
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA MXene-supported single atom catalyst selectively converts CO2 into methanol and methane†
Hasan
Al-Mahayni
,
Rongyu
Yuan
and
Ali
Seifitokaldani
 *
*
Department of Chemical Engineering, McGill University, Montréal, QC H3A0C5, Canada. E-mail: ali.seifitokaldani@mcgill.ca; Tel: +1-514-398-4866
First published on 20th February 2025
Abstract
Single atom catalysts (SACs) have emerged as new-generation catalysts that exhibit unique properties and catalytic activity due to their tunable coordination environment and uniform catalytic active sites. MXenes are two-dimensional inorganic materials composed of thin layers of nitrides, carbides or carbonitrides of transition metals, which have been recently used as supports for single metal atoms (SMAs) due to their superior electronic, thermal, and mechanical properties. The catalytic active sites in SACs are too far from each other to enable H–H and C–C coupling through the Tafel process, suggesting that both H2 production—via the hydrogen evolution reaction—and multi-carbon product (C2+) formation—via the CO2 reduction reaction—are significantly suppressed on these catalysts. Therefore, these catalysts are expected to be selective towards single carbon (C1) products in electrochemical CO2RR. However, there are few computational studies that have investigated MXene-supported SACs towards the CO2RR, especially for C1 products such as methane and methanol. In the present study, density functional theory (DFT) is used to systematically evaluate the stability of the MXene support and SAC, and to screen different MXene structures for selective CO2RR to C1 products. Among a combination of ten metals and four supports screened, five catalysts exhibit low limiting potentials for C1 products, especially methanol: Ni/Pd@Ti3C2O2 and Ru/Fe/Co@Mo2CO2. Ni exhibits an exceptionally low reaction energy of 0.27 eV towards methane, while all others exhibit low reaction energy toward methanol ranging from 0.3 to 0.60 eV. The novel and in-depth understanding attained in this systematic high throughput DFT study guides the experimentalist to synthesize SACs based on MXene materials, with exceptional activity and selectivity for highly reduced C1 products.
Sustainability spotlightCO2 Capture, Storage and Utilization (CCSU) is an exciting and emerging strategy to mitigate the rising CO2 concentration in the atmosphere. Electrochemical carbon dioxide reduction to valuable products is a green pathway to achieve this, but suffers from poor selectivity, high applied potential and low energy efficiency. Single atom catalysts (SACs) can suppress undesired side reactions, such as the hydrogen evolution reaction and C–C coupling. In this work, we offer a systematic computational framework to screen different SAC MXenes, a structure that has superior electronic structure, conductivity, and stability to traditional SACs, to find candidate catalysts that selectively reduce CO2 to valuable C1 products such as methane and methanol. The proposed efficient catalysts in this investigation could bring the CO2 conversion technology closer to its practical realization which aligns with the UN's Sustainable Development Goals for waste recycling (SDG 12) and contributes to the industrialization of efficient clean energy consumption (SDG 7 and 9), thus combating climate change (SDG 13). |
Introduction
Global warming is an ongoing crisis due to excessive CO2 emissions into the atmosphere.1–3 One emerging strategy to lower CO2 concentration in the atmosphere is through CO2 Capture, Storage and Utilization (CCSU).4–6 Specifically, the utilization part of this strategy is the newest and has attracted much research attention7–9 as it aims to close the carbon cycle. One pathway to utilize and convert CO2 into valuable feedstocks and fuels such as alcohols is through an electrochemical route: the CO2 reduction reaction (CO2RR).10,11 However, for commercial viability it requires meeting certain criteria. The energy conversion efficiency of the electrochemical system must be comparable to or better than that of the existing fossil fuel driven chemical synthesis processes, and that requires a low applied potential. In addition, the selectivity of the reaction must be high to reduce the post-reaction separation costs.12–15 Thus, one of the main challenges in the CO2RR is achieving a high faradaic efficiency (FE), i.e., reaction selectivity, and at a low applied potential, which is the driving force of the reaction.There are several products that can emerge from the CO2RR, which can be seen in Fig. 1a, thereby contributing to low FE attained for each product. The main single carbon (C1) products are carbon monoxide (CO),16,17 formate (HCOO−),17–19 formaldehyde20 (OCH2), methanol21–23 (CH3OH) and methane24–28 (CH4). There are numerous multi-carbon (C2+)29,30 products such as ethylene29,31 (C2H4), ethane32 (C2H6), ethanol30,33,34 (C2H5OH), acetate (CH3COO−),35–37 propanol38,39 (C3H7OH), etc. Additionally, one of the main reasons for the existing low FEs is the competing hydrogen evolution reaction (HER). Catalysts that efficiently suppress the HER are needed to improve the FE of the CO2RR. Some of the products such as CO and HCOO− can be produced with near unity FE.12 However, FEs for methanol and methane are still too low for commercialization, most being in the range of 30–70% and at a low current density which is not suitable for industrial high-throughput production.12 Increasing the selectivity of methane and methanol is important for their large market and applications. Methanol, particularly, is a clean fuel, a reagent in emerging direct methanol fuel cells (DMFCs),21 and is also utilized as an important intermediate for daily use products such as silicone, paint, and plastics.21 In addition, its liquid form facilitates its use and transportation. Similarly, methane makes up the majority of natural gas, a high energy density fuel at 55.2 MJ kg−1.40 Methanation is a green method of producing renewable natural gas (RNG), but remains costly, making the electrochemical approach an appealing alternative.41 However, bringing the CO2RR technology closer to the large-scale commercialization stage is impossible if we do not learn how to achieve a high selectivity for a single CO2RR product, how to suppress the HER, how to lower the overpotential of the reaction for optimal cell energy efficiency, and how to promote the formation of highly reduced products such as methanol and methane.12
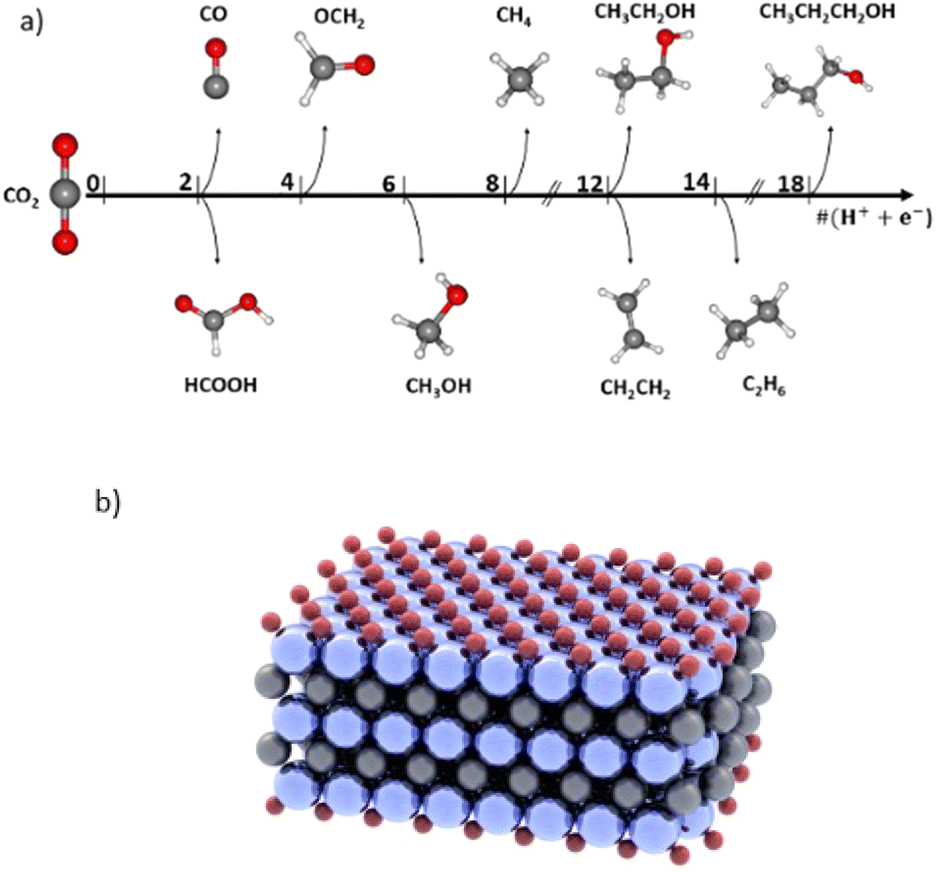 | ||
| Fig. 1 (a) Products emerging from the electrochemical reduction of CO2 with multiple proton-coupled electron transfer (PCET) steps. (b) Schematic of an MXene support used in this study: Ti3C2O2. | ||
Single atom catalysts (SACs) have recently emerged as promising electrocatalysts to enable achieving these goals of technology commercialization.42 As suggested by their name, SACs are catalysts that contain isolated metal atoms that are stabilized by a conductive substrate, in the case of electrocatalysis. SACs are distinguished by their unique unsaturated and tuneable coordination environment. Since the catalytic metal species is at the atomic level, the electronic structure of SACs is drastically different from that of nanoparticles and bulk metals, which leads to their exceptional reactivity in addition to their high atomic utilization.42 Another advantage they hold is the easy tuning of their properties, which can be done by changing the coordination number and environment of the metal species. For the CO2RR, SACs and their nanoparticle counterparts exhibit different behaviours. For instance, nanoparticles of Fe and Ni are selective towards the HER, while SACs Fe and Ni are selective towards the CO2RR to CO.42 In fact, the inability to have an adjacent adsorbed hydrogen (*H), or adsorbed carbonaceous intermediate (*C), heavily underpromotes H–H and C–C coupling, and thus not only suppresses the HER, but also suppresses multi-carbon product formation. For the HER, the reaction must undergo the Heyrovsky mechanism instead of the preferred Tafel mechanism:
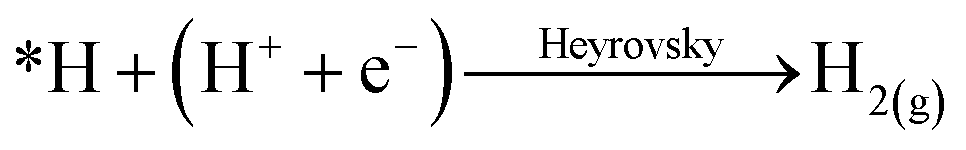
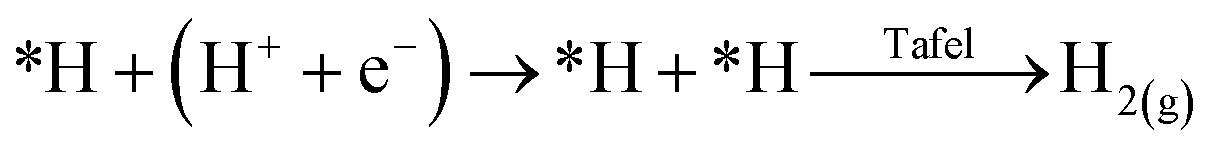
However, from the reaction kinetics perspective, it is known that the Heyrovsky mechanism is less favourable than the Tafel mechanism; yielding a reaction energy of almost twice the value compared to the Tafel on Pt (111) for instance.42
There exist many types of SACs. They vary based on the substrate or support: metal organic frameworks (MOFs), graphene, molecular (Metal-Phthalocyanine or MPc) or metal-on-metal.43 Discovered in 2011,44 MXenes are 2-dimensional materials comprising 2, 3 or 4 layers of a transition metal M (Ti, Mo, Sc, V, …),45,46 and with an element X (C or N) between each layer of metal. The MXene can also be surface terminated by an element T (O, H, F, Cl, etc.).45 Fig. S1a and b† are an example of two and three layered MXenes with M = Ti (a), Mo (b); X = C and T = O. An example of Ti3C2O2 can also be seen in Fig. 1b.
