
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Leveraging molybdenum sulfur compounds as catalysts for the synthesis of biobased poly(ethylene 2,5-furandicarboxylate) and recycling†
Dmitrii
Razinkov
 a,
Beatriz
Agostinho
b,
Sigridur G.
Suman
*a and
Andreia F.
Sousa
*bc
a,
Beatriz
Agostinho
b,
Sigridur G.
Suman
*a and
Andreia F.
Sousa
*bc
aScience Institute, University of Iceland, Dunhagi 3, 107, Reykjavik, Iceland
bCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal. E-mail: andreiafs@ua.pt
cCentre for Mechanical Engineering, Materials and Processes, Department of Chemical Engineering, University of Coimbra Rua Sílvio Lima – Polo II, 3030-790 Coimbra, Portugal
First published on 27th September 2024
Abstract
The uncovered potential of two safe dinuclear molybdenum complexes with non-rigid bidentate phosphinoyldithio formate ligands, each distinguished by their phosphorus atom substituents, was demonstrated for the first time in both the synthesis of poly(ethylene furan 2,5-dicarboxylate) (PEF) and its depolymerization. An in-depth evaluation of the reaction conditions, in terms of time and temperature, showcases the air and water stable handling of catalysts and their suitability to mediate PEF synthesis in a solvent-free, bulk polycondensation reaction, resulting in high efficiency and comparable properties as obtained with a benchmark titanium-based catalyst. Notably, excellent thermal properties and optically transparent polymers with over 89% transmittance in the visible region were achieved. Furthermore, these innovative molybdenum complexes were also able to efficiently prompt the chemical recycling of PEF through a glycolysis pathway under very mild conditions and in short reaction times.
Sustainability spotlightLeveraging molybdenum sulfur compounds as catalysts for retrosynthesis of biobased poly(ethylene 2,5-furandicarboxylate) and recycling the vast majority of globally produced polymers is fossil-based and heavily associated with greenhouse gas emissions. Developing more sustainable alternatives anchored in bio-based plastics is, thus, urgently needed. Beyond their greener origin, their synthesis and end-of-life should not be overlooked to move towards more sustainable polymers. In this work, the synthesis of a biobased furanic polyester, poly(ethylene 2,5-furandicarboxylate) (PEF), in a one-pot, solvent-free, bulk polycondensation reaction employing non-toxic, air and water-stable catalysts is reported. Additionally, these catalysts were revealed to be useful for PEF's end-of-life chemical recycling by prompting its depolymerization through glycolysis under mild conditions. This work contributes to the following UN sustainable development goals: responsible consumption and production (SDG 12) and climate action (SDG 13). |
Introduction
Over the last few decades, extensive search for more sustainable chemicals, polymers, and materials has witnessed interest in bio-based derivatives. Furan-based ones are among the most spotlighted1–3 due to their potential to replace their conventional fossil-based counterparts. In this regard, poly(ethylene 2,5-furandicarboxylate) (PEF) can compete with fossil-based poly(ethylene terephthalate) (PET) due to a unique set of similar or even enhanced thermal, mechanical, and barrier properties.4,5 For example, PEF has superior gas barrier properties towards carbon dioxide (≈15×) and oxygen (≈10×) compared to PET, which spans this polymer applications into, for example, high-barrier packaging films in the food sector.3,6–9 Also, PEF has a melting temperature of 30 °C lower than PET, allowing PEF processing into different objects at lower temperatures and, hence, saving energy. However, some technical barriers persist that hamper its full market introduction and the limited availability of the 5-hydroxymethylfurfural (HMF) intermediate for 2,5-furandicarboxylic acid (FDCA) synthesis. Other relevant issues concern the sustainability aspects, such as the need to implement green polymerization synthetic paths and to develop advanced recycling strategies to manage future PEF waste according to circular and sustainable principles.10Very often, greener approaches for PEF synthesis, using milder temperatures, with negligible use of solvents, and/or using safe catalysts are thoroughly overlooked. The current typical procedure for synthesizing furan-based polymers is still a two-stage bulk polycondensation reaction carried out at high temperatures (up to 220–260 °C) over several hours and using oxygen- and moisture-sensitive titanium alkoxide complexes or hazardous Sb2O3 as a catalyst.2,11–25 Other alternative catalysts, including Zn(AcO)2 or Al(acac)3, require additional steps to achieve high molecular weight PEF, reaching an overall process time of 78 hours.26
Notably, a few studies address the mild enzymatic catalysis of furan-based polymers, though the reaction time typically exceeds days and has been revealed to be inefficient for PEF synthesis27–30 Nevertheless, an interesting study by Loos et al. introduced the combined use of Candida Antarctica lipase B and innovative solvents such as deep eutectic solvents (DESs) or ionic liquids for the polymerization of several furan-based polyesters.27 The results are promising, but still, products were isolated in very moderate yields (0.9–56%) with a number-average molecular weight of 1000–3000 g mol−1, and the approach was not applied in the case of PEF. Imidazolium-based ionic liquids were successfully used as catalysts for the synthesis of PEF with good molecular weight and thermal properties.25 However, besides poor biodegradability, ionic liquids can be toxic and harmful to the environment.31–35
As mentioned previously, titanium alkoxide complexes, such as titanium(IV)butoxide (TBT) and titanium(IV)isopropoxide, are still the most used catalysts to attain high molecular weight PEF polymers. These complexes are air- and water-sensitive, requiring extra care in handling. They are also decomposed in the reaction, preventing potential catalyst recycling and reuse.36,37 For example, by exposure to the atmosphere or in the presence of traces of water, they readily form dioxides.38,39 In this vein, catalysts that are air- and water-stable and exhibit higher stability under the reaction conditions may offer alternative routes to PEF synthesis and its recycling, thereby improving the process efficiency and sustainability.
