DOI:
10.1039/D4SU00781F
(Paper)
RSC Sustainability, 2025, Advance Article
Efficient capture of carbon dioxide to form organic crystals at low pressure and room temperature†
Received
9th December 2024
, Accepted 30th April 2025
First published on 1st May 2025
Abstract
Carbon dioxide capture and storage (CCS) and carbon dioxide capture and utilization (CCU) are potential solutions for mitigating the greenhouse effect. Organic amines had been used for CCS. However, there are still scarce reports on the formation of solids after the capture of CO2, especially in the crystalline form. This study explores a novel method for capturing and storing carbon dioxide using organic superbases 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and tetrahydropyrrole (Py) to form stable carbamate crystals DBU–Py–CO2 at room temperature and low pressure. Interestingly, these crystals also exhibited exceptional stability when stored in a closed centrifuge tube at room temperature for over 500 days. The process involves a nucleophilic attack on CO2 by an amino anion generated from these materials, forming well-ordered carbamate crystals as confirmed by single-crystal diffraction. The captured CO2 can be reversibly released upon low dose heating. This approach presents promise for mitigating greenhouse gas emissions and utilizing CO2 as a crystalline material without the need for metal catalysts or solvents.
Sustainability spotlight
Efficient capture of CO2 under low pressure at room temperature is still an attractive topic today, especially for solid production, which is easily stored and transported. Here, we find that CO2 can be captured by using organic superbases 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and tetrahydropyrrole (Py) to form stable carbonate crystals DBU–Py–CO2, which are very stable at room temperature for long time storage and can release CO2 again efficiently on heating. This provides a simple method for the efficient conversion of CO2 to crystals for various applications.
|
Introduction
The reduction of carbon dioxide emissions is an urgent global imperative and an indispensable component of sustainable development.1–5 Carbon capture, utilization, and storage has been widely employed in the past and is regarded as a promising strategy for mitigating CO2 emissions.6–8 The technical methods for CO2 capture include adsorption, absorption, membrane separation, cryogenic separation, and hydration.9–18 It is widely acknowledged that the absorption method of chemical substances is one of the most competitive CCU and CCS methods.19,20 The current industrial technology for CO2 involves the utilization of monoethanolamine aqueous solution, which demonstrates the practical feasibility of chemical absorption of CO2.21 The utilization of ionic liquids for CO2 absorption has been increasingly reported in order to achieve a balance between energy consumption and environmental sustainability.22,23 As a class of organic compounds with distinctive functionalities, organic superbases (DBU, DBN, and TMG) are commonly employed in the investigation of CO2 absorption.24–32 It is widely recognized that upon acquiring hydrogen protons, organic superbases undergo stabilization of conjugated structures, thereby exhibiting a pronounced capacity for extracting active hydrogen from the system.31,33–35 When an organic superbase is present, the amino group undergoes deprotonation and forms the amino anion, resulting in a significant enhancement of nucleophilicity.34,36,37 Although carbon dioxide is chemically inert, the electron density of the carbon–oxygen double bond affects the carbonyl carbon atoms in carbon dioxide, resulting in its overall electrophilic properties.38–43 Consequently, even at lower temperatures and pressures, the combination of an organic superbase and an amino anion can effectively capture and activate carbon dioxide.44,45 An organic superbase (DBU) and tetrahydropyrrole (Py) are combined as an absorbent in this study, under room temperature and pressure conditions. Under these conditions, DBU converts the secondary amine functional group on Py into an amino anion, which effectively captures carbon dioxide to produce carbamate (DBU–Py–CO2). Interestingly, DPU–Py–CO2 is a robust and crystalline white solid. Upon heating, the entrapped CO2 is liberated while the residual DPU–Py reverts back to its initial ionic liquid state, rendering it suitable for subsequent adsorption–desorption cycles (Scheme 1). This reversible absorption process can undergo multiple iterations without significant reduction in both quantity and efficacy of absorption. The three-dimensional spatial structure of the DPU–Py–CO2 crystal is confirmed through the characterization of a four-circle diffractometer and computational simulation, thereby validating the process of DPU–Py absorbing CO2 to form crystals. In the field of CCU, the research on CO2 absorption for crystal production has garnered significant attention from many researchers. Notably, the relatively short time required to obtain organic crystals and their ability to reversibly release CO2 at temperatures ranging from 80 to 120 °C are particularly intriguing.46–48 It is noteworthy that this study successfully achieves a controllable and reversible transition between the absorbent and CO2 as well as the crystal phase.
 |
| | Scheme 1 Schematic representation of carbon dioxide being converted to crystals by adsorption in the DBU–Py system. | |
Results and discussion
When two organic compounds DBU and Py are combined (in the liquid form), they exhibit enhanced CO2 absorption capabilities, surpassing their individual capacities, with a molar ratio close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 1). Notably, even under mild temperature and pressure conditions (including room temperature and atmospheric pressure), the reaction yields a white solid material upon CO2 absorption in the reactor (Fig. 2a). The white solid is indeed a mixture, as it contains unreacted monomers; however, the phenomenon of phase transition continues to captivate researchers. This phenomenon has been thoroughly investigated and discussed in this study. This intriguing phenomenon has been thoroughly documented in a video provided in the ESI (Video S1).† The organic superbase DBU initially abstracts the active hydrogen proton from Py in the system, resulting in the formation of [DBUH] and amino anions. The amino anion exhibits remarkable nucleophilicity, facilitating its facile capture of carbon dioxide even under mild conditions. Despite the relatively weak electrophilicity of carbon dioxide, this deficiency is compensated by the strong nucleophilicity of amino anions. Subsequently, electrostatic forces drive the attraction between the negative charge on the carboxyl group and the positive charge on tertiary amine within both carbamate and [DBUH]. This orderly arrangement gives rise to a long-range crystal structure.
