
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of an 8-membered oxygen-containing benzo-fused heterocycle using flow technologies – an exercise in undertaking research with sustainability as a driver†
Bernice M.
Currie
a,
Nicole C.
Neyt-Galetti
a,
Tanya
Olivier
a,
Petra
Van der Merwe
a,
Lerato S.
Dibokwane
a,
A.
Michelle Reinhardt
a,
Lorinda T.
van Wyk
a,
Jenny-Lee
Panayides
 *b and
Darren L.
Riley
*a
*b and
Darren L.
Riley
*a
aDepartment Chemistry, University of Pretoria, Pretoria, 0028, South Africa. E-mail: darren.riley@up.ac.za; Tel: +2712-420-3097
bPharmaceutical Technologies, Chemicals Cluster, CSIR, Pretoria Campus, Pretoria, 0001, South Africa. E-mail: jpanayides@csir.co.za; Tel: +2712-841-2878
First published on 29th October 2024
Abstract
Due to their natural abundance and biological properties, benzo-fused heterocycles are attractive targets in the field of drug discovery. Previously, a synthetic strategy for accessing 5-, 6-, 7- and 8-membered oxygen-containing benzo-fused heterocycles with the oxygen atom in the less commonly encountered 2-position was reported, however, the approach was hindered by long reaction times and a reliance on high boiling point solvents such as DMF. Targeting an 8-membered analogue as an exemplar, we highlighted that the adoption of basic green chemistry principles coupled with the use of flow chemistry techniques could be utilised (with limited development time) to improve day-to-day sustainability when performing synthetic research. In the case in hand, several key improvements were noted including (i) a higher overall yield (37% vs. 26%), (ii) a significantly reduced reaction time (110 min vs. 136 h) and (iii) the avoidance of the undesirable solvent DMF.
Sustainability spotlightA driver in sustainable laboratories thinking is to improve day-to-day sustainability through changes in daily actions and design planning. Aligning with this mindset, and UN sustainable development goals linked to responsible consumption and production patterns (SDG12), we've highlighted how flow technologies, employing green chemistry principles can improve sustainability. Flow chemistry, despite offering performance advantages remains underutilised for daily synthetic needs, arguably due to perceptions that it's complex, requires significant time investment and is better suited for niche' applications. To inspire a change in this thinking, we present an exemplar of how we have been able to rapidly and with limited development time adopt flow approaches to improve sustainability and greenness as part of our daily synthetic endeavours. |
Introduction
Benzo-fused heterocycles are a class of compounds which in their simplest form are comprised of a benzene ring fused with a heterocyclic ring containing at least one atom of nitrogen, oxygen or sulphur. Due to their natural abundance and biological properties, oxygen-containing heterocycles have historically been targets of interest in drug discovery.1 Notable, 5-, 6-, 7- and 8-membered oxygen-containing benzo-fused heterocycles with the oxygen atom in the one-position are commonly reported in literature to display antibiotic and antimicrobial activities.2 Comparatively, compounds with the oxygen atom in the two-position, although also of interest in drug discovery, are far less common, occurring mostly in higher oxidized forms.3Previously, van Otterlo and co-workers developed a novel approach to prepare various benzo-fused heterocycles with the oxygen in the 2-position. The approach reported employed the use of ring closing metathesis in conjunction with alkene isomerization as key steps to access related 5-, 6-, 7- and 8-membered oxygen and nitrogen-containing ring systems.3
An exemplar of the approach showing the synthesis of the oxygen containing 8-membered analogue 1 (Scheme 1) reveals an elegant approach with modest to high yields, however, there are several drawbacks. The most notable being the high-temperature Claisen rearrangement (Stage 2) which required extensive heating (64 h) at high temperatures (>150 °C). To access the desired temperatures, undesirable dimethyl formamide (DMF) was employed, but the prolonged heating caused significant solvent degradation resulting in a difficult to process crude reaction matrix. The reaction time could be reduced significantly using microwave irradiation; however, this alternative approach had associated safety issues as the irradiation had to be carefully pulsed due to the extreme exotherm that resulted.4 In addition, the approach requires three O-alkylations (Stages 1, 3 and 5) and in all cases these required heating in the range of 18 to 20 hours to afford modest to good yields.
 | ||
| Scheme 1 (i) K2CO3, 60 °C, 20 hours, DMF (99%); (ii) 150–160 °C, DMF, 64 hours; (iii) K2CO3, 60 °C, 18 hours, DMF (75% over 2 steps); (iv) LiAlH4, 0 °C, 12 hours, THF, N2 (86%); (v) NaH, reflux, 20 hours, THF, N2 (77%); (vi) Grubbs II catalyst (5 mol%), 60 °C, 2 hours, toluene (52%).3,13 | ||
In recent times, there has been a paradigm shift in the chemical sciences field with many researchers acknowledging the importance of focusing their research to reduce the environmental impact of their labs. In 2022, the Royal Society of Chemistry undertook a Sustainable Laboratories survey the results of which showed that 84% of respondents wished to reduce their impact and 63% had already taken measures to do so.5 The resulting report highlighted that changes could be divided into daily actions (turning off equipment, reusing laboratory disposables, reducing water consumption etc.) and research and design planning (adoption of green chemistry principles, use of greener solvents, reducing solvent volume etc.). In addition, at a broader level, the United Nations sustainable development goals, point to a growing drive to improve sustainability across all fields.6 In the case of synthetic chemistry the “ensure sustainable consumption and production patterns” goal (SDG12) is arguably most relevant, and we believe that alignment and adoption of the ethos of this goal should not only be limited to production level synthesis but also to the daily activities in basic research laboratories. Wishing to align with this mindset of responsible and sustainable research on a day-to-day basis we elected to re-look at the van Otterlo approach to benzo-fused heterocycles (which we had identified for use in an in-house drug discovery project). The goal of the exercise being to see if the application of some basic green chemistry principles and the adoption of flow chemistry could be utilised (with limited development time) to afford robust, general approaches to some of the key reactions in the synthetic sequence.
Flow technology has previously been identified as a key emerging technology with the potential to make our planet more sustainable.7 That being noted, the technology is often perceived to be technically challenging with niche’ applications and requiring significant time investment to successfully translate and optimise reactions.8 As a result, the technology is arguably not the go-to option for general day-to-day synthetic operations, particularly when batch-based alternatives are available. In this regard, we believe a change in mindset is needed. The technology by today's standard is well developed with numerous reaction classes having already been reported as flow variants. As a result, adoption of flow approaches for day-to-day synthetic needs should, where feasible, be considered to improve sustainability. To speak to this, we herein show an exemplar highlighting that with synthetic and green chemistry knowhow one can adopt some basic flow techniques/principles to improve your day-to-day sustainability.