MXene structure is an ideal support for SACs due to its superior electronic structure, conductivity, and stability,45–48 compared to other SAC supports such as graphene or MOFs. Consequently, MXene supports have been used in a plethora of electrocatalytic applications such as the nitrogen reduction reaction (NRR),49 the oxygen reduction reaction (ORR),50 the HER,51 the oxygen evolution reaction (OER)52 and the CO2RR.48,53–55 The latest literature review shows that a maximum FE of 59% was achieved towards methanol, at an applied potential of −1.4 V vs. RHE, using Cu MXene-based (Cu@Ti3C2O2) SAC structures.56 Other groups have used CoPc/CNT57 to achieve 44% FE at −0.9 V vs. RHE, and Cu SAs/TCNFs (Cu–N4)58 to achieve the same FE at the same potential. For methane production, Xin et al. used a Zn–N4 (ref. 27) to achieve 85% FE at −1.8 V vs. SCE; Yang et al. used Cu–CeO2 (ref. 59) to achieve 58% FE at −1.8 V vs. RHE. These catalysts, however, suffer from poor stability and/or high overpotentials.
While the attained results so far are impressive, a systematic discovery of new MXene-based SACs is still lacking to find a superior catalyst that exhibits an even lower thermodynamic energy barrier towards C1 products such as methanol and methane. In addition, the existing studies either do not evaluate the stability of the catalysts, or the reaction mechanism does not cover the full pathways for different possible products.54,60–63 Overall, there is a lack of in-depth theoretical studies on SAC MXene catalysts for the CO2RR which focus on stability, scaling relations and a full reaction pathway that considers numerous products including C–C coupling and the HER.
Here, we aim to investigate Ti- and Mo-based MXenes (the most common transition metals in MXenes) that promote the full reduction of CO2 to C1 products, in hopes of finding a novel catalyst that increases the low selectivity. Oxygen-terminated MXenes will be used as they have been found to promote methanol and methane formation.54,56,62,64 However, these studies are on pure MXene substrates without SACs or are purely experimental without satisfactory performance as explained above. Specifically, the effect of the type of support (Ti or Mo), the number of layers (2 or 3) and the transition metal SAC will be evaluated using theoretical tools in density functional theory (DFT). The goal is to unravel which structure has the lowest reaction energy towards C1 products, while simultaneously unveiling the unique mechanism that leads to the product.
Computational details
The MXene structures were built from MAX phase bulk structures to imitate the synthesis process.65 For example, Ti2AlC (MAX) was first optimized to get the lattice vectors, then Ti2C (MXene) surfaces were created, followed by Ti2CO2 (oxygen terminated MXene). The four support structures considered are Ti2C, Ti3C2, Mo2C, and Mo3C2. Then, the single metal atom (Ag, Au, Co, Cu, Fe, Ni, Ru, Pd, Pt, Zn) was added on top of the oxygen layer. A vacuum region in the z-direction (i.e., perpendicular to the surface) was set to be 20 Å to avoid interaction between the layers in periodic imaginary cells. Ti2CO2 slab structures were constructed using 18 Ti atoms, 9 C atoms, and 18 O atoms for a final formula of Ti18C9O18. Ti3C2O2 structures were constructed using 27 Ti atoms, 18 C atoms, and 18 O atoms for a final formula of Ti27C18O18. The same was done for the Mo MXenes, as demonstrated in Fig. 2.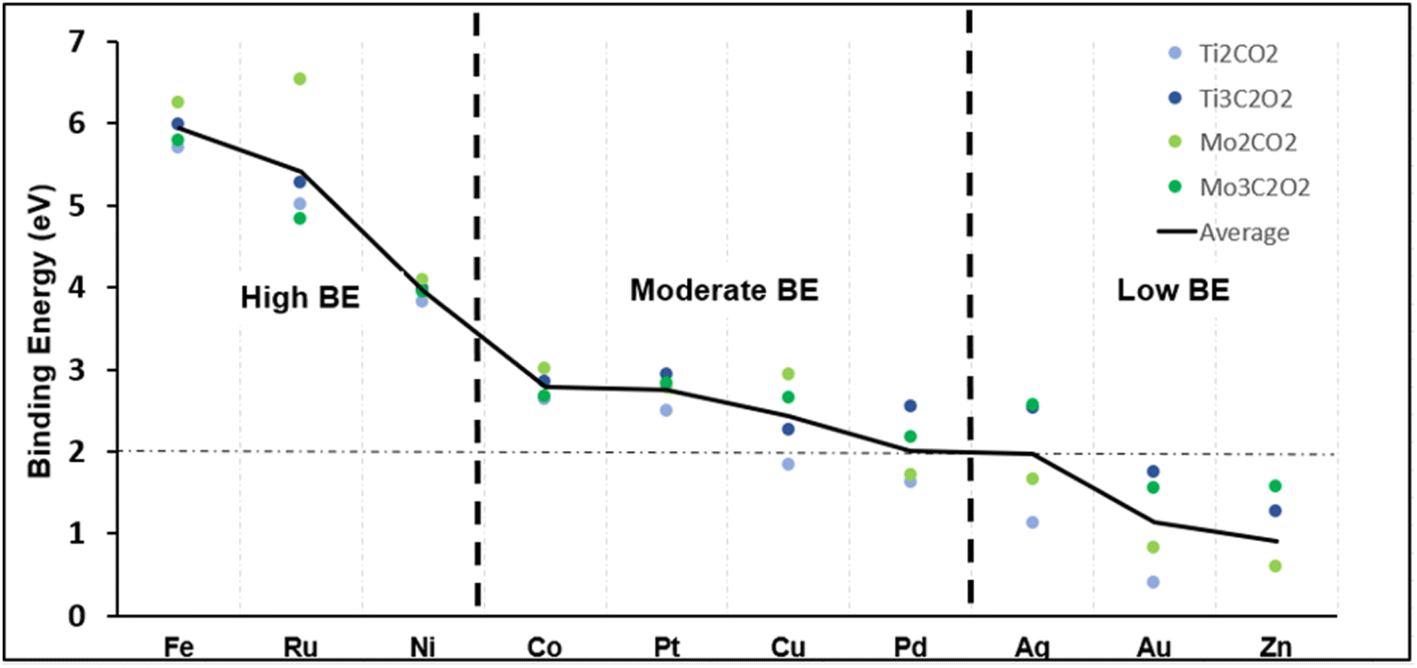 | ||
| Fig. 2 Binding energy of single metals adsorbed on four different structures. | ||
All first principles calculations were done using the CP2K package.66 To obtain the optimal cut-off energy, a standard convergence test was performed.67 The energy cut-off used was thus 550 Ry. The force convergence was taken to be 3 × 10−4 (Bohr−1 × Hartree). A 5 × 5 × 1 Monkhorst–Pack mesh was used for k-point sampling for geometric optimization, while an 8 × 8 × 8 mesh was used for unit cell optimization. van der Waals corrections enabled by the DFT-D3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 68 method are incorporated to calculate the long-range interactions. The dipole moment was considered but did not affect the energy by a significant amount. The PBE functional was used to describe the exchange–correlation functional.69 While other functionals such as RPBE, BEEF-vdW and PBE0 were used in previous computational studies on SACs, there is still no clear consensus on what the best Functional for this system is.70–75 PBE has been widely used in electrocatalytic systems, and corrections for certain compounds have been calculated.76,77 Refer to the ESI† for further computational information.
