
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetically induced drug release from niosome-based nanocarriers loaded with doxorubicin†
Philip Drake *a,
Ilma Amalinab,
Retno Saric,
Amalia Ruizd,
Saliha Ramazana,
Gordon Hopea,
Dharmisthaben Pancholia and
Andang Miatmokoc
*a,
Ilma Amalinab,
Retno Saric,
Amalia Ruizd,
Saliha Ramazana,
Gordon Hopea,
Dharmisthaben Pancholia and
Andang Miatmokoc
aSchool of Chemistry and Biosciences, Faculty of Life Sciences, University of Bradford, Bradford, Richmond Rd BD7 1DP, UK. E-mail: pdrake@bradford.ac.uk
bNanotechnology Engineering, Faculty of Advanced Technology and Multidiscipline, Universitas Airlangga, Surabaya 60115, Indonesia
cDepartment of Pharmaceutical Sciences, Faculty of Pharmacy, Universitas Airlangga, Surabaya 60115, Indonesia
dInstitute of Cancer Therapeutics, University of Bradford, Bradford, Richmond Rd BD7 1DP, UK
First published on 1st July 2025
Abstract
Niosomes co-loaded with doxorubicin and magnetic nanoparticles were synthesised using the thin film hydration method. The loading efficiency of the doxorubicin was between 60–70%. The hydrodynamic diameter measured as the average number (mean ± standard deviation), using dynamic light scattering, was found to be 188 ± 68 nm, 141 ± 86 nm and 169 ± 69 nm for the plain niosomes, niosomes loaded with doxorubicin and niosomes loaded with doxorubicin and magnetic nanoparticles, respectively. The zeta potential for all three niosome samples was determined to be −26.4 mV ± 1.9 mV. The thermally mediated release of doxorubicin was monitored using fluorescence spectroscopy and found to follow 1st order kinetics. The rate constant for the thermal release was 1.2 × 10−6, 1.0 × 10−4 and 5.1 × 10−4 min−1 at 298, 313 and 333 K, respectively. The doxorubicin was also released using an alternating magnetic field, this also followed 1st order kinetics and had a rate constant of 1.7 × 10−2 min−1. This is four orders of magnitude greater than the thermal release at the same temperature (298 K). The work shows the magnetically controlled, burst release from a drug-loaded niosome delivery system. The release was triggered on demand by the application of the alternating magnetic field, resulting in 86% doxorubicin release within 3 hours compared to 3% release in 30 days via thermal release.
Introduction
Niosomes are spherical bilayer structures made from non-ionic surfactants.1 They have a hydrophilic core and a hydrophobic region within the bilayer and can be used as drug delivery systems for hydrophilic or hydrophobic drugs.2 An illustration of the niosome structure can be seen in Fig. 1. Nanocarriers such as liposomes,3 niosomes4 and polymeric particles5 have many advantages as drug delivery agents.6,7 Their small size, between 10–200 nm, enhances tissue penetration and cellular uptake.8 They have a large surface-to-volume ratio, and they can be modified with a variety of ligands to target specific cell types and to modulate their uptake.9,10 There are several nanocarrier-based drug delivery agents currently on the market,11 with the majority focusing on cancer therapeutics.12 This is an important area as the nanocarrier drug delivery system can help to reduce side effects and improve tumour targeting. They can target tumour sites through several mechanisms, including passive and active targeting. Passive targeting involves enhanced permeability and retention,13 leading to the increased uptake of the nanocarriers in the tumour microenvironment.14 Active targeting involves the modification of the nanocarrier surface with small molecules,15 proteins and biopolymers, such as antibodies,16 peptides,17 and nucleic acids.18 These active targeting options are based on specific binding to receptors overexpressed in the tumour cells.19 Within the general class of nanocarriers, niosomes have some unique properties that are being exploited in the next generation of drug delivery agents.20 The non-ionic surfactants used to assemble the niosomes are biocompatible and biodegradable and do not induce an immune response in the body.2 They are cheaper to produce than liposomes, which makes them more attractive for commercialisation.21 Niosomes also offer increased blood circulation time compared to liposomes and protect the drug cargo from degradation.22 The surfactants used to form the niosomes are both physically and chemically more stable than their liposome counterparts, which leads to easier formulation and production.23 | ||
| Fig. 1 An illustration of the niosome structure composed of sorbitane monostearate (Span60) and cholesterol. | ||
When loaded with a drug molecule, the niosomes have been shown to slowly release the drug, giving rise to a sustained release that can be modulated by the formulation.24,25 This controlled release of a targeted drug is of great interest as it can lead to better therapeutic response and reduced side effects.26 Niosomes release their drug cargo by three main mechanisms: (1) diffusion or Fickian release, where the drug diffuses out due to concentration gradients,27 (2) reorganisation of the bilayer leading to increased diffusion of the drug,28 and (3) degradation of the noisome, leading to drug release.29 These different release mechanisms lead to complex models that can be used to investigate the release kinetics and give insights into the processes involved.30 Traditional drug delivery systems and controlled release aim to maintain a constant level of the active pharmaceutical ingredient (API) in the plasma or a specific area of the body.26 An alternative to this is the ‘release on demand’ system, where the drug is released quickly when activated. To this end, niosomes have been designed to give an initial burst of API followed by slow release, which can take over a few hours or a few days. Alemi et al., developed a PEGylated niosome formulation that delivered curcumin and paclitaxel with 20–30% release after 72 hours.25 Ugorji et al., used sorbitane monostearate (Span60) based niosomes loaded with 5-fluorouracil that showed 50–60% release after 3 hours.27 More recently stimuli-responsive drug release systems have been developed that release their drug payload under specific physiological or chemical conditions such as redox activation,31 enzyme levels,32 near IR irradiation,33,34 ultrasound,35 in low pH environments36,37 and temperature changes.10,38 Wang et al., developed a thermally-responsive polymer-modified liposome carrier that could release its drug content at temperatures above 37 °C.39 Their system showed 60% API release after only 5 minutes at temperatures above 40 °C.39 Unlike traditional drug release systems, these stimuli-responsive nanocarriers are designed to protect their drug cargo until a specific stimulus is present and then release the drug in a sudden burst. This can see 100% release in minutes after stimulation, compared to traditional controlled release that can give 30% release in 72 hours. A specific class of stimuli-responsive nanocarriers are the magnetoliposomes.40 These incorporate magnetic nanoparticles (MNPs) into the liposome structure and have been used for MRI contrast agents41 or the targeting of tumours through magnetic field trapping.42 Ma et al., developed thermally sensitive liposomes with hydrophobic MNPs trapped in the lipid bilayer and the hydrophilic core loaded with doxorubicin (DOX), a chemotherapeutic drug.43 They showed the thermally controlled release of DOX in vitro and in vivo using murine colon cancer cells (CT26) and female BALB/c mice models. The thermal release was triggered by a fibre-coupled laser system operating at 808 nm. The MNPs absorbed the laser energy and heated up the samples to release the DOX.43 Magnetoliposomes have also been used in combined magnetic field hyperthermia (MFH) and chemotherapy.44,45 In this dual therapy approach, the MNPs are used to induce the thermal release of the drug from the liposome structure and, at the same time, heat the tumour cells and induce cell death through MFH.46,47
Here, we report, for the first time, a stimuli-responsive niosome-based system that releases its drug cargo in response to an alternating magnetic field (AMF). The system is based on Span60 and cholesterol niosomes with dextran-coated iron oxide nanoparticles (MNPs) and DOX loaded into the hydrophilic core. On exposure to an AMF, the NPs produce heat that induces the niosomes to release the DOX. Fluorescent spectroscopy is used to monitor the DOX release. The work covers the synthesis and characterisation of the MNPs and niosomes and the kinetic studies for the DOX release at different temperatures and induced by the AMF.