Results and discussion
Stage 1: phenolic allylation
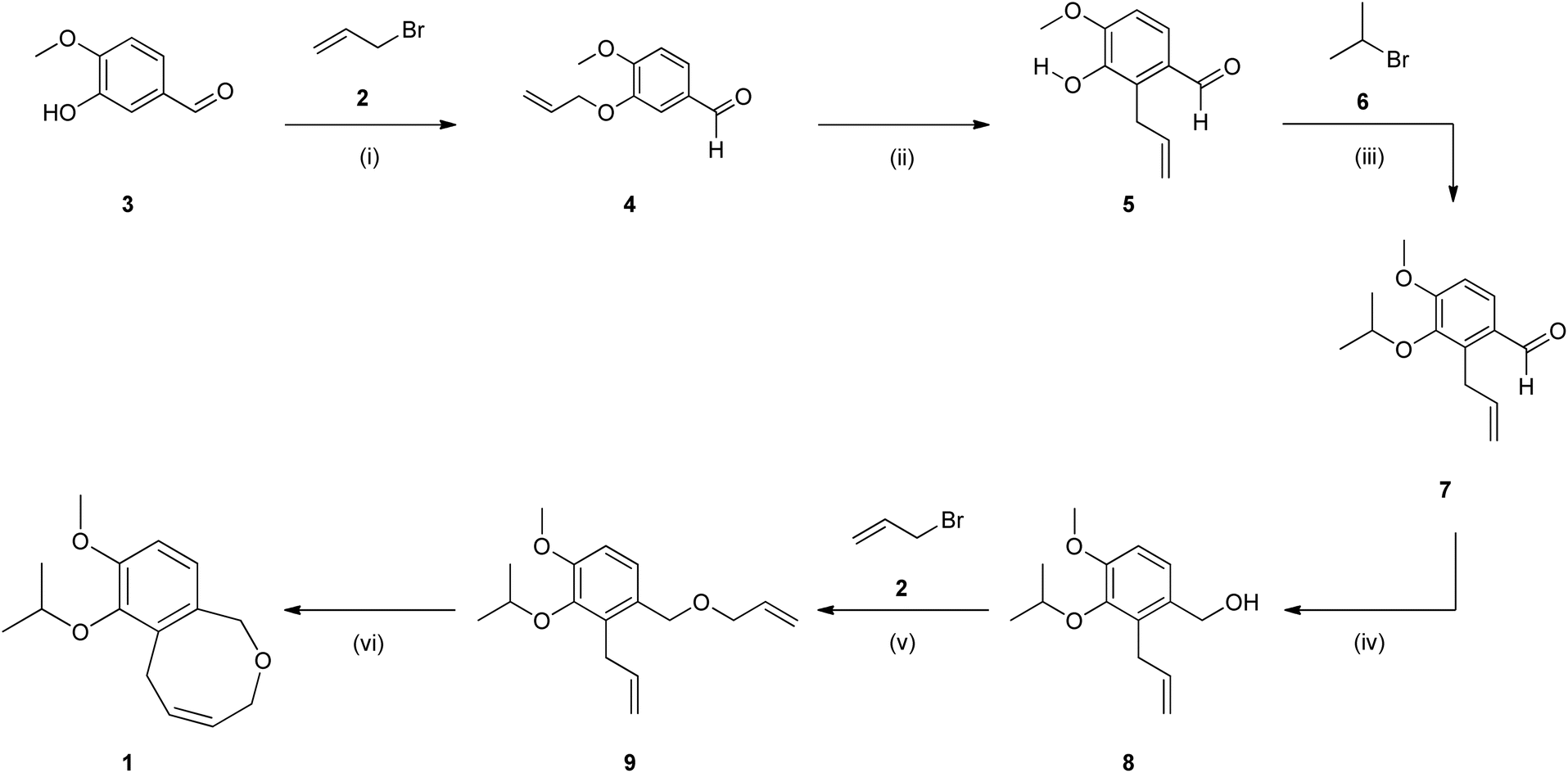
The approach outlined by van Otterlo and co-workers using allyl bromide 2 and potassium carbonate as a base in dimethylformamide (0.50 M) to afford the allylation of iso-vanillin 3 was validated in-house (Scheme 1).3 In our hands the approach afforded an 82% isolated yield of 4 after heating at 60 °C for 20 h.We had previously translated this approach directly to flow employing the use of a packed-bed reactor (PBR) to house the insoluble potassium carbonate.9 In this instance we used a machine learning approach to optimise the reaction in terms of productivity rate. In the context of the work reported herein, we employed a similar setup (see ESI Section 2.1† for more information) and performed a basic re-optimization of the conditions identified previously, focusing instead on increasing the overall yield. In this instance the reaction matrix (0.50 M in DMF) was heated to 100 °C in the PBR with a 20 min residence time to afford the desired product 4 in 92% conversion (by HPLC analysis) which translated to an 85% isolated yield.
Critically speaking, the use of DMF is undesirable from an environmental and safety point-of-view, and its high boiling point means that its removal is both time-consuming and energetically unfavourable. As such we considered toluene or acetonitrile as a potential greener replacement. Under comparable batch conditions, toluene proved to be largely ineffective affording a poor yield of only 2.1% after 20 h, this was not completely unexpected as substitution reactions prefer polar solvent systems. Employing the use of acetonitrile under analogous batch conditions afforded a 66% isolated yield after 20 hours, which could be increased to 88% by extending the heating to 32 hours. Unfortunately, under flow conditions the use of acetonitrile was characterized by the formation of precipitates post-PBR which led to reactor fouling.
An alternative approach using a basic solution of potassium hydroxide (0.60 M) in methanol/water (95/5) was reported by Cortés-Borda and co-workers.10 In this instance, under flow conditions, the authors employed the use of allyl iodide as opposed to allyl bromide 2, because potassium iodide, which is generated as a by-product, was appreciably more soluble in the reaction matrix than the analogous potassium bromide.
Although an efficient solution, the use of allyl iodide is not ideal as it is significantly more expensive than allyl bromide (∼22× as per prices from Merck).11 As such we elected to screen the reaction under batch conditions at 70 °C in aqueous methanol and aqueous ethanol while increasing the ratio of water to see if we could improve the solubility of the reaction matrix. We found that a 70/30 ratio of either methanol or ethanol to water afforded complete solubilization without perceivable deleterious effects. In both instances comparable yields of 78 and 83% respectively were obtained.
The approach at this stage was deemed amenable for flow translation. The flow setup (Scheme 2) was compromised of two HPLC pumps connected downstream relative to stock solutions of (i) isovanillin 3 (0.50 M) and potassium hydroxide in methanol/water (70/30), and (ii) allyl bromide 2 in methanol. As allyl bromide 2 and water are not miscible it was important to only use methanol for the second stock solution. This ensured that there was no phase separation in the reservoir and that a consistent concentration of the allyl bromide was introduced into the reactor. The two stock solutions were then combined at a T-piece mixer followed by a 0.5 mL static mixer to facilitate rapid mixing of the resulting biphasic matrix. This was followed by a 25 mL PTFE coil reactor (internal diameter = 1.5 mm) connected in series with an 8 bar back pressure regulator (BPR).
 | ||
| Scheme 2 Flow synthesis for the allylation of isovanillin 3 using potassium hydroxide in methanol/water. | ||
An initial reaction screen was performed under similar conditions to those reported by Cortés-Borda and co-workers using 1.80 equivalents of allyl bromide and 1.50 equivalents of potassium hydroxide with a residence time of 15 minutes.10 Under these conditions, a modest conversion of 68% was realised (Table 1, entry 1). Thereafter, increasing the residence time to 22.5 minutes and the temperature to 85 °C afforded an increase in conversion to 75.5% (entry 2). It was then decided to increase the equivalents of allyl bromide to 2.0 and a similar conversion of 78% was observed at 80 °C for both 15 and 10 min residence times (entries 3 and 4). Additionally, it was found that increasing the temperature to 100 °C, had little effect on the observed conversion (entry 5).
| Exp. no. | KOH (equiv.) | Allyl Br (equiv.) | Temp. (°C) | T res (min) | Conv. (%) |
|---|---|---|---|---|---|
| a 88% Isolated yield. Reaction carried out on 1 g scale. | |||||
| 1 | 1.50 | 1.8 | 75 | 15 | 68.0 |
| 2 | 1.50 | 1.8 | 85 | 22.5 | 75.5 |
| 3 | 1.50 | 2.0 | 80 | 15 | 78.0 |
| 4 | 1.50 | 2.0 | 80 | 10 | 78.4 |
| 5 | 1.50 | 2.0 | 100 | 10 | 76.5 |
| 6 | 1.80 | 2.0 | 75 | 15 | 76.9 |
| 7 | 1.80 | 2.0 | 80 | 15 | 80.3 |
| 8 | 1.80 | 2.0 | 85 | 15 | 79.3 |
| 9 | 1.80 | 2.0 | 90 | 15 | 77.3 |
| 10 | 1.85 | 2.0 | 80 | 15 | 78.1 |
| 11 | 1.85 | 2.1 | 80 | 15 | 80.1 |
| 12 | 1.80 | 2.0 | 80 | 15 | 92.9a |
The reaction performed best with temperatures ranging between 80 and 85 °C coupled with a shorter residence time of 15 minutes. As the reaction is biphasic the shorter residence time (higher flow rate) is preferred as it improves mixing and mass transfer by facilitating the formation of smaller reaction slugs in-line. Several additional screens led to the identification of optimal conditions employing the use of 1.80 equivalents potassium hydroxide and 2.00 equivalents allyl bromide at 80 °C with a 15 min residence time when using the 70/30 of methanol/water solvent mix (entries 6–11). On a 1 g scale, a conversion of 92.9% was observed which translated to an isolated yield of 88% (entry 12).