68 method are incorporated to calculate the long-range interactions. The dipole moment was considered but did not affect the energy by a significant amount. The PBE functional was used to describe the exchange–correlation functional.69 While other functionals such as RPBE, BEEF-vdW and PBE0 were used in previous computational studies on SACs, there is still no clear consensus on what the best Functional for this system is.70–75 PBE has been widely used in electrocatalytic systems, and corrections for certain compounds have been calculated.76,77 Refer to the ESI† for further computational information.
To evaluate the stability of the MXene products, the formation energy (FE) was used as formulated in eqn (1):
| FEMxCyOz = EMxCyOz − (xEM + yEC + zEO) | (1) |
To evaluate the binding strength of the SMA to the MXene support, the binding energy (BE) was used.
| BESMA@MxCyOz = ESMA@MxCyOz − (ESMA,vacuum + EMxCyOz) | (2) |
| ΔEads = E*adsorbate − Emolecule − Eslab | (3) |
| ΔEdes = −E*product + Eproduct + Eslab | (4) |
The asterisk denotes species that are adsorbed on the surface. Corrections were made for the gas-phase energies of CO and CO2 following Nørskov's work.76 The corrections for CO and CO2 were −0.51 and +0.13 eV, respectively. These corrections were applied for the Gibbs free energy calculations done later.
Results and discussion
To evaluate the stability of the MXene structure in the closest method possible to experimental conditions, we start with the bulk material: Ti2AlC for Ti MXene and Mo2C for Mo MXenes. The formation energy (eqn (1)) is calculated for this bulk material, then metal carbide 2D layers are made. The formation energy is calculated again before making the MXene by adding oxygen layers at the top and bottom. A correction of +0.57 eV was applied to get the accurate energy of oxygen,77 and this is described in eqn (S3) and (S4) in the ESI.† This methodology was applied to mimic experimental synthesis of the oxygen terminated MXene. The results are summarized in Table 1 for all four supports.| Synthesis stage/type | Ti2 | Ti3 | Mo2 | Mo3 |
|---|---|---|---|---|
| Bulk | −0.710 | −0.771 | −0.410 | −0.303 |
| Metal carbide | −0.134 | −0.013 | 0.369 | 0.003 |
| O-terminated MXene | −2.163 | −1.842 | −1.268 | −1.047 |
All four O-terminated MXenes have negative formation energies indicating they are stable, and their synthesis is thermodynamically favourable. Starting from the bulk, making the metal carbide layers is slightly endothermic, which is expected as energy input is needed to convert a 3D bulk into a 2D metal carbide layer. Importantly, the O-terminated MXenes exhibited significantly lower formation energies than the metal carbides, indicating that the terminal groups play a major role in the stability of MXenes. This is consistent with how MXenes are synthesised experimentally for catalysis use, according to many previous studies.78–84 Many of these studies suggest that the SMA deposits on top of the oxygen layer or takes the position of an oxygen vacancy. In this study, we consider the former.
We considered ten total transition metals as SMAs in this study, for they were previously being used as MXene SMC, graphene-based SMC, or as bulk transition metals that have been shown to have considerable CO2RR performance.56,57,76,85–87 A DFT study looking at graphene-based SACs86 suggested that the rate determining step for these catalysts is mostly *CO hydrogenation to *CHO, the usual rate determining step on bulk copper. However, we observe in this work, due to the unique electronic structure of MXenes, *OCHO will be favoured to *COOH adsorption and will lead to formic acid instead of CO. This can thus lead to other rate determining steps and the breaking of scaling relations.54,88–90
Bulk transition metals such as platinum, nickel and iron do not show any activity towards the CO2RR, producing hydrogen. However, this differs in the SAC form and the HER is suppressed. Additionally, iron and nickel are particularly interesting to research as they are much cheaper than other transition metals like cobalt or copper. A relatively cheap metal SAC can decrease cost and loading masses which is crucial to commercialization.12
Metals that favour two electron transfer products in their bulk, such as carbon monoxide and formate, should also be considered as it is hypothesized that SACs will hold on to intermediates better than their bulk counterpart, leading to more electron transfer and easier full reduction of products. This is the case with silver and gold, that mainly produces CO.12 Copper was included since it is the most researched and effective bulk metal for the CO2RR.76 Zinc is included for it has been shown to be a potential catalyst for ethanol production, promoting C–C coupling.91 It would be then interesting to see how zinc and copper perform as a SAC, since C–C coupling is inhibited. Altogether, the metals chosen were Fe, Ru, Ni, Co, Pt, Cu, Ag, Au, Pd and Zn.
The binding energy of the selected SACs was calculated (eqn (2)) to first find the most stable SAC, and then to see which support each metal prefers. We hypothesize that SACs with a high binding energy (BE) to their metals are more stable and thus will be more performant when it comes to the CO2RR. Fig. 2 shows the binding energy in eV for each metal and support. Since the absolute value of the BE means little, the plot serves as a comparison point to separate the metals into three distinct categories. Fe, Ru and Ni held “high BEs” compared to Co, Pt, Cu and Pd, which held “moderate BEs”. The remaining metals had an average BE of less than 2 eV and were deemed too unstable for their role as a catalyst. This value was chosen based on our generated results in Fig. 2, and previous similar works,92,93 where the rationale is a high BE suggests strong affinity and the suppression of diffusion, leading to a more stable structure. It is worth noting that silver and palladium had an average binding energy equal to 2 eV which falls where the cut-off was deducted. Thus, we pick only the best support for each: Ag@Ti3 and Pd@Ti3.
For the supports, there are clear trends when it comes to which support is most viable based on binding strength. Fe, Ru, Ni, Co, and Cu are more stable on Mo2 (light green circles in Fig. 2), although some more than others. Specifically, Ru@Mo2 has the highest binding energy, and attaches to the metal atom significantly better than the other three supports. Furthermore, Ti3 (dark blue circles in Fig. 2) is systematically more stable than Ti2 (light blue circles in Fig. 2), while Mo2 is more stable than Mo3 (dark green in Fig. 2). Thus, we predict Mo2 and Ti3 structures to be the most favourable as the SAC support, assuming BE plays a role in the catalytic activity in the CO2RR, which is investigated below. The rest of the catalysts, Ag, Au, Pd and Zn, are omitted.
To evaluate the performance of each catalyst for the CO2RR to C1 products, different pathways are evaluated, as demonstrated in Fig. 3. The black pathway leading to CH3OH in Fig. 3 is used for the second stage of screening. This pathway is suggested based on the results for all SACs considered. A high throughput computation was performed, and the results are summarized in Table S1.† It is important to note that the adsorption energy of CO2 (ΔECO2,ads) was evaluated on the oxygen hollow site to compare it to the adsorption energy of CO2 on the SMA. ΔECO2,ads on the oxygen hollow sites for Ti3 and Mo2 was −0.12 and −0.13 eV, respectively. Since all the later selected catalysts had ΔECO2,ads < −0.22 eV, we hypothesize that the only active site is the SMA.