Molybdenum sulfides are well known for their catalytic performance in the desulfurization of petrochemical mixtures and stability at elevated temperatures in related processes.40,41 Furthermore, molybdenum clusters and complexes are air-stable and water-tolerant. These molecular catalysts have proven to be efficient in the hydrogen evolution reaction and sulfur transfer reactions.42–49 Furthermore, molybdenum complexes with disulfide and α-amino acid ligands were shown to have very high IC50 values well above 100 μM in various cell lines.50,51 Molybdenum complex toxicity has been directly related to the ligands coordinated to the molybdenum centers. Sn(IV) complexes with phosphinoyldithio formate ligands show efficiency against cancer cells, although the same study showed that the ligands themselves are not cytotoxic.52
One important example is dinuclear molybdenum complexes with phosphinoyldithio formate ligands [Mo2O2(μ-S)2[R2P(O)CS2]2] (1, R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Bn; 2, RPh) (Scheme 2).53 These complexes differ in the organic substituents of the phosphorus atom; detailed spectroscopic studies confirmed that they are non-rigid and very flexible molecules and the ligands are ambidentate possessing S,S- or S,O-bidentate, or S-monodentate coordination abilities (Fig. S1 in the ESI†).53–55 Their non-rigid properties in solution offer easy access to the Mo centers displaying catalytic activity in desulfurization reactions of thiiranes where the catalysts remained intact. Both complexes showed high conversion in short reaction time, with the highest catalytic activity achieved with the Mo-complex 1.53 These complexes are stable in oxygen- and moisture-rich atmospheres compared to well-known oxophilic titanium alkoxide.53 Despite these complexes' advantages, their catalytic activity has never been explored in transesterification reactions. Therefore, to close this gap, two innovative dinuclear molybdenum complexes with phosphinoyldithio formate ligands with different substituents on the phosphorus atom were used in both PEF synthesis and its chemical recycling. Depolymerization reactions were also studied to prove their dual role and the need to find greener recycling strategies for PEF.
Bn; 2, RPh) (Scheme 2).53 These complexes differ in the organic substituents of the phosphorus atom; detailed spectroscopic studies confirmed that they are non-rigid and very flexible molecules and the ligands are ambidentate possessing S,S- or S,O-bidentate, or S-monodentate coordination abilities (Fig. S1 in the ESI†).53–55 Their non-rigid properties in solution offer easy access to the Mo centers displaying catalytic activity in desulfurization reactions of thiiranes where the catalysts remained intact. Both complexes showed high conversion in short reaction time, with the highest catalytic activity achieved with the Mo-complex 1.53 These complexes are stable in oxygen- and moisture-rich atmospheres compared to well-known oxophilic titanium alkoxide.53 Despite these complexes' advantages, their catalytic activity has never been explored in transesterification reactions. Therefore, to close this gap, two innovative dinuclear molybdenum complexes with phosphinoyldithio formate ligands with different substituents on the phosphorus atom were used in both PEF synthesis and its chemical recycling. Depolymerization reactions were also studied to prove their dual role and the need to find greener recycling strategies for PEF.
Results and discussion
PEF synthesis was carried out by a bulk polyesterification reaction approach (Scheme 1) exploring the ability of non-toxic dinuclear molybdenum complexes with benzyl substituted ligand 1 and phenyl substituted ligand 2 (Scheme 2), under a range of different temperature and time conditions. For comparison reasons, non-catalyzed reactions and PEF synthesis using the conventional TBT catalyst were also studied. | ||
| Scheme 1 Synthesis and depolymerization of PEF. | ||
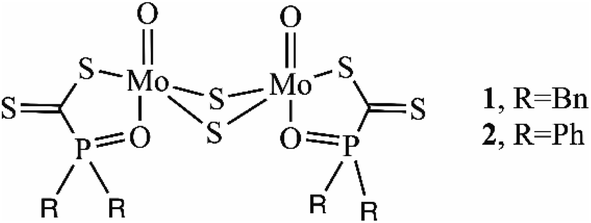 | ||
| Scheme 2 Dinuclear molybdenum complexes of phosphinoyldithio formate with benzyl substituted ligand 1 and phenyl substituted ligand 2. | ||
Screening the efficiency of the dinuclear molybdenum complexes for polytransesterification reactions
Both the molybdenum complexes 1 and 2 proved to be able to catalyze a polytransesterification reaction, although demonstrating different catalytic activities toward PEF synthesis under conventional fixed conditions (heating ramp ranging 190–230 °C for 9 h and applying vacuum). Employing 1 as the catalyst led to the highest PEF isolation yield, number-average molecular weight (Mn), and intrinsic viscosity (81%, 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, and 0.25 dL.g−1, respectively), compared to TBT and 2 (Table 1, entries 1, 5 and 8). TBT proved to be an efficient catalyst for the synthesis of PEF in good to excellent yields and high molecular weights.14 Similar results were achieved using molybdenum complex 1 (entry 1 vs. 8 of Table 1). Moreover, complex 1 is not sensitive to moisture and oxygen, simplifying the handling and storage procedures.
000, and 0.25 dL.g−1, respectively), compared to TBT and 2 (Table 1, entries 1, 5 and 8). TBT proved to be an efficient catalyst for the synthesis of PEF in good to excellent yields and high molecular weights.14 Similar results were achieved using molybdenum complex 1 (entry 1 vs. 8 of Table 1). Moreover, complex 1 is not sensitive to moisture and oxygen, simplifying the handling and storage procedures.
| Entry | Experimental conditions (temperature, time) | Catalyst | Isolation yield (%) | M n | Intrinsic viscosity, dL.g−1 | |
|---|---|---|---|---|---|---|
| 1st stage a | 2nd stage | |||||
| a Catalysts were introduced in the monomer slurry at 120 °C before raising the reaction temperature in the first stage. b Intense sublimation. No polymer was isolated at the end of the reaction. c Complex 2 was introduced in the monomer slurry before heating the reaction. d M n was determined using 1H NMR data (Appendix S1). | ||||||
| 1 (PEF_1) | 190 °C, 5h | 170–230 °C, 4h | 1 | 81 | 19000 |
0.25 |
| 2 (PEF_2) | 150–190 °C, 6h | 170–230 °C, 4h | 1 | 73 | 6400 | 0.16 |
| 3 | 150/170 °C, 3/2h | —b | 1 | No product | ||
| 4 | 150/170 °C, 5/4 h | —b | 1 | No product | ||
| 5 (PEF_5) | 190 °C, 5h | 170–230 °C, 4h | 2 | 54 | 4300 | 0.14 |
| 6 (PEF_6) | 190 °C, 5h c | 170–230 °C, 4h | 2 | 46 | 4800 | — |
| 7 | 150/170 °C, 3/2h | —b | 2 | No product | ||
| 8 (PEF_8) | 190 °C, 5h | 170–230 °C, 4h | TBT | 91 | 12700 |
0.18 |
| 9 (PEF_9) | 190 °C, 5h | 170 – 230 °C, 4h | no catalyst | 33 | n.d. | — |
The success of PEF formation employing the dinuclear molybdenum complexes 1 and 2 was confirmed by FTIR and proton and carbon 13 nuclear magnetic resonance (1H and 13C NMR) analyses (Fig. S2–4 in the ESI†). The typical ATR FTIR spectrum of PEF_1 (Fig. 3a and S2 in the ESI†) displays the expected vibrational features of PEF: symmetric and asymmetric C–H stretching (2970 cm−1); CO stretching of the ester group (1718 cm−1); symmetric and asymmetric stretching of C–O–C (1266 cm−1); and several vibrations associated with 2,5-disubstituted furans (1578, 1132, 967, 834, and 762 cm−1, including two weak C–H stretches at 3156 and 3122 cm−1).15 The absence of any detectable band ascribed to the O–H stretching mode is in accordance with the high molecular weight attained. The 1H and 13C NMR spectra (Fig. S3 and 4 in the ESI†) also corroborate PEF's successful formation.