Stage 2: Claisen rearrangement
The Claisen rearrangement was initially screened in-house under batch conditions using dimethylformamide as solvent, affording 5 in a low yield of 27% despite refluxing for 65 h. Notably, visible decomposition in the form of solution darkening and the deposition of black solids was observed due to DMF degradation under refluxing conditions. Under similar conditions, van Otterlo and co-workers were able to perform the rearrangement and telescope the material directly into the next stage achieving a 75% yield over both stages. The difference in yields was initially perplexing, however, upon investigation it was revealed that van Otterlo and co-workers performed the reaction by heating the mixture in a silicon oil bath at temperatures >200 °C.3 In doing so, although the reflux temperature of the bulk of the reaction matrix was limited by the boiling point of dimethylformamide (153 °C) material in contact with the glass walls of the reactor experienced elevated temperatures of more than 200 °C.We turned our attention to the use of toluene as a greener alternative which would afford simpler downstream processing and be more amenable to solvent recycling. As expected, because toluene has a significantly lower boiling point its use only afforded a poor conversion of 6.9% after refluxing for 72 h. Despite this, the reaction matrix remained homogeneous, and no decomposition was apparent. As such, we felt that translation to flow was feasible and would provide the opportunity to access the higher temperatures required for good conversions.
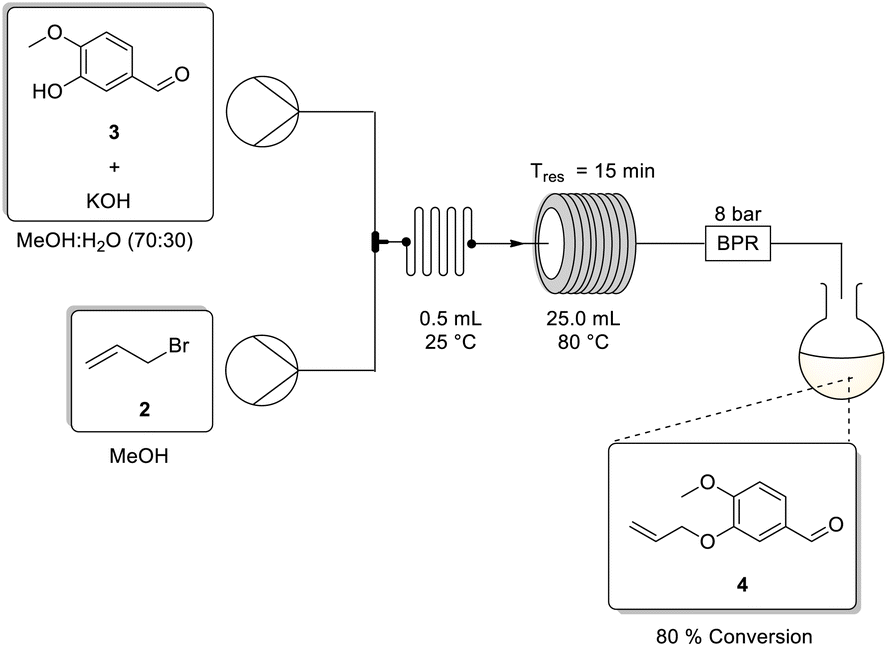
To screen the approach under flow conditions a single HPLC pump from a Uniqsis Binary Pump module was connected upstream to a stock solution of 4 in toluene, and thereafter downstream to a heated 20 mL stainless steel coil (240 °C), followed by a 2 mL stainless steel cooling coil (0 °C) (Scheme 3). The latter was necessary to (i) ensure that the reaction stopped rapidly on exiting the heating coil, preventing additional conversion from occurring in the collection flask and (ii) as a safety measure to prevent rapid uncontrolled gassing out upon exiting the reactor.
 | ||
| Scheme 3 Flow preparation for the Claisen rearrangement 5 using toluene. | ||
Several residence times were screened using stock solutions of 4 in toluene at different concentrations (Table 2). Immediately, when employing a 45 min residence time we observed near quantitative conversions (entries 1–4), regardless of the concentration. We had expected concentration to have little to no effect on conversion as the process is a unimolecular rearrangement, however, the experiments served to confirm that we could work up to 1.00 M concentration without any deleterious precipitation events. Furthermore, when the reaction was performed on a larger scale (10 g) at a concentration of 1.00 M, there was no significant variation in product yield (entry 5).
| Exp. no. | Conc. [M] | T res (min) | Flow rate (mL min−1) | Conv. (%) |
|---|---|---|---|---|
| a 2 g scale. b 10 g scale. c 0.38 g scale. | ||||
| 1a | 0.25 | 45 | 0.44 | 96 |
| 2a | 0.50 | 45 | 0.44 | 98 |
| 3a | 0.75 | 45 | 0.44 | 95 |
| 4a | 1.00 | 45 | 0.44 | 94 |
| 5b | 1.00 | 45 | 0.44 | 99 |
| 6c | 1.00 | 45 | 0.44 | 93 |
| 7c | 1.00 | 30 | 0.67 | 96 |
| 8c | 1.00 | 20 | 1.00 | 100 |
| 9c | 1.00 | 15 | 1.33 | 98 |
| 10c | 1.00 | 10 | 2.00 | 96 |
| 11c | 1.00 | 5 | 4.00 | 95 |
Finally, reducing the residence time from 45 to 5 min while maintaining the temperature at 240 °C (entries 6–11) showed that near quantitative to quantitative conversions could be realized with a residence time of only 15–20 min. Interestingly, the approach can be replicated using dimethylformamide as the solvent under flow conditions with comparable yields, but as with the batch approach the reaction is characterized by the degradation of DMF which commonly leads to reactor fouling (see ESI Section 2.2† for additional information).
Stage 3: alcohol protection
The third stage protection of the alcohol as an isopropyl ether was validated using 2-bromopropane 6 and potassium carbonate.3 The reaction was performed in dimethylformamide (0.50 M) by heating at 60 °C for 20 h affording the desired product 7 in a 78% isolated yield. Envisaging the potential to telescope stages 2 and 3 additional screens were carried out to test reagent/product solubility and reactivity in toluene. Disappointingly, though again (see Stage 1) not unexpectedly, toluene performed poorly with a best conversion of only 2.8%. As a result, to achieve a flow translation we elected to adopt two approaches. The first approach was to directly translate the van Otterlo approach using dimethylformamide and the second was to use potassium hydroxide in aqueous methanol (see Stage 1 allylation).The first approach was less desirable due to its reliance on dimethylformamide. That being noted, in this case we needed to displace a bulky secondary alkyl halide instead of a primary alkyl halide. As such we anticipated that the use of the polar aprotic solvent which would enhance the nucleophilicity of the phenolic anion (in contrast to the use of a polar protic solvent which would reduce the nucleophilicity) may have been critical.
