
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bifunctionality of supported metal hydrodeoxygenation catalysts†
Raiven I. Balderasab,
Logan Myersab,
Jacob Millerb,
Cody J. Wrasman b,
Derek Vardonc and
Ryan M. Richards*ab
b,
Derek Vardonc and
Ryan M. Richards*ab
aDepartment of Chemistry, Colorado School of Mines, 1500 Illinois St, Golden, CO 8040, USA. E-mail: rrichard@mines.edu
bCatalytic Carbon Transformation and Scale-up Center, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, 80401, CO, USA
cAlder Renewables, 2755 Canyon Boulevard, Boulder, CO 80302, USA
First published on 19th May 2025
Abstract
The transition to sustainable energy relies on innovative methods to convert biomass-derived compounds into viable biofuels. In this study, the hydrodeoxygenation (HDO) of 6-undecanone is used as a model reaction to screen bifunctional catalysts, where metal sites facilitate hydrogenation and the support promotes deoxygenation, enabling high conversion and selectivity toward desirable alkanes for biofuel production. This reaction is particularly relevant as it represents a critical step in upgrading volatile fatty acids, derived from biomass, into long-chain hydrocarbons suitable for fuel applications. By examining a range of metals (nickel, cobalt, and tin) on different supports, it is revealed that the choice of metal–support combination is critical to catalyst performance. Zeolite beta's 3D microporous structure and adjustable acidity provide an ideal environment for fine-tuning metal–support interactions (MSIs), which are essential for balancing deoxygenation with alkane isomerization, a desirable trait for biofuels.
Sustainability spotlightThis research advances sustainability by addressing the United Nations Sustainable Development Goals (SDGs) of affordable and clean energy (SDG 7) and climate action (SDG 13). By developing cost-efficient catalysts for hydrodeoxygenation (HDO) of biomass-derived intermediates, this work enables the production of long-chain alkanes, essential for sustainable aviation fuels (SAFs). Unlike conventional approaches relying on platinum-group metals, this study uses earth abundant, non-precious metals, reducing costs and resource dependence. The research highlights strategies to enhance catalyst performance, achieving selective conversion of biomass into valuable hydrocarbons while minimizing byproducts. These innovations support circular economy principles, valorizing waste biomass, and provide scalable solutions for transitioning to renewable energy systems, lowering greenhouse gas emissions from fossil fuels. |
Introduction
The global demand for sustainable energy solutions has prompted extensive research into the conversion of biomass-derived compounds to produce biofuels. Biomass represents a potential and abundant feedstock derived from a non-fossil origin, capable of producing sustainable fuels and other additives and reducing CO2 levels.1 In recent years, there has been a growing emphasis on developing advanced technologies and processes for biomass conversion, including the integration of biorefinery concepts, the development of novel catalysts, and the exploration of emerging conversion pathways.2–8 Additionally, there is a strong focus on improving the efficiency, scalability, and economic viability of biomass conversion processes.9,10Wet waste, as a renewable feedstock, can be converted into volatile fatty acids (VFAs) via arrested methanogenesis. Among the various sources of wet waste, slaughterhouse waste streams are particularly relevant, as they are rich in lipid- and protein-derived fatty acids, including hexanoic acid. The ketonization of these wet waste-derived VFAs (primarily aliphatic C3–C8 acids) produces longer chain ketone (C5–C15) molecules. C8–C15 ketones can undergo direct hydrodeoxygenation while C5–C7 ketones require aldol condensation prior to hydrodeoxygenation to produce carbon chains long enough to for use as diesel/jet fuels.4 Both conversion pathways yield alkanes suitable for biofuel production. For instance, the ketonization of hexanoic acid yields 6-undecanone, water, and carbon dioxide.5,7,11–13 6-undecanone (C-11), the product of hexanoic acid ketonization, is particularly attractive for hydrodeoxygenation (HDO) processes because it yields undecane, a straight-chain alkane with a carbon chain length centrally positioned within the optimal range for sustainable aviation fuel (SAF). This makes it a relevant model compound for studying the production of biofuels tailored for aviation applications.
However, achieving efficient and selective hydrodeoxygenation requires the careful selection and optimization of catalysts, which can profoundly influence reaction rates, product distributions, and overall process viability.14 In addition to achieving high conversion and selectivity in HDO reactions, controlling product distribution through isomerization is crucial for optimizing biofuel properties. Branched alkanes, formed through isomerization, enhance fuel performance by improving cold flow properties and increasing energy density – key factors for SAF.15 Therefore, understanding how catalyst design influences isomerization pathways is essential for developing efficient bifunctional catalysts. This study not only investigates the conversion of 6-undecanone to undecane but also explores how metal–acid site balance affects the formation of isomerized products, providing insights into catalyst design for selective hydrocarbon production.
The HDO reaction is the removal of oxygen from oxygen-containing compounds. This reaction occurs in three steps: (1) the reduction of ketones to alcohols, (2) the dehydration of the alcohols to alkenes, and (3) the hydrogenation of the alkene to the alkanes as shown in Scheme 1. This reaction requires the use of a bifunctional catalyst with both metal, often a platinum group metal (PGM), and acid sites (often supports such as Al2O3, TiO2, or zeolites).16 In the first step an acid site binds the ketone carbonyl group and H2 dissociates on the metal. In the second step the dissociated hydrogen is transferred from the metal to hydrogenate the bound ketone and form an alcohol.16 Next, the alcohol is dehydrated on the acid site to form an alkene. And lastly, the alkenes are further hydrogenated on the metal site to form the desired 6-undecane product.16 These alkanes can later be distilled into different types of biofuels such as naphtha, jet, and diesel. Considering the mechanism explained above, the importance of the proximity of the metal site to the acid site, porosity of the support, and acid site density of the catalyst is hypothesized to be crucial in tuning production rate, selectivity, and yield of the desired product.
 | ||
| Scheme 1 Hydrodeoxygenation of 6-undecanone. | ||
We evaluated different low-cost, non-precious metals – nickel (Ni), cobalt (Co), tin (Sn) at varying weight percentages (3%, 6%, and 9%), and precious metal platinum (Pt) at weight percentage (3%) as a control – on supports with varied acid site densities and characteristics. The supports selected for this study included zeolite beta (with SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3 ratios of 25:1 and 300:1), TiO2, and SiO2 (control). Zeolite beta offers a high surface area and tunable acidity, featuring both Brønsted and Lewis acid sites, which are critical for facilitating hydrodeoxygenation reactions. Additionally, its 3D microporous structure, with interconnected channels and pores approximately 6–7 Å in size, enhances catalytic activity by promoting close proximity between acid and metal sites, facilitating bifunctional reactions.17 However, accessing internal sites in these micropores may be more challenging compared to external sites on non-porous supports like TiO2, where reactants and intermediates directly interact with active surface sites without requiring diffusion through a porous network. Despite this potential limitation, the data in this study indicate that diffusion limitations are minimal, as reactants (e.g., 6-undecanone) and intermediates are sufficiently small (∼5–6 Å wide) to effectively access internal sites within zeolite beta's micropores. Commercial TiO2 is generally considered nonporous. The overall material may exhibit some mesoporosity (typically defined as pores within diameters 2–50 nm) due to the spaces between aggregated particles.18 These larger pores do not provide the same level of confinement for metal particles within the pores as zeolite beta does, potentially affecting its ability to control catalytic reactions in the same way. SiO2 inherently does not possess acid sites, so it is not expected to be active for dehydration reactions. Nickel, cobalt, and tin are not platinum group metals (PGMs), which presents significant advantages in terms of cost and availability. These metals are more abundant and less expensive, making them attractive alternatives for industrial applications. Their use can potentially lower the overall process cost and enhance the economic feasibility of producing biofuels via HDO. Nickel based catalysts are known to exhibit high hydrogenation activity whereas cobalt and tin offer distinct catalytic behaviors that could be beneficial under specific conditions.14,19 Cobalt catalysts are noted for their ability to balance hydrogenation with deoxygenation, making them particularly suitable for bifunctional reactions that require both metal and acid site synergy, such as the hydrodeoxygenation of oxygen-rich feedstocks. Their moderate hydrogenation strength can reduce over-hydrogenation, preserving olefin intermediates in some cases. For instance, a study demonstrated that cobalt catalysts supported on silica exhibited high selectivity towards aromatic hydrocarbons during the HDO of phenolic compounds, highlighting their effectiveness in deoxygenation processes.20 Tin catalysts demonstrate unique selectivity for deoxygenation pathways and olefin formation, which can be advantageous when olefins are desired intermediates or final products. These properties make tin catalysts intriguing choices for conditions where mild hydrogenation is required to avoid excessive saturation or where the feedstock contains highly oxygenated molecules necessitating selective oxygen removal without over-reduction. Research has shown that tin-modified nickel catalysts can enhance activity, but also lead to lower selectivity of desired products during the HDO of bio-oil model compounds.20 Additionally, the variation in acid site densities among the supports plays a significant role in facilitating the deoxygenation step, influencing both the activity and selectivity of the catalysts. The interplay between metal type, metal loading, and support characteristics is integral for determining the optimal conditions required for the HDO process.