Optimization of polytransesterification conditions
A moderately low temperature range, between 150 and 190 °C, for 5–9 hours was tested in the first stage (Table 1). The results show that the synthesis of PEF under these conditions is inadequate (entries 2–4 and 7), even if the reaction time of the first stage was increased from the conventional 5 to 9 hours (entries 3 and 4, respectively). The best conditions in terms of both yield and molecular weight for synthesizing PEF were revealed to be with a temperature range between 190 and 230 °C for 9 hours for both dinuclear molybdenum complexes (entries 1 and 5, Table 1), which is in accordance with previous results obtained when using TBT and this work (PEF_8, entry 8, Table 1).11,15–18,24 The Mo-complex catalysts are active in the homogeneous desulfuration of episulfides according to a proposed mechanism where the substrate is activated by coordination to the Mo center;53 a similar mechanism may take place here. Adding the catalyst at the beginning of the reaction to the monomers' slurry, without pre-melting of the monomers, led to a lower isolation yield (entry 6, Table 1), which is probably associated with lower homogenization of the reaction mixture.PEF thermal and crystallinity properties
The thermal properties of the synthesized PEF polymers were studied using TGA, DMA, and DSC analyses (Table 2 and S5–7 in the ESI†). The obtained polymers employing 1 and 2 showed high thermal stability: 5% weight loss was observed at 341–349 °C and a maximum weight loss at 378–387 °C (PEF_1,2,5). The results are closely aligned with those obtained for TBT-catalyzed PEF (PEF_8) and similar to previously reported values.15–18,56| Sample | TGA | DMA | DSC | ||
|---|---|---|---|---|---|
| T d,5%/°C | T d, max/°C | T g/°C | T cc/°C | T m/°C | |
| PEF_1 | 348.3 | 380.0 | 122 | 152.3 | 201.0 |
| PEF_2 | 348.7 | 386.9 | 121 | 155.1 | 199.6 |
| PEF_5 | 341.9 | 378.4 | 115 | — | 189.0 |
| PEF_8 | 350.6 | 382.3 | 109 | 152.8 | 211.2 |
| PEF_9 | 370.6 | 413.4 | 110 | 138.8 | 199.1 |
The glass transition temperature (Tg) was determined as the maximum peak of tan δ of the DMA thermogram (Fig. S5 in the ESI†). The Tg was observed at 115–122 °C for the polymers synthesized employing catalysts 1–2 (PEF_1,2,5). Slightly lower values of Tg were observed for the polymers synthesized without a catalyst (110 °C, PEF_9) and employing TBT (109 °C, PEF_8). The cold crystallization (Tcc) and the melting (Tm) temperatures of the PEF polymers were determined by DSC (Fig. S6 in the ESI†). The Tcc was detected at approximately 152–155 °C, and the Tm varied between 152 and 155 °C. Slightly higher values were obtained for PEF_2, and lower thermal features were observed for the non-catalyzed PEF_9. Overall, as expected, the thermal properties of PEF catalyzed by the Mo-complexes are in accordance with the literature data.7,57,58
The expected semi-crystalline nature of PEF was confirmed by X-ray diffraction. The obtained patterns displayed three sharp signals (Fig. 1) at 2θ ∼15.98, 20.58 and 27.96° (PEF_1). These results are in close agreement with previously reported data for PEF.15,59
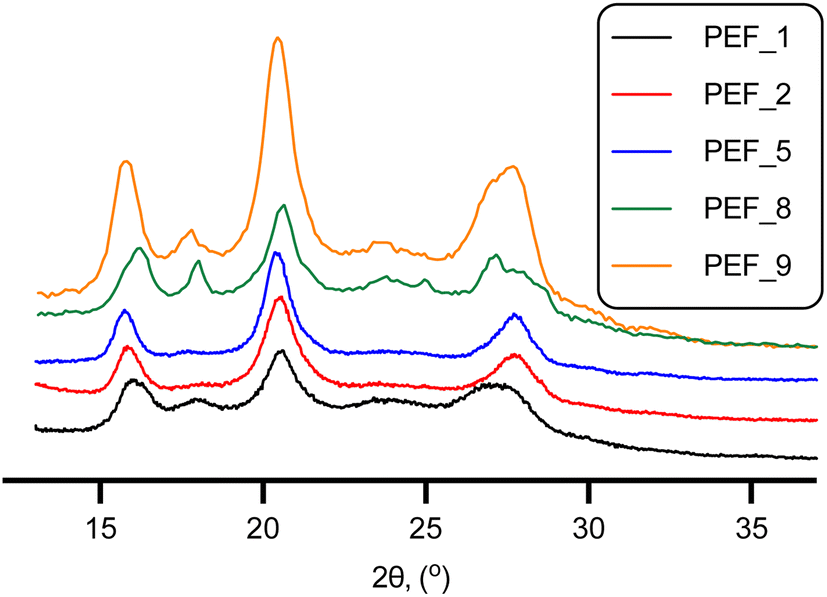 | ||
| Fig. 1 XRD patterns of PEF. | ||
PEF optical properties
High temperatures and certain catalysts such as cobalt, germanium, and manganese have been reported to yield colored PEF (typical yellowing of some PEF-related products).60,61 However, this is an important property that may hamper market introduction in relevant applications where color plays a crucial role, such as packaging. Therefore, in this work, the UV-visible spectra of PEF synthesized with the new Mo complexes (Fig. 2 and Table 3) were recorded and compared with those of TBT-catalyzed PEF. Importantly, Mo-mediated catalysis of PEF leads to optically transparent polymers with over 89% transmittance in the visible region (400–700 nm).63 The optical transmittance of semi-crystalline PEF_1 is 89.0 at 400 nm, higher than those reported for PET and its copolymers.64,65 Also, there is a correlation between coloring and absorbance at 400 nm: a lower extinction coefficient at 400 nm results in a more transparent polymer.60,62 Importantly, comparable results between those obtained for 1 and reported for TBT62 were obtained (e.g., 3.16 and 2.55 M−1 cm−1 at 400 nm, respectively), supporting the suitability of the Mo complex 1. Slightly higher values were obtained when complex 2 was adopted instead (4.50 M−1 cm−1 at 400 nm).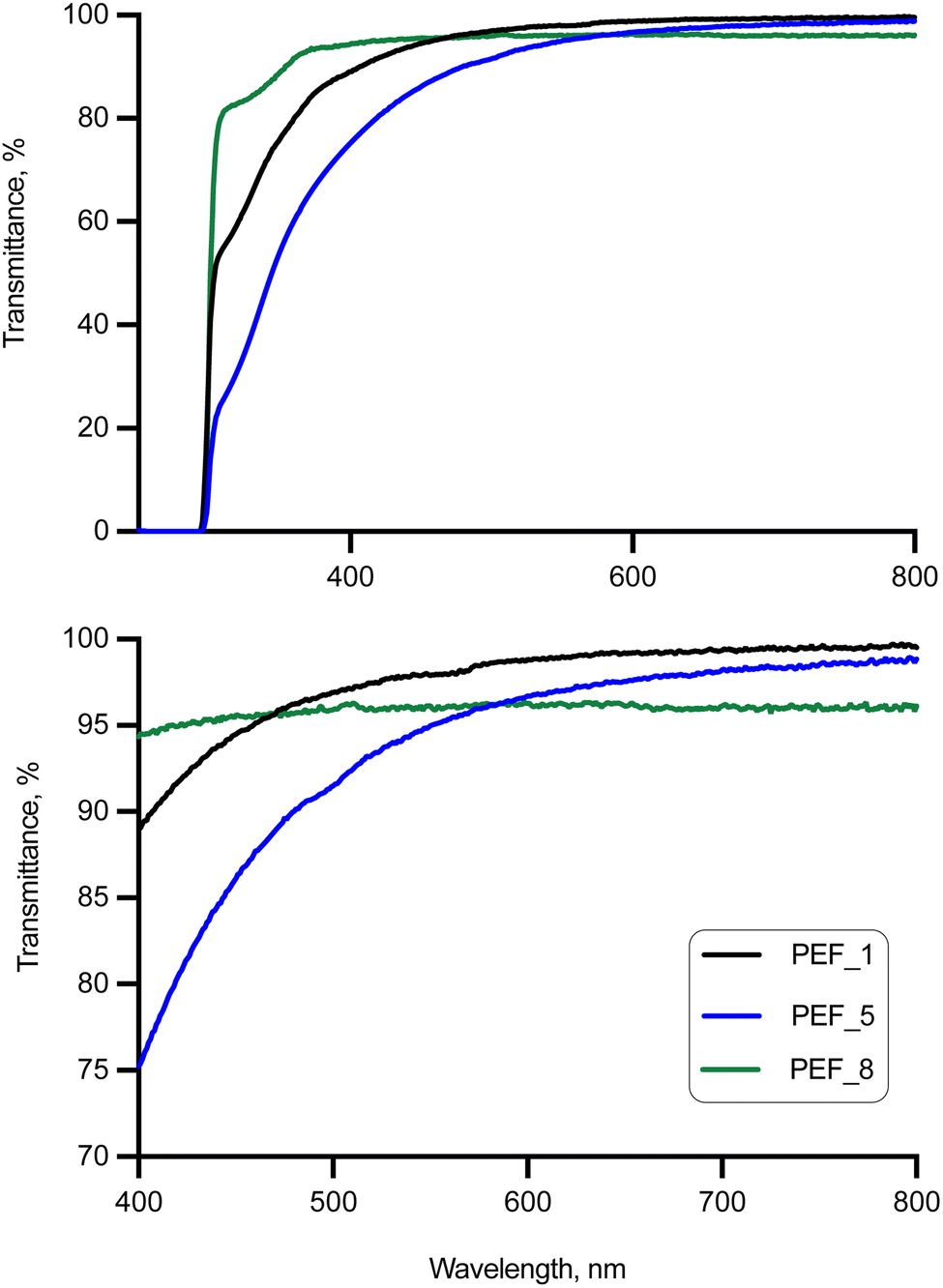 | ||