Iso-propyl protection using 2-bromopropane in dimethylformamide
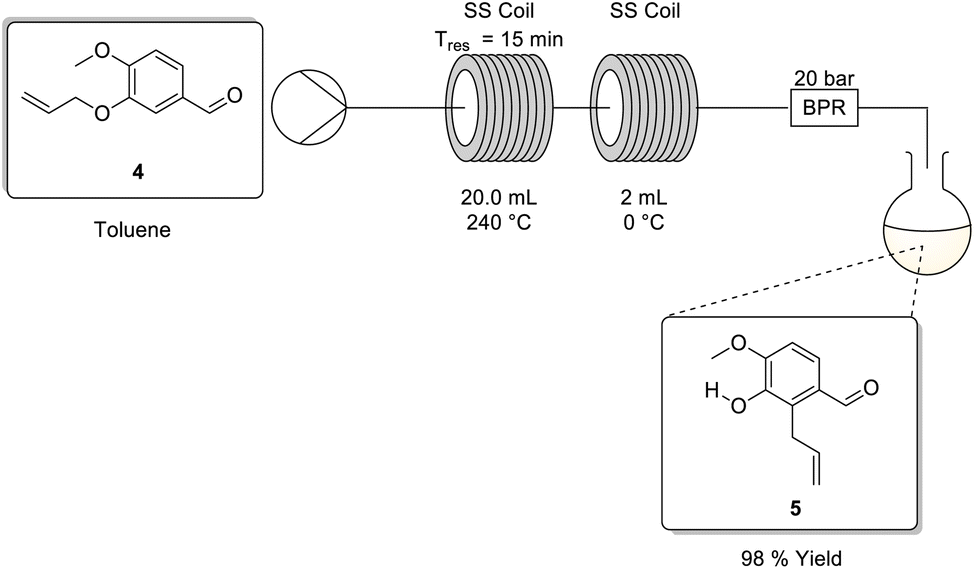
To screen the protection in dimethylformamide we adopted the flow setup shown in Scheme 4. The setup was comprised of two HPLC pumps connected downstream relative to two reservoirs containing stock solutions of 5, and 2-bromopropane 6 both in dimethylformamide. The flow streams were then combined downstream at a T-piece mixer prior to passage through an Omnifit™ glass column packed bed reactor (PBR) housing crushed potassium carbonate, followed by an 8 bar BPR and finally product collection. Preliminary reaction conditions adopted involved performing the reaction at a 0.13 M concentration with a 20 min residence time at 80 °C affording the product in a 92% isolated yield (Table 3, entry 1).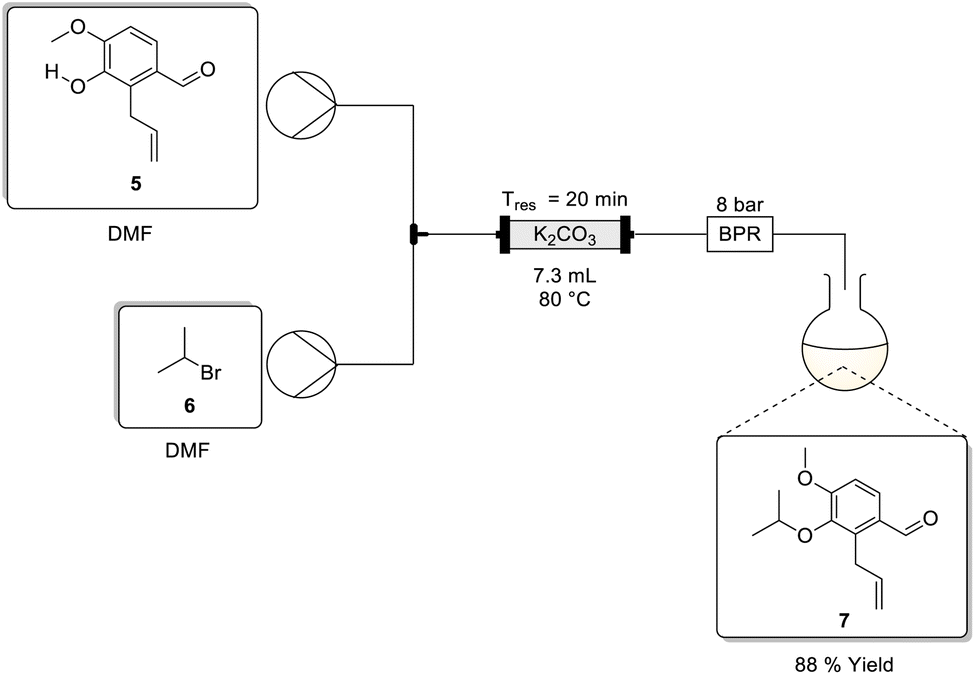 | ||
| Scheme 4 Flow synthesis of the protection of 5 using potassium carbonate. | ||
| Exp. no. | Conc. [M] | Iso-propyl Br (equiv.) | T res (min) | Yield (%) |
|---|---|---|---|---|
| 1 | 0.13 | 2.5 | 20 | 92 |
| 2 | 0.40 | 2.5 | 20 | 88 |
| 3 | 0.50 | 2.5 | 20 | 77 |
| 4 | 0.50 | 2.0 | 20 | 68 |
| 5 | 0.50 | 1.5 | 20 | 64 |
| 6 | 0.50 | 1.0 | 20 | 60 |
The effect of concentration and the equivalents of 2-bromopropane 6 were then assessed while maintaining the temperature at 80 °C and residence time at 20 min. When performed at 0.40 M concentration using 2.5 equivalents of 2-bromopropane 6 the desired product was formed in a comparable high isolated yield of 88% (Table 3, entry 2). Interestingly, as the concentration was increased, the product yield decreased to 77% (entries 1 to 3). Furthermore, decreasing the equivalents of 2-bromopropane also resulted in a sharp drop-off in yield (entries 4–6). Although counterintuitive, increasing the concentration while retaining the residence time at 20 minutes may have negatively impacted the overall mass transfer occurring in the packed-bed reactor. We hypothesised that at lower concentrations the mass transfer is more efficient as there is less competition at the liquid–solid interface and as the concentration increases a saturation point is reached resulting in the mass transfer plateauing.
Iso-propyl protection using potassium hydroxide in aqueous methanol
To screen the use of potassium hydroxide in aqueous methanol we adopted an analogous flow setup to that described for stage 1 (Scheme 2). Using the optimised conditions from stage 1 as a starting point (temp. = 80 °C, Tres = 15 min, conc. = 0.4 M, 6 2.0 eq., KOH 1.80 eq.) we only achieved a low conversion of 24%. Several additional screens (see ESI Section 2.3†) revealed that increasing the temperature (120 °C), residence time (25 min) and stoichiometric excess of iso-propyl bromide 6 (2.50 eq.) while reducing the concentration to 0.2 M afforded 7 in a 69% yield. Interestingly, exchanging the methanol for acetonitrile (to increase the nucleophilicity of the phenolic anion) afforded an increase in yield to 86% (temp. = 100 °C, Tres = 25 min, 6 2.50 eq. and KOH 1.80 eq.).Stage 4: aldehyde reduction
van Otterlo and co-workers performed the aldehyde reduction using lithium aluminium hydride in tetrahydrofuran at 0 °C over a period of 12 h to afford the product 8 in an isolated yield of 86%.3 We elected to screen the reaction in-house using Red-Al (65% toluene) in toluene (0.15 M) instead, at a temperature of 0–20 °C over 20 h affording 8 in a 76% isolated yield (Table 4, entry 2). Alternatively, if performed in dichloromethane the yield can be increased to 93%, however, we elected to continue with toluene prioritising working in a greener manner.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:Unfortunately, due to its high viscosity the use of Red-Al is not ideally suited for flow translation. As such sodium borohydride and sodium acetoxyborohydride were also screened producing the desired product 8 in 62 and 59% yields respectively, under comparable reaction conditions (entries 3 and 4). Alarmingly, in both instances precipitation was observed, as such we changed strategy, testing the use of sodium borohydride in aqueous methanol as described previously by Watts and co-workers.12 Under batch conditions the approach afforded the desired product 8 in a 47% yield after 20 h (entry 5). Despite the lower yield, we elected to translate this approach to flow as it avoided the viscosity issues associated with Red-Al and the precipitation issues noted above.
