripon, M. Gajdács, M. U. K. Sahibzada, Md. J. Hossain and N. Koirala, J. Infection Public Health, 2021, 14, 1750–1766 CrossRef PubMed .
- X. Luo, H. Chen, Y. Song, Z. Qin, L. Xu, N. He, Y. Tan and W. Dessie, Eur. J. Pharm. Sci., 2023, 181, 106363 CrossRef CAS PubMed .
- A. Moretta, C. Scieuzo, A. M. Petrone, R. Salvia, M. D. Manniello, A. Franco, D. Lucchetti, A. Vassallo, H. Vogel, A. Sgambato and P. Falabella, Front. Cell. Infect. Microbiol., 2021, 11, 668632 CrossRef CAS PubMed .
- D. I. Duarte-Mata and M. C. Salinas-Carmona, Front. Immunol., 2023, 14, 1119574 CrossRef CAS .
- M. E. Falagas, S. K. Kasiakou and L. D. Saravolatz, Clin. Infect. Dis., 2005, 40, 1333–1341 CrossRef CAS PubMed .
- F. N. Ballı, P. B. Ekinci, M. Kurtaran, E. Kara, G. T. Dizman, M. Ç. Sönmezer, M. Hayran, K. Demirkan and G. Metan, Int. J. Antimicrob. Agents, 2024, 63, 107035 CrossRef PubMed .
- A. C. Dijkmans, E. B. Wilms, I. M. C. Kamerling, W. Birkhoff, N. V. Ortiz-Zacarías, C. van Nieuwkoop, H. A. Verbrugh and D. J. Touw, Ther. Drug Monit., 2015, 37, 419 CrossRef CAS .
- A. H. Mondal, K. Khare, P. Saxena, P. Debnath, K. Mukhopadhyay and D. Yadav, Microorganisms, 2024, 12, 772 CrossRef CAS .
- M. A. E.-G. El-Sayed Ahmed, L.-L. Zhong, C. Shen, Y. Yang, Y. Doi and G.-B. Tian, Emerging Microbes Infect., 2020, 9, 868–885 CrossRef CAS .
- Y.-H. Liu, S.-C. Kuo, B.-Y. Yao, Z.-S. Fang, Y.-T. Lee, Y.-C. Chang, T.-L. Chen and C.-M. J. Hu, Acta Biomater., 2018, 82, 133–142 CrossRef CAS PubMed .
- T. D. Vogelaar, A. E. Agger, J. E. Reseland, D. Linke, H. Jenssen and R. Lund, Biomacromolecules, 2024, 25, 4267–4280 CrossRef CAS .
- I. K. Voets, A. de Keizer and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2009, 147–148, 300–318 CrossRef CAS .
- A. Kakkar, G. Traverso, O. C. Farokhzad, R. Weissleder and R. Langer, Nat. Rev. Chem., 2017, 1, 1–17 CrossRef .
- A. E. Marras, J. M. Ting, K. C. Stevens and M. V. Tirrell, J. Phys. Chem. B, 2021, 125, 7076–7089 CrossRef CAS .
- J. R. Magana, C. C. M. Sproncken and I. K. Voets, Polymers, 2020, 12, 1953 CrossRef CAS .
- C. Zheng, L. Niu, W. Pan, J. Zhou, H. Lv, J. Cheng and D. Liang, Polymer, 2014, 55, 2464–2471 CrossRef CAS .
- I. Bos, E. Brink, L. Michels and J. Sprakel, Soft Matter, 2022, 18, 2012–2027 RSC .
- A. C. Obermeyer, C. E. Mills, X.-H. Dong, R. J. Flores and B. D. Olsen, Soft Matter, 2016, 12, 3570–3581 RSC .
- C. G. de Kruif, F. Weinbreck and R. de Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340–349 CrossRef CAS .
- M. Abbas, W. P. Lipiński, J. Wang and E. Spruijt, Chem. Soc. Rev., 2021, 50, 3690–3705 RSC .
- J. Kalkowski, C. Liu, P. Leon-Plata, M. Szymusiak, P. Zhang, T. Irving, W. Shang, O. Bilsel and Y. Liu, Macromolecules, 2019, 52, 3151–3157 CrossRef CAS .