| Fig. 2 UV-visible spectra of PEF samples (top) and magnification inset (bottom). | ||
| Sample | C, mol L−1 10−2 | A 400nm | E/M−1 cm−1 | % T400nm | % T450nm | % T700nm |
|---|---|---|---|---|---|---|
| a PEF synthesized employing TBT as a catalyst. b Results reported in ref. 62. | ||||||
| PEF_1 | 1.60 | 0.0505 | 3.16 | 89.0 | 94.5 | 99.3 |
| PEF_5 | 2.74 | 0.1236 | 4.50 | 75.2 | 86.1 | 98.2 |
| PEF_81 | 2.75 | 0.0253 | 0.92 | 94.3 | 95.6 | 96.1 |
| PEF_82 | 2.75 | 0.0700 | 2.55 | 85.1 | — | — |
| PEF_9 | 2.13 | 0.0434 | 2.04 | 90.5 | 88.0 | 98.2 |
Proof-of-concept for PEF recycling
As a proof-of-concept, within the imperative need to address the end-of-life of future commercial polymers such as biobased PEF,4,66 its chemical recycling employing 1 or 2 and a glycolysis pathway67 at moderate temperature and ambient room pressure was performed (Scheme 1). This allowed us to assess the versatility of these complexes to catalyze glycolysis reactions at moderate temperature, at ambient room pressure, and without the need for an inert atmosphere. Nevertheless, both complexes depolymerized a semi-crystalline PEF film with an intrinsic viscosity of 0.25 dL.g−1. Once more, 1 was revealed to be a more efficient catalyst also for PEF recycling, converting PEF into its building-block derivatives. The weight loss of PEF was followed gravimetrically and calculated as described elsewhere,67 reaching 51% weight loss when using complex 1 and slightly less for 2 (34%). The results suggest that 1 is a more efficient catalyst than 2 also for the chemical recycling of PEF. The depolymerization product was confirmed by both FTIR and 1H and 13C NMR spectroscopies (Fig. 3b, S8–10 in the ESI†) to be a bis(2-hydroxyethyl) furan-2,5-dicarboxylate derivative, most probably an oligomer as determined using 1H NMR integration areas (calculations are described in Appendix S1† and elsewhere).68 The 1H NMR spectra (Fig. S9 in the ESI†) also suggest the presence of typical observed ether bridges with relevant resonances at 4.00 and 4.75 ppm.69 As expected,15,67 the FTIR spectrum of this product displays the typical bands of the 2,5-disubstituted furans (1148; 967; 834 and 766 cm−1) and the CO stretching vibrations of the esters (1728 cm−1); and it also displays a broad band near 3544 cm−1 ascribed to the O–H stretching mode of alcohols. The chemical nature of the depolymerization products was further elucidated using electrospray ionization mass spectrometry and the ensuing spectra compared with simulated patterns (Fig. S11–13 in the ESI†). The mass spectrum revealed two major signals at m/z = 545 and m/z = 641. The measured and simulated patterns corroborate the oligomeric nature of the recycled products, essentially composed of 3 repeating units, suitable for PEF synthesis. Comparable methods of chemical recycling of PEF have been reported before.67 The urea: zinc acetate deep eutectic solvent showed high activity in the mild glycolysis of PEF in a reaction time of one hour. However, only 17% of PEF was depolymerized through glycolysis for each 1 mol% of deep eutectic solvent after one hour, whereas employing a similar amount of 1 or 2 under similar conditions showed better results (35 and 27%, respectively). It has been reported before that glycolysis depolymerization products, PEF or PET homologous, can be repolymerized independently of being oligomers.67,70
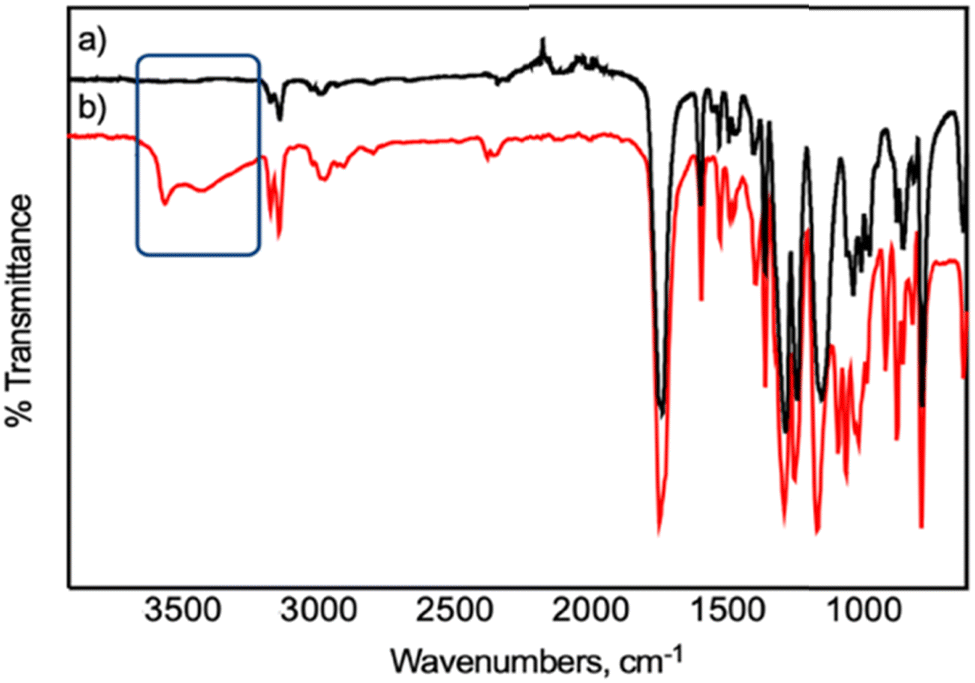 | ||
| Fig. 3 ATR FTIR spectra of (a) the initial PEF_1 and (b) its depolymerisation products. | ||
Conclusions