:Al2O3 ratios of 25:1 and 300:1), TiO2, and SiO2 (control). Zeolite beta offers a high surface area and tunable acidity, featuring both Brønsted and Lewis acid sites, which are critical for facilitating hydrodeoxygenation reactions. Additionally, its 3D microporous structure, with interconnected channels and pores approximately 6–7 Å in size, enhances catalytic activity by promoting close proximity between acid and metal sites, facilitating bifunctional reactions.17 However, accessing internal sites in these micropores may be more challenging compared to external sites on non-porous supports like TiO2, where reactants and intermediates directly interact with active surface sites without requiring diffusion through a porous network. Despite this potential limitation, the data in this study indicate that diffusion limitations are minimal, as reactants (e.g., 6-undecanone) and intermediates are sufficiently small (∼5–6 Å wide) to effectively access internal sites within zeolite beta's micropores. Commercial TiO2 is generally considered nonporous. The overall material may exhibit some mesoporosity (typically defined as pores within diameters 2–50 nm) due to the spaces between aggregated particles.18 These larger pores do not provide the same level of confinement for metal particles within the pores as zeolite beta does, potentially affecting its ability to control catalytic reactions in the same way. SiO2 inherently does not possess acid sites, so it is not expected to be active for dehydration reactions. Nickel, cobalt, and tin are not platinum group metals (PGMs), which presents significant advantages in terms of cost and availability. These metals are more abundant and less expensive, making them attractive alternatives for industrial applications. Their use can potentially lower the overall process cost and enhance the economic feasibility of producing biofuels via HDO. Nickel based catalysts are known to exhibit high hydrogenation activity whereas cobalt and tin offer distinct catalytic behaviors that could be beneficial under specific conditions.14,19 Cobalt catalysts are noted for their ability to balance hydrogenation with deoxygenation, making them particularly suitable for bifunctional reactions that require both metal and acid site synergy, such as the hydrodeoxygenation of oxygen-rich feedstocks. Their moderate hydrogenation strength can reduce over-hydrogenation, preserving olefin intermediates in some cases. For instance, a study demonstrated that cobalt catalysts supported on silica exhibited high selectivity towards aromatic hydrocarbons during the HDO of phenolic compounds, highlighting their effectiveness in deoxygenation processes.20 Tin catalysts demonstrate unique selectivity for deoxygenation pathways and olefin formation, which can be advantageous when olefins are desired intermediates or final products. These properties make tin catalysts intriguing choices for conditions where mild hydrogenation is required to avoid excessive saturation or where the feedstock contains highly oxygenated molecules necessitating selective oxygen removal without over-reduction. Research has shown that tin-modified nickel catalysts can enhance activity, but also lead to lower selectivity of desired products during the HDO of bio-oil model compounds.20 Additionally, the variation in acid site densities among the supports plays a significant role in facilitating the deoxygenation step, influencing both the activity and selectivity of the catalysts. The interplay between metal type, metal loading, and support characteristics is integral for determining the optimal conditions required for the HDO process.
Previous research has extensively utilized (PGMs) such as platinum (Pt) and ruthenium (Ru) for HDO reactions due to their excellent catalytic properties and stability under reaction conditions.21 For instance, platinum metal on various supports created via atomic layer deposition have demonstrated high conversion and selectivity towards the desired alkane.16 Similarly, Ru-based catalysts have shown promising results in both activity and resistance to deactivation.21 However, the high cost and limited availability of PGMs pose significant challenges for their widespread industrial application.
In addition to PGMs, sulfided catalysts, such as sulfided nickel-molybdenum (NiMoS) and cobalt-molybdenum (CoMoS), have also been widely studied for HDO processes and are used at large scale in refinery hydrotreating.14 These catalysts are effective in promoting deoxygenation reactions, particularly in the presence of sulfur-containing feedstocks, which help maintain the sulfided state of the catalyst and prevent deactivation. However, the use of sulfided catalysts introduces challenges related to sulfur management, including the need for sulfiding agents and the production of sulfur-containing byproducts, which require additional handling and processing steps.14,21,22
Further optimization of the HDO process includes the need to achieve high conversion and selectivity towards desired hydrocarbons while minimizing the formation of undesirable byproducts, such as incomplete deoxygenation that produces partially oxygenated compounds like 6-undecanol. This research aims to elucidate the optimal catalyst formulations for the HDO of 6-undecanone and contribute to the development of efficient, scalable processes for producing biofuels by systematically modulating these variables. The findings from this study are expected to provide valuable insights into the design of next-generation catalysts for biomass conversion and support the broader adoption of renewable energy technologies.
Experimental
Catalyst preparation
All catalysts were made through an incipient wetness impregnation technique. Four supports were used: Zeolite Beta(25:1, SiO2:Al2O3, Alfa Aesar), Zeolite Beta(300:1, SiO2:Al2O3, Zeolyst), 25 nm anatase TiO2 (Sigma Aldrich), and SiO2 (Perlkat 97–0). Herein, zeolite beta will be termed ZB(25:1) and ZB(300:1). Initially, 1 gram of each support was subjected to nanopure water with dissolved metal salts until the incipient wetness point was reached. The metal salts used for impregnation included cobalt(II) nitrate hexahydrate, nickel(II) nitrate hexahydrate, tin(II) chloride dihydrate, and platinum(IV) chloride (Sigma-Aldrich). After thorough mixing on a watch glass, the paste was dried at 80 °C overnight. Reduction of the catalysts was conducted in a tube furnace at 400 °C, 4 °C per min ramp rate, for 5 h, under 1sccm of 10%H2/Ar flowing gas to reduce the metal salts to metals. An additional reduction was performed on 3%Ni–ZB(25:1) at 850 °C, 4 °C per min ramp rate, for 5 h, under 1sccm of 10%H2/Ar flowing gas to understand metal particle sintering effects on the catalyst. The catalysts were used as is post-calcination with no further treatment prior to catalytic testing.
BET surface area & BJH pore size distribution
Nitrogen physisorption experiments were conducted using the Micromeritics ASAP 2020 instrument using a 5-point nitrogen adsorption/desorption curve at 77 K. Prior to analysis, samples are degassed under vacuum at 90 °C at a ramp rate of 10 °C min−1 and held for 4 h. Data for BET surface areas were determined using the Brunauer–Emmett–Teller (BET) method over a relative pressure range of 0.050 to 0.300 P/P0. BJH pore size distributions were calculated from the desorption branch of the isotherms using the Barrett–Joyner–Halenda (BJH) method, as implemented in the Micromeritics ASAP 2020 software.Determination of active metal sites via chemisorption
Surface metal site density was determined using Altamira Instruments AMI-300. Carbon monoxide chemisorption experiments were performed by utilizing an established procedure for TiO2 coated Pt–Al2O3 catalysts using 100 mg of sample.16 The samples were first heated to 150 °C at 10 °C min−1 under 25 sccm Helium and held for 30 min. The gas flow was then switched to 25 sccm of 10%H2/Ar and the sample was heated to 200 °C at 10 °C min−1 and reduced for 2 h. Next, the carrier gas was switched 25 sccm helium and the sample was cooled to 50 °C. Analysis occurred by injecting 25 pulses of 10%CO/He into the carrier gas with 2 min delays between the pulses with a 515 mL pulse loop size. The post-break through pulse areas were averaged to calculate the CO uptake.Ammonia temperature-programmed desorption (NH3-TPD)
NH3-TPD was performed using an Altamira Instruments AMI-300 equipped with a thermal conductivity detector (TCD) operated at 135 mA and a gain of 10. Approximately 100 mg of catalyst was loaded into a quartz U-tube reactor and pretreated under helium flow (30 cm3 min−1) at 600 °C for 60 min (10 °C per min ramp), followed by cooling to 50 °C. Ammonia adsorption was conducted by exposing the sample to 10% NH3/He (30 cm3 min−1) at 50 °C for 1 min, followed by a helium purge for 60 min at the same temperature. Desorption was performed by ramping the temperature from 50 to 600 °C at 10 °C min−1 and holding for 60 min under helium flow. The TCD signal was recorded continuously at 1 Hz. Ammonia uptake was quantified by integrating the area under the TCD curve. Final acid site densities are reported in mol NH3 per gram of catalyst.Catalytic testing
Hydrodeoxygenation of the model ketone, 6-undecanone (TCI >98% purity), was performed in a Parr 5000 multi reactor heating system. To start, 180 mg of catalyst was added to a glass liner with 15 mL of 10% 6-undecanone in cyclohexane and sealed tightly in the reactor vessel. Next, nitrogen was flushed through the system three times followed by pressurizing the reactor vessel with hydrogen at 35 bars. The reaction was then heated to 250 °C with a stirring rate of 600 rpm and held for 1 h followed by quenching using a water bath.Control tests
Control tests were conducted using commercial SiO2, ZB(25:1), 3%Pt–SiO2, and 3%Pt–SiO2 physically combined with ZB(25:1). SiO2 was chosen as a control due to its lack of acid sites. ZB(25:1) was chosen to assess the role of the support alone, without metal incorporation. 3%Pt–SiO2 was chosen as platinum is a highly active metal used in various industries. 3%Pt–SiO2 physically combined with ZB(25:1) was chosen to elucidate whether the proximity of the acid site to the metal site is important. Additionally, a hot-filtration test was conducted to determine if metals leach post-reaction. Furthermore, 3%Ni–ZB(25:1) was reduced at 850 °C to verify that agglomeration can hinder catalytic activity.