- H. Wu, J. M. Ting, B. Yu, N. E. Jackson, S. Meng, J. J. de Pablo and M. V. Tirrell, ACS Macro Lett., 2020, 9, 1674–1680 CrossRef CAS .
- J. Zhang, S. Chen, Z. Zhu and S. Liu, Phys. Chem. Chem. Phys., 2014, 16, 117–127 RSC .
- M. Amann, J. S. Diget, J. Lyngsø, J. S. Pedersen, T. Narayanan and R. Lund, Macromolecules, 2019, 52, 8227–8237 CrossRef CAS .
- H. Wu, J. M. Ting and M. V. Tirrell, Macromolecules, 2020, 53, 102–111 CrossRef CAS .
- X. Liu, M. Haddou, I. Grillo, Z. Mana, J.-P. Chapel and C. Schatz, Soft Matter, 2016, 12, 9030–9038 RSC .
- R. Takahashi, T. Narayanan and T. Sato, J. Phys. Chem. Lett., 2017, 8, 737–741 CrossRef CAS PubMed .
- T. D. Vogelaar, H. Torjusen, N. Theyencheri and R. Lund, Macromolecules, 2024 DOI:10.1021/acs.macromol.4c02077 .
- I. Bos and J. Sprakel, Macromolecules, 2019, 52, 8923–8931 CrossRef CAS PubMed .
- T.-Y. Heo, S. Kim, L. Chen, A. Sokolova, S. Lee and S.-H. Choi, ACS Macro Lett., 2021, 10, 1138–1144 CrossRef CAS PubMed .
- I. Bos, M. Timmerman and J. Sprakel, Macromolecules, 2021, 54, 398–411 CrossRef CAS PubMed .
- G. Debais, L. L. Missoni, Y. A. P. Sirkin and M. Tagliazucchi, Soft Matter, 2023, 19, 7602–7612 RSC .
- T. D. Vogelaar, S. M. Szostak and R. Lund, Mol. Pharmaceutics, 2024, 21, 4157–4168 CrossRef CAS PubMed .
- C. Facciotti, V. Saggiomo, A. Bunschoten, R. Fokkink, J. B. ten Hove, J. Wang and A. H. Velders, Soft Matter, 2018, 14, 9542–9549 RSC .
- S. Shah and L. Leon, Curr. Opin. Colloid Interface Sci., 2021, 53, 101424 CrossRef CAS .
- H. M. van der Kooij, E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. van der Gucht, Langmuir, 2012, 28, 14180–14191 CrossRef CAS PubMed .
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS .
- W. C. Blocher and S. L. Perry, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1442 Search PubMed .
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed .
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS .
- D. L. Jasinski, H. Li and P. Guo, Mol. Ther., 2018, 26, 784–792 CrossRef CAS PubMed .
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed .
- D. Priftis, X. Xia, K. O. Margossian, S. L. Perry, L. Leon, J. Qin, J. J. de Pablo and M. Tirrell, Macromolecules, 2014, 47, 3076–3085 CrossRef CAS .
- M. Zhao, X. Xia, J. Mao, C. Wang, M. B. Dawadi, D. A. Modarelli and N. S. Zacharia, Mol. Syst. Des. Eng., 2019, 4, 110–121 RSC .
- R. Kembaren, R. Fokkink, A. H. Westphal, M. Kamperman, J. M. Kleijn and J. W. Borst, Langmuir, 2020, 36, 8494–8502 CrossRef CAS .
- M. D. Tully, J. Kieffer, M. E. Brennich, R. Cohen Aberdam, J. B. Florial, S. Hutin, M. Oscarsson, A. Beteva, A. Popov, D. Moussaoui, P. Theveneau, G. Papp, J. Gigmes, F. Cipriani, A. McCarthy, C. Zubieta, C. Mueller-Dieckmann, G. Leonard and P. Pernot, J. Synchrotron Radiat., 2023, 30, 258–266 CrossRef PubMed .
- S. Hutin, M. D. Tully and M. Brennich, Adv. Exp. Med. Biol., 2024, 3234, 163–172 CrossRef PubMed .