Non-toxic and safe phosphinoyldithio formate molybdenum complexes with Bn and Ph substituents (1 and 2) were tested in the synthesis of biobased PEF, highlighting their suitability to catalyze polycondensation reactions and advantageously offering easy handling and use. Complex 1 was revealed to be a more effective catalyst, enabling the formation of PEF (PEF_1) with the highest number-average molecular weight (19000), intrinsic viscosity (0.25 dL.g−1), and yield (81%). The benchmark results using TBT for PEF synthesis are in accordance with these values; however, a lower number-average molecular weight PEF (12700) was attained. Employing 1 resulted in a 49% higher number-average molecular weight of the polyester (19000 vs. 12700, respectively, for 1 and TBT) and 38% higher intrinsic viscosity (0.25 vs. 0.18 dL.g−1, respectively, for 1 and TBT). Mo-mediated catalysis of PEF leads to optically transparent polymers with over 89% transmittance in the visible region (400–700 nm). In-depth characterization of PEF polymers synthesized using the innovative catalytic system compared to benchmark TBT showed that Tg and Tm were in close agreement, including with previously published results. All polyesters were shown to be highly thermally stable materials since 5% weight loss was determined at 341–371 °C.
The reversibility of this reaction employing these air-tolerant and innovative molybdenum complexes was also examined due to its relevance for the future recycling of this polyester waste. Both complexes demonstrated the ability to convert PEF into its building-block derivatives under mild glycolysis and in short reaction times, confirming that they are efficient dual-direction catalysts. The results show that 1 is a more effective catalyst for the chemical recycling of PEF compared to 2 and previously reported deep eutectic solvents. The non-rigid behavior of the complex ligands may explain the high catalytic activity of the complexes, whereas complex 1 likely has more degrees of freedom, whereas the stability of symmetric coordination of the ligand in solution possibly causes lower catalytic activity of 2.
Experimental
Chemicals and materials
Dimethyl furan-2,5-dicarboxylate (99.9%) was purchased from Sarchem Laboratories, Inc. Ethylene glycol (EG, anhydrous, 99.8%), titanium(IV) butoxide (TBT, 97%), trifluoroacetic acid (TFA, 99%), deuterated trifluoroacetic acid (TFA-d, 99.5%), 1,1,2,2-tetrachloroethane (TCE, 98%), deuterated 1,1,2,2-tetrachloroethane (TCE-d2, 99.5%), phenol (99%), methanol (99%), chloroform (99.8%) and deuterated chloroform (CDCl3, 99.8%) were purchased from Sigma-Aldrich.Analytical techniques
Infrared spectra were recorded with a PARAGON 1000 PerkinElmer FTIR spectrophotometer equipped with a single horizontal Golden Gate ATR cell in the mid-IR range. 1H and 13C NMR spectra were obtained on a Brucker AMX 300 spectrometer at 300 and 75 MHz, respectively. UV-visible spectra were recorded on a Cary UV-Vis Multicell Peltier employing a solvent mixture of chloroform and TFA (200 ml chloroform and 14 ml TFA). X-Ray Diffraction (XRD) patterns were recorded on a Philips X'Pert MPD instrument operating with Cu Kα radiation (λ = 1.5405980 Å) at 45 kV and 20 mA. Samples were scanned in the 2θ range of 5 to 60°, with a step size of 0.105° and a step time of 400 s. Differential Scanning Calorimetry (DSC) analysis thermograms were obtained with a Pyris Diamond DSC calorimeter from PerkinElmer (Waltham, MA, USA), using nitrogen as purging gas (20 ml min−1) and aluminum pans to encapsulate the samples (ca. 5 mg). Scans were carried out under nitrogen with a heating rate of 10 °C min−1 in the temperature range from 20 to 250 °C. Two heating/cooling cycles were repeated. Dynamic Mechanical Thermal Analyses (DMTA) were performed in a material pocket accessory with a Tritec 2000 DMA Triton, operating in the single cantilever mode. Tests were performed at 1 and 10 Hz and the temperature was varied from −100 to 250 °C, at a 2°C min−1 heating rate. Tg was determined as the maximum peak of tan δ. Thermogravimetric analyses (TGA) were carried out using a METTLER TOLEDO TGA 2 analyzer equipped with a platinum cell, using platinum pans to encapsulate the samples (ca. 5 mg). Thermograms were recorded under a nitrogen flow (50 ml min−1) and heating at a constant rate of 20 °C min−1 from room temperature up to 800 °C. Intrinsic viscosities were measured on a Ubbelohde-type viscometer employing a mixture of phenol/1,1,2,2-tetrachloroethane (50/50) (wt%/wt%) as a solvent at 25 °C. The polymer was dissolved in that solvent mixture (0.1 g per 20 ml). The intrinsic viscosity was determined using the ratio of specific viscosity and polymer solution concentration (ηsp/C where ηsp = (t1 − t0)/t0 and t0 and t1 are the solvent mixture elution times of the solvent mixture and polyester solution, respectively). Number average molecular weight (Mn) was determined using 1H NMR data (Appendix S1 and Fig. S11†).68 Mass spectra (MS) were recorded using a micrOTOF-Q spectrometer, equipped with an E-spray atmospheric pressure ionization chamber (ESI). 