Product analysis
Liquid products obtained post catalytic reaction were analyzed using Agilent Technologies gas chromatograph model 7890A with mass spectrometer model 5975C (GC-MS) and Polyarc system. The PolyARC system (Activated Research Company) was used as an add-on device for GC/FID analysis that completely converts all organic compounds to methane via a two-step combustion–reduction reaction for a uniform carbon response in the FID. This allows for more accurate quantification, increased sensitivity for oxygenated compounds, compound independent calibration, and overall simpler analysis. Sample preparation included injecting 20 mL of sample, 1 mL of nonane as the internal standard, and 1 mL of acetone into the GC vial.Field emission scanning electron microscopy (FE-SEM) & electron dispersive spectroscopy (EDS)
Images were obtained using an SEMTech Solutions Amray 3300 Field Emission Scanning Electron Microscope (FESEM) equipped with WIN10™ SEMView8000 Technology and an Energy-Dispersive X-ray Spectroscopy (EDS) detector. Sample preparation included attaching carbon tape to a mount and flaking powder on top.Scanning transmission electron microscopy (STEM) & EDS
Images were obtained on an FEI Talos 200X Scanning Transmission Electron Microscope (STEM), equipped with high-resolution (HRTEM) and an Energy Dispersive X-ray Spectroscopy (EDS detector). Preparation of the sample slide consisted of a small amount of the sample powder (∼5–10 mg) being dissolved in ethanol and sonicated for 1 minute before being drop cast onto a copper TEM grid with a carbon mesh.Inductively coupled plasma atomic emission spectroscopy (ICP-AES)
Samples were prepared for ICP-AES by firstly measuring out 100 mg of sample and adding concentrated nitric acid and hydrochloric acid. Once the samples were prepared, they were subjected to microwave digestion (MARS) to ensure efficient leaching of the metals. Next the sample container was washed 3 times with nanopure water corresponding to a total volume of 0.042 L. 1 mL of this solution was extracted and diluted with 9 mL of nanopure water and submitted for ICP-AES analysis.X-ray diffraction (XRD)
All samples were analysed via XRD to determine phase purity and the presence of metal. Samples were scanned between 10 and 80° two-theta and plotted against their parent support XRD patterns as well as individual metal XRD patterns sourced from The Materials Project Database.23Results and discussion
Catalyst characterization – as prepared
The XRD patterns presented for Fig. 1 and 2 show the diffraction data for 3, 6, and 9% metal loadings of cobalt (Co), nickel (Ni), and tin (Sn) supported on ZB(25:1) and ZB(300:1). These X-ray diffractograms (XRD) are crucial for determining the crystalline phases and confirming the presence of the metal species, alongside the structural integrity of the zeolite framework. All samples exhibit characteristic peaks of the zeolite beta structure, confirming that the framework remains intact post metal incorporation. The lack of visible tin peaks in the XRD patterns likely indicates that tin is highly dispersed as small nanoparticles or amorphous clusters. In Tables 1–3, the BET surface area of most samples decreases as metal loading increases; however, pore volume remains relatively consistent. Additionally, the ICP-AES results confirm the presence of cobalt, nickel, and tin. Interestingly, the ICP-AES results for tin catalysts showed significantly lower loadings than targeted. This may be due to precursor volatility. Tin chloride precursors are more volatile than cobalt or nickel nitrates. During calcination, volatile species like SnCl4 can evaporate before being anchored onto the zeolite, leading to a lower final tin content.24 Cloudiness on the quartz tube furnace was visible post-calcination for tin samples indicating this might be the potential reason for lower tin content. For the Ni–ZB(25:1 and 300:1) series, the diffraction peaks can be attributed to metallic nickel (Ni0). Metallic nickel typically shows its strongest reflection at around 44.5°(2θ) corresponding to the (111) plane of the FCC structure, with weaker peaks at approximately 51.8°(2θ) and 76.4°(2θ) corresponding to the (200) and (220) planes, respectively. The increase in peak intensity with higher loadings suggests larger or more crystalline nickel particles. Cobalt on ZB(25:1) and ZB(300:1) has similar reflections to nickel albeit with reduced intensity and peak broadening suggesting smaller dispersed metal particles.
 | ||
| Fig. 1 XRD patterns of 3, 6, 9 wt% Co, Ni, and Sn on ZB(25:1). | ||
 | ||
| Fig. 2 XRD patterns of 3, 6, 9 wt% Co, Ni, and Sn on ZB(300:1). | ||
:1) supports
| Targeted catalyst | Surface area (m2 g−1) | BJH ads. pore volume (cm3 g−1) | ICP-AES (metal wt%) |
|---|---|---|---|
| 3%Co–ZB(25:1) |
550 | 0.75 | 4.48 |
| 6%Co–ZB(25:1) |
470 | 0.64 | 6.69 |
| 9%Co–ZB(25:1) |
480 | 0.66 | 11.99 |
| 3%Ni–ZB(25:1) |
530 | 0.65 | 3.53 |
| 6%Ni–ZB(25:1) |
500 | 0.63 | 7.27 |
| 9%Ni–ZB(25:1) |
490 | 0.64 | 10.77 |
| 3%Sn–ZB(25:1) |
520 | 0.67 | 3.54 |
| 6%Sn–ZB(25:1) |
490 | 0.66 | 5.00 |
| 9%Sn–ZB(25:1) |
470 | 0.60 | 6.52 |
:1) supports
| Targeted catalyst | Surface area (m2 g−1) | BJH ads. pore volume (cm3 g−1) | ICP-AES (metal wt%) |
|---|---|---|---|
| 3%Co–ZB(300:1) |
530 | 0.12 | 3.13 |
| 6%Co–ZB(300:1) |
480 | 0.12 | 6.64 |
| 9%Co–ZB(300:1) |
450 | 0.10 | 8.70 |
| 3%Ni–ZB(300:1) |
580 | 0.13 | 2.82 |
| 6%Ni–ZB(300:1) |
560 | 0.12 | 5.83 |
| 9%Ni–ZB(300:1) |
560 | 0.12 | 8.43 |
| 3%Sn–ZB(300:1) |
570 | 0.13 | 2.57 |
| 6%Sn–ZB(300:1) |
550 | 0.13 | 5.00 |
| 9%Sn–ZB(300:1) |
550 | 0.14 | 5.83 |
| Targeted catalyst | Surface area (m2 g−1) | BJH ads. pore volume (cm3 g−1) | ICP-AES (metal wt%) |
|---|---|---|---|
| 3%Co–TiO2 | 63 | 0.32 | 2.66 |
| 6%Co–TiO2 | 60 | 0.30 | 6.70 |
| 9%Co–TiO2 | 60 | 0.35 | 12.19 |
| 3%Ni–TiO2 | 70 | 0.35 | 3.69 |
| 6%Ni–TiO2 | 60 | 0.33 | 6.40 |
| 9%Ni–TiO2 | 60 | 0.31 | 12.29 |
| 3%Sn–TiO2 | 60 | 0.33 | 3.75 |
| 6%Sn–TiO2 | 50 | 0.32 | 5.31 |
| 9%Sn–TiO2 | 50 | 0.32 | 7.98 |
The XRD patterns shown in Fig. 3 illustrate the diffraction data for cobalt (Co), nickel (Ni), and tin (Sn) supported on TiO2 (anatase phase) with metal loadings of 3, 6, and 9 wt%. The anatase TiO2 diffraction peaks are present in all samples, confirming that the TiO2 support remains intact across all metal loadings and metals. Cobalt reflections are absent across all TiO2 supports, indicating high dispersion or amorphous Co domains. For 6%Sn–TiO2, faint reflections assignable to a crystalline Sn-containing phase begin to emerge, suggesting partial crystallization at higher loading, although the majority of tin likely remains amorphous or highly dispersed. In the Ni–TiO2 series, Ni reflections become more apparent as loading increases from 6% to 9%, though they remain relatively broad and less intense than those observed for Ni supported on zeolite beta.