Experimental
Phosphate buffer saline (PBS) tablets were purchased from Sigma-Aldrich (P4417) and used to prepare a solution of 0.01 M phosphate buffer, 2.7 mM potassium chloride, and 0.137 M sodium chloride. Sorbitane monostearate and cholesterol was purchased from Sigma-Aldrich (S7010) and (C8667) respectively. Doxorubicin was purchased from Biosynth (AD15377). The gel filtration columns were PD10 Cytiva columns with Sephadex G-25 M stationary phase. Iron(II) chloride tetrahydrate and iron(III) chloride hexahydrate were also purchased from Sigma-Aldrich. Deionised water was produced using SLS LAB PRO – PURA-Q+, 18 MΩ cm. Dextran (D9260) and dimethyl sulfoxide (472301) were purchased from Sigma, and chloroform (10784143) was purchased from Fisher Scientific.Magnetic nanoparticle synthesis
The magnetic iron-oxide nanoparticles (MNPs) were synthesized under a nitrogen atmosphere. A schematic representation is shown in Fig. 2, briefly, iron(II) chloride tetrahydrate (0.0085 moles), iron(III) chloride hexahydrate (0.0175 moles) and deionized water (75 mL) were combined in a reaction flask. Nitrogen gas was bubbled through for 10 minutes to remove any dissolved oxygen. NaOH (4 mol dm−3) was added to adjust the pH of the mixture. The solution was subjected to continuous stirring during the reaction until the mixture became basic (pH 11). The black precipitate formed was washed with deionized water until pH 7 and dispersed in 30 mL of fresh deionized water. The dextran (MW = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) coating was then added following the procedure below. Dextran (5 g) was mixed with deionised water (20 mL) resulting in a 25% solution by weight which was added to a flask along with all the MNPs (30 mL). This was mixed in an ultrasonic bath for 5 minutes. Ammonium hydroxide solution (2 M) was added dropwise until a pH of more than 10 was achieved. The flask was then set to heat in an oil bath at 75 °C for 1 hour with stirring. Once cool, the solution was decanted into a sample vial and washed with deionized water using magnetic precipitation until a pH of 7 was achieved. This resulted in a black solution. A sample (1 mL) was removed and magnetically precipitated, the clear liquid was decanted, and the remaining black magnetic precipitate was dried under nitrogen until constant mass. This gave a MNP concentration of 17.3 mg mL−1. The ATR-FTIR spectra of the clear liquid removed showed no dextran was present in this wash. The ATR-FTIR spectra of the black precipitate showed the presence of magnetite and dextran. This observation supports the synthesis of dextran coated iron-oxide nanoparticles. The MNPs were subjected to TEM analysis with EDX and electron diffraction (FEI Titan3 Themis 300: X-FEG 300 kV S/TEM with S-TWIN objective lens, monochromator (energy spread approx. 0.25 eV), multiple HAADF/ADF/BF STEM detectors, FEI Super-X 4-detector EDX system).
000 Da) coating was then added following the procedure below. Dextran (5 g) was mixed with deionised water (20 mL) resulting in a 25% solution by weight which was added to a flask along with all the MNPs (30 mL). This was mixed in an ultrasonic bath for 5 minutes. Ammonium hydroxide solution (2 M) was added dropwise until a pH of more than 10 was achieved. The flask was then set to heat in an oil bath at 75 °C for 1 hour with stirring. Once cool, the solution was decanted into a sample vial and washed with deionized water using magnetic precipitation until a pH of 7 was achieved. This resulted in a black solution. A sample (1 mL) was removed and magnetically precipitated, the clear liquid was decanted, and the remaining black magnetic precipitate was dried under nitrogen until constant mass. This gave a MNP concentration of 17.3 mg mL−1. The ATR-FTIR spectra of the clear liquid removed showed no dextran was present in this wash. The ATR-FTIR spectra of the black precipitate showed the presence of magnetite and dextran. This observation supports the synthesis of dextran coated iron-oxide nanoparticles. The MNPs were subjected to TEM analysis with EDX and electron diffraction (FEI Titan3 Themis 300: X-FEG 300 kV S/TEM with S-TWIN objective lens, monochromator (energy spread approx. 0.25 eV), multiple HAADF/ADF/BF STEM detectors, FEI Super-X 4-detector EDX system).
 | ||
| Fig. 2 A schematic representation of the magnetic nanoparticle (MNP) synthesis from iron chlorides followed by dextran coating. The final MNP sample appeared as a black opaque solution in water at pH 7. | ||
Niosome synthesis
The niosomes were synthesised using the thin film hydration method, Fig. 3. Sorbitane monostearate (8.82 mg) and cholesterol (5.88 mg) were dissolved in chloroform (2 mL) in a 25 mL round bottomed flask. The chloroform was slowly removed under reduced pressure using a rotary evaporator. Once dried the sample was maintained on the rotary evaporator under vacuum for 30 minutes at 60 °C to form a thin film. The film was hydrated in 3 mL of the MNPs with an iron oxide concentration of 0.15 mg mL−1 in deionised water and 140 μL of DOX (5 mg mL−1 in DMSO) for 1 h at 60 °C with stirring. The niosome suspension was then sonicated for 5 minutes resulting in a cloudy pink solution with some obvious larger precipitates, this is referred to as the raw niosome solution. Finally, a 1 mL sample of the raw niosome solution was cleaned by passing through a PD10 column using PBS buffer as the mobile phase. The column eluent was collected in 1 mL fractions immediately after the raw noisome solution was loaded (1 mL). These were labelled fraction 1–25. The 4th, 5th and 6th fractions appeared cloudy and were combined to give a final 3 mL sample of the cleaned niosomes. This final solution was analysed with UV-vis spectroscopy and DLS to confirm the encapsulation efficiency of doxorubicin and the colloidal properties of the suspension. This synthesis produced the niosomes with the hydrophilic core loaded with MNPs and DOX (NS–DOX–MNP). | ||
| Fig. 3 A schematic representation of the niosome synthesis. NS, DOX, MNP and SEC stand for niosome, doxorubicin, magnetic nanoparticles and size exclusion respectively. | ||
The synthesis was repeated but with the MNP solution replaced with deionised water (3 mL) plus 140 μL of DOX (5 mg mL−1 in DMSO) to produce the DOX-loaded niosomes (NS–DOX) and a sample with no MNPs and no DOX was produced by hydrating with just deionised water (3 mL) and DMSO (140 μL) to produce unloaded niosomes (NS). An illustration of the samples are shown in Fig. 4.