5 mg of the sample was dissolved in 1 ml of chloroform with a few drops of trifluoroacetic acid. 2 μL of the solution were dissolved in 0.5 ml of methanol and injected directly into the ESI ionization unit of the mass spectrometer. Predictive spectra were obtained and attributions were performed using Compass HyStar software from Bruker.Synthesis of dinuclear molybdenum complexes
[Mo2O2(μ-S)2[R2P(O)CS2]2] (1; RBn, 2; RPh) complexes were prepared as reported elsewhere.53 Briefly, the complexes were synthesized in ligand exchange reactions of [Me4N][R2P(O)CS2] (RBn or Ph) (0.64 mmol) in DMF (5 ml) by dropwise addition of a solution of (Me4N)2[Mo2O2(μ-S)2(Cl)4] (0.32 mmol) in DMF (5 ml), under stirring. The solution was stirred for one hour. Diethyl ether (25 ml) was added to induce precipitation of the byproduct (Me4NCl) which was filtered off. Solvents were removed under reduced pressure; the solids were washed with diethyl ether (20 ml) and dried in vacuo. The analytically pure compound was obtained in 60–70% yield. Purity was verified using 1H NMR and elemental analysis. Both complexes are air-and-water stable and do not require extra care in handling compared to TBT.
PEF synthesis
PEF synthesis was carried out using an adapted procedure from a previously described report.24,71 Ethylene glycol (0.03 mol) was added to DMFDC (0.0136 mol) under a nitrogen atmosphere. The mixture was pre-heated at 120 °C for several minutes until it melted to add the catalyst (400 ppm of 1, 2, or TBT) (Table 1, entries 1–5,7,8). The addition of the catalyst (400 ppm of 2) before starting to heat the reaction was also tested (Table 1, entry 6). Subsequently, in the first stage, the reaction mixture was heated under nitrogen for several hours until a theoretical amount of methanol was collected in the trap. In the case of using TBT as a catalyst, a nitrogen flux was used to avoid its decomposition. In the second stage, a high vacuum was applied to remove the excess ethylene glycol, and then the temperature was gradually increased. The reactions continued at evaluated temperatures for several hours. At the end of the reaction, the reaction mixture was dissolved in 100 ml of chloroform with a few drops of trifluoroacetic acid, and the polymer was precipitated by pouring this solution into excess cold methanol, followed by filtration and drying at 40 °C overnight. Yield: 46–91%. An additional PEF synthesis carried out with no catalyst was also performed (Table 1, entry 9). Yield: 33%. The experimental conditions are summarized in Table 1. FT-IR, cm−1: 3122 (C–H, furan ring), 2970 (C–Hsym/asym), 1718 (CO), 1578 (CC), 1266 (C–Osym/asym), 1132 (=C–O–C, furan ring), 967, 834, 762 (C–H, furan ring). 1H NMR (CDCl3 + TFA-d), δ, ppm: 7.30 (s, 2H, C–H (furan ring)), 4.71 (s, 4H, CH2), 4.56 (t, traces, CH2), 4.13 (t, traces, CH2). 13C NMR (CDCl3 + TFA-d), δ, ppm: 159.58 (s, CO), 146.33 (s, C2, C5 furan ring), 120.33 (s, C3, C4 furan ring), 63.94 (s, CH2). XRD, 2θ: 15.98°, 20.58°, 27.96° (PEF_1); 16.29°, 20.76°, 27.59° (PEF_2); 15.85°, 20.52°, 27.90° (PEF_5); 16.26°, 20.69°, 27.21° (PEF_8); 15.93°, 20.56°, 27.80° (PEF_9).
Depolymerization of PEF
PEF (0.0011 mol), ethylene glycol (0.018 mol), and a catalyst 1 or 2 (1 mol%) were heated at 180 °C for one or two hours. After the reaction, the mixture was cooled down and water (150 ml) was added. The white precipitate was filtered off, washed with 20 ml of water, and dried at 40 °C overnight. FT-IR, cm−1: 3543, 3408 (O–H), 3122 (C–H, furan ring), 2957 (C–Hsym/asym), 1728 (CO), 1573 (CC), 1269 (C–Osym/asym), 1148 (=C–O–C, furan ring), 966, 834, 766 (C–H, furan ring). 1H NMR (TCE-d2 + TFA-d), δ, ppm: 7.36 (m, 8H, C–H (furan ring)), 4.75, 4.69 (s, 12H, CH2), 4.56 (t, 4H, CH2), 4.12 (t, 4H, CH2), 4.00 (s, 2H, CH2). 13C NMR (TCE-d2 + TFA-d), δ, ppm: 158.90, 158.79 (s, CO), 145.69, 145.32, 145.27 (s, C2, C5 furan ring), 119.63 (s, C3, C4 furan ring), 63.30, 64.51, 63.13, 62.40, 60.22 (s, CH2).