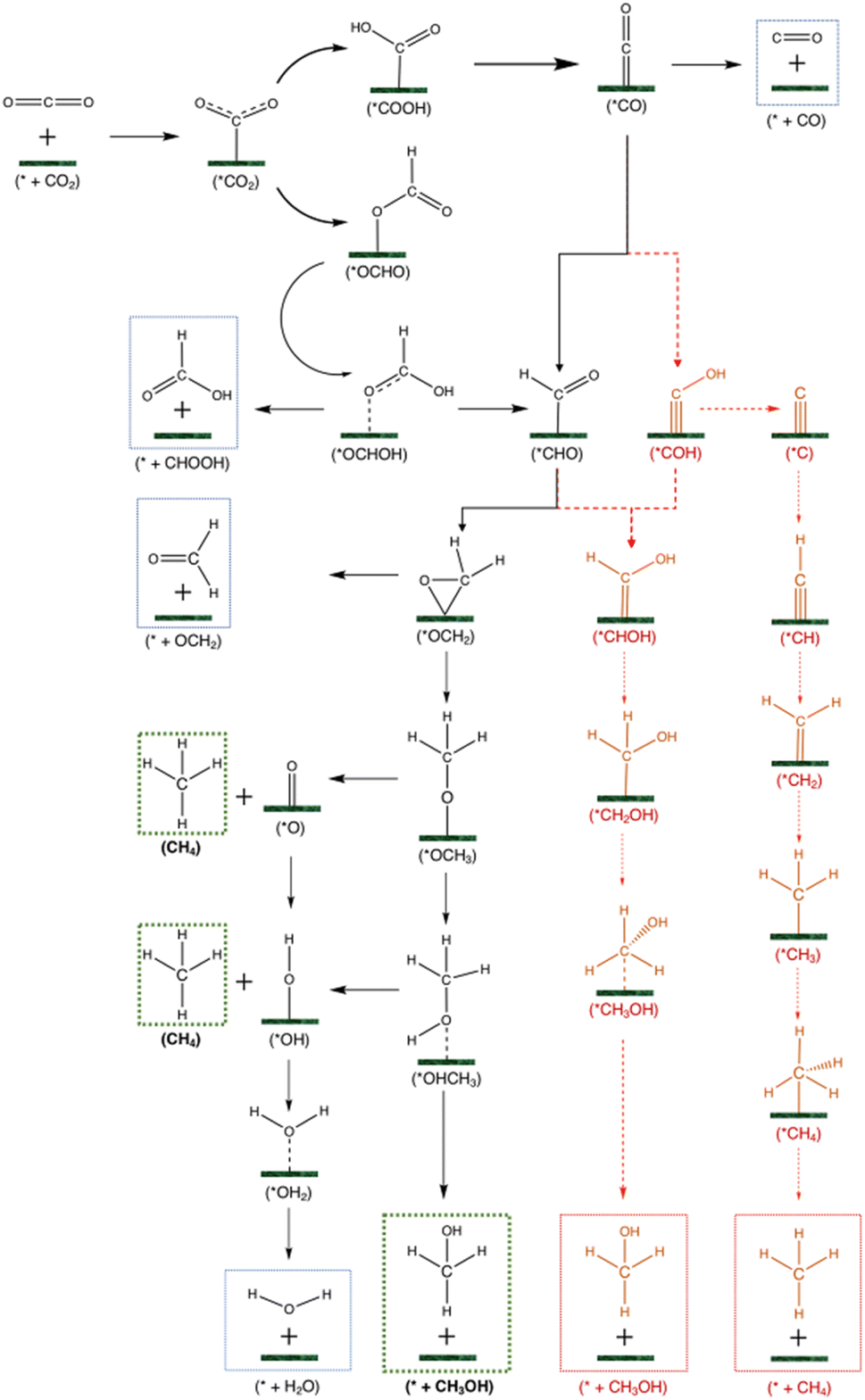 | ||
| Fig. 3 Different pathways for the CO2RR leading to carbon monoxide, formic acid, methane, and methanol. Compounds with an asterisk * are adsorbed species, while compounds with no asterisk are either reactants or desorbed products. Red pathways are deemed unfavourable on most catalysts. Species in dashed boxes are final products. Products in blue boxes are intermediate, unfavourable products. Products in green boxes are the targeted products in this study: methanol and methane. | ||
The following criteria are taken to rationalize how we pick the catalysts that will go through to the next stage of screening:
• We first look at the HER reaction energy compared to the CO2RR. The first point to look at is the CO2 and H adsorption on each catalyst. Active catalysts will have a large H adsorption energy (inhibiting the HER) and a negative CO2 adsorption energy that promotes CO2 activation and thus the CO2RR.
• Active catalysts will have a larger H adsorption energy than the reaction energy of the thermodynamic limiting step (TLS) of the CO2RR.
• Any catalysts with high reaction energy for the TLS (>0.8 eV) are omitted for low activity, regardless of the above points.
• We pick the best performing support for every metal. For example, Ni@Ti2 and Ni@Ti3 both exhibit a low reaction energy for the CO2RR while suppressing the HER. However, since the TLS of Ni@Ti3 has a reaction energy of 0.35 eV compared to 0.6 eV for Ni@Ti2, only Ni@Ti3 is considered.
The main products that are investigated are carbon monoxide (CO), formic acid (HCOOH), methanol (CH3OH), methane (CH4) and hydrogen (H2). The HER is the first thing to consider since catalysts that are more selective towards hydrogen production than the CO2RR are undesirable. After CO2 adsorption and the first hydrogenation step, the two possible intermediates are *OCHO and *COOH, as seen in Fig. S2.† The *COOH is considered as the more favourable reaction intermediate in most CO2RR studies; however, on MXene-based SACs we observed that the *OCHO is the more favourable compound (see Table S2†). For example, Cu@Ti3 has a reaction energy of ∼0 eV towards the formation of *OCHO and 0.52 eV towards *COOH.
Subsequently, *CO can only be produced from *COOH since the carbon in the latter is not hydrogenated. On the other hand, from *OCHO, we expect only *HCOOH to be produced since the carbon in the latter is already hydrogenated. *HCOOH can either desorb as a final product in formate or be further protonated to release water and leave *CHO or *COH. However, *COH is unstable on a SAC as the carbon in an isolated COH form is bonded once to oxygen, meaning it is heavily unsaturated, and a one-atom catalyst is not enough to provide adequate electrons to the carbon of *COH to share 4 of its valence electrons. On the other hand, the carbon in CHO is bonded three times, meaning *CHO has a better chance to be stabilized by a SAC. Thus, the stability of *CHO/*COH is investigated on Ni@Ti2, and *CHO is found to be 0.95 eV more stable than *COH. On Ni@Ti3, *CHO is 0.77 eV more stable than *COH (see Fig. S3†). To investigate this difference in stability, Bader charge and charge delocalization calculations were performed. The charge density difference can be seen in Fig. S3.† As hypothesized, on *COH, there are two blue areas around the carbon, representing electron depletion, forming a dumbbell shape. On the other hand, on *CHO, there is only a small blue area around the carbon and the electron distribution between the metal and carbon (the yellow area) is more uniform than that on *COH, indicating that the carbon in *CHO is more saturated than on *COH. Thus, the *COH pathway is shown in red as unfavourable, in Fig. 3. Chemisorption of intermediates after *CHO is shown to be slightly more stable through *O than *C (see Table S4†), which is why the pathway involving *CHOH and *CH2OH is in red. This was observed in 21 out of 24 catalysts. Previous works have shown that *COH is more likely to lead to methane while *CHO leads to methanol formation.94,95 Further protonation steps can either be through intermediates adsorbed by the *C atom or the *O atom. From *OCH3, CH4 production can occur leaving behind *O which needs to be protonated to form water. The other possibility is *CH2 formation from *CH2OH which also leads to methane. Finally, methanol can be produced from *OCH3 or *CH2OH.
Applying the above criteria in the analysis of Table S1† leads to the conclusion that the six following catalysts are chosen for subsequent steps: Pt@Ti3, Ni@Ti3, Co@Mo2, Fe@Mo2, Ru@Mo2, and Pd@Ti3. Fig. 4 shows how the first criterion point is used to screen the catalysts.
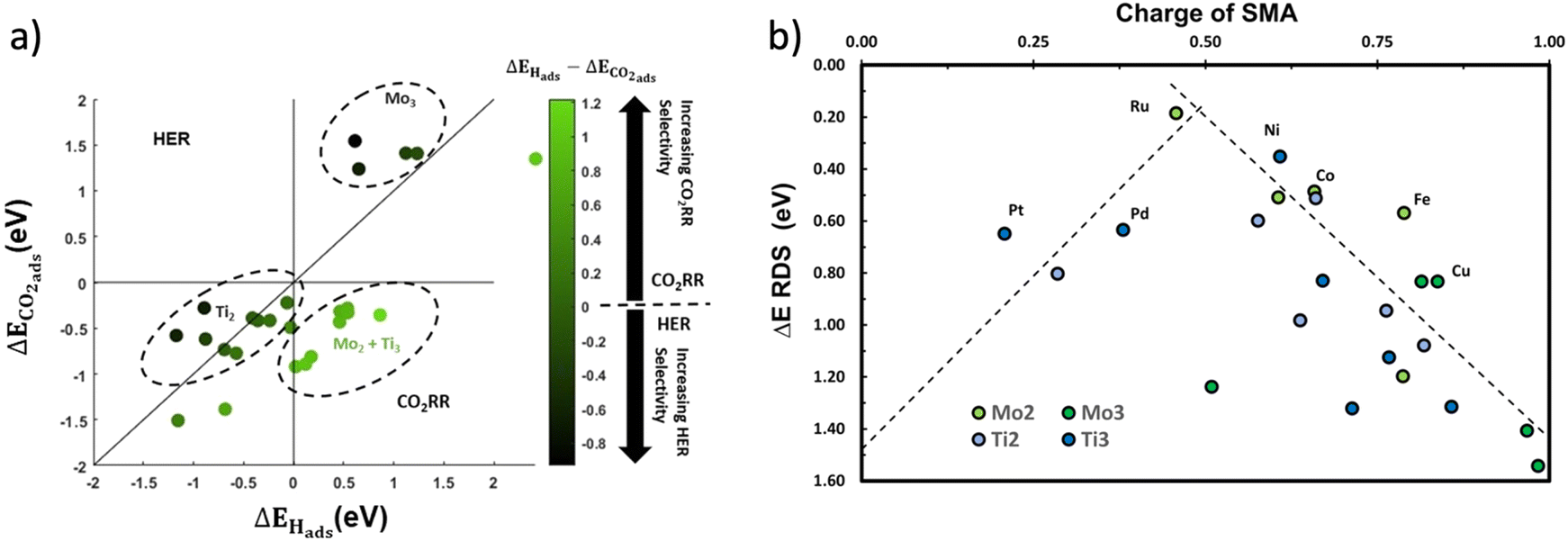 | ||
| Fig. 4 (a) The adsorption of CO2 against the adsorption of H on all 26 catalysts. The figure is split into four regions: the HER-favorable region in the top left, the CO2RR-favorable region in the bottom right, and mixed regions in the top right and bottom left, (b) Volcano plot showing the reaction energy of the rate-determining step for all catalysts versus the charge of the single metal atom of the catalyst calculated based on the Bader charge analysis.96,97 All atoms displayed on the chart correspond to the best performing catalyst for that transition metal. | ||
The colour bar in Fig. 4a effectively highlights the selectivity of each catalyst by performing X − Y = ΔEH ads − ΔECO2 ads. The more positive this value is, the more selective the catalyst is for the CO2RR. Negative values denote catalysts that prefer the HER to the CO2RR. There are various key regions, which are delimited by dashed lines. Every catalyst in the top right circle has Mo3 as support, effectively showing that is not an active catalyst for the CO2RR. This is similar in the bottom left quadrant with Ti2. The bottom right quadrant which is the area for selective catalysts holds a mix of Ti3 and Mo2 catalysts. Applying criterion 4 with this graph, we reduce transition metals that appear twice in the bottom right quadrant. This leaves two points in the bottom left quadrant which correspond to Pt@Ti3 and Pt@Mo2, and one point in the upper right quadrant corresponding to Cu@Mo3. Since Pt@Mo2 holds a large maximum reaction energy of 1.5 eV (towards CH3OH production, see Table S1†), while Pt@Ti3 has a maximum reaction energy of 0.65 eV (towards *CHO), only the latter is kept. For Cu@Mo3, even though its colour suggests high selectivity, the adsorption of CO2 has a large energy barrier of 1.35 eV, making it an unviable catalyst based on criterion 3. The preliminary CO2RR mechanism was still conducted for this catalyst, and in Table S1† we see the catalyst is selective towards formic acid, as its desorption energy is −0.2 eV compared to the subsequent *CHO formation step evaluated at 1.48 eV (see Table S3†). To summarize, there are thus a total of six catalysts that are selected from this stage.