 | ||
| Fig. 4 An illustration of the niosome samples prepared. NS, DOX and MNP stand for niosome, doxorubicin and magnetic nanoparticles respectively. | ||
The synthesised niosome samples were characterised using dynamic light scattering (DLS) carried out on a Zetasizer NanoZS (Malvern Instruments) equipped with a 4.0 mW He–Ne laser operating at 633 nm with a photodiode detector at a 90° detection angle. The NS solutions were used directly from the PD10 column (3 mL) with a 500 μL disposable cuvette. The Z-average and polydispersity values are quoted as well as the number average distribution and standard deviation (SD). The zeta-potential of the samples was analysed using a folded capillary zeta cell. Values are reported as the mean ± SD of three measurements.
DOX loading calculation
The DOX loading efficiency (EE%) is the percentage of the available DOX that was loaded into the core of the niosomes (eqn (1)). This is based on the fluorescent emission of the DOX molecule. The loaded niosome sample (100 μL) is dispersed in DMSO (900 μL) and the fluorescent intensity recorded at λ 590 nm with λ 480 nm excitation. This was compared to the same sample made from the raw niosome solution (100 μL) prior to going through the column, dispersed in DMSO (900 μL). The raw niosome solution has all the DOX present, either inside the niosomes or free in solution so this represents 100% of the DOX present. The sample that is passed through the PD10 column has the free DOX removed, leaving only the DOX loaded inside the niosome core.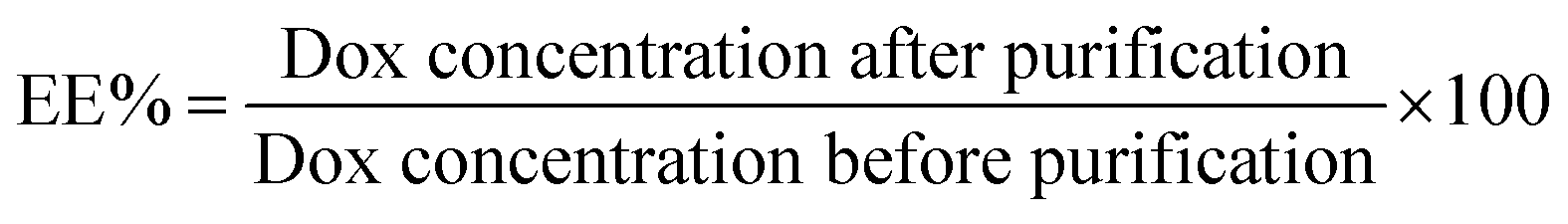
DOX release study
The release of DOX from the niosomes was monitored using fluorescent spectroscopy (Horiba Fluoromax-4P TCSPC). The excitation source was 544 nm, and the fluorescent emission recorded from 570–800 nm. Once eluted from the PD10 column, the 3 mL sample was sealed in a quartz cuvette, and the fluorescent spectra were recorded in the spectrometer. The sample was then stored at a set temperature (333 K, 313 K or 298 K) for the required period of time (up to 30 days). The fluorescent spectra were recorded for the samples as a function of time and compared.For the AMF-initiated release, an induction heating coil was fitted around the fluorescent cuvette. The coil operated at 125 kHz and had a field intensity of 1000 A m−1 at its centre. The coil was made from 2.2 mm wide hollow copper tubing with a 1.1 mm internal diameter. Cooling water was pumped through the coil at a rate of 10 mL per minute to maintain room temperature at all times. The coil was 41 mm in height with an internal diameter of 17 mm and an external diameter of 20 mm and consisted of 6.5 turns. The drive voltage was 12 V, resulting in a current of 10.5 A. The coil design was based on that reported by Drake et al. and shared the same drive circuit.48
Results and discussion
The MNP synthesis produced a black, magnetically responsive dispersion of nanoparticles with an average diameter of 5.80 nm and a standard deviation of 2.08 nm, estimated from TEM image analysis. Fig. 5 shows the TEM data (A–D), along with the electron diffraction pattern indexed to magnetite (H). Image 5D shows a close-up view of lattice lines with a d-spacing of 0.296 nm corresponding to the 220-crystal face in magnetite. EDX data showed the presence of Fe in all samples. Vibrational spectroscopy analysis of the MNPs dried at 40 °C under a N2 flow are shown in Fig. 5E and F. The ATR-FTIR spectra (F) shows the expected peaks for dextran.49 Magnetite shows no peaks in the range 2000–1000 cm−1 the magnetite spectra should be dominated by the Fe–O bands at 621 and 574 cm−1.50,51 The peak at 1632 cm−1 seen in (F) can be assigned to the water v2 bending mode, and the two peaks at 1497 cm−1 and 1392 cm−1 are characteristic of dextran,49 they can be assigned to the v(H–C–OH) and v(C–O–C) respectively.52 The MNPs were washed with water until the washings showed no evidence of the presence of free dextran, as shown by the FTIR spectra for the washings. The first wash showed the expected dextran peaks; subsequent washes showed a reduction in these peaks, showing that free dextran was being removed from the MNPs. Eventually, the washings showed no evidence for the presence of dextran. The MNPs were then redispersed in fresh water, and a sample was removed for drying at 40 °C under N2 flow to constant mass. The fact that the ATR-FTIR spectra for these dried MNPs still show characteristic peaks for dextran provides evidence to support the synthesis of dextran-coated MNPs. The Raman spectra for the same sample, Fig. 5(E) shows a peak at 680 cm−1 characteristic of magnetite. The absence of hematite Raman peaks at 219 cm−1 and 280 cm−1 of equal intensity supports the assumption that the MNPs are the magnetite phase. The presence of maghemite phase would give a second peak or a shoulder at 710 cm−1.53 The Raman spectra, combined with the observation that the material was a black colour and magnetically responsive, along with the electron diffraction pattern (H), all confirm the synthesis of the magnetite phase. | ||
| Fig. 5 Showing the TEM data for the magnetic nanoparticles (MNPs). (A) Standard TEM showing a larger area view, scale bar 200 nm. (B) Zoomed in TEM image, scale bar 50 nm. (C) High resolution TEM showing individual MNPs and the lattice lines, scale bar 10 nm. (D) Close up view of lattice lines with a d-spacing of 0.296 nm corresponding to the 220 crystal face in magnetite. (E) Raman spectra the dried MNP taken with a 633 nm laser. (F) ATR-FTIR spectra of the dried MNPs. (G) Size distribution of the MNPs estimated from TEM image analysis. The line is a normal distribution curve fitted to the histogram data (dots) with an average of 5.80 nm and a standard deviation of 2.08 nm. (H) electron diffraction data collected from image A, with the rings indexed to the (hkl) lattice parameters for magnetite. | ||
The niosomes were synthesised following the thin film hydration method (Fig. 3).54,55 The surfactants were dissolved in chloroform with a mass ratio of 3:2 (Span60:cholesterol) and a thin film formed on the inside of the round bottom flask by the slow evaporation of the solvent. The thin film was rehydrated at 60 °C with water, water and DOX or water, DOX and MNP to produce the plain niosomes (NS), niosomes loaded with DOX (NS–DOX) or niosomes loaded with DOX and MNPs (NS–DOX–MNP) respectively. The initial rehydration resulted in a raw niosome samples that appeared as a cloudy white solution for the NS and a cloudy pink solution for the NS–DOX and NS–DOX–MNP. The raw niosome solutions showed evidence of aggregation and some large precipitates could be seen. The DLS analysis for the raw NS–DOX–MNP sample gave Z-average = 533 nm and a PDI of 0.848. In order to clean the solutions, they were passed through a PD10 size exclusion column using PBS as the mobile phase. The raw niosome sample (1 mL) was added to the column and eluted with PBS. The initial eluent was a clear colourless solution, as expected. Eluent volume 4–6 mL appeared cloudy, after this volume the eluent became clear again. A PD10 column with PBS was also run loaded with DOX only. Using