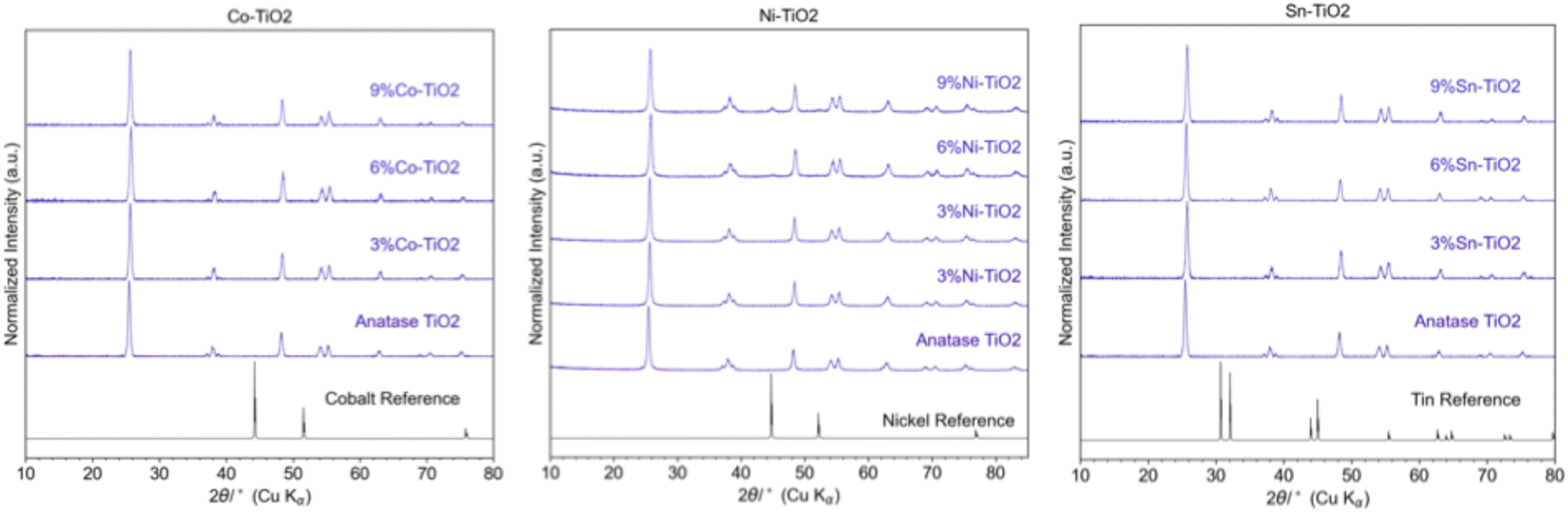 | ||
| Fig. 3 XRD patterns of 3, 6, 9 wt% Co, Ni, and Sn on TiO2. | ||
The differing crystalline behaviors of Ni, Co, and Sn on zeolite beta and TiO2 can be attributed to metal–support interaction strength, precursor properties, and reduction tendencies. Ni and Co exhibit strong interactions with the acidic framework of zeolite beta, which promote anchoring and spatial confinement within the micropores. This anchoring supports the formation of moderate crystalline metal domains. For Ni, distinct reflections are observed, while Co also produces diffraction peaks, albeit broader and of lower intensity-indicating smaller or less crystalline domains. These differences likely reflect variations in crystallization behavior or reduction extent between the two metals. The confined pore structure of zeolite beta limits particle growth, but still allows ordered domain formation under certain conditions.
In contrast, TiO2 provides weaker metal–support interaction, resulting in poorer nucleation control and less ordering, which explains the broader and less intense Ni reflections seen in Fig. 3. While broad XRD peaks generally indicate smaller crystallite sizes, in the case of TiO2, the poor crystallinity more likely reflects limited nucleation efficiency and weak anchoring. The lack of well-defined Co reflections on TiO2 suggests that cobalt remains highly dispersed or amorphous. For Sn, weak interactions with both supports combined with precursor volatility likely suppresses crystalline formation altogether on zeolite beta and limit it on TiO2, where only faint reflections are seen on 6%Sn–TiO2. This behavior is consistent with the lower tin loadings measured by ICP (Tables 1–3). These comparisons reinforce that it is not just the strength, but also the nature of the metal–support interaction- acidic versus non-acidic, porous versus non-porous-that influence crystallinity and XRD visibility of supported metal phases.
Fig. 4–6 display SEM and EDS mapping for 9% metal-loaded catalysts supported on ZB(25:1), ZB(300:1), and TiO2, respectively. In all cases, the metal particles appear uniformly distributed across the surface, indicating good dispersion despite the relatively high metal loading. This uniformity is especially notable on zeolite beta supports, where the microporous structure likely promotes enhanced metal anchoring through interactions with acidic sites. Fig. 7 provides further insight into how support acidity influences metal dispersion by comparing 9%Ni–ZB(25:1) and 9%Ni–ZB(300:1) via STEM and EDS mapping. In 9%Ni–ZB(25:1), nickel nanoparticles are finely dispersed and appear embedded within the zeolite framework. This distribution is consistent with stronger metal–support interactions, likely facilitated by the higher aluminum content and greater acid site density of the ZB(25:1). In contrast, 9%Ni–ZB(300:1) shows larger, more crystalline nickel particles concentrated along the external surfaces of the zeolite. Elemental mapping reveals minimal overlap between nickel and aluminum, indicating weaker interactions and less incorporation of nickel into the support. This result demonstrates that metal dispersion is strongly influenced by the acid site density of the support and is further supported by the increased peak intensities of the nickel reflections observed in the XRD patterns shown in Fig. 2.
 | ||
| Fig. 4 SEM and EDS mapping of 9%metal–ZB(25:1). (A) = 9%Co–ZB(25:1), (B) = 9%Ni–ZB(25:1), (C) = 9%Sn–ZB(25:1). | ||
 | ||
| Fig. 5 SEM and EDS mapping of 9%metal–ZB(300:1). (A) = 9%Co–ZB(300:1), (B) = 9%Ni–ZB(300:1), (C) = 9%Sn–ZB(300:1). | ||
 | ||
| Fig. 6 SEM and EDS mapping of 9%metal–TiO2. (A) = 9%Co–TiO2, (B) = 9%Ni–TiO2 (C) = 9%Sn–TiO2. | ||
 | ||
| Fig. 7 STEM and EDS analysis of 9%Ni–ZB catalysts. (A) = 9%Ni–ZB(25:1), (B) = 9%Ni–ZB(300:1), (C and D) = EDS mapping of 9%Ni–ZB(300:1). | ||
To further investigate the role of support acidity in catalyst function, NH3 temperature-programmed desorption (NH3-TPD) was conducted to quantify the acid site density across the same set of materials. As expected in Table 4, bare zeolite beta with a lower Si/Al ratio exhibited significantly higher NH3 uptake: ZB(25:1) adsorbed 8.59 × 10−4 molNH3 g−1, while ZB(300:1) showed a reduced uptake of 6.66 × 10−5 mol g−1, consistent with its lower framework aluminum content. These differences in acid site density reflect the anticipated variation in Brønsted acidity and are in good agreement with previously reported values for zeolite beta. Upon metal loading, acid site accessibility decreased, with 3%Co–ZB(25:1) displaying a markedly low uptake of 4.29 × 10−4 mol g−1, likely due to cobalt occupying or blocking acid sites. In contrast, nickel catalysts exhibited higher acid site accessibility: 9%Ni–ZB(25:1) adsorbed 5.72 × 10−3 mol g−1 and 9%Ni–ZB(300:1) adsorbed 4.39 × 10−3 mol g−1. These values reflect both the influence of metal loading and the underlying framework acidity from the Si/Al ratio and suggest that nickel interacts less strongly with acid sites compared to cobalt, allowing for greater preservation of acid function. This trend is consistent with the CO chemisorption data in Table 5, which show that while cobalt catalysts exhibit higher total metal loading, their CO-accessible site density is low, likely due to agglomeration or inaccessibility of Co sites. Meanwhile, nickel catalysts maintain higher dispersion, as indicated by greater CO accessibility and higher acid site density.
| Catalyst | Acid site density (mol g−1) – fresh |
|---|---|
| ZB(25:1) |
8.59 × 10−4 |
| ZB(300:1) |
6.66 × 10−5 |
| 3%Co–ZB(25:1) |
4.29 × 10−4 |
| 9%Ni–ZB(25:1) |
5.72 × 10−3 |
| 9%Ni–ZB(300:1) |
4.39 × 10−3 |
| Catalyst | Metal site density (mol g−1) – fresh | Metal site density (mol g−1) – spent | Retention of metal site density (%) | BET surface area (m2 g−1) – fresh | BET surface area (m2 g−1) – spent | ΔBET surface area (m2 g−1) |
|---|---|---|---|---|---|---|
| 3%Co–ZB(25:1) |
9.70 × 10−6 | 1.00 × 10−7 | 1.03 | 550 | 384 | 166 |
| 3%Ni–ZB(25:1) |
2.83 × 10−6 | 2.11 × 10−6 | 74.6 | 527 | 342 | 185 |
| 9%Ni–ZB(25:1) |
2.54 × 10−6 | 1.40 × 10−6 | 55.1 | 491 | 389 | 102 |
| 9%Ni–ZB(300:1) |
3.42 × 10−6 | 2.00 × 10−6 | 58.5 | 564 | 443 | 121 |
Notably, the higher Brønsted acid site density of ZB(25:1) correlates directly with increased selectivity toward isomerized products, particularly iso-alkenes and iso-alkanes, as shown in Table 6. This relationship confirms that acid site density is not only essential for dehydration, but also plays a critical role in enabling the branching reactions necessary for isomerization. Overall, NH3-TPD and CO chemisorption data together support the hypothesis that acid site density—tuned by Si/Al ratio and metal incorporation—plays a critical role in the bifunctionality of these catalysts for hydrodeoxygenation, particularly in enabling dehydration and isomerization pathways.