To further justify our findings, Bader charge analysis is conducted on all above catalysts to see the effect of the charge of the SMA on the activity of the catalyst. By taking two lines of best fits, a volcano plot is generated, as can be seen in Fig. 4b. The catalyst with the best activity towards C1 products is Ru@Mo2 with ΔEmax = 0.18 eV, with the Ruthenium atom having a charge of +0.46. When plotting the volcano curve, Ruthenium does sit close at the top, implying that a charge of around +0.5 is optimal. Most Mo3 supported catalysts in dark green have too high of a charge, lowering their activity. Most catalysts are above that optimal charge of +0.5, except Pt and Pd, with their optimal configuration having charges of +0.21 and +0.38, respectively.
To highlight the significant difference in properties between SACs and their bulk counterpart, we compared the adsorption of H and CO2, and the desorption of CH3OH and CH4, on both MXene supported SACs and their most stable counterpart slab structure. Fig. 5 displays the results. Several trends and conclusions can be derived:
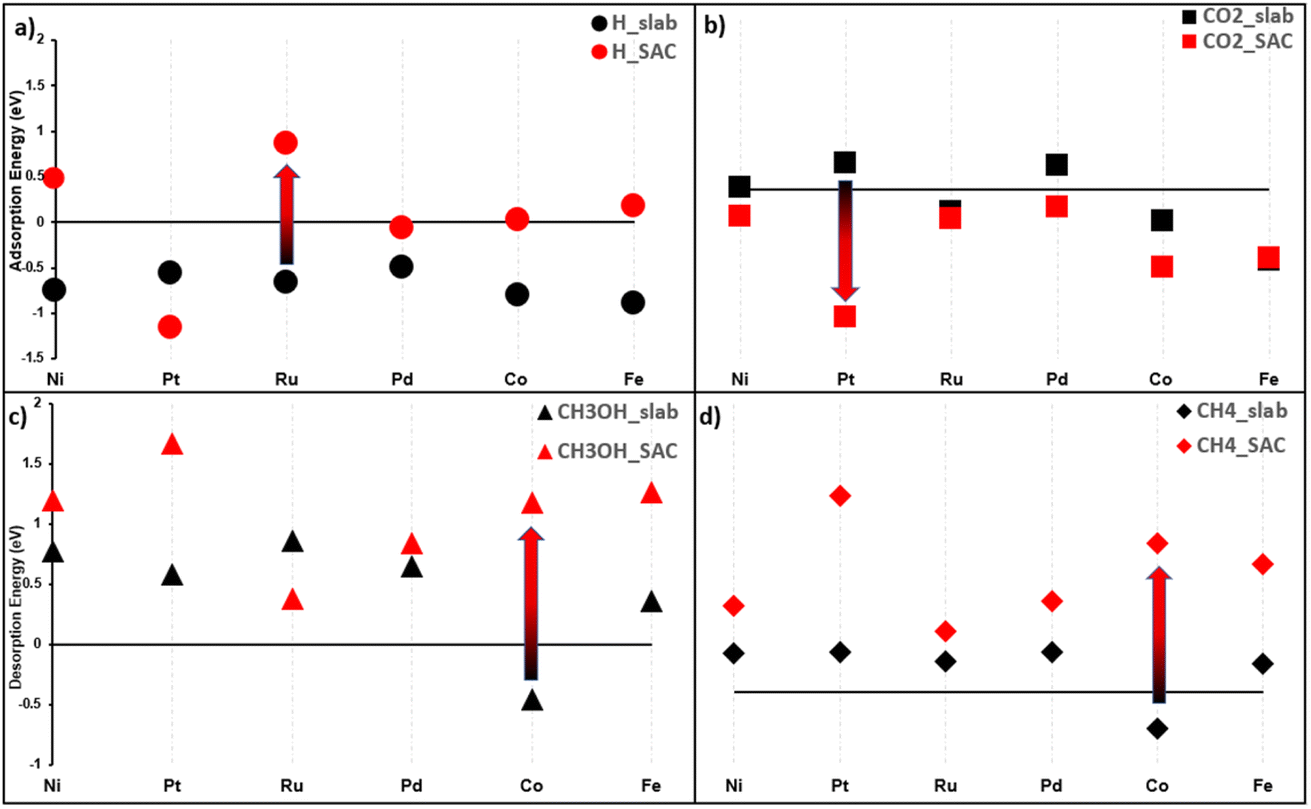 | ||
| Fig. 5 Adsorption energies of: (a) H and (b) CO2 on a slab and SAC, for the six chosen transition metals. Similarly, desorption energies of: (c) CH3OH and (d) CH4. Arrows show the biggest difference between the slab and SAC for each sub-figure. | ||
(i) Five out of six metals exhibit the worst H adsorption on the SAC than slabs (Fig. 5a), aiding in the HER suppression. Note that the bigger the distance between the red and black dot, for a given metal, the bigger the difference between the slab and MXene SAC.
(ii) The exception to (1) is Pt@Ti3 which has a H adsorption value of −1.15 eV. It shows that the adsorption of the H atom is facilitated; however, the HER overall reaction energy is the absolute value of that i.e., 1.15 eV, a high barrier. Thus, we cannot make a conclusion on Pt based on ΔE values only until we calculate the Gibbs reaction energy (ΔG) values.
(iii) All metals have stronger CO2 adsorption on SACs than on the slab, as seen in Fig. 5b.
(iv) While the desorption step of products (ΔEdes) is small (0–0.3 eV) on slabs, we do not observe the same easy desorption step on SACs. Take methane as an example in Fig. 5d, slab desorption values are all lower than SAC desorption values. The same can be said for methanol in Fig. 5c.
(v) One exception to (4) is the Ru:Ru@Mo2 has a lower methanol desorption energy than bulk Ru while not replicating the same trend on methane. Ru@Mo2 is thus hypothesized to be an active catalyst for methanol production.
Although the selected catalysts possess great potential to be selective for C1 products such as methane and methanol, the reaction mechanism towards C–C coupling needs to be investigated too. The unique active site of SACs makes it difficult to achieve C–C coupling and obtain C2+ products. The reaction energy of C–C coupling was investigated via two different pathways which are mostly studied in the literature98–102 for the six catalysts.
| 2*CO → *OCCO | (Rx1) |
| 2*CHO → *OCHCHO | (Rx2) |
Fig. 6a demonstrates that the distance between two *CO on a SMA increases and the coupling is not favourable, as expected. C–C coupling by the *CHO intermediate can occur as can be seen in Fig. 6b, however at each instance the reaction energy is higher than it would be for the TLS of methanol or methane production through the CO2RR. It is worth nothing that iron is the exception, where C–C coupling is at 0.1 eV compared to CH3OH production at 0.56 eV. *CHO protonation in this catalyst has a ΔE = −0.3 eV which is lower than that for the C–C coupling. Thus, it is inconclusive whether C–C coupling will occur on Fe@Mo2 or the *CHO protonation. On the other five catalysts, the C–C coupling energy is too high, suppressing the multi-carbon product formation. It is important to note that at high metal loadings, SMA can agglomerate either forming nanoparticles/nanoclusters on the support or dual sites which are called Dual Atom Catalysts (DACs). While thermodynamically,79 DACs can be more favourable than SACs, appropriate control of the loading % of the SMA can ensure only SACs are synthesized.56,79,81,103 One challenge to note is achieving high loading of the SMA in SACs is challenging due to agglomeration, which can lead to a decrease in performance.56 One reason for this is that C–C coupling on DACs can be more favourable than on SACs. However, this is out of the scope of this study.