UV-vis analysis of the eluent it could be seen that the DOX eluted between 8 mL and 30 mL. This is consistent with the size exclusion principle. Using the PD10 column it is possible to separate the niosomes loaded with DOX from any free DOX in solution (not inside the niosomes). The DOX loading efficiency is the percentage of the available DOX that was loaded into the core of the niosomes. This is based on the fluorescent emission of the DOX molecule. If no DOX was inside the niosomes then the elution volume 4–6 mL would not have DOX present. This would simply be the niosomes. Any free DOX would pass down the column slower and elute after 8 mL of PBS. A sample of the eluted niosomes (100 μL) was dispersed in 900 μL of DMSO. This destroys the niosome structure and releases DOX in the DMSO solvent. The fluorescent intensity of this solution can then be used to estimate the encapsulation efficiency (EE) of the DOX (the % of available DOX that was encapsulated inside the niosomes). This was found to be 67% for the NS–DOX sample and 61% for the NS–DOX–MNP sample, demonstrating that the presence of MNP doesn’t interfere with the loading of the chemotherapeutic drug.
Dynamic light scattering was used to characterise the average diameter and size distribution of the niosomes in solution. The data can be seen in Table 1. The Z-average niosome size is the intensity weighted hydrodynamic size determined by the cumulants method, the polydispersity index (PDI) represents the width of this distribution.56 This is the ISO recommend method for representing DLS data. It can be seen that the Z-average niosome diameter was around 310–360 nm and PDI ranged from 0.4–0.6. The intensity average particle size can be dominated by large particles. The scattering data can be transformed into an average number of niosome sizes by applying Mie's theory.57 This gives a more accurate size distribution for complex samples with a small amount of large particles (>600 nm). The number average size distribution for the niosome samples can be seen in Table 1, along with the standard deviation. This shows that the samples are mostly composed of niosomes with an average diameter below 200 nm (96–99%) and a few larger niosomes. The zeta potential for all three niosomes was determined to be −26.4 mV ± 1.9 mV. This compares well with the 212 nm diameter and zeta potential −27 to 24 mV quoted by Miatmoko et al. for the same surfactants and synthesis method but a different API.58
| Sample | Z-Average/nm | PDI | Number average/nm | Standard deviation/nm | % of NS below 200 nm |
|---|---|---|---|---|---|
| NS | 363.6 | 0.57 | 187.8 | 68.18 | 98 |
| NS–DOX | 309.4 | 0.335 | 140.7 | 86.22 | 99 |
| NS–DOX–MNP | 358.3 | 0.403 | 169.3 | 69.02 | 96 |
The stability of the niosomes was investigated by measuring the change in Z-average and PDI over the course of 1 month with the samples stored under standard conditions and room temperature. This revealed only a 5% variation in both values over the time period, suggesting the samples are stable under these conditions.
AMF release study
The NS sample appeared as a cloudy white solution whereas the DOX loaded samples (NS–DOX and NS–DOX–MNP) showed the expected red tinge associated with the DOX. The fluorescent spectra from these samples were recorded (480 nm excitation, 500–800 nm emission) with the spectra matching that expected for pure DOX.59 These showed a fluorescent peak centred on λmax 590 nm with a shoulder at λmax 560 nm and a weak shoulder at λmax 650 nm, see Fig. 6A. The two DOX-loaded samples were placed inside a magnetic coil operating at 125 kHz with a field intensity of 1000 A m−1 at its centre. The coil design was based on that reported by Drake et al. and shared the same drive circuit.48 Inside the niosomes the DOX is self-quenching with lower fluorescent intensity.60 The fluorescent intensity should increase over time if the DOX is released from the niosome core (see Fig. S1–S4, ESI†). If the DOX remains inside the niosome then the fluorescent intensity should remain constant. It can be seen (Fig. 6B), that on the activation of the AMF the fluorescent intensity at 590 nm for the NS–DOX–MNP sample started to increase, reaching a ΔI590 of 10k CPS after 10 minutes and eventually plateauing at around a ΔI590 of 90k CPS after 200 min. Conversely, the NS–DOX sample showed only a small increase over time, reaching a ΔI590 of 1k CPS after 10 minutes and plateauing at around a ΔI590 of 10k CPS after 200 min. | ||
| Fig. 6 (A) Fluorescent spectra of NS–DOX–MNP recorded with 480 nm excitation and 500–800 nm emission. (B) Time dependent plot of the change in fluorescent intensity at 590 nm (ΔI590) where ΔI590 = ((tI590) − (tI800)) − ((0I590) − (0I800)), tI590 and tI800 are the fluorescent intensity for time t at 590 nm and 800 nm respectively and 0I590 and 0I800 are the fluorescent intensity for time = 0 at 590 nm and 800 nm respectively. The best fit line shown in B for the NS–DOX–MNP data is a 1st order kinetic plot with a rate constant (k) = 0.017 min−1 and a final ΔI590 value (ΔImax) = 90000 CPS. The fluorescent intensity at 200 min for the NS–DOX–MNP sample shows 86% of the total DOX present has been released. | ||
The release of DOX from the niosomes is a measure of the diffusion of the DOX across the Span60 bilayer. Assuming that the plateau region seen in Fig. 6B represents the final concentration of free DOX then the 1st order rate equation of the diffusion of DOX can be written as:
 | (1) |
000 CPS, k is the 1st order rate constant (min−1) and t is the reaction time (min). Plotting Ln[1 − (ΔtI590/ΔImax)] against time (t) should produce a straight line fit with gradient = −k. These kinetic plots for NS–DOX–MNP and NS–DOX are shown in Fig. 7. They show that the 1st order kinetic plot is a good fit to the NS–DOX–MNP sample (R2 = 0.9803). This suggests that the release of the DOX out of the niosome follows a Fickian mechanism and is diffusion controlled with a high initial rate tailing off as the concentration of DOX inside the niosomes drops.29 The rate constant for the DOX release in the NS–DOX–MNP sample is k = 1.7 × 10−2 min−1. The same plot can be seen for the NS–DOX sample. This has no MNPs present and so is expected to show no AMF induced heating. The plot shows very little change in the fluorescent emission over time suggesting very little DOX release is occurring. The 1st order plot does not fit as well to this sample (R2 = 0.7459). This poor fit is mostly for the early stage of the reaction with time less than 20 minutes. The overall rate constant for the NS–DOX sample is k = 2.0 × 10−3 min−1. The difference between the two samples is the addition of the MNPs. In the presence of the AMF these heat the inside of the niosome structures. This heating appears to increase the rate of diffusion of the DOX across the bilayer, with over 60% of the total DOX released in just 60 minutes compared to only 7% released for the NS–DOX sample. The NS–DOX–MNP sample went on to release 100% of the loaded DOX in 200 minutes, whereas the NS–DOX sample only released 20% of the DOX at 200 minutes and showed no further release with time.