:1) catalysts
| Catalyst | Conversion (%) | Selectivity iso-alcohol (%) | Selectivity iso-alkene (%) | Selectivity iso-alkane (%) | Yield iso-alcohol (%) | Yield iso-alkene (%) | Yield iso-alkane (%) |
|---|---|---|---|---|---|---|---|
| 3%Co–ZB(25:1) |
83.32 | 0.44 | 9.60 | 7.52 | 0.37 | 8.00 | 6.26 |
| 6%Co–ZB(25:1) |
99.99 | — | — | 7.04 | — | — | 7.04 |
| 9%Co–ZB(25:1) |
100 | — | — | 3.74 | — | — | 3.74 |
| 3%Ni–ZB(25:1) |
100 | — | — | 3.39 | — | — | 3.39 |
| 6%Ni–ZB(25:1) |
100 | — | — | 1.22 | — | — | 1.22 |
| 9%Ni–ZB(25:1) |
100 | — | — | 0.79 | — | — | 0.79 |
| 3%Sn–ZB(25:1) |
0.79 | — | — | — | — | — | — |
| 6%Sn–ZB(25:1) |
0.76 | — | — | — | — | — | — |
| 9%Sn–ZB(25:1) |
0.60 | — | — | — | — | — | — |
Catalyst characterization – spent samples
Table 5 provides both metal site densities (mol g−1) and BET surface areas (m2 g−1) for a subset of fresh and spent catalysts. A subset of catalysts was chosen strategically to represent key variables and trends in the dataset, ensuring meaningful insights could still be drawn. The selected catalysts include 3%Co–ZB(25:1), which represents the impact of a low metal loading and the influence of cobalt as an active metal while highlighting the behavior of a catalyst supported on ZB(25:1). Similarly, 3%Ni–ZB(25:1) was chosen to provide a comparison to cobalt at the same low metal loading but with nickel, a metal known for its strong hydrogenation activity. To examine the effect of increased metal loading on the same support, 9%Ni–ZB(25:1) was included. Finally, 9%Ni–ZB(300:1) was selected to contrast the 25:1 support by representing the influence of lower acidity (300:1 Si/Al ratio) on metal site density and catalyst stability.
These data shed light on both the accessible active metal sites and the overall surface area of the catalyst, offering insights into catalyst stability and structural changes after the HDO reaction. The metal site density of all catalysts decreased after the HDO reaction, with the most significant loss observed for 3%Co–ZB(25:1), which retained only 1.03% of its initial site density. This sharp decrease suggests severe deactivation, possibly due to cobalt leaching, migration, or agglomeration under reaction conditions. In contrast, 3%Ni–ZB(25:1) retained 74.6% of its initial site density, indicating that nickel is substantially more stable than cobalt under identical conditions. This stability may reflect stronger metal–support interactions or the inherent robustness of nickel in the reaction environment.
For 9%Ni–ZB(25:1), the catalyst retains 55.1% of its initial metal site density after reaction, suggesting moderate deactivation despite the higher nickel loading. This level of retention may indicate some loss of accessible sites due to sintering, migration, or pore blockage, but still reflects relatively good stability under reaction conditions. The 9%Ni–ZB(300:1) catalyst, supported on the lower-acidity zeolite beta framework, shows comparable behavior, retaining 58.5% of its initial metal site density. This result suggests that decreasing support acidity does not significantly impair metal site stability. Overall, across both supports and loadings, nickel demonstrates stronger retention and structural stability than cobalt, reinforcing its suitability as an active metal for hydrodeoxygenation catalysts.
The BET surface area measurements show reductions across all catalysts after the reaction. 3%Co–ZB(25:1) decreases from 550 to 384 m2 g−1 (Δ166 m2 g−1), indicating a loss in surface area that may suggest structural changes in the support. Similarly, 3%Ni–ZB(25:1) decreases from 527 to 342 m2 g−1 (Δ185 m2 g−1), indicating considerable losses in surface area which aligns with the reduced metal site density. 9%Ni–ZB(25:1) sees the smallest surface area reduction from 491 to 389 m2 g−1 (Δ102 m2 g−1), suggesting that the catalyst retains more of its initial surface area despite the reaction conditions. 9%Ni–ZB(300:1) shows a reduction from 564 to 443 m2 g−1 (Δ121 m2 g−1). While still moderate, this reduction in surface area is greater than that seen in 9%Ni–ZB(25:1), possibly due to the lower acid site density of ZB(300:1). The lower acidity might provide weaker metal–support interactions, which could lead to a slightly higher susceptibility to structural changes in the reaction environment. These BET surface area reductions indicate that while all supports experience some degree of surface area loss, 9%Ni–ZB(25:1) shows the highest resistance to such changes, suggesting that both metal–support interactions and acid site density contribute to the structural stability of the catalyst.
Control tests
In this study, conversion refers to the transformation of 6-undecanone into any reaction products, including partially deoxygenated intermediates (such as 6-undecanol) and fully deoxygenated hydrocarbons (such as 5-undecene, undecane and their isomers).Fig. 8 presents control experiments designated to investigate critical factors affecting catalytic performance in the hydrodeoxygenation (HDO) of 6-undecanone. These experiments provide essential insights into the roles of acid sites, metal sites, proximity between active sites, and the effects of high-temperature treatment on catalyst activity.
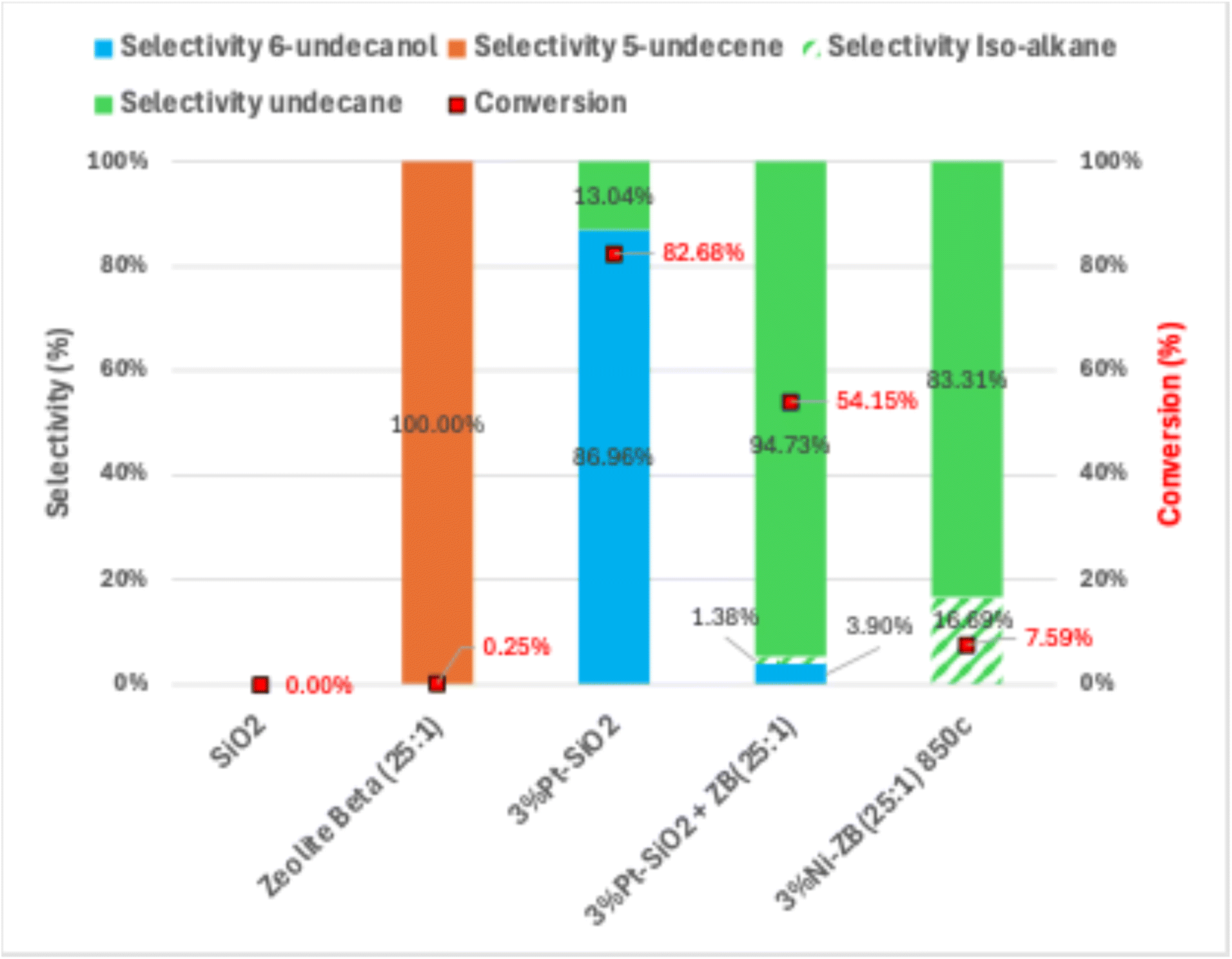 | ||
| Fig. 8 Conversion and selectivity of control experiments. Green = undecane and isomers associated. Blue = 6-undecanol and isomers associated. | ||
SiO2, lacking any inherent acid sites, should be inactive in HDO reactions. This is confirmed in Fig. 8. SiO2 by itself shows no catalytic activity, resulting in no conversion nor selectivity towards deoxygenated products (alkenes or alkanes). This highlights the essential role of acid sites in facilitating key steps in the HDO pathway, particularly the dehydration of alcohol intermediates. Without acid sites or metal sites, SiO2 cannot advance the reaction. The inclusion of ZB(25:1) as a control experiment allows us to isolate and evaluate the contribution of the acidic support in the hydrodeoxygenation (HDO) process. The results show that zeolite beta alone exhibits 100% selectivity to 5-undecene but achieves 0% conversion to undecane, indicating that the acidic sites of the zeolite beta are sufficient to promote dehydration (alkene formation) but lack the necessary hydrogenation activity to saturate the hydrocarbon.