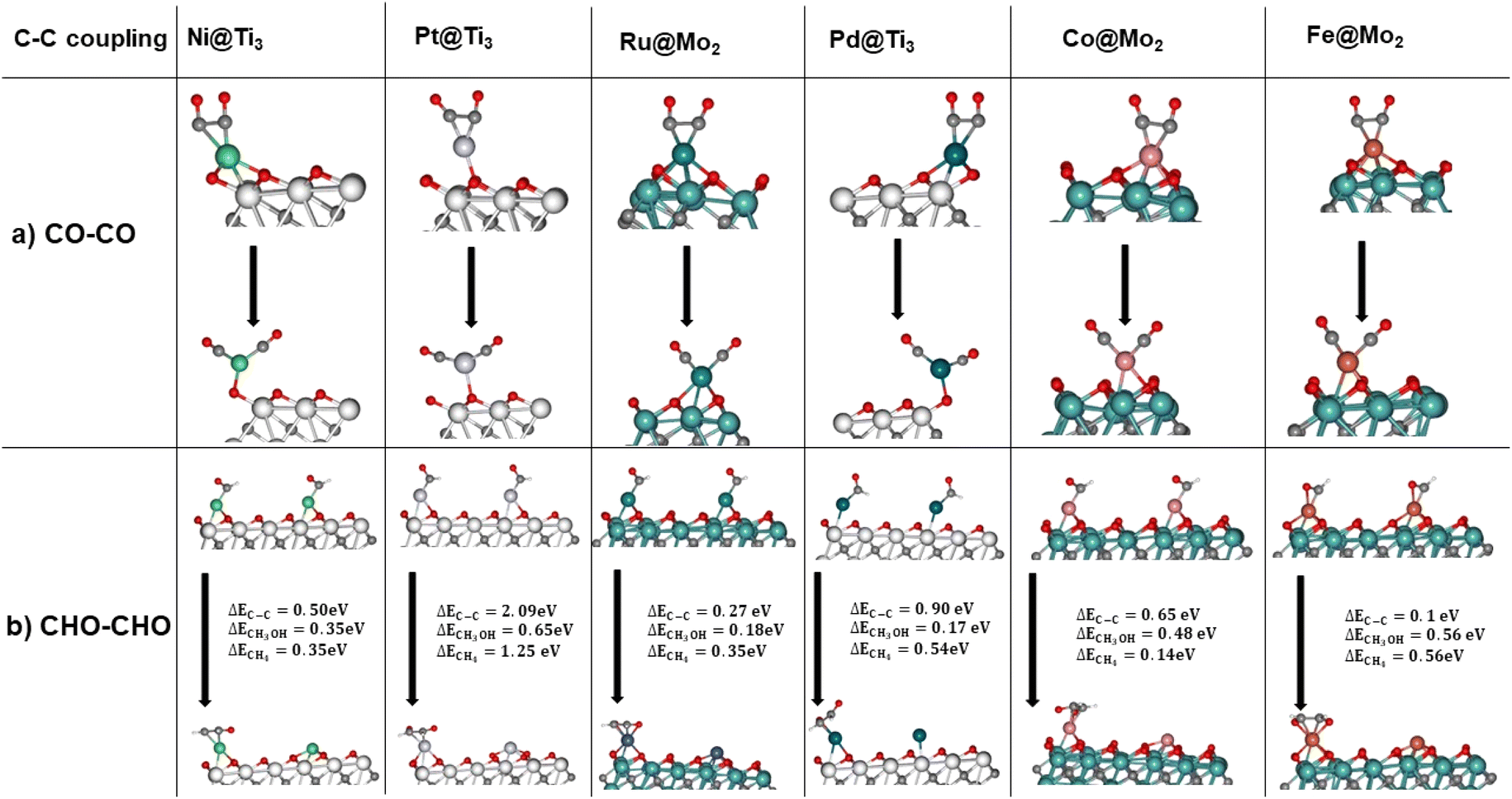 | ||
| Fig. 6 (a) *CO + *CO coupling. The first image is before optimization while the bottom image is the optimized structure. (b) *CHO + *CHO coupling and the reaction energy comparatively to methanol and methane production. | ||
Having screened 25 catalysts, six are selected as favourable C1 producers. The DFT energies were converted to the Gibbs free energies to include the temperature and vibrational effects on the energy (Table S5†). The Gibbs free energy of the reactants and products that are not adsorbed was calculated using the ideal gas model.104 One notable difference that arose after performing the Gibbs calculations is the shift in the Pt@Ti3 reaction energy. In Table S6,† we see that Pt prefers the *CO2 conversion into *COOH and subsequently to the *CO pathway than the *OCHO pathway. However, the new TLS is through *CO conversion into *CHO which is at a ΔG = 1.16 eV. Furthermore, when looking at CO desorption values in Fig. S4,† Pt has a remarkably high *CO desorption energy of 2.76 eV. Other catalysts all have similar *CO desorption values ranging from 0.92 to 1.16 eV, except Ru@Mo2 with a value of 0 eV. For the four catalysts in the ∼1 eV region, a modest value for *CO affinity leads to the best CO2RR performance. For Ru@Mo2, to produce CO, *COOH must be made instead of *OCHO, however *CO2 hydrogenation to *COOH has a ΔG = 0.78 eV while its hydrogenation to *OCHO has a ΔG = 0.59 eV and is the TLS. Thus, we conclude that Pt is unfavourable for methanol or methane formation, while the five others are selective for either methanol or methane formation.
Looking back at the mechanism in Fig. 3, to produce methane, water must be released through *O hydrogenation to *OH, subsequent hydrogenation to *H2O, and eventually desorption of H2O. We observed that for products such as water, methane, and methanol, it is imperative to include the desorption step as SACs tend to have significant affinity for products, unlike metal slabs. Therefore, the following reactions in the later steps of the mechanism are considered:
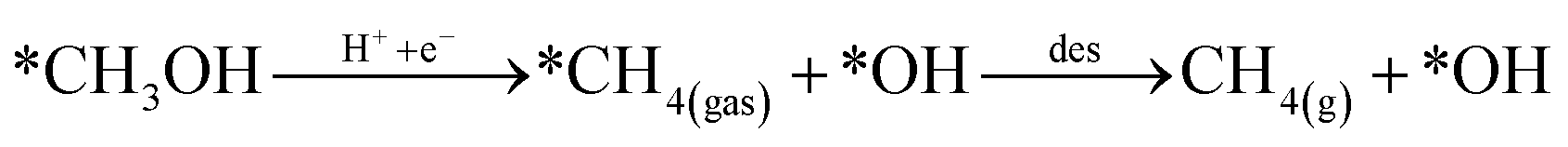 | (Rx3) |
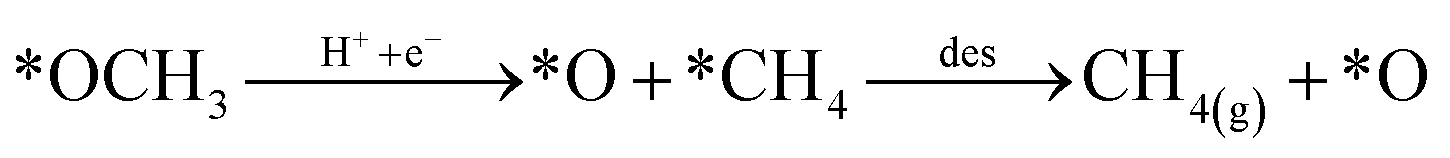 | (Rx4) |
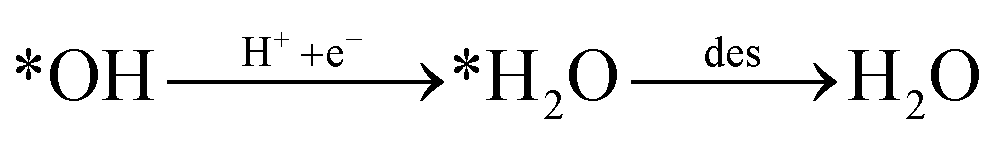 | (Rx5) |
Desorption energy of water and methane compared to methanol determines which product will be formed. If either the desorption of methane or water is higher than methanol, then the latter will be formed. Table S6† shows the desorption energies for these compounds as well as for intermediate products like formate and formaldehyde. Table 2 summarizes the conclusion drawn from each of these six selected catalysts.