 | ||
| Fig. 7 1st order kinetic plots for NS–DOX–MNP sample (left) and the NS–DOX sample (right). ΔtI590 is the change in fluorescent intensity at 590 nm = ((tI590) − (tI800)) − ((0I590) − (0I800)), where tI590 and tI800 are the fluorescent intensity for time t at 590 nm and 800 nm respectively and 0I590 and 0I800 are the fluorescent intensity for time = 0 at 590 nm and 800 nm respectively. | ||
The final absolute fluorescent intensity recorded at λ = 590 nm represents the concentration of DOX in the PBS solution after being released from the niosomes (see Fig. S5, ESI†). Using the calibration curve for pure DOX in PBS (see Fig. S3, ESI†), this can be estimated to be 0.04 mg mL−1 [DOX], in the 3 mL volume. This means a total of 0.12 mg of free DOX. The total amount of DOX added at the start of the synthesis was 0.14 mg (assuming 60% encapsulation). This gives an estimate of 86% of total DOX released over 200 minutes during the AMF exposure.
Thermal release of DOX
The diffusion-controlled release of DOX from the niosomes would be expected to show a temperature dependence. At higher temperatures the diffusion process should occur at a faster rate due to the surfactant molecules having increased thermal motion and increased bilayer fluidity.61 It has been shown that the permeability of surfactant bilayers reaches a maximum at the surfactant chain-melting temperature, this is the solid–liquid phase transition for the bilayer structure, in the case of Span60 this transition occurs around 58 °C.62 See Blicher et al. for a more in-depth discussion of the temperature dependence of lipid membrane permeability.63 In order to investigate the temperature dependence of the diffusion and permeability of DOX across the niosome bilayer NS–DOX–MNP samples were prepared and incubated at different temperatures. In order to investigate this, three more NS–DOX–MNP samples were made following the same method. The samples were sealed in a 3 mL fluorescent cuvette and stored at different temperatures. The fluorescent signal was recorded as a function of time. Three temperatures were used (298 K, 313 K and 333 K). Immediately after the SEC column the three samples all showed the same fluorescent spectra that matched that observed before, the major peak centred on λmax 590 nm with a shoulder at λ 560 nm and a weak shoulder at λ 650 nm and is characteristic of DOX. As expected, over time the fluorescent peak at 590 nm showed an increase in intensity. This was most pronounced at the higher temperatures. The NS–DOX–MNP sample at 333 K showed a steady increase in fluorescence that reached a plateau at 100% release after 6 days. The 313 K, NS–DOX–MNP sample reached 100% after 20 days. The 298 K sample showed the slowest release, reaching 1% released after 6 days, 2% released after 13 days and about 3% released after 20 days. The release profiles can be fitted with 1st order kinetic plots using eqn (1) and the data can be seen in Fig. 8 and Table 2. The modelling of the thermal release at physiological temperature (37 °C) shows that 10% of the DOX will be released after about 5 days with 100% release reached at about 200 days at this temperature (see Fig. S6 for the release profile, ESI†).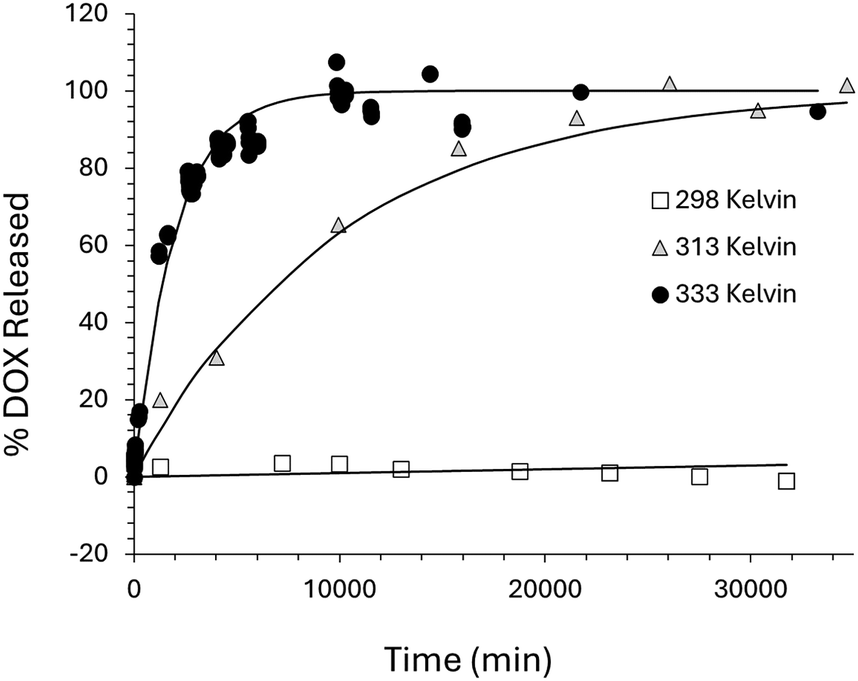 | ||
| Fig. 8 Thermal release of DOX from NS–DOX–MNP system maintained at a constant temperature inside an oven at 298, 313 and 333 K along with the 1st order kinetic fits to the data based on the rate constants shown in Table 2. The rate constants were calculated from eqn (1). | ||
| Temperature (K) | 1st order rate constant (min−1) | R2 |
|---|---|---|
| 298 | 1.2 × 10−6 | 0.8841 |
| 313 | 1.0 × 10−4 | 0.9952 |
| 333 | 5.1 × 10−4 | 0.9884 |
| AMF | 1.7 × 10−2 | 0.9803 |
The rate of thermal release of DOX is controlled by the temperature of the system and is independent of the MNP presence. At 333 K the rate constant k is 5.1 × 10−4 min−1, leading to 100% DOX released after approximately 6 days. At room temperature (298 K) the rate constant k is 1.2 × 10−6 min−1, this is 400 times smaller. The 1st order fit to the release data at 298 K is poor, but suggest that only 3% of the DOX is released after 20 days. By contrast, the magnetically (AMF) controlled release of DOX from the same niosome system has a rate constant k of 1.7 × 10−2 min−1; this is four orders of magnitude quicker than the thermal release measured at 298 K and leads to 100% DOX release after just 3 hours. It is worth pointing out that during the magnetic release, the overall temperature of the sample does not change, remaining at about 298 K (room temperature in the spectrometer). Using an Arrhenius plot, we can use the thermal release data to predict what the temperature would be for the system with an equivalent rate constant to the magnetic release, a plot of Lnk against 1/T, (where k is the 1st order rate constant and T is temperature), should produce a straight line fit.64 This can be seen in Fig. 9.