- L. Matthews and T. Narayanan, Colloid Polym. Sci., 2023, 301, 721–728 CrossRef CAS .
- I. Berndt, J. S. Pedersen, P. Lindner and W. Richtering, Langmuir, 2006, 22, 459–468 CrossRef CAS PubMed .
- I. Berndt, J. S. Pedersen and W. Richtering, Angew. Chem., 2006, 118, 1769–1773 CrossRef .
- P. Debye, J. Phys. Chem., 1947, 51, 18–32 CrossRef CAS PubMed .
- A. Nolles, E. Hooiveld, A. H. Westphal, W. J. H. van Berkel, J. M. Kleijn and J. W. Borst, Langmuir, 2018, 34, 12083–12092 CrossRef CAS PubMed .
- A. E. Neitzel, Y. N. Fang, B. Yu, A. M. Rumyantsev, J. J. de Pablo and M. V. Tirrell, Macromolecules, 2021, 54, 6878–6890 CrossRef CAS .
- D. J. Adams, S. H. Rogers and P. Schuetz, J. Colloid Interface Sci., 2008, 322, 448–456 CrossRef CAS PubMed .
- A. E. Marras, T. R. Campagna, J. R. Vieregg and M. V. Tirrell, Macromolecules, 2021, 54, 6585–6594 CrossRef CAS .
- B. Hofs, I. K. Voets, A. de Keizer and M. A. C. Stuart, Phys. Chem. Chem. Phys., 2006, 8, 4242–4251 RSC .
- I. K. Voets, P. M. Moll, A. Aqil, C. Jérôme, C. Detrembleur, P. de Waard, A. de Keizer and M. A. C. Stuart, J. Phys. Chem. B, 2008, 112, 10833–10840 CrossRef CAS .
- S. van der Burgh, A. de Keizer and M. A. Cohen Stuart, Langmuir, 2004, 20, 1073–1084 CrossRef CAS .
- A. B. Kayitmazer, Adv. Colloid Interface Sci., 2017, 239, 169–177 CrossRef CAS PubMed .
- W. C. B. McTigue, E. Voke, L.-W. Chang and S. L. Perry, Phys. Chem. Chem. Phys., 2020, 22, 20643–20657 RSC .
- M. Lueckheide, J. R. Vieregg, A. J. Bologna, L. Leon and M. V. Tirrell, Nano Lett., 2018, 18, 7111–7117 CrossRef CAS .
- Y. N. Fang, A. M. Rumyantsev, A. E. Neitzel, H. Liang, W. T. Heller, P. F. Nealey, M. V. Tirrell and J. J. de Pablo, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2302151120 CrossRef CAS .
- S. K. Filippov, R. Khusnutdinov, A. Murmiliuk, W. Inam, L. Y. Zakharova, H. Zhang and V. V. Khutoryanskiy, Mater. Horiz., 2023, 10, 5354–5370 RSC .
- E. Y. Kramarenko, A. R. Khokhlov and P. Reineker, J. Chem. Phys., 2006, 125, 194902 CrossRef .
- E. E. Dormidontova, Macromolecules, 1999, 32, 7630–7644 CrossRef CAS .
- C. E. Sing, Adv. Colloid Interface Sci., 2017, 239, 2–16 CrossRef CAS PubMed .
- R. De, M. K. Mahata and K.-T. Kim, Adv. Sci., 2022, 9, 2105373 CrossRef CAS .
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed .
- M. Makowski, Í. C. Silva, C. Pais do Amaral, S. Gonçalves and N. C. Santos, Pharmaceutics, 2019, 11, 588 CrossRef CAS PubMed .
- C. J. Rijcken, C. J. Snel, R. M. Schiffelers, C. F. van Nostrum and W. E. Hennink, Biomaterials, 2007, 28, 5581–5593 CrossRef CAS PubMed .
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6, 100–116 CrossRef CAS PubMed .
- E. Kizilay, A. B. Kayitmazer and P. L. Dubin, Adv. Colloid Interface Sci., 2011, 167, 24–37 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Henrik
Torjusen
a,
Henrik
Torjusen