| Ni@Ti3 | Ru@Mo2 | Fe@Mo2 | Co@Mo2 | Pd@Ti3 | Pt@Ti3 | |
|---|---|---|---|---|---|---|
| Product formed | Methane | Methanol | Methanol | Methanol | Methanol | H2/CO |
| TLS | *OCH2 → *OCH3 | *CO2 → *OCHO | *CH3OH → CH3OH | *OCHO → *HCOOH | CO2 ads | *H → H2 |
| Highest reaction energy (eV) | 0.267 | 0.594 | 0.411 | 0.369 | 0.296 | 0.88 |
| Activity (UCO2) (eV) | 0.267 | 0.594 | 0.411 | 0.369 | 0.296 | 0.880 |
| Selectivity (UCO2 − UH2) (eV) | −1.062 | −0.537 | −0.024 | 0.098 | 0.116 | −0.008 |
| Activity & selectivity (eV) | −0.795 | 0.057 | 0.387 | 0.467 | 0.412 | 0.872 |
| Ranking | 1 | 2 | 3 | 5 | 4 | 6 |
Based on the results in Table S6,† all catalysts but Pt SAC, which is not active for the CO2RR, had low methanol desorption values compared to water and methane, making them selective towards methanol. The exception is Ni@Ti3, which has a lower methane and water desorption value, making it a more active catalyst for methane production. Interestingly, we did not observe any similarity among the TLS steps for the different catalysts studied here. It is worth noting that Ti3 catalysts have the adsorption of CO2 as the TLS, while for the other Mo2 catalysts different protonation steps are the TLS. The reaction energies range from 0.27 to 0.6 eV, as seen in Fig. 7, with Ni@Ti3 exhibiting the lowest reaction energy. To create a ranking system, the activity and selectivity of each catalyst was calculated. The activity is defined as the CO2RR reaction energy while the selectivity is defined as the CO2RR reaction energy minus the HER reaction energy. Note that for both parameters, lower values mean superior performance. Finally, we sum both values as the final number to classify each catalyst. Fig. 7 depicts the energy diagram of the best 3 catalysts based on this ranking system, and a full energy diagram is available in Fig. S5.† The proposed five catalysts have been shown in previous studies to be synthesizable experimentally,78,81–84,103,105 but none have been used for the CO2RR, to our knowledge. More information can be found in Table S7 in the ESI.†
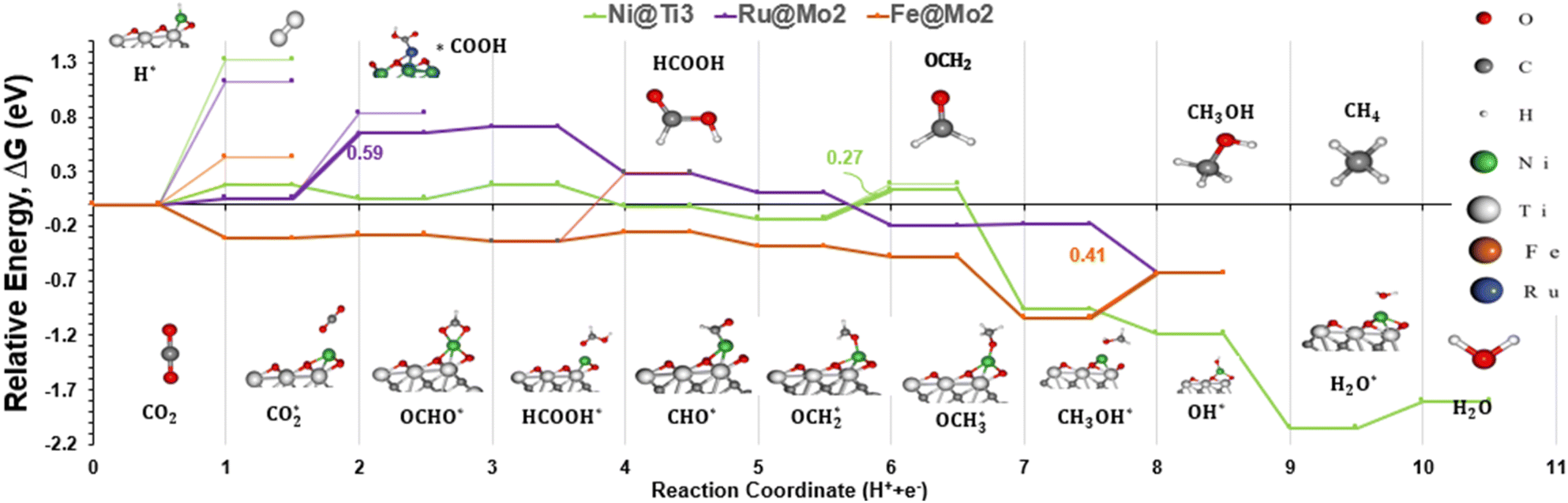 | ||
| Fig. 7 Energy diagram for Ni@Ti3, Ru@Mo2, and Fe@Mo2. The diagram includes the pathways to methane (CH4) and methanol (CH3OH). On the bottom, the main intermediates are presented and match the curves above. On top, the final products, side products or intermediates are shown and match the thin curves below them, if applicable. The reaction coordinate number represents the number of PCET steps, except for adsorption and desorption steps. | ||
Conclusions
We have performed systematic DFT computations to screen and investigate potential single atom MXene catalysts that exhibit high selectivity and low overpotentials towards the CO2RR, specifically for highly reduced C1 products such as methanol and methane. After screening based on formation energy, binding energy, activity, and selectivity, five catalysts were found to exhibit exceptional performance. These catalysts are, in order of performance: Ni@Ti3, Ru@Mo2, Fe@Mo2, Co@Mo2 and Pd@Ti3. Specifically, nickel had the lowest overall reaction energy barrier at 0.27 eV while effectively suppressing the HER. Additionally, iron had an overall reaction energy barrier of 0.4 eV, making these two low-cost transition metals attractive catalysts to synthesize and test in experiments. Finally, we observe that Mo2-based SACs exhibit high performance, opening an avenue for further experimental investigation on them, as they have not been widely exploited for the CO2RR, although they have been synthesized previously. For further computation, kinetic barriers should be calculated to confirm the trends observed.Data availability
The data supporting this article have been included as part of the ESI.† Other computational information, including the DFT input and output files, are available via request from the authors.Author contributions
AS and HA conceived the project. HA performed all the computations, while AS supervised the project. RY helped with the code for high throughput DFT computations. The manuscript was written through contributions of all authors. All authors have given approval to final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NSERC Discovery Grant (RGPIN-2020-04960), Canada Research Chair (950-23288), and FRQNT-NOVA grant (ALLRP 577180 and 2023-NOVA-329854). Hasan Al-Mahayni was financially supported by the Fonds de Recherche du Québec – Nature et technologies (FRQNT) and the NSERC Master Scholarships. The DFT computations carried out in this study were supported by Calcul Quebec, Compute Canada, and the Digital Research Alliance of Canada. Authors would like to thank Mahdi Salehi, a PhD candidate in the Electrocatalysis Lab at McGill University for his comments and help with figure preparation in this study.Notes and references
- L. Al-Ghussain, Environ. Prog. Sustain. Energy, 2019, 38, 13–21 CrossRef CAS.
- A. Ç. Köne and T. Büke, Renew. Sustain. Energy Rev., 2010, 14, 2906–2915 CrossRef.
- P. Nejat, F. Jomehzadeh, M. M. Taheri, M. Gohari and M. Z. A. Majid, Renew. Sustain. Energy Rev., 2015, 43, 843–862 CrossRef CAS.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- C.-H. Huang and C.-S. Tan, Aerosol Air Qual. Res., 2014, 14, 480–499 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- X. Zhang, S.-X. Guo, K. A. Gandionco, A. M. Bond and J. Zhang, Mater. Today Adv., 2020, 7, 100074 CrossRef.
- G. O. Larrazábal, A. J. Martín and J. Pérez-Ramírez, J. Phys. Chem. Lett., 2017, 8, 3933–3944 CrossRef PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O'Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- A. Seifitokaldani, C. M. Gabardo, T. Burdyny, C.-T. Dinh, J. P. Edwards, M. G. Kibria, O. S. Bushuyev, S. O. Kelley, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 3833–3837 CrossRef CAS.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- P. Ding, H. Zhao, T. Li, Y. Luo, G. Fan, G. Chen, S. Gao, X. Shi, S. Lu and X. Sun, J. Mater. Chem. A, 2020, 8, 21947–21960 RSC.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871–874 CrossRef CAS PubMed.
- S. Zhang, X. Jing, Y. Wang and F. Li, ChemNanoMat, 2021, 7, 728–736 CrossRef CAS.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS PubMed.
- K. Rossi and R. Buonsanti, Acc. Chem. Res., 2022, 55, 629–637 CrossRef CAS PubMed.
- M. E. Ahmed, S. Adam, D. Saha, J. Fize, V. Artero, A. Dey and C. Duboc, ACS Energy Lett., 2020, 5, 3837–3842 CrossRef CAS.
- Y. Zheng, Y. Wang, Y. Yuan and H. Huang, ChemNanoMat, 2021, 7, 502–514 CrossRef CAS.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- M. Salehi, H. Al-Mahayni, A. Farzi, M. McKee, S. Kaviani, E. Pajootan, R. Lin, N. Kornienko and A. Seifitokaldani, Appl. Catal., B, 2024, 353, 124061 CrossRef CAS.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- T. N. Nguyen, J. Guo, A. Sachindran, F. Li, A. Seifitokaldani and C.-T. Dinh, J. Mater. Chem. A, 2021, 9, 12474–12494 RSC.