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, D. R., B. A., S. G. S. and A. F. S.; data curation, D. R., B. A., S. G. S. and A. F. S.; formal analysis, D. R. and B. A.; funding acquisition, S. G. S., and A. F. S.; investigation, D. R., B. A., S. G. S., and A. F. S.; methodology D. R. and B. A; project administration, A. F. S.; resources, D. R., B. A., S. G. S. and A. F. S.; software, D. R. and B. A.; supervision, S. G. S. and A. F. S.; validation, D. R., B. A., S. G. S and A. F. S.; visualization, D. R. and A. F. S.; writing – original draft, D. R. and A. F. S.; writing – review and editing, D. R., B. A., S. G. S and A. F. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by COST Action FUR4Sustain (CA18220) supported by COST (European Cooperation in Science and Technology). This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI 10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI 10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI 10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). The FCT is acknowledged for the research contract under Scientific Employment Stimulus to AFS (CEECIND/02322/2020 and DOI 10.54499/2020.02322.CEECIND/CP1589/CT0008) and for a doctorate grant to BA (2020.04495.BD). DR thanks for the STSM grant under the framework of COST Action CA18220, and DR and SGS acknowledge the Icelandic Centre of Research for support (grant no.173667). Ira Volkova is thanked for the illustration of the graphical abstract of this article.Notes and references
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 6096 RSC.
- N. Lotti, A. Munari, M. Gigli, M. Gazzano, V. Tsanaktsis, D. N. Bikiaris and G. Z. Papageorgiou, Polymer, 2016, 103, 288–298 CrossRef CAS.
- E. de Jong, H. A. Visser, A. S. Dias, C. Harvey and G. J. M. Gruter, Polymers, 2022, 14, 943 CrossRef CAS PubMed.
- A. F. Sousa, R. Patricio, Z. Terzopoulou, D. N. Bikiaris, T. Stern, J. Wenger, K. Loos, N. Lotti, V. Siracusa, A. Szymczyk, S. Paszkiewicz, K. S. Triantafyllidis, A. Zamboulis, M. S. Nikolic, P. Spasojevic, S. Thiyagarajan, D. S. van Es and N. Guigo, Green Chem., 2021, 23, 8795–8820 RSC.
- P. Sahu, A. Thorbole and R. K. Gupta, ACS Sustain. Chem. Eng., 2024, 12, 6811–6826 CrossRef CAS.
- S. K. Burgess, G. B. Wenz, R. M. Kriegel and W. J. Koros, Polymer, 2016, 98, 305–310 CrossRef CAS.
- S. K. Burgess, O. Karvan, J. R. Johnson, R. M. Kriegel and W. J. Koros, Polymer, 2014, 55, 4748–4756 CrossRef CAS.
- S. K. Burgess, R. M. Kriegel and W. J. Koros, Macromolecules, 2015, 48, 2184–2193 CrossRef CAS.
- C. F. Araujo, M. M. Nolasco, P. J. A. Ribeiro-Claro, S. Rudic, A. J. D. Silvestre, P. D. Vaz and A. F. Sousa, Macromolecules, 2018, 51, 3515–3526 CrossRef CAS.
- L. Carlsen and R. Bruggemann, Int. J. Sustain. Dev. World, 2022, 29, 219–229 CrossRef.
- S. Zaidi, M. J. Soares, A. Bougarech, S. Thiyagarajan, N. Guigo, S. Abid, M. Abid, A. J. D. Silvestre and A. F. Sousa, Eur. Polym. J., 2021, 150, 5324–5332 CrossRef.
- M. J. Soares, P. K. Dannecker, C. Vilela, J. Bastos, M. A. R. Meier and A. F. Sousa, Eur. Polym. J., 2017, 90, 301–311 CrossRef CAS.
- J. P. Ma, X. F. Yu, J. Xu and Y. Pang, Polymer, 2012, 53, 4145–4151 CrossRef CAS.
- J. P. Ma, Y. Pang, M. Wang, J. Xu, H. Ma and X. Nie, J. Mater. Chem., 2012, 22, 3457–3461 RSC.
- A. Gandini, A. J. D. Silvestre, C. P. Neto, A. F. Sousa and M. Gomes, J. Polym. Sci., 2009, 47, 295–298 CrossRef CAS.
- M. Gomes, A. Gandini, A. J. D. Silvestre and B. Reis, J. Polym. Sci., 2011, 49, 3759–3768 CrossRef CAS.
- P. Gopalakrishnan, S. Narayan-Sarathy, T. Ghosh, K. Mahajan and M. N. Belgacem, J. Polym. Res., 2013, 21, 340 CrossRef.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Y. Zhou, J. Polym. Sci., 2012, 50, 1026–1036 CrossRef CAS.
- R. J. I. Knoop, W. Vogelzang, J. van Haveren and D. S. van Es, J. Polym. Sci., 2013, 51, 4191–4199 CrossRef CAS.
- G. Z. Papageorgiou, D. G. Papageorgiou, Z. Terzopoulou and D. N. Bikiaris, Eur. Polym. J., 2016, 83, 202–229 CrossRef CAS.
- Z. Terzopoulou, N. Kasmi, V. Tsanaktsis, N. Doulakas, D. N. Bikiaris, D. S. Achilias and G. Z. Papageorgiou, Materials, 2017, 10, 801 CrossRef.
- M. Matos, A. F. Sousa, N. H. C. S. Silva, C. S. R. Freire, M. Andrade, A. Mendes and A. J. D. Silvestre, Polymers, 2018, 10, 810 CrossRef.
- M. Matos, A. F. Sousa, P. V. Mendonca and A. J. D. Silvestre, Materials, 2019, 12, 328 CrossRef CAS.
- Z. Terzopoulou, L. Papadopoulos, A. Zamboulis, D. G. Papageorgiou, G. Z. Papageorgiou and D. N. Bikiaris, Polymers, 2020, 12, 1209 CrossRef CAS PubMed.
- Q. Zhou, Y. Y. Zhao, Y. F. Shi, R. R. Zheng and L. Y. Guo, Polymers, 2024, 16, 103 CrossRef CAS PubMed.
- M. B. Banella, J. Bonucci, M. Vannini, P. Marchese, C. Lorenzetti and A. Celli, Ind. Eng. Chem. Res., 2019, 58, 8955–8962 CrossRef CAS.
- F. Silvianti, D. Maniar, L. Boetje, A. J. J. Woortman, J. van Dijken and K. Loos, ACS Polym. Au, 2022, 3, 82–95 CrossRef.
- D. Maniar, F. Silvianti, V. M. Ospina, A. J. J. Woortman, J. van Dijken and K. Loos, Polymer, 2020, 205, 122662 CrossRef CAS.
- J. Meimoun, Y. Bernhard, L. Pelinski, T. Bousquet, S. Pellegrini, J. M. Raquez, J. De Winter, P. Gerbaux, F. Cazaux, J. F. Tahon, V. Gaucher, T. Chenal, A. Favrelle-Huret and P. Zinck, Polym. Chem., 2020, 11, 7506–7514 RSC.
- Y. Jiang and K. Loos, Polymers, 2016, 8, 243 CrossRef.
- M. C. Bubalo, K. Radosevic, I. R. Redovnikovic, I. Slivac and V. G. Srcek, Arh. Hig. Rad. Toksikol., 2017, 68, 171–179 CrossRef CAS.