Upon adopting the reported flow approach,12 we noted that the HPLC pumps that we employed were struggling to pump the sodium borohydride solution and we were only able to isolate 8 in a best yield of 11% using this approach. After this setback, we elected to re-screen the reaction under batch conditions using isopropanol/water and methanol/water solvent mixtures without the addition of the sodium hydroxide as suggested by Watts and co-workers.12
Encouragingly, both instances yielded good results after 20 hours, with the aqueous isopropanol solvent mix performing best with an isolated yield of 87%. We subsequently re-screened the approach with sodium hydroxide as an additive (25%) achieving comparable yields. Moving forward, we elected to continue development using aqueous isopropanol as it afforded a higher overall yield and from a safety perspective did not suffer from the toxicity issues associated with the use of methanol.
In translating to flow we elected to use two peristaltic pumps connected downstream relative to two reservoirs containing stock solutions of (i) 7 in isopropyl alcohol, and (ii) a stirred suspension of aqueous sodium borohydride (Scheme 5). Thereafter, the two flow streams were combined at a T-piece mixer, followed by an in-line static mixer (0.5 mL) and a 25 mL heated PTFE coil reactor (1.5 mm internal diameter) prior to passage through a 3 bar BPR. The reaction stream was quenched directly in a stirred solution of saturated aqueous sodium bicarbonate. Initially the reaction was screened at a 1.00 M concentration affording an 83% conversion (temp. = 60 °C, Tres = 30 min) (Table 5, entry 1). Despite this being an improvement over the batch results obtained over 20 h, the solution was fairly viscous, and the pumps struggled to keep the reaction mixture moving consistently. As such we elected to reduce the concentration to 0.25 and 0.5 M for further screens.
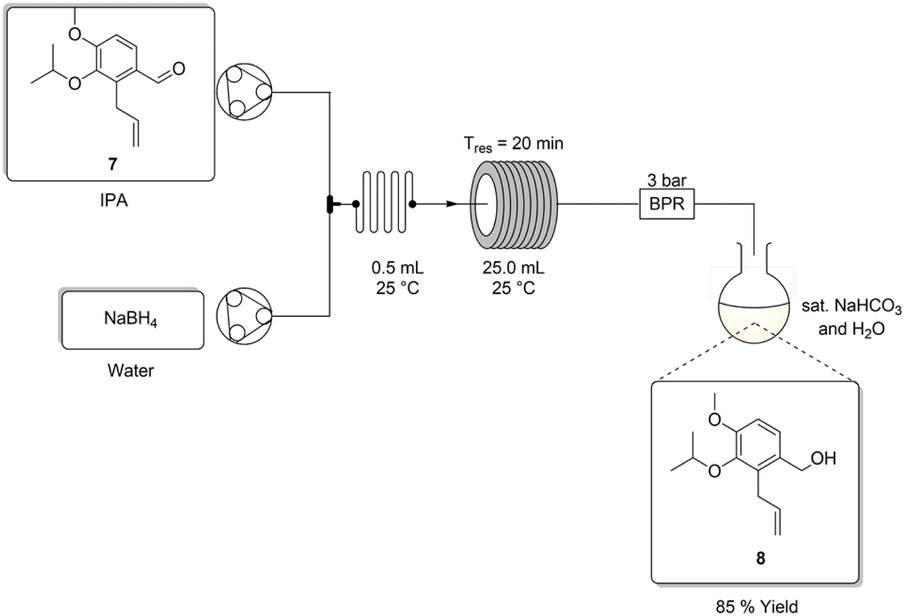 | ||
| Scheme 5 Flow synthesis of aldehyde reduction using sodium borohydride in water. | ||
| Exp. no. | Conc. [M] | T res (min) | Temp. (°C) | Conv. (%) |
|---|---|---|---|---|
| a Viscous sodium borohydride solution. b Scale 0.15 g. c Scale 2.94 g, Isolated yield after workup 85%. | ||||
| 1a,b | 1.00 | 30 | 60 | 83.0 |
| 2b | 0.25 | 5 | 30 | 79.0 |
| 3b | 0.25 | 10 | 30 | 82.0 |
| 4b | 0.25 | 20 | 30 | 93.0 |
| 5b | 0.50 | 20 | 30 | 84.0 |
| 6b | 0.50 | 30 | 30 | 83.0 |
| 7b | 0.50 | 20 | 60 | 79.0 |
| 8c | 0.50 | 20 | 25 | 88.0 |
When performed at 0.25 M concentration the conversion was improved to 93% (temp. = 30 °C, Tres = 20 min) at a reduced temperature and residence time (Table 5, entry 4). Increasing the concentration to 0.5 M afforded a similar conversion of 88% (isolated yield = 85%) again without the viscosity becoming an issue. Alternatively, the transformation can also be achieved in a comparable yield using polymer supported sodium borohydride housed in a packed-bed reactor (see ESI Section 2.4† for more information).
Stage 5: second allylation
The reduced acidity of the benzylic alcohol (Stage 5) vs. the phenolic alcohol (Stage 1) meant that the previously employed potassium carbonate in aqueous methanol afforded no conversion (Table 6, entry 1). As a result, we screened several stronger bases including sodium methoxide, potassium tert-butoxide, DBU, and DMAP in combination with different solvents (entries 2 to 9) with only DMAP affording the desired product 9, albeit in low yields (entries 8 and 9). Switching to the more commonly utilized sodium hydride in tetrahydrofuran afforded a highest conversion of 85% when performed at 60 °C, at 0.2 M concentration (entry 12). Notably, the approach required the use of excess sodium hydride in the range of 2.5 to 5.0 eq., and the removal of the mineral oil that the sodium hydride is supplied in was essential under batch conditions to afford good conversions (see entry 10 vs. 11 and 12).| Exp. no. | Solvent | Base | Base pka | Base equiv. | Conv. (%) |
|---|---|---|---|---|---|
| a Refluxed at 80 °C,2 heated to 60 °C. b Unwashed NaH (containing mineral oil) used. c Washed NaH with hexane prior to reaction. d Heated to 60 °C. | |||||
| 1 | DMF | K2CO3 | 10.3 | 5.0 | 0 |
| 2 | DMSO | TEA | 10.8 | 3.0 | 0 |
| 3a | ACN | TEA | 10.8 | 3.0 | 0 |
| 4 | MeOH | NaOCH3 | 15–16 | 10.0 | 0 |
| 5 | THF | KtOBu | 17.0 | 5.0 | 0 |
| 6 | ACN | DBU | 24.0 | 3.0 | 0 |
| 7 | ACN | DMAP | 28.1 | 1.2 | 0 |
| 8a | ACN | DMAP | 28.1 | 2.5 | 8 |
| 9a | DMSO | DMAP | 28.1 | 2.5 | 27 |
| 10b | THF | NaH | 35.0 | 5.0 | 41 |
| 11c | THF | NaH | 35.0 | 5.0 | 80 |
| 12c,d | THF | NaH | 35.0 | 2.5 | 85 |
Considering these results, we elected to employ the use of a sodium hydride slurry when translating the reaction to flow. The setup employed the use of a peristaltic pump connected downstream relative to a single reservoir containing a stock solution of 8, allyl bromide 3 and sodium hydride in tetrahydrofuran. The use of a single stock solution was deemed feasible as offline testing revealed no reaction occurred at ambient temperature (Scheme 6). Note, (i) for the flow translation the NaH was used as supplied, and the aforementioned hexane wash to remove the mineral oil was found to be unnecessary and (ii) the stock solution was rapidly stirred to limit settling of the sodium hydride to try keep its stoichiometric delivery as uniform as possible. Downstream, the reaction mixture was passed through an in-line static mixer (0.5 mL) followed by a 10 mL stainless steel coil reactor (temp. = 100 °C). Thereafter, a peristaltic pump was used as an active back-pressure regulator, maintaining a system pressure of ∼7 bar. Finally, the reaction was quenched in water directly upon collection. The entire system was kept inert by purging the flow lines with nitrogen gas.