 | ||
| Fig. 9 Arrhenius plot for the thermal release data showing the inverse linear dependence of the rate constant for the diffusion of DOX from inside the NS–DOX–MNP samples, on the temperature of the system (k is the 1st order rate constant (min−1) and T is temperature in Kelvin). The linear fit is for the thermal release data only. The AMF data point shows the predicted temperature of the magnetic release (AMF) rate constant based on the linear fit. | ||
The linear fit is for the thermal release data only. Based on the measured 1st order rate constant for the magnetic release data, the Arrhenius plot suggests that the diffusion of DOX out of the niosomes under magnetic control is occurring at a rate equivalent to the rate at 353 K (80 °C). As the actual temperature of the sample remains at 298 K this data supports the assumption that the MNPs are heating the internal core of the niosome to around 80 °C due to induction heating via the AMF, this leads to increased diffusion of the DOX across the Span60 bilayer and release into the surrounding solution. The heating is limited to the hydrophilic core containing the MNPs and so the overall temperature of the sample remains unchanged. The MNPs couple to the AMF and produce heat through Brownian and Néel relaxation mechanism.65 This heats the surrounding liquid. Since the MNPs are in the internal core of the niosomes, this heat is restricted to this small volume and the internal core quickly reaches a temperature of 80 °C. The heat increases diffusion of DOX and promotes thermal relaxations within the Span60 bilayer, resulting in increased DOX release.
Conclusions
The synthesis of the niosomes went as planned as evidenced by the appearance of the samples (cloudy white solution for the NS sample and cloudy red solution for the DOX loaded samples, NS–DOX and NS–DOX–MNP). The zeta potential for all three niosome samples was determined to be −26.4 mV ± 1.9 mV and the Z-average niosome diameter as measured by DLS was around 310–360 nm with a PDI ranging from 0.4–0.6. The number average diameter was found to be (average ± standard deviation) 188 ± 68 nm, 141 ± 86 nm and 169 ± 69 nm for the NS, NS–DOX and NS–DOX–MNP samples, respectively. The loaded DOX could be released from the core of the niosomes by diffusion across the surfactant bilayer and this could be modelled using 1st order kinetics with rate constants of 1.2 × 10−6 min−1, 1.0 × 10−4 min−1 and 5.1 × 10−4 min−1 at 298 K, 313 K and 333 K respectively. The release of DOX from the NS–DOX–MNP sample could also be triggered by the application of an external, alternating magnetic field. With the magnetic field activated the rate constant increased to 1.7 × 10−2 min−1. This is four orders of magnitude greater than the thermal release at the same temperature (298 K). The work shows the magnetically controlled, burst release from a drug loaded niosome delivery system. The release was triggered, on demand, by the application of the alternating magnetic field resulting in 100% doxorubicin release within 3 hours compared to 3% release in 30 days with no magnetic field. For comparison, in the DOX–magnetoliposome system developed by Hardiansyah et al., the thermal release and the magnetic release showed a five times difference in rate and when the magnetic system reached 100% release of DOX the thermal only system (no MNPs) showed 80% release of DOX.45Future work will focus improving the niosome synthesis to lower the average size to around 200 nm and to improve the size distribution. We will also modify the niosome structure and niosome surface using different surfactant formulations and polymer systems to control the surface charge and drug diffusion kinetics, as well as looking at the drug release in the presence of live cells and combining the magnetically induced drug release with magnetic field hyperthermia in a dual-modality therapy.
Author contributions
Philip Drake wrote the manuscript and was responsible for the nanoparticle synthesis and AMF design. He was also the principal investigator for the work and along with Ilma Amalina, was responsible for the conceptualization, grant writing and data collection in all areas. Ilma Amalina was responsible for most of the experimental work and data collection. Retno Sari and Andang Miatmoko contributed to the grant writing and data reviewing as well as experimental aspects of niosome synthesis. Amalia Ruiz contributed to the experimental design of the niosome synthesis, supervising aspects of the synthesis and reviewing the manuscript. Saliha Ramazan, Gordon Hope and Dharmisthaben Pancholi were project students that contributed to the data collection.Conflicts of interest
There are no conflicts to declare.Data availability
All data supporting the findings of this study are available within the paper and its ESI.†Acknowledgements
This research was supported by the Royal Society of Chemistry (E22-5590558249), the University of Bradford and Airlangga University. We also acknowledge support from the University of Leeds, Leeds electron microscopy and spectroscopy centre in running the TEM, EDX and electron diffraction analysis.References
- K. M. Kazi, A. S. Mandal, N. Biswas, A. Guha, S. Chatterjee, M. Behera and K. Kuotsu, J. Adv. Pharm. Technol. Res., 2010, 1, 374–380, DOI:10.4103/0110-5558.76435.
- M. P. Izhar, A. Hafeez, P. Kushwaha and Simrah, J. Cluster Sci., 2023, 34, 2257–2273 CrossRef CAS.
- T. M. Allen, Drugs, 1998, 56, 747–756, DOI:10.2165/00003495-199856050-00001.
- S. Moghassemi and A. Hadjizadeh, J. Controlled Release, 2014, 185, 22–36, DOI:10.1016/j.jconrel.2014.04.015.