- J. Flieger and M. Flieger, Int. J. Mol. Sci., 2020, 21, 6267 CrossRef CAS.
- L. E. Hallas, J. S. Thayer and J. J. Cooney, Appl. Environ. Microbiol., 1982, 44, 193–197 CrossRef CAS PubMed.
- A. Yamamoto, R. Honma and M. Sumita, J. Biomed. Mater. Res., 1998, 39, 331–340 CrossRef CAS PubMed.
- M. L. Wu, W. J. Tsai, J. Ger and J. F. Deng, Vet. Hum. Toxicol., 2003, 45, 243–246 CAS.
- M. S. Alsuhybani and E. M. Alosime, Polymers, 2021, 13, 2109 CrossRef CAS PubMed.
- M. Manssen and L. L. Schafer, Chem. Soc. Rev., 2020, 49, 6947–6994 RSC.
- F. A. Cotton and G. Wilkinson, Adv. Inorg. Chem., 1962, 670–671 Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, 1984, pp. 1128–1129 Search PubMed.
- E. I. Stiefel, T. R. Halbert, C. L. Coyle, L. Wei, W. H. Pan, T. C. Ho, R. R. Chianelli and M. Daage, Polyhedron, 1989, 8, 1625–1629 CrossRef CAS.
- P. C. H. Mitchell, J. Inorg. Biochem., 1986, 28, 107–123 CrossRef CAS PubMed.
- Z. J. Huang, W. J. Luo, L. Ma, M. Z. Yu, X. D. Ren, M. F. He, S. Polen, K. Click, B. Garrett, J. Lu, K. Amine, C. Hadad, W. L. Chen, A. Asthagiri and Y. Y. Wu, Angew. Chem. Int. Ed., 2015, 54, 15181–15185 CrossRef CAS PubMed.
- B. O. Birgisson, L. J. Monger, K. K. Damodaran and S. G. Suman, Inorg. Chim. Acta, 2020, 501, 119272 CrossRef CAS.
- D. N. Chirdon, R. F. Lalisse, J. N. Sun, S. W. Zhang, B. R. Garrett, C. M. Hadad and Y. Y. Wu, SN Appl. Sci., 2020, 2, 889 CrossRef CAS.
- B. R. Garrett, S. M. Polen, K. A. Click, M. F. He, Z. J. Huang, C. M. Hadad and Y. Y. Wu, Inorg. Chem., 2016, 55, 3960–3966 CrossRef CAS PubMed.
- B. R. Garrett, K. A. Click, C. B. Durr, C. M. Hadad and Y. Y. Wu, J. Am. Chem. Soc., 2016, 138, 13726–13731 CrossRef CAS PubMed.
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- W. Adam and R. M. Bargon, Chem. Rev., 2004, 104, 251–261 CrossRef CAS PubMed.
- J. M. Gretarsdottir, S. Jonsdottir, W. Lewis, T. W. Hambley and S. G. Suman, Inorg. Chem., 2020, 59, 18190–18204 CrossRef CAS PubMed.
- J. M. Gretarsdottir, I. H. Lambert, S. Sturup and S. G. Suman, ACS Pharmacol. Transl. Sci., 2022, 5, 907–918 CrossRef CAS PubMed.
- M. Balogová, S. Sharma, P. Cherek, S. N. Olafsson, S. Jónsdóttir, H. M. Ögmundsdóttir and K. K. Damodaran, Dalton Trans., 2022, 51, 13119–13128 RSC.
- D. Razinkov, H. I. Ingvarsdottir, A. Kvaran, S. Jonsdottir and S. G. Suman, Catalysts, 2021, 11, 593–611 CrossRef CAS.
- S. N. Olafsson, A. Kvaran, S. Jonsdottir and S. G. Suman, J. Organomet. Chem., 2018, 854, 38–48 CrossRef CAS.
- S. N. Olafsson, R. Bjornsson, O. Helgason, S. Jonsdottir and S. G. Suman, J. Organomet. Chem., 2016, 825, 125–138 CrossRef.
- B. G. Girija, R. R. N. Sailaja and G. Madras, Polym. Degrad. Stabil., 2005, 90, 147–153 CrossRef CAS.
- A. Codou, M. Moncel, J. G. van Berkel, N. Guigo and N. Sbirrazzuoli, Phys. Chem. Chem. Phys., 2016, 18, 16647–16658 RSC.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- G. D. Ji, H. M. Ni, C. Wang, G. Xue and Y. T. Liao, Macromolecules, 1996, 29, 2691–2693 CrossRef CAS.
- G. J. M. Gruter, L. Sipos and M. A. Dam, Comb. Chem. High Throughput Screen., 2012, 15, 180–188 CrossRef CAS PubMed.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, V. Tsanaktsis, N. Nikolaidis, M. Kostoglou, G. Z. Papageorgiou, D. A. Lambropoulou and D. N. Bikiaris, Polym. Chem., 2017, 8, 6895–6908 RSC.
- E. DeJong, R. Dam, L. Sipos, D. Den Ouden and G. J. Gruter, Abstr. Pap. Am. Chem. Soc., 2011, 241, 1–13 Search PubMed.
- S. Zaidi, S. Thiyagarajan, A. Bougarech, F. Sebti, S. Abid, A. Majdi, A. J. D. Silvestre and A. F. Sousa, Polym. Chem., 2019, 10, 5324–5332 RSC.
- Y. Tsai, C. H. Fan and J. H. Wu, J. Polym. Res., 2016, 23, 42 CrossRef.
- Y. Tsai, C. H. Fan, C. Y. Hung and F. J. Tsai, J. Appl. Polym. Sci., 2007, 104, 279–285 CrossRef CAS.
- A. Chamas, H. Moon, J. J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustain. Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- B. Agostinho, A. J. D. Silvestre and A. F. Sousa, Green Chem., 2022, 24, 3045–3360 RSC.
- J. U. Izunobi and C. L. Higginbotham, J. Chem. Educ., 2011, 88, 1098–1104 CrossRef CAS.
- E. Gabirondo, B. Melendez-Rodriguez, C. Arnal, J. M. Lagaron, A. M. de Ilarduya, H. Sardon and S. Torres-Giner, Polym. Chem., 2021, 12, 1571–1580 RSC.
- S. Mohammadi, M. G. Bouldo and M. Enayati, ACS Appl. Polym. Mater., 2023, 5, 6574–6584 CrossRef CAS PubMed.
- A. Bourdet, A. Esposito, S. Thiyagarajan, L. Delbreilh, F. Affouard, R. J. I. Knoop and E. Dargent, Macromolecules, 2018, 51, 1937–1945 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00474d |
| This journal is © The Royal Society of Chemistry 2025 |