The experiment with 3%Pt–SiO2, tests whether the presence of metal sites alone, without acid sites, can generate catalytic activity. Fig. 8 shows that while 3%Pt–SiO2 enables some level of hydrogenation, it does not proceed through the entire deoxygenation pathway to fully convert 6-undecanone to alkanes. A high selectivity (86.96%) and yield (71.90%) towards 6-undecanol proves this. This limited activity suggests that, although platinum can initiate reduction of the ketone, the absence of acid sites prevents further reaction steps such as dehydration of intermediates.
The experiment with 3%Pt–SiO2 physically combined with ZB(25:1) tests the effect of spatial proximity between metal and acid sites. Interestingly, this combined setup does show partial activity, with the formation of some isomerized alkanes and a noticeable selectivity shift towards the alkane product rather than the alcohol product. This suggests that while the physical separation limits full catalytic efficiency, some intermediates generated on the metal sites of Pt–SiO2 can interact with the acid sites of ZB(25:1) to undergo structural rearrangement, producing isomers. However, the overall catalytic activity and selectivity are lower compared to integrated catalysts (Fig. 11 and 12), indicating that close proximity between acid and metal sites is necessary for efficient conversion and complete deoxygenation. This result demonstrates that while physical combinations can introduce some level of functionality, they fall short of effectiveness achieved when metal and acid sites are closely integrated.
The hot-filtration control test was conducted by introducing 5 mL of the post-reaction filtrate from 3%Ni–ZB(25:1) (which achieved 100% conversion) into a fresh solution containing 10 mL of 10% 6-undecanone in cyclohexane. The results show no further conversion which aligns with the design and intended functionality of zeolite-supported nickel catalysts, where the active metal sites are retained within the solid structure rather than leaching into the reaction medium.
The final control test, involving 3%Ni–ZB(25:1) calcined at 850 °C, evaluates the effects of high-temperature calcination on catalyst structure and performance. Elevated calcination temperatures are known to promote metal particle sintering and agglomeration, which can diminish metal dispersion and active site accessibility. As shown in Fig. 9, sharper diffraction peaks in the XRD pattern of the 850 °C sample indicate the emergence of larger, more crystalline nickel domains. This observation is further supported by EDS elemental mapping, which reveals regions of nickel agglomeration across the zeolite surface. Together, these structural changes align with the observed decrease in catalytic activity and selectivity (Fig. 8), confirming that thermal sintering compromises performance. These results emphasize the importance of maintaining a high degree of metal dispersion to preserve catalytic efficiency in hydrodeoxygenation reactions.
 | ||
| Fig. 9 (A) XRD patterns of 3%Ni–ZB(25:1) calcined at 400 °C and 850 °C. (B) EDS mapping of 3%Ni–ZB(25:1) calcined at 850 °C. | ||
Influence of metal and acid site density on HDO performance
Several catalysts tested showcased remarkable activity towards the hydrodeoxygenation reaction. Fig. 10–12 illustrate conversion and selectivity across anatase TiO2, ZB(25:1), and ZB(300:1) respectively. Tables 6 and 7 showcase conversion, selectivity, and yield of isomers for ZB(25:1), ZB(300:1).
 | ||
| Fig. 10 Conversion and selectivity of all TiO2 catalysts. Green = undecane and isomers associated. Orange = undecene and isomers associated. Blue = 6-undecanol and isomers associated. | ||
 | ||
| Fig. 11 Conversion and selectivity of all ZB(25:1) catalysts. Green = undecane and isomers associated. Orange = undecene and isomers associated. Blue = 6-undecanol and isomers associated. | ||
 | ||
| Fig. 12 Conversion and selectivity of all ZB(300:1) catalysts. Green = undecane and isomers associated. Orange = undecene and isomers associated. Blue = 6-undecanol and isomers associated. | ||
:1) catalysts
| Catalyst | Conversion (%) | Selectivity iso-alkene (%) | Selectivity iso-alkane (%) | Yield iso-alkene (%) | Yield iso-alkane (%) |
|---|---|---|---|---|---|
| 3%Co–ZB(300:1) |
100 | — | 11.61 | — | 11.61 |
| 6%Co–ZB(300:1) |
100 | — | 2.26 | — | 2.26 |
| 9%Co–ZB(300:1) |
100 | — | 5.38 | — | 5.38 |
| 3%Ni–ZB(300:1) |
100 | — | 2.41 | — | 2.41 |
| 6%Ni–ZB(300:1) |
100 | — | 1.05 | — | 1.05 |
| 9%Ni–ZB(300:1) |
100 | — | 1.08 | — | 1.08 |
| 3%Sn–ZB(300:1) |
0.77 | 16.52 | — | 0.43 | — |
| 6%Sn–ZB(300:1) |
0.55 | 8.98 | — | 0.31 | — |
| 9%Sn–ZB(300:1) |
0.98 | 11.20 | — | 0.61 | — |
Fig. 10 presents the conversion and selectivity data for all TiO2 support catalysts in the hydrodeoxygenation of 6-undecanone. Compared to zeolite beta supports shown in Fig. 11 and 12, TiO2 exhibits notably lower selectivity and conversion to alkanes. Commercial TiO2 is generally considered nonporous with some degree of mesoporosity.18 With this in mind, these differences are attributed not only to TiO2's lower acid density, but also its nonporous or mesoporous structure, which lacks the confinement effects present in the microporous framework of zeolite beta. This difference in porosity affects how reactants and intermediates interact with active sites within the support. In zeolite beta, the micropores create a confined environment that enhances interactions between metal sites and acid sites, facilitating sequential deoxygenation steps and supporting more complex pathways, such as isomerization.25 However, the TiO2 support provides a less confined environment, reducing the proximity between metal particles and available acid sites. This can lead to lower conversion efficiency, absence of isomerization, and an increased likelihood of incomplete deoxygenation, as seen by the significant presence of 6-undecanol and alkene products in Fig. 10.
Fig. 11 and 12 illustrate a clear trend in the catalytic behavior of metal-supported catalysts in the HDO of 6-undecanone, specifically highlighting superior catalytic activity with cobalt and nickel on zeolite supports. The data indicates that as metal loading increases, the extent of alkene and alkane isomerization decreases (Tables 6 and 7). This suggests a correlation between metal concentration on the support and the catalyst's ability to promote branching in the alkane products. At lower metal loadings, the catalysts exhibit higher isomerization, producing a more branched alkane product distribution, as well as a small degree of alcohol and alkene isomerization seen in 3%Co–ZB(25:1). This trend likely stems from the increased availability of active sites for secondary reactions, such as isomerization, when the metal is more finely dispersed. Additionally, lower metal loadings leave more acid sites exposed, which are primarily responsible for catalyzing isomerization reactions. However, as metal loading increases, the catalysts tend to favor direct hydrogenation over isomerization, resulting in a higher proportion of straight-chain alkanes. This shift could be due to reduced surface area at higher metal loadings, which could limit the catalyst's ability to facilitate complex rearrangement reactions such as isomerization.
Fig. 12 reveals once again notable trends in isomerization and selectivity across various metal-supported ZB(300:1) catalysts in the hydrodeoxygenation (HDO) of 6-undecanone. Among these catalysts, nickel and cobalt exhibit strong performance in promoting both full conversion to alkanes and moderate levels of alkane isomerization, while tin demonstrated comparatively weaker performance, favoring alkene formation and isomerization. The observed isomerization levels within the alkane product for nickel and cobalt suggest that, despite the lower acid site density of ZB(300:1), these metals can effectively catalyze branching reactions (Table 7). This isomerization likely results from an interaction between the metal sites, which promote hydrogenation, and the limited acid sites on the support, facilitating rearrangement. Nickel and cobalt's robust hydrogenation activity allows them to fully utilize the available acid sites, enabling both the deoxygenation to undecane and subsequent isomerization to produce branched alkane products. A noticeable trend in Fig. 11, 12, Tables 6 and 7 can be witnessed by looking at the increasing metal loading in Ni–ZB(25:1) and Ni–ZB(300:1). As nickel loading increases, isomerization decreases. The same trend can be seen with Co–ZB(25:1) and Co–ZB(300:1) albeit to a smaller degree (Tables 6 and 7). Specifically for 3%Co–ZB(300:1), compared to 3%Co–ZB(25:1), there is a selectivity shift towards undecane isomerization rather than alkene isomerization. This can be explained by the lower acidity of ZB(300:1) resulting in weaker interactions between cobalt and the support. This difference may alter the distribution of active sites, favoring direct hydrogenation to undecane over dehydration and isomerization pathways that require acid site participation. As seen in Fig. 9, the metal appears as distinct clusters directly on the support for 9%Ni–ZB(300:1), which could lead to pore blockage and limit the accessibility of acid sites necessary for alkene isomerization. In contrast 9%Ni–ZB(25:1) in Fig. 9 shows a more dispersed metal distribution, where it is difficult to differentiate metal from the support. This suggests that in ZB(300:1), metal clustering may reduce the number of available active sites for dehydration and alkene isomerization, further driving the selectivity shift toward undecane isomerization.