- M. Cetin, A. Atila and Y. Kadioglu, AAPS PharmSciTech, 2010, 11, 1250–1256, DOI:10.1208/s12249-010-9489-6.
- A. Miatmoko, R. T. Octavia, T. Araki, T. Annoura and R. Sari, Saudi Pharm. J., 2024, 32, 102085, DOI:10.1016/j.jsps.2024.102085.
- S. Saraf, A. Jain, A. Tiwari, A. Verma, P. K. Panda and S. K. Jain, J. Drug Delivery Sci. Technol., 2020, 56, 101549, DOI:10.1016/j.jddst.2020.101549.
- S. D’Souza, Adv. Pharm., 2014, 2014, 304757, DOI:10.1155/2014/304757.
- S. Z. Alshawwa, A. A. Kassem, R. M. Farid, S. K. Mostafa and G. S. Labib, Pharmaceutics, 2022, 14, 883, DOI:10.3390/pharmaceutics14040883.
- N. AlSawaftah, W. G. Pitt and G. A. Husseini, ACS Pharmacol. Transl. Sci., 2021, 4, 1028–1049, DOI:10.1021/acsptsci.1c00066.
- R. K. Thapa and J. O. Kim, J. Pharm. Invest., 2023, 53, 19–33, DOI:10.1007/s40005-022-00607-6.
- X. Shan, X. Gong, J. Li, J. Wen, Y. Li and Z. Zhang, Acta Pharm. Sin. B, 2022, 12, 3028–3048, DOI:10.1016/j.apsb.2022.02.025.
- W. Islam, T. Niidome and T. Sawa, J. Pers. Med., 2022, 12, 1964 CrossRef.
- K. Bromma, A. Bannister, A. Kowalewski, L. Cicon and D. B. Chithrani, Cancer Nanotechnol., 2020, 11, 8, DOI:10.1186/s12645-020-00064-6.
- M. F. Attia, N. Anton, J. Wallyn, Z. Omran and T. F. Vandamme, J. Pharm. Pharmacol., 2019, 71, 1185–1198, DOI:10.1111/jphp.13098.
- N. Wathoni, L. E. Puluhulawa, I. M. Joni, M. Muchtaridi, A. F. A. Mohammed, K. M. Elamin, T. Milanda and D. Gozali, Drug Delivery, 2022, 29, 2959–2970, DOI:10.1080/10717544.2022.2120566.
- S. Guo, J. Wang, Q. Wang, J. Wang, S. Qin and W. Li, Heliyon, 2024, 10, e26009, DOI:10.1016/j.heliyon.2024.e26009.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010, 10.1039/C2CS15344K.
- F. Oroojalian, M. Beygi, B. Baradaran, A. Mokhtarzadeh and M.-A. Shahbazi, Small, 2021, 17, 2006484, DOI:10.1002/smll.202006484.
- B. A. Witika, K. E. Bassey, P. H. Demana, X. Siwe-Noundou and M. S. Poka, Int. J. Mol. Sci., 2022, 23, 9668, DOI:10.3390/ijms23179668.
- I. F. Uchegbu and S. P. Vyas, Int. J. Pharm., 1998, 172, 33–70, DOI:10.1016/S0378-5173(98)00169-0.
- P. Bhardwaj, P. Tripathi, R. Gupta and S. Pandey, J. Drug Delivery Sci. Technol., 2020, 56, 101581, DOI:10.1016/j.jddst.2020.101581.
- X. Ge, M. Wei, S. He and W. E. Yuan, Pharmaceutics, 2019, 11, 55, DOI:10.3390/pharmaceutics11020055.
- A. A. Targhi, A. Moammeri, E. Jamshidifar, K. Abbaspour, S. Sadeghi, L. Lamakani and I. Akbarzadeh, Bioorg. Chem., 2021, 115, 105116, DOI:10.1016/j.bioorg.2021.105116.
- A. Alemi, J. Zavar Reza, F. Haghiralsadat, H. Zarei Jaliani, M. Haghi Karamallah, S. A. Hosseini and S. Haghi Karamallah, J. Nanobiotechnol., 2018, 16, 28, DOI:10.1186/s12951-018-0351-4.
- S. Adepu and S. Ramakrishna, Molecules, 2021, 26, 5905, DOI:10.3390/molecules26195905.
- O. L. Ugorji, O. N. C. Umeh, C. O. Agubata, D. Adah, N. C. Obitte and A. Chukwu, Heliyon, 2022, 8, e12369, DOI:10.1016/j.heliyon.2022.e12369.
- S. Sangkana, K. Eawsakul, T. Ongtanasup, R. Boonhok, W. Mitsuwan, S. Chimplee, A. K. Paul, S. S. Saravanabhavan, T. Mahboob, M. Nawaz, M. L. Pereira, P. Wilairatana, C. Wiart and V. Nissapatorn, Nanoscale Adv., 2024, 6, 1467–1479, 10.1039/D3NA01016C.
- N. S. Heredia, K. Vizuete, M. Flores-Calero, V. K. Pazmiño, F. Pilaquinga, B. Kumar and A. Debut, PLoS One, 2022, 17, e0264825, DOI:10.1371/journal.pone.0264825.
- A. Jain and S. K. Jain, Chem. Phys. Lipids, 2016, 201, 28–40, DOI:10.1016/j.chemphyslip.2016.10.005.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12, DOI:10.1016/j.jconrel.2011.01.030.
- J. Li, Y. J. Ma, Y. Wang, B. Z. Chen, X. D. Guo and C. Y. Zhang, Chem. Eng. J., 2018, 341, 450–461, DOI:10.1016/j.cej.2018.02.055.
- A. Raza, T. Rasheed, F. Nabeel, U. Hayat, M. Bilal and H. M. N. Iqbal, Molecules, 2019, 24, 1117 CrossRef CAS.
- Q. Yu, T. Huang, C. Liu, M. Zhao, M. Xie, G. Li, S. Liu, W. Huang and Q. Zhao, Chem. Sci., 2019, 10, 9091–9098, 10.1039/C9SC03161H.
- S. Hossen, M. K. Hossain, M. K. Basher, M. N. H. Mia, M. T. Rahman and M. J. Uddin, J. Adv. Res., 2019, 15, 1–18, DOI:10.1016/j.jare.2018.06.005.
- M. C. Pereira, M. Pianella, D. Wei, A. Moshnikova, C. Marianecci, M. Carafa, O. A. Andreev and Y. K. Reshetnyak, Mol. Membr. Biol., 2016, 33, 51–63, DOI:10.1080/09687688.2017.1342969.
- N. M. AlSawaftah, N. S. Awad, W. G. Pitt and G. A. Husseini, Polymers, 2022, 14, 936 CrossRef CAS.