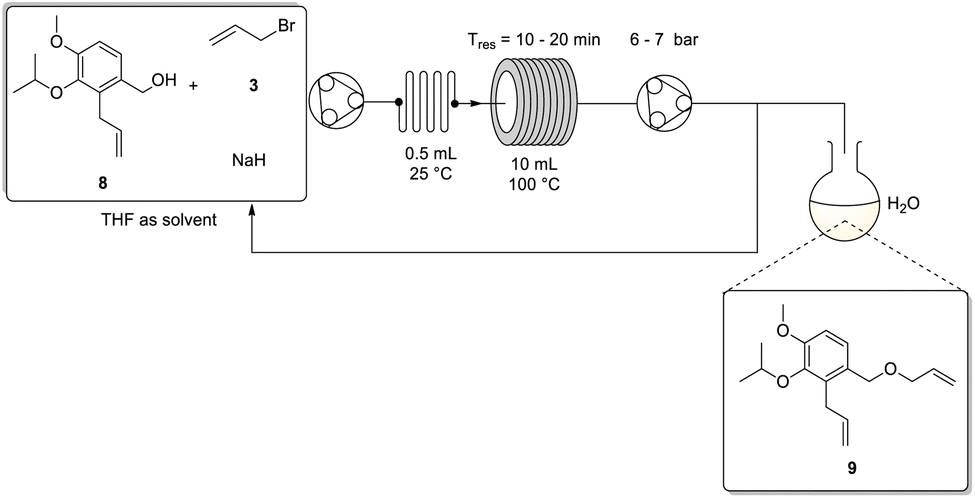 | ||
| Scheme 6 Flow synthesis for recycling of the second allylation using one peristaltic pump and a static mixer with sodium hydride slurry. | ||
We noted that at low flow rates the sodium hydride slurry began to settle in the reactor and adhere to the tubing walls. To overcome this issue an overall flow rate of ≥0.5 mL min−1 needed to be employed, however, this presented a challenge as at these rates the residence time in the 10 mL coil proved insufficient for reasonable conversion. Unfortunately, we did not have access to a longer coil reactor of comparable internal diameter and connecting multiple coils in series resulted in an accumulation of sodium hydride at the connection points leading to reactor blockages. Alternative testing involved the use of a 52 mL wider bore coil (2.10 mm ID vs. 1.00 mm ID), however, in this instance the coil needed to be mounted horizontally which exacerbated the settling issue leading to poor conversions. As a result, we retained the original 10 mL coil (mounted vertically on a VapourTec R4 heater) and elected to insert a manual selector valve directly after the second peristaltic pump which was acting as the BPR. The selector valve was plumbed with a line feeding back into the reagent reservoir (recycle loop) and a second line into the collection/quench flask (collection loop) (Scheme 6). The overall residence time was then increased by simply recycling the flow stream for several cycles before manually switching the valve to the collection loop. In hindsight, although the flow rate was selected to minimise NaH settling it is not unreasonable to expect that its distribution over the flow path would vary, as such the use of a recycling system may be preferential as it would be more forgiving of these variations.
In this instance we employed a Design of Experiment (DoE) approach for optimising the stage using the MODDE 13 software package. A full-factorial screen was chosen with 2 replicates and 18 experiments. The concentration of the starting material (0.20 M) and the reaction temperature (100 °C) were maintained across all experiments. The sodium hydride stoichiometric excess was screened in the range of 2.5 to 7.5 equivalents and the allyl bromide stoichiometric excess in the range of 1.5 to 3.0 equivalents. These ranges were selected based on chemical intuition gained during previous experience with similar systems. In addition, the residence time was screened in a range of 10 to 20 minutes and the number of system recycles in a range 3 to 9 cycles. To ensure there was no experimental bias linked to the isolation and purification of the product the process was monitored by conversion using HPLC (For more information on the DoE screen see ESI Section 2.5†). The DoE screening results are summarised in Table 7.
| Exp. no. | NaH (eq.) | 3 eq. | T res (min) | Cycles | Conv. (%) |
|---|---|---|---|---|---|
| a q-NMR revealed 49.66% product conversion. b q-NMR revealed 99.09% product conversion, 69% isolated product. | |||||
| 1 | 2.5 | 1.5 | 10 | 3 | 61.0 |
| 2 | 7.5 | 1.5 | 10 | 3 | 89.2 |
| 3 | 2.5 | 1.5 | 20 | 3 | 30.8 |
| 4 | 7.5 | 1.5 | 20 | 3 | 79.7 |
| 5 | 2.5 | 1.5 | 10 | 9 | 62.7 |
| 6 | 7.5 | 1.5 | 10 | 9 | 89.9 |
| 7 | 2.5 | 1.5 | 20 | 9 | 37.7 |
| 8 | 7.5 | 1.5 | 20 | 9 | 85.7 |
| 9 | 2.5 | 3.0 | 10 | 3 | 45.7 |
| 10 | 7.5 | 3.0 | 10 | 3 | 79.6 |
| 11a | 2.5 | 3.0 | 20 | 3 | 39.5 |
| 12 | 7.5 | 3.0 | 20 | 3 | 82.3 |
| 13 | 2.5 | 3.0 | 10 | 9 | 59.0 |
| 14 | 7.5 | 3.0 | 10 | 9 | 84.0 |
| 15 | 2.5 | 3.0 | 20 | 9 | 30.4 |
| 16b | 7.5 | 3.0 | 20 | 9 | 92.0 |
| 17 | 5.0 | 2.3 | 15 | 6 | 71.7 |
| 18 | 5.0 | 2.3 | 15 | 6 | 65.9 |
The stoichiometric excess of sodium hydride was shown to have the greatest effect on the overall conversion with response contour plots (Fig. 1) revealing that within the ranges studied higher conversions were favoured at higher sodium hydride stoichiometric excesses, lower allyl bromide stoichiometric excesses and lower residence times (few recycles). A maximum conversion of 92% (Table 7, entry 16) was achieved when employing 3 system recycles (60 min total residence time), while using 7.5 equivalents of sodium hydride and 1.5 equivalents of allyl bromide. The optimal red area of the bottom right plot (Fig. 1) was screened further without additional improvement in the overall conversion (see ESI†).