The activity difference between ZB(25:1) and ZB(300:1) can be attributed to the balance between metal and acid sites, which significantly influences both conversion and selectivity. Zeolite beta with a lower Si/Al ratio ZB(25:1) contains a higher density of Brønsted acid sites (BAS), which strongly promote dehydration and isomerization reactions, while the higher Si/Al ratio in ZB(300:1) results in weaker acidity, shifting selectivity towards direct hydrogenation. Previous studies have shown that dehydration reactions catalyzed by strong Brønsted acid sites can be two orders of magnitude faster than metal-catalyzed hydrogenation reactions, highlighting the critical role of acid sites in determining selectivity trends.26 Additionally, the balance between metal and acid sites can significantly impact hydrodeoxygenation (HDO) performance, where stronger acid sites in ZB(25:1) facilitate dehydration and alkene formation, while the lower acidity of ZB(300:1) results in greater hydrogenation selectivity towards undecane.27 These differences explain why ZB(25:1) supports higher selectivity to 5-undecene and isomerized alkenes, while ZB(300:1) favors undecane isomerization with minimal alkene intermediates.
As seen in Fig. 10–12, tin catalysts exhibit consistent limitations on both supports. In Fig. 11, where Sn is supported on ZB(25:1), no isomerized products are observed, indicating that this high-acid density support primarily directs tin towards straightforward deoxygenation without significant branching. The high acidity of ZB(25:1) likely promotes strong metal–support interactions, which may limit tin's ability to facilitate hydrocarbon rearrangements and restricts the reaction pathway to simple deoxygenation. In contrast, Fig. 12 where tin is supported on ZB(300:1), shows isomerized alkene products, suggesting that the lower acidity allows for additional catalytic pathways. The reduced acid density in ZB(300:1) may create an environment where tin can interact with available acid sites without strong confinement effects, thus promoting isomerization. This shift in activity highlights the influence of acid site density on tin's catalytic behavior, where moderate acidity appears to enable more flexibility in hydrocarbon rearrangement. This incomplete hydrogenation on ZB(300:1) suggests that tin's weaker hydrogenation ability becomes a limiting factor, as the catalyst is unable to keep up with the faster reaction rate of the support, resulting in higher selectivity towards alkene formation and isomerization.
The choice of support also plays a significant role in the product distribution. Supports with higher acid site density, such as ZB(25:1) and ZB(300:1) compared to TiO2, provide the necessary acid functionality for both deoxygenation and isomerization steps, particularly at lower metal loadings. This relationship highlights the bifunctional nature of the HDO process, where both metal and acid sites must be present to achieve selective isomerization along with complete deoxygenation. These findings suggest that modulating metal loading and support acidity can tune the selectivity of HDO toward either branched or straight-chain alkanes.
These experimental observations, when considered collectively, provide mechanistic insights regarding the influence of support acidity and metal dispersion on bifunctional catalytic behavior. The performances observed across the tested systems arises from a complex interplay between metal dispersion, support acidity, and spatial proximity of active sites. For nickel, XRD, STEM, and CO chemisorption data collectivity indicate high metal dispersion and internal incorporation within ZB(25:1), likely facilitated by the support's higher Brønsted acid site density. This close association between Ni and acid sites enables efficient sequential reactions-ketone reduction, alcohol dehydration, and alkene hydrogenation – and maintains site accessibility after reaction, as evidenced by a 74.6% retention in CO-active sites (Table 5). In contrast, cobalt exhibits an order of magnitude lower retention and nearly complete loss of CO accessibility after reaction, which may result from weaker anchoring, increased susceptibility to metal migration or sintering, or leaching of cobalt species into the reaction medium. Tin presents yet another case: its lack of detectable XRD reflections suggest either highly dispersed or amorphous tin species, which may still reside on or within the support without forming distinct crystalline phases. However, when considered alongside the notably low ICP-measured loading and the minimal catalytic activity, these results together imply weak anchoring or inefficient deposition-likely exacerbated by the volatility of the tin precursor during calcination. The resulting poor reducibility and low hydrogenation activity under reaction conditions further support this interpretation.
Support acidity, quantified via NH3-TPD (Table 5), further modulates these interactions by influencing where and how metals anchor to the framework. In ZB(25:1), stronger acidity not only promotes initial metal dispersion but also stabilizes metal particles within the internal pore network – a conclusion supported by EDS mapping of 9%Ni–ZB(25:1), where metals are more uniformly distributed compared to their external clustering in ZB(300:1). This internal anchoring enhances proximity between hydrogenation and dehydration sites, a prerequisite for bifunctional HDO reactions. In contrast, lower acidity of ZB(300:1) limits metal incorporation and shifts the balance toward external surface acidity as seen in Fig. 7. This spatial decoupling likely explains the diminished formation of alkene isomers, which require a close handoff between acid and metal sites for tandem reaction steps.17
These findings are consistent with broader models of metal–support interactions reported in recent HDO literature, which describe how electronic effects, anchoring strength, and chemical cooperativity between acid and metal sites define both catalyst stability and reaction pathways.28 In the present work, the enhanced CO retention observed for Ni–ZB(25:1) suggest that strong acid–metal proximity stabilizes metal dispersion through geometric confinement and acid coordination. The metal–support framework also suggests that such proximity alters the energetic landscape of the reaction by enabling cooperative activation steps – such as acid-catalyzed dehydration adjacent to metal sites – which may account for increased isomerization in the ZB(25:1) supported catalysts. Conversely, the lower isomer selectivity on ZB(300:1) may stem from external metal deposition, where the spatial separation of acid and metal sites limits intermediate migration and cooperative catalysis. These mechanistic interpretations are in line with recent findings that show how metal–support interactions, especially in bifunctional systems, governs not only site retention but also product distribution by facilitating hydrogen spillover, transition state stabilization, and reactant channeling through confined environments.28,29 Mechanistic models outlined by Strapasson et al., who demonstrated that Brønsted vs. Lewis-rich supports govern selective C–O vs. C–C cleavage, reinforce that acidity not only modulates activity but also directs distinct reaction pathways in HDO.29 Similarly, Yan et al., showed that low Si/Al zeolite supports with strong Brønsted acidity facilitated deoxygenation and ring opening of biocrude components, emphasizing the critical synergy between acidity and pore accessibility.30
Taken together, these findings illustrate that acidity and pore structure function not only as passive supports, but as active design levers controlling reactivity, site accessibility, and product selectivity in bifunctional HDO catalysts.
Benchmarking against literature catalysts
To contextualize the performance of our catalysts, we compiled a comprehensive comparison table (Table 8) summarizing conversion and alkane selectivity across reported catalysts. The Ni–ZB catalysts in this work show high selectivity to linear (n-) alkanes, achieving 100% conversion and up to 99.2% selectivity to undecane with less than 1.1% iso-alkanes under batch conditions at 250 °C. Meanwhile, the Co–ZB catalysts in this work also achieve full conversion, with slightly lower undecane selectivity (94–96%) and greater isomerization (3.7–5.4%), offering flexibility for tailoring fuel properties.| Catalyst | Reactant | Reactor type | Temp. °C | Conversion (%) | Selectivity to alkane (%) | Selectivity to iso-alkane |
|---|---|---|---|---|---|---|
| 9%Ni–ZB(25:1) (this work) |
6-Undecanone | Batch | 250 | 100 | 99.21 | 0.79 |
| 9%Ni–ZB(300:1) (this work) |
6-Undecanone | Batch | 250 | 100 | 98.92 | 1.08 |
| 9%Co–ZB(25:1) (this work) |
6-Undecanone | Batch | 250 | 100 | 96.26 | 3.74 |
| 9%Co–ZB(300:1) (this work) |
6-Undecanone | Batch | 250 | 100 | 94.62 | 5.38 |
| 5cTiO2–0.5%Pt–Al2O3 (McNeary et al., 2023)16 | 6-Undecanone | Batch | 250 | 95 | 70 | Not reported |
| 10cTiO2–0.5%Pt–Al2O3 (McNeary et al., 2023)16 | 6-Undecanone | Batch | 250 | 100 | 100 | Not reported |
| Ni-7/ZSM-5 (Yang et al., 2019)32 | 5-Nonanone | Flow | 250 | 65 | 88 | 15 |
| Co-11/ZSM-5 (Yang et al., 2019)32 | 5-Nonanone | Flow | 250 | 70 | 84 | 45 |
| 10%Ni-ZSM-5 (Hu et al., 2023)33 | Fatty ketones (C11–C17) | Batch | 140 | Not reported | 92–96 | Not reported |
| 20%Cu–Al2O3 (Al-Auda 2019)34 | 5-Methyl-3-heptanone | Flow | 220 | 100 | 54 | Not reported |
| 1%Cu–Al2O3 (Al-Auda 2019)34 | 5-Methyl-3-heptanone | Flow | 220 | 51 | 10 | Not reported |
| 1%Pt–Al2O3 (Al-Auda 2019)34 | 5-Methyl-3-heptanone | Flow | 220 | 100 | 98 | Not reported |
In contrast, Pt-based catalysts, though effective, rely on expensive, scarce metals. For instance, the 2025 spot price of platinum is approximately $32,000 per kilogram, while more abundant metals like nickel and cobalt are priced at roughly $15–20 per kilogram and $25–30 per kilogram, respectively.31 This substantial cost difference highlights the economic advantage of using nickel and cobalt for large-scale catalyst deployment. Cu-based systems, such as 20%Cu–Al2O3 have shown only moderate selectivity (54% to alkanes) despite full conversion, indicating less favorable performance. ZSM-5 supported systems, while active, result in excessive isomerization (up to 45%) and a poor carbon balance. In contrast, our zeolite beta-supported catalysts offer high linearity, minimal branching, and precise control over product distribution, highlighting their superior bifunctional tuning for selective alkane production.