- H. Zhang, W. Gong, Z.-Y. Wang, S.-J. Yuan, X.-Y. Xie, Y.-F. Yang, Y. Yang, S.-S. Wang, D.-X. Yang, Z.-X. Xuan and X.-G. Mei, J. Pharm. Sci., 2014, 103, 2177–2183, DOI:10.1002/jps.24019.
- J. Wang, E. Ayano, Y. Maitani and H. Kanazawa, ACS Omega, 2017, 2, 316–325, DOI:10.1021/acsomega.6b00342.
- A. Hardiansyah, M.-C. Yang, T.-Y. Liu, C.-Y. Kuo, L.-Y. Huang and T.-Y. Chan, Nanoscale Res. Lett., 2017, 12, 355, DOI:10.1186/s11671-017-2119-4.
- M.-S. Martina, J.-P. Fortin, C. Ménager, O. Clément, G. Barratt, C. Grabielle-Madelmont, F. Gazeau, V. Cabuil and S. Lesieur, J. Am. Chem. Soc., 2005, 127, 10676–10685, DOI:10.1021/ja0516460.
- T. Kubo, T. Sugita, S. Shimose, Y. Nitta, Y. Ikuta and T. Murakami, Int. J. Oncol., 2000, 17, 309–315, DOI:10.3892/ijo.17.2.309.
- G. Ma, N. Kostevšek, I. Monaco, A. Ruiz, B. Markelc, C. C. L. Cheung, S. Hudoklin, M. E. Kreft, H. A. F. M. Hassan, M. Barker, J. Conyard, C. Hall, S. Meech, A. G. Mayes, I. Serša, M. Čemažar, K. Marković, J. Ščančar, M. C. Franchini and W. T. Al-Jamal, J. Controlled Release, 2021, 332, 419–433, DOI:10.1016/j.jconrel.2021.03.002.
- A. Hardiansyah, L.-Y. Huang, M.-C. Yang, T.-Y. Liu, S.-C. Tsai, C.-Y. Yang, C.-Y. Kuo, T.-Y. Chan, H.-M. Zou, W.-N. Lian and C.-H. Lin, Nanoscale Res. Lett., 2014, 9, 497, DOI:10.1186/1556-276X-9-497.
- A. Hardiansyah, F. Destyorini, Y. Irmawati, M.-C. Yang, C.-M. Liu, E. R. Chaldun, M.-C. Yung and T. Y. Liu, J. Polym. Res., 2019, 26, 282, DOI:10.1007/s10965-019-1964-5.
- B. D. Cardoso, D. E. M. Fernandes, C. O. Amorim, V. S. Amaral, P. J. G. Coutinho, A. R. O. Rodrigues and E. M. S. Castanheira, Nanomaterials, 2023, 13, 2597 CrossRef CAS.
- M. T. Luiz, J. A. P. Dutra, J. S. R. Viegas, J. T. C. de Araújo, A. G. Tavares Junior and M. Chorilli, Pharmaceutics, 2023, 15, 751 CrossRef CAS.
- P. Drake, A. Algaddafi, T. Swift and R. A. Abd-Alhameed, BioMedInformatics, 2024, 4, 1006–1018 CrossRef.
- S. Zhou, H. Dou, Z. Zhang, K. Sun, Y. Jin, T. Dai, G. Zhou and Z. Shen, Polym. Chem., 2013, 4, 4103–4112, 10.1039/C3PY00522D.
- A. P. Herrera, C. Barrera and C. Rinaldi, J. Mater. Chem., 2008, 18, 3650–3654, 10.1039/B805256E.
- R. A. Bini, R. F. C. Marques, F. J. Santos, J. A. Chaker and M. Jafelicci, J. Magn. Magn. Mater., 2012, 324, 534–539, DOI:10.1016/j.jmmm.2011.08.035.
- P. H. Linh, N. X. Phuc, L. V. Hong, L. L. Uyen, N. V. Chien, P. H. Nam, N. T. Quy, H. T. M. Nhung, P. T. Phong and I.-J. Lee, J. Magn. Magn. Mater., 2018, 460, 128–136, DOI:10.1016/j.jmmm.2018.03.065.
- M. Testa-Anta, M. A. Ramos-Docampo, M. Comesaña-Hermo, B. Rivas-Murias and V. Salgueiriño, Nanoscale Adv., 2019, 1, 2086–2103, 10.1039/C9NA00064J.
- S. Zolghadri, A. G. Asad, F. Farzi, F. Ghajarzadeh, Z. Habibi, M. Rahban, S. Zolghadri and A. Stanek, Pharmaceuticals, 2023, 16, 1680 CrossRef CAS.
- A. Miatmoko, S. A. Safitri, F. Aquila, D. M. Cahyani, B. S. Hariawan, E. Hendrianto, E. Hendradi and R. Sari, Res. Pharm. Sci., 2021, 16, 660–673, DOI:10.4103/1735-5362.327512.
- B. J. Frisken, Appl. Opt., 2001, 40, 4087–4091, DOI:10.1364/AO.40.004087.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427, DOI:10.1007/s12551-016-0218-6.
- A. Miatmoko, A. A. Faradisa, A. A. Jauhari, B. S. Hariawan, D. M. Cahyani, H. Plumeriastuti, R. Sari and E. Hendradi, Sci. Rep., 2022, 12, 21397, DOI:10.1038/s41598-022-26085-2.
- S. Shah, A. Chandra, A. Kaur, N. Sabnis, A. Lacko, Z. Gryczynski, R. Fudala and I. Gryczynski, J. Photochem. Photobiol., B, 2017, 170, 65–69, DOI:10.1016/j.jphotobiol.2017.03.024.
- N. S. Gjerde, A. N. Nardi, C. G. Chen, P. Di Gianvincenzo, M. D’Abramo, A. Scipioni, L. Galantini, S. E. Moya and M. Giustini, Phys. Chem. Chem. Phys., 2022, 24, 25990–25998, 10.1039/D2CP02714C.
- D. Papahadjopoulos, K. Jacobson, S. Nir and I. Isac, Biochim. Biophys. Acta, Biomembr., 1973, 311, 330–348, DOI:10.1016/0005-2736(73)90314-3.
- E. Mazzotta, M. Romeo, Z. Hafidi, L. Perez, I. D. Perrotta and R. Muzzalupo, Pharmaceutics, 2024, 16, 909 CrossRef CAS.
- A. Blicher, K. Wodzinska, M. Fidorra, M. Winterhalter and T. Heimburg, Biophys. J., 2009, 96, 4581–4591, DOI:10.1016/j.bpj.2009.01.062.
- P. Banerjee, S. Yashonath and B. Bagchi, J. Chem. Phys., 2017, 146, 164502, DOI:10.1063/1.4981257.
- J.-H. Lee, B. Kim, Y. Kim and S.-K. Kim, Sci. Rep., 2021, 11, 4969, DOI:10.1038/s41598-021-84424-1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00411j |
| This journal is © The Royal Society of Chemistry 2025 |