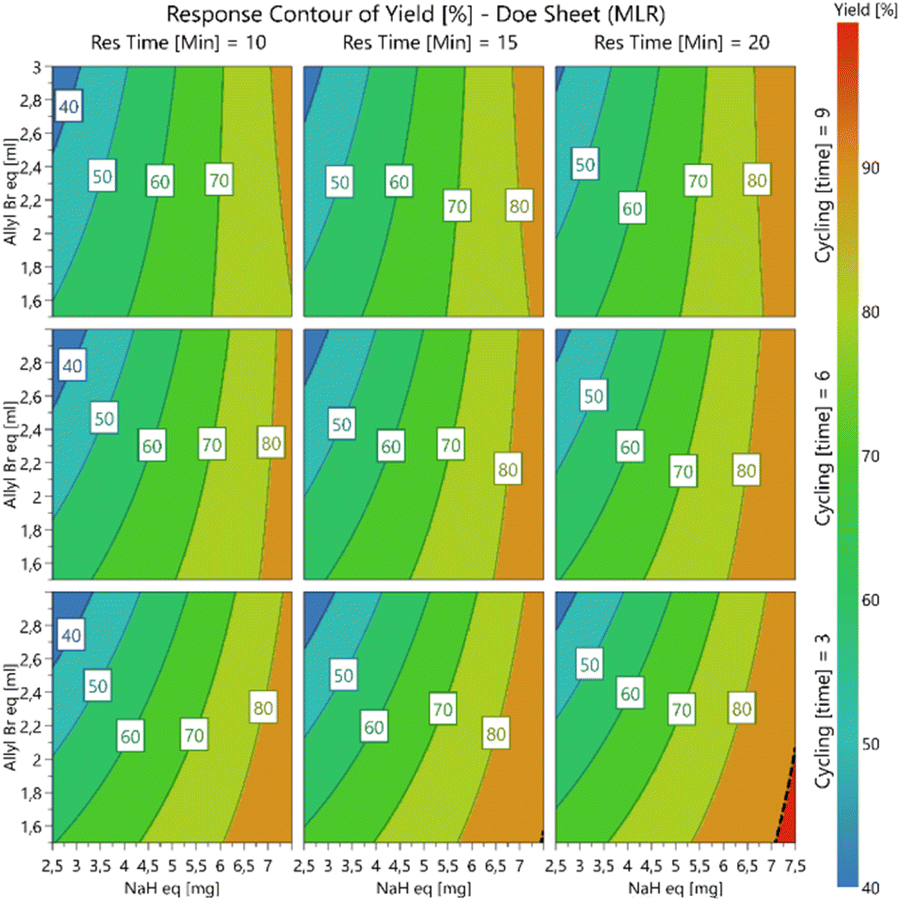 | ||
| Fig. 1 Contour plots for stage 5 DoE optimisation. | ||
Stage 6: ring-closing metathesis (RCM)
The final ring-closing metathesis stage involved performing the reaction in the presence of the Grubbs II catalyst (5 mol%) in toluene at 60 °C for 2 h affording the product 1 in a 52% isolated yield.3 Under comparable conditions we were able to prepare 1 in a 46% isolated yield.We again elected to employ a DoE approach to evaluate and optimise the flow translation. A full-factorial DoE screen was selected with 19 experiments including 3 replicates. The DoE screening parameters included 4 variable factors namely: temperature, residence time, solvent (ethyl acetate vs. toluene) and catalyst (Grubbs II vs. Hoveda Grubbs II) and one constant factor (catalyst loading – 3 mol%).
The flow setup (Scheme 7) employed the use of a Uniqsis Binary Pump equipped with 2 mL sample loops, The loops were washed with degassed solvent prior to loading with stock solutions of 9 and the ruthenium catalyst respectively. The HPLC pumps were connected downstream relative to a solvent reservoir containing either ethyl acetate or toluene (as required) as a pushing solvent and they were combined upstream in a 20 mL heated glass mixing chip prior to passage through a 4 bar BPR.
 | ||
| Scheme 7 Flow preparation of the ring closing metathesis of 1. | ||
The DoE results are summarized in Table 8, for more information see the ESI (Section 2.6).† The study revealed a complex series of interactions with the solvent, catalyst, temperature and residence times all affecting one another. Interestingly, the contour plots generated (Fig. 2) revealed that the use of GII catalyst was mainly dependent on temperature with only a limited variation in the yields at different residence times and in the two different solvents. In contrast, when using the HGII catalyst a clear dependence on both residence time and temperature was observed in both solvents screened. In the case of ethyl acetate, the yields increased as the residence time and temperature decreased but surprisingly in the case of toluene the yields increased as the residence time decreased and the temperature increased.
| Exp. no. | Solvent | Catalyst | Temp. (°C) | T res (min) | Conv. (%) |
|---|---|---|---|---|---|
| 1 | EtOAc | GII | 60 | 15 | 45.6 |
| 2 | Toluene | GII | 60 | 15 | 43.3 |
| 3 | EtOAc | HGII | 60 | 15 | 48.0 |
| 4 | Toluene | HGII | 60 | 15 | 40.9 |
| 5 | EtOAc | GII | 90 | 15 | 24.7 |
| 6 | Toluene | GII | 90 | 15 | 35.9 |
| 7 | EtOAc | HGII | 90 | 15 | 46.5 |
| 8 | Toluene | HGII | 90 | 15 | 54.8 |
| 9 | EtOAc | GII | 60 | 45 | 43.3 |
| 10 | Toluene | GII | 60 | 45 | 45.4 |
| 11 | EtOAc | HGII | 60 | 45 | 46.9 |
| 12 | Toluene | HGII | 60 | 45 | 37.0 |
| 13 | EtOAc | GII | 90 | 45 | 15.0 |
| 14 | Toluene | GII | 90 | 45 | 23.4 |
| 15 | EtOAc | HGII | 90 | 45 | 26.9 |
| 16 | Toluene | HGII | 90 | 45 | 37.0 |
| 17 | EtOAc | GII | 75 | 30 | 34.7 |
| 18 | EtOAc | GII | 75 | 30 | 36.3 |
| 19 | EtOAc | GII | 75 | 30 | 34.4 |
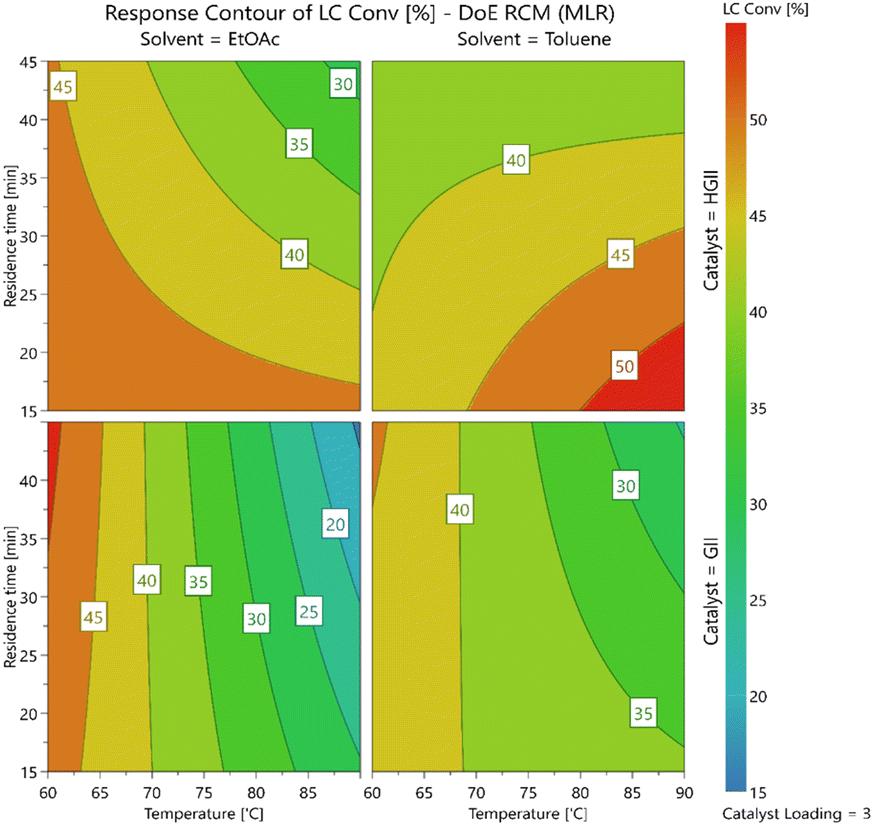 | ||
| Fig. 2 Contour plots for stage 6 RCM. | ||
Unfortunately, the overall conversions remained modest in the ranges studied. We elected to screen a few additional reactions using HGII focusing on the areas predicted to have higher conversions (Table 9). We initially screened the reaction in both ethyl acetate and toluene with the residence time reduced to 10 min (entries 1 and 2). Based on the trends shown in the contour plots the reaction in ethyl acetate was screened at 60 °C and the reaction in toluene at 100 °C affording the desired product 1 in 53.6 and 35.6% product conversion respectively. The conversion in toluene appeared to be unexpectedly low so we decided to replicate the optimal conditions (for toluene) from Table 8 while changing the equivalents of the catalyst (entries 3–6). Notably, as the catalyst loading increased, we observed a drop-off in conversion, this could possibly be linked to unwanted polymerization and/or cross-metathesis occurring.