- M. Abdinejad, A. Farzi, R. Möller-Gulland, F. Mulder, C. Liu, J. Shao, J. Biemolt, M. Robert, A. Seifitokaldani and T. Burdyny, Nat. Catal., 2024, 7, 1109–1119 CrossRef CAS.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- X. Wang, Z. Wang, T.-T. Zhuang, C.-T. Dinh, J. Li, D.-H. Nam, F. Li, C.-W. Huang, C.-S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S.-C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- P. Zhu and H. Wang, Nat. Catal., 2021, 4, 943–951 CrossRef CAS.
- O. Talu, in Studies in Surface Science and Catalysis, ed. M. Suzuki, Elsevier, 1993, vol. 80, pp. 655–662 Search PubMed.
- B. van der Zwaan, R. Detz, N. Meulendijks and P. Buskens, Fuel, 2022, 311, 122547 CrossRef CAS.
- T. N. Nguyen, M. Salehi, Q. V. Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- D. Gao, T. Liu, G. Wang and X. Bao, ACS Energy Lett., 2021, 6, 713–727 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- S. Ponnada, M. S. Kiai, D. B. Gorle, R. S. C. Bose, V. Rajagopal, B. Saini, M. Kathiresan, A. Nowduri, R. Singhal, F. Marken, M. A. Kulandainathan, K. K. Nanda and R. K. Sharma, Catal. Sci. Technol., 2022, 12, 4413–4441 RSC.
- A. Liu, X. Liang, X. Ren, W. Guan, M. Gao, Y. Yang, Q. Yang, L. Gao, Y. Li and T. Ma, Adv. Funct. Mater., 2020, 30, 2003437 CrossRef CAS.
- H. Zhu, Z. Liang, S. Xue, X. Ren, X. Liang, W. Xiong, L. Gao and A. Liu, Ceram. Int., 2022, 48, 27217–27239 CrossRef CAS.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.-C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- L.-X. Li, W.-J. Sun, H.-Y. Zhang, J.-L. Wei, S.-X. Wang, J.-H. He, N.-J. Li, Q.-F. Xu, D.-Y. Chen, H. Li and J.-M. Lu, J. Mater. Chem. A, 2021, 9, 21771–21778 RSC.
- L. Jiang, J. Duan, J. Zhu, S. Chen and M. Antonietti, ACS Nano, 2020, 14, 2436–2444 CrossRef CAS PubMed.
- M. Yu, Z. Wang, J. Liu, F. Sun, P. Yang and J. Qiu, Nano Energy, 2019, 63, 103880 CrossRef CAS.
- T. Y. Ma, J. L. Cao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 1138–1142 CrossRef CAS.
- N. Li, X. Chen, W.-J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS.
- A. D. Handoko, K. H. Khoo, T. L. Tan, H. Jin and Z. W. Seh, J. Mater. Chem. A, 2018, 6, 21885–21890 RSC.
- K. Eid, Q. Lu, S. Abdel-Azeim, A. Soliman, A. M. Abdullah, A. M. Abdelgwad, R. P. Forbes, K. I. Ozoemena, R. S. Varma and M. F. Shibl, J. Mater. Chem. A, 2022, 10, 1965–1975 RSC.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu, W. Chen, C. Liu, J. Shang, L. Song and S. Yang, ACS Nano, 2021, 15, 4927–4936 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- Á. Morales-García, A. Fernández-Fernández, F. Viñes and F. Illas, J. Mater. Chem. A, 2018, 6, 3381–3385 RSC.
- Y. Xiao and W. Zhang, Nanoscale, 2020, 12, 7660–7673 RSC.
- H. Chen, A. D. Handoko, T. Wang, J. Qu, J. Xiao, X. Liu, D. Legut, Z. Wei Seh and Q. Zhang, ChemSusChem, 2020, 13, 5690–5698 CrossRef CAS PubMed.
- H. Chen, A. D. Handoko, J. Xiao, X. Feng, Y. Fan, T. Wang, D. Legut, Z. W. Seh and Q. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 36571–36579 CrossRef CAS PubMed.
- T.-Y. Shuai, Q.-N. Zhan, H.-M. Xu, C.-J. Huang, Z.-J. Zhang and G.-R. Li, Chem. Commun., 2023, 59(27), 3968–3999 RSC.
- J. Gonzalez-Julian, J. Am. Ceram. Soc., 2021, 104, 659–690 CrossRef CAS.
- T. D. Kühne, M. Iannuzzi, M. D. Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- H. Al-Mahayni, X. Wang, J.-P. Harvey, G. S. Patience and A. Seifitokaldani, Can. J. Chem. Eng., 2021, 99, 1885–1911 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- X. Liao, R. Lu, L. Xia, Q. Liu, H. Wang, K. Zhao, Z. Wang and Y. Zhao, Energy Environ. Mater., 2022, 5, 157–185 CrossRef CAS.
- J. Scaranto and H. Idriss, Chem. Phys. Lett., 2019, 737, 100008 CrossRef.
- S. Lu, H. L. Huynh, F. Lou, K. Guo and Z. Yu, Nanoscale, 2021, 13, 12885–12895 RSC.
- I. Barlocco, L. A. Cipriano, G. Di Liberto and G. Pacchioni, Adv. Theory Simul., 2023, 6, 2200513 CrossRef CAS.
- A. M. Patel, S. Ringe, S. Siahrostami, M. Bajdich, J. K. Nørskov and A. R. Kulkarni, J. Phys. Chem. C, 2018, 122, 29307–29318 CrossRef CAS.
- G. Di Liberto and G. Pacchioni, Adv. Mater., 2023, 35, 2307150 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- E. Sargeant, F. Illas, P. Rodríguez and F. Calle-Vallejo, J. Electroanal. Chem., 2021, 896, 115178 CrossRef CAS.
- Y. Zou, S. A. Kazemi, G. Shi, J. Liu, Y. Yang, N. M. Bedford, K. Fan, Y. Xu, H. Fu, M. Dong, M. Al-Mamun, Y. L. Zhong, H. Yin, Y. Wang, P. Liu and H. Zhao, EcoMat, 2023, 5, e12274 CrossRef CAS.
- M. A. U. Din, S. S. A. Shah, M. S. Javed, M. Sohail, A. u. Rehman, M. A. Nazir, M. A. Assiri, T. Najam and N. Cheng, Chem. Eng. J., 2023, 474, 145700 CrossRef CAS.
- O. P. Nanda, A. G. Prince, L. Durai and S. Badhulika, Energy Fuels, 2023, 37, 4701–4710 CrossRef CAS.
- S. Zhou, Y. Zhao, R. Shi, Y. Wang, A. Ashok, F. Héraly, T. Zhang and J. Yuan, Adv. Mater., 2022, 34, 2204388 CrossRef CAS PubMed.
- L. Jin, S. You, N. Ren, B. Ding and Y. Liu, Environ. Sci. Technol., 2022, 56, 11750–11759 CrossRef CAS PubMed.
- W. Peng, M. Luo, X. Xu, K. Jiang, M. Peng, D. Chen, T.-S. Chan and Y. Tan, Adv. Energy Mater., 2020, 10, 2001364 CrossRef CAS.
- Z. Kou, W. Zang, W. Pei, L. Zheng, S. Zhou, S. Zhang, L. Zhang and J. Wang, J. Mater. Chem. A, 2020, 8, 3071–3082 RSC.
- G. Bharath, K. Rambabu, A. Hai, I. Othman, N. Ponpandian, F. Banat and P. Loke Show, Chem. Eng. J., 2021, 414, 128869 CrossRef CAS.
- X. Cui, W. An, X. Liu, H. Wang, Y. Men and J. Wang, Nanoscale, 2018, 10, 15262–15272 RSC.
- S. Back, J. Lim, N.-Y. Kim, Y.-H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC.
- J. Zeng, W. Zhang, Y. Yang, D. Li, X. Yu and Q. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 33074–33081 CrossRef CAS PubMed.
- Z. W. Chen, Z. Gariepy, L. Chen, X. Yao, A. Anand, S.-J. Liu, C. G. Tetsassi Feugmo, I. Tamblyn and C. V. Singh, ACS Catal., 2022, 12, 14864–14871 CrossRef CAS.
- F. Li and Q. Tang, J. Mater. Chem. A, 2021, 9, 8761–8771 RSC.
- Y. Baek, H. Song, D. Hong, S. Wang, S. Lee, Y.-C. Joo, G.-D. Lee and J. Oh, J. Mater. Chem. A, 2022, 10, 9393–9401 RSC.
- B. Huang, N. Li, W.-J. Ong and N. Zhou, J. Mater. Chem. A, 2019, 7, 27620–27631 RSC.
- Y. Cheng, J. Dai, Y. Song and Y. Zhang, ACS Appl. Energy Mater., 2019, 2, 6851–6859 CrossRef CAS.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.:Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- R. Bader, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 13348 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- H. Xiao, T. Cheng and W. A. Goddard III, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed.
- J. A. Gauthier, Z. Lin, M. Head-Gordon and A. T. Bell, ACS Energy Lett., 2022, 7, 1679–1686 CrossRef CAS.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 6638–6645 RSC.
- J. Zhang, E. Wang, S. Cui, S. Yang, X. Zou and Y. Gong, Nano Lett., 2022, 22, 1398–1405 CrossRef CAS PubMed.
- A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 Search PubMed.
- D. Kan, D. Wang, X. Zhang, R. Lian, J. Xu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 3097–3108 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00747f |
| This journal is © The Royal Society of Chemistry 2025 |