Additionally, many HDO studies report isomerized products as “iso-alkanes, iso-alkenes, or iso-alcohols” as lumped categories without structural detail. In contrast, this work identifies and distinguishes the specific isomeric products formed during reaction. These detailed assignments are included in the ESI,† enabling a better understanding of branched pathways and offering a higher level of product characterization than is typically reported.
Conclusions
The bifunctional nature of these catalysts-combining metal sites for hydrogenation with acidic sites on the support for deoxygenation and isomerization-plays a crucial role in achieving efficient conversion of biomass-derived intermediates to hydrocarbons. By examining the impact of metal type, metal loading, support acidity, and structural properties such as microporosity versus mesoporosity, this study clarifies how these factors influence catalyst performance, site accessibility, and structural stability after reaction.This study highlights that catalyst performance is influenced by the interplay between metal type, loading, support acidity, and structural properties such as microporosity. Both nickel and cobalt showed strong catalytic activity across zeolite beta supports, with trends indicating that modulating the relative loading of acid sites and metals enables engineers to tailor the catalyst toward producing either iso-alkanes or n-alkanes. These hydrocarbons typically compose approximately 55–60% of conventional jet fuel and offer opportunities to optimize fuel properties.15 Tin, while demonstrating unique selectivity, exhibited limited catalytic activity, likely due to weaker hydrogenation capabilities and less effective interactions with the support.
The acidity of zeolite beta, particularly the high-acid-density ZB(25:1), was found to be essential for promoting deoxygenation and isomerization, while the lower-acid-density ZB(300:1) performed comparably well, suggesting that moderate acidity is sufficient for achieving full deoxygenation. The microporous structure of zeolite beta enabled efficient bifunctionality by ensuring close proximity between metal and acid sites, enhancing sequential reaction steps. In contrast, TiO2, demonstrated lower conversions and higher selectivity to partially deoxygenated products, emphasizing the importance of support acidity and pore structure.
Overall, the findings suggest that bifunctional catalysts supported on microporous, acidic supports can be tuned to achieve selective hydrocarbon production from biomass feedstocks. Further studies should focus on long-term stability, catalyst reusability and regeneration, time on stream testing, and optimizing metal–support interactions to selectively produce desired branched alkanes or straight chain alkanes.
Data availability
All data will be provided upon request.Author contributions
Writing – original draft: Raiven I. Balderas. Writing – review and editing: Logan Myers, Jacob Miller, Cody Wrasman. Supervision: Ryan M. Richards, Jacob Miller, Cody Wrasman, Derek Vardon. Methodology: Raiven I. Balderas, Logan Myers. Investigation: Raiven I. Balderas. Formal analysis: Raiven I. Balderas. Project administration: Ryan M. Richards, Jacob Miller, Cody Wrasman, Derek Vardon. Funding acquisition: Ryan M. Richards, Derek Vardon.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the University of New Mexico for providing spent catalyst characterization. As well as the NEXUS collaboration between Colorado School of Mines and the National Renewable Energy Laboratory for making this research possible. R. I. B. and R. M. R. acknowledge DOE Contract No. DE-AC36-08GO28308. Additionally, R. M. R. acknowledges NSF CBET Grant No. 2139971 for support.Notes and references
- J. Ethiraj, D. Wagh and H. Manyar, Energy Fuels, 2022, 36, 1189–1204 CrossRef CAS.
- S. Mondal, S. Ruidas, S. Chongdar, B. Saha and A. Bhaumik, ACS Sustainable Resour. Manage., 2024, 1, 1672–1704 CrossRef CAS.
- R. Ahorsu, M. Constanti and F. Medina, Ind. Eng. Chem. Res., 2021, 60, 18612–18626 CrossRef CAS.
- N. A. Huq, G. R. Hafenstine, X. C. Huo, H. Nguyen, S. M. Tifft, D. R. Conklin, D. Stuck, J. Stunkel, Z. B. Yang, J. S. Heyne, M. R. Wiatrowski, Y. M. Zhang, L. Tao, J. Q. Zhu, C. S. McEnally, E. D. Christensen, C. Hays, K. M. Van Allsburg, K. A. Unocic, H. M. Meyer, Z. Abdullah and D. R. Vardon, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 1–11 CrossRef PubMed.
- C. Gaertner, J. Serrano-Ruiz, D. Braden and J. Dumesic, Ind. Eng. Chem. Res., 2010, 49, 6027–6033 CrossRef CAS.
- C. Gärtner, J. Serrano-Ruiz, D. Braden and J. Dumesic, CHEMSUSCHEM, 2009, 2, 1121–1124 CrossRef PubMed.
- C. Gaertner, J. Serrano-Ruiz, D. Braden and J. Dumesic, J. Catal., 2009, 266, 71–78 CrossRef CAS.
- T. Pham, T. Sooknoi, S. Crossley and D. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- M. Wiatrowski, J. Miller, A. Bhatt, S. Tifft, Z. Abdullah and L. Tao, RENEWABLE ENERGY, 2024, 232, 1–18 CrossRef.
- J. Miller, C. Nimlos, Y. Li, A. Young, P. Ciesielski, L. Chapman, T. Foust and C. Mukarakate, ACS Sustainable Chem. Eng., 2023, 12, 666–679 CrossRef PubMed.
- J. Miller, G. Hafenstine, H. Nguyen and D. Vardon, Ind. Eng. Chem. Res., 2022, 61, 2997–3010 CrossRef CAS.
- C. A. Gartner, J. C. Serrano-Ruiz, D. J. Braden and J. A. Dumesic, Chemsuschem, 2009, 2, 1121–1124 CrossRef CAS PubMed.
- B. Boekaerts and B. Sels, Appl. Catal., B, 2021, 283, 1–37 CrossRef.
- Y. Hong, A. Hensley, J. McEwen and Y. Wang, Catal. Lett., 2016, 146, 1621–1633 CrossRef CAS.
- J. Holladay, Z. Abdullah and J. Heyne, Sustainable Aviation Fuel : Review on Technical Pathways Report, O. O. E. E. R. Energy, Bioenergy Technologies Office, 2020 Search PubMed.
- W. W. McNeary, J. H. Miller, S. A. Tacey, J. Travis, G. D. Lahti, M. B. Griffin, K. L. Jungjohann, G. Teeter, T. E. Erden, C. A. Farberow, L. Tuxworth, M. J. Watson, A. A. Dameron and D. R. Vardon, Chem. Eng. J., 2023, 467, 1–11 Search PubMed.
- A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef CAS PubMed.
- T. Dontsova, O. Yanushevska, S. Nahirniak, A. Kutuzova, G. Krymets and P. Smertenko, J. Chem., 2021, 2021 Search PubMed.
- A. Davidson, D. Lennon, P. Webb, P. Albers, M. Berweiler, R. Poss, M. Roos, A. Reinsdorf, D. Wolf and S. Parker, Top. Catal., 2021, 64, 644–659 CrossRef CAS.
- L. Pan, Y. He, M. Niu, Y. Dan and W. Li, RSC Adv., 2019, 9, 21175–21185 RSC.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- B. Donnis, R. Egeberg, P. Blom and K. Knudsen, Top. Catal., 2009, 52, 229–240 CrossRef CAS.
- T. M. Project, Computed XRD patterns for metal phases were obtained from the Materials Project database, https://legacy.materialsproject.org/.
- A. Ponomaryov, A. Smirnov, M. Shostakovsky, E. Pisarenko, A. Popov and M. Kashkina, Pet. Chem., 2024, 1147–1158 CrossRef CAS.
- X. Li, S. Ma and H. Li, Green Chem., 2022, 24, 7243–7280 RSC.
- Q. Sun, G. Chen, H. Wang, X. Liu, J. Han, Q. Ge and X. Zhu, CHEMCATCHEM, 2016, 8, 551–561 CrossRef CAS.
- X. Wu, Q. Sun, H. Wang, J. Han, Q. Ge and X. Zhu, Catal. Today, 2020, 355, 43–50 CrossRef CAS.
- T. Li, D. Jia, S. Zhou, Z. Liu, J. Chen, T. Ban, A. Li, H. Li and H. Gao, Fuel, 2024, 373 Search PubMed.
- G. B. Strapasson, G. B. Bafero, D. S. Leite, D. B. L. Santos, A. Albuquerque, I. D. Barcelos, L. M. Rossi, C. B. Rodella and D. Zanchet, Appl. Catal., B, 2025, 373, 125316 CrossRef CAS.
- P. Yan, J. Mensah, M. Drewery, E. Kennedy, T. Maschmeyer and M. Stockenhuber, Appl. Catal., B, 2021, 281 CAS.
- Daily Metal Price, https://www.dailymetalprice.com/.
- X. K. Yang, R. W. Jenkins, J. H. Leal, C. M. Moore, E. J. Judge, T. A. Semelsberger and A. D. Sutton, ACS Sustainable Chem. Eng., 2019, 7, 14521–14530 CrossRef CAS.
- Y. Hu, X. Li, M. Liu, S. Bartling, H. Lund, P. Dyson, M. Beller and R. Jagadeesh, ACS Sustainable Chem. Eng., 2023, 11, 15302–15314 CrossRef CAS.
- Z. Al-Auda, H. Al-Atabi, X. Li, P. Thapa and K. Hohn, Catalysts, 2019, 9, 1–16 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00773e |
| This journal is © The Royal Society of Chemistry 2025 |