| Exp. no. | Solvent | Cat. | Temp. (°C) | T res (min) | Cat. (mol%) | Conv. (%) |
|---|---|---|---|---|---|---|
| 1 | EtOAc | HGII | 60 | 10 | 3 | 53.6 |
| 2 | Toluene | HGII | 100 | 10 | 3 | 35.6 |
| 3 | Toluene | HGII | 90 | 15 | 1 | 47.5 |
| 4 | Toluene | HGII | 90 | 15 | 3 | 43.0 |
| 5 | Toluene | HGII | 90 | 15 | 5 | 37.7 |
| 6 | Toluene | HGII | 90 | 15 | 8 | 26.6 |
Conclusions
The synthesis of (Z)-7-isopropoxy-8-methoxy-3,6-dihydro-1H-benzo[c]oxocine 1 was successfully translated to and optimised under flow conditions. The approach notably avoided the use of problematic dimethylformamide which had previously been employed in three of six stages. In addition, the overall yield was improved from 26 to 37% (44% improvement relative to batch approach) while the reaction time was reduced dramatically from 136 h to a residence time of only 110 min (98.7% reduction) (Table 10). We undertook this research due to an interest in benzo-fused heterocycles as a class of compounds from a drug discovery point-of-view, but also to demonstrate that the adoption of flow chemistry on a day-to-day basis could allow research to be conducted in a more sustainable and responsible manner. The exercise highlighted that flow technologies (in conjunction with general principles of green chemistry) certainly allowed the development of more sustainable and/or greener approaches to each stage in the synthesis in our case. That being noted, if one wished to adopt this approach for their day-to-day synthetic needs it would be important to holistically assess the suitability of the reactions for flow translation and if the benefits outweigh the drawbacks.| Stage: reaction name | Batch resultsa | Flow results | ||
|---|---|---|---|---|
| Time (hours) | Yield% | Time (minutes) | Yield% | |
| a van Otterlo and co-workers. | ||||
| (1) Phenolic allylation | 20 | 99 | 15 | 93 |
| (2) Claisen rearrangement | 64 | 75 | 15 | 98 |
| (3) Alcohol protection | 18 | 25 | 86 | |
| (4) Aldehyde reduction (NaBH4 method) | 12 | 86 | 20 | 93 |
| (5) Benzylic allylation | 20 | 77 | 20 | 92 |
| (6) RCM | 2 | 52 | 15 | 55 |
| Overall | 136 h | 110 min | ||
| 25.6% | 36.9% | |||
Notably, we would argue that switching to flow approaches (in the absence of an existing flow protocol) on a day-to-day basis should be performed for reactions or reaction classes that will be used routinely, as any translation and optimisation requires an investment in time and resources. Furthermore, we would also advise that one carefully consider if the integration of downstream processing/purification and/or telescoping of reactions; which is often targeted in flow syntheses; offers appreciable advantages as the implementation of these can prove challenging and greatly increase complexity. In our case, we were able to develop a flow protocol quite rapidly for each stage only requiring an investment of between a dozen and two dozen experiments depending on the degree of optimisation required. We would argue further that in several of the stages the optimisation that we performed may be more excessive than one might generally require. We hope that this work will inspire others, where feasible and appropriate, to look at the adoption of flow chemistry as a means of improving your research sustainability on a day-to-day basis.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. L. R and J.-L. P. conceptualization, formal analysis, funding acquisition project administration, supervision, writing – original draft, writing – review & editing; B. M. C. conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft, writing – review and editing; N. C. N. conceptualization, data curation, formal analysis, investigation, methodology; T. O., P. v. D. M., L. S. D., A. M. R. and L. T. V. data curation, formal analysis, investigation, methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Council for Scientific and Industrial Research (CSIR) and the University of Pretoria (University, Science Faculty Research Councils and Research and Development Program). The authors would like to gratefully acknowledge Mamoalosi Selepe for NMR spectroscopy services, Madelien Wooding for mass spectroscopy services, Prof Willem van Otterlo for discussions and Uniqsis Ltd and Vapourtec Ltd for flow equipment.References
- H. Venkatachalam, N. Venkatesh and A. Kumar, The Oxygen-Containing Fused Heterocyclic Compounds, in Heterocycles – Synthesis and Biological Activities, ed. B. P. Nandeshwarappa and S. O. Sadashiv, IntechOpen, 2020, DOI:10.5772/intechopen.78709.
- L. Gavin Madeley, Synthesis of Benzo-Fused Heterocycles Using Isomerization and Ring-Closing Metathesis Reactions, 2010 Search PubMed.
- R. Pathak, J.-L. Panayides, T. D. Jeftic, C. B. De Koning and W. A. L. van Otterlo, The Synthesis of 5-, 6-, 7-and 8-Membered Oxygen-Containing Benzo-Fused Rings Using Alkene Isomerization and Ring-Closing Metathesis Reactions Search PubMed.
- G. Nordmann and S. L. Buchwald, A domino copper-catalyzed C-O coupling-Claisen rearrangement process, J. Am. Chem. Soc., 2003, 125, 4978–4979 CrossRef CAS PubMed.
- G. Reid, H. Pain and Sustainability Team, Sustainable Laboratories A Community-wide Movement toward Sustainable Laboratory Practices, 2022 Search PubMed.
- United Nations Department of Economic and Social Affairs, THE 17 GOALS, https://sdgs.un.org/goals, accessed 5 August 2024.
- F. Gomollón-Bel, Ten Chemical Innovations that Will Change Our World: IUPAC Identifies Emerging Technologies in Chemistry with Potential to Make Our Planet More Sustainable, 2019, vol. 41, pp. 12–17 Search PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- C. J. van der Westhuizen, J. du Toit, N. Neyt, D. Riley and J.-L. Panayides, Use of open-source software platform to develop dashboards for control and automation of flow chemistry equipment, Digit. Discov., 2022, 1, 596–604 RSC.
- D. Cortés-Borda, E. Wimmer, B. Gouilleux, E. Barré, N. Oger, L. Goulamaly, L. Peault, B. Charrier, C. Truchet, P. Giraudeau, M. Rodriguez-Zubiri, E. Le Grognec and F. X. Felpin, An Autonomous Self-Optimizing Flow Reactor for the Synthesis of Natural Product Carpanone, J. Org. Chem., 2018, 83, 14286–14289 CrossRef PubMed.
- Merck, Allyl iodide (CAS 556-56-9-25 g) and Allyl bromide (CAS 106-95-6-250 mL), https://www.sigmaaldrich.com/ZA/en/product/aldrich/337528, accessed 15 May 2024.
- D. Mandala, S. Chada and P. Watts, Semi-continuous multi-step synthesis of lamivudine, Org. Biomol. Chem., 2017, 15, 3444–3454 RSC.
- J.-L. Panayides, Investigations into the Use of Ring Closing Metathesis to Form 5-, 6-, 7-and 8-membered Benzo-Fused Heterocycles, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00528g |
| This journal is © The Royal Society of Chemistry 2025 |