:1 (Fig. 1). Notably, even under mild temperature and pressure conditions (including room temperature and atmospheric pressure), the reaction yields a white solid material upon CO2 absorption in the reactor (Fig. 2a). The white solid is indeed a mixture, as it contains unreacted monomers; however, the phenomenon of phase transition continues to captivate researchers. This phenomenon has been thoroughly investigated and discussed in this study. This intriguing phenomenon has been thoroughly documented in a video provided in the ESI (Video S1).† The organic superbase DBU initially abstracts the active hydrogen proton from Py in the system, resulting in the formation of [DBUH] and amino anions. The amino anion exhibits remarkable nucleophilicity, facilitating its facile capture of carbon dioxide even under mild conditions. Despite the relatively weak electrophilicity of carbon dioxide, this deficiency is compensated by the strong nucleophilicity of amino anions. Subsequently, electrostatic forces drive the attraction between the negative charge on the carboxyl group and the positive charge on tertiary amine within both carbamate and [DBUH]. This orderly arrangement gives rise to a long-range crystal structure.
 |
| | Fig. 1 Possible mechanism of capturing carbon dioxide and producing crystals (DBU–Py–CO2). | |
 |
| | Fig. 2 The solid carbamate (DBU–Py–CO2): (a) unpurified, (b) purified single crystal, (c) structure was determined using a four-circle single crystal diffractometer, and (d) stored at room temperature for 500 days. | |
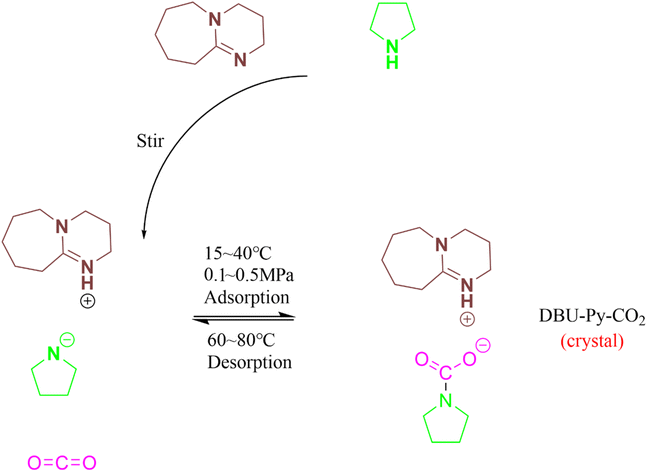
The subsequent four-circle single-crystal diffraction was employed to determine the spatial three-dimensional structure of DPU–Py–CO2 and validate the conjecture. First, DBU–Py–CO2 was dissolved in a mixture of dioxane and ethyl acetate, followed by slow evaporation of the solvent at room temperature to obtain a single crystal (Fig. 2b). The results obtained from diffraction and simulation using single crystals revealed that the DBU–Py–CO2 crystal belongs to the P21/n space group in the monoclinic crystal system. It is evident from the ball and stick model and the ORTEP diagram (Fig. 2c) derived from the diffraction of a single crystal that the positive hydrogen charge on the tertiary amine of the DBU ring attracts the negative charge of the carbamate carboxyl group. Detailed information regarding crystallography can be found in the ESI.†
The DPU–Py–CO2 crystal belongs to the monoclinic crystal system, which is classified as a lower class of crystals with relatively low symmetry, resulting in its thermodynamic instability. When the temperature exceeds 60 °C, the crystal structure will be disrupted and CO2 trapped within it will be released, leaving behind DPU–Py in the form of an ionic liquid. Interestingly, this heat instability can also serve as an advantage since the remaining DPU–Py ionic liquid after heating retains its ability to capture CO2 once again. In other words, the absorber can be considered as a carrier for CO2; it forms crystals upon adsorption and only requires heating for release. The thermal instability is also usable for the reversible release of CO2 captured by the DBU–Py absorber. Furthermore, under normal temperature and pressure conditions, these crystals maintain their original morphological characteristics for at least 500 days (Fig. 2d). As evident from the XRD pattern (Fig. 3b), each peak of newly synthesized crystals corresponds to those stored for 500 days; however, individual peak intensities are reduced in samples stored for extended periods indicating that minimal structural changes have occurred. Thus, DPU–Py–CO2 crystals possess significant potential for CO2 storage.
 |
| | Fig. 3 (a) ORTEP diagram of the DBU–Py–CO2 crystal obtained by four-circle single crystal diffraction, (b) comparison of XRD patterns of newly made and DBU–Py–CO2 crystals stored for 500 days, (c) influence of temperature on DBU:Py = 1:1 absorption of CO2, 0.5 MPa, and (d) influence of pressure on DBU:Py = 1:1 absorption of CO2, 293 K. | |
Next, the impact of reaction temperature, carbon dioxide pressure, and molar ratio on absorption is investigated, as depicted in Fig. 3c, d, and Table 1. As the reaction temperature increases from 293 K to 313 K, the CO2 absorption capacity gradually increases until reaching a peak value of 0.87 mol per mol absorber. Because the adsorption process is an equimolar absorption mechanism, 0.87mol per mol absorber is obviously a relatively objective molar absorption capacity compared with that reported in recent literature.49,50 However, when approaching normal temperatures, the enhancement in absorption capacity becomes less pronounced. This can be attributed to higher temperatures prolonging the phase transition process and promoting liquid-state formation, thereby enhancing mass transfer efficiency. Nevertheless, at temperatures exceeding 313 K, an increase in temperature leads to a decrease in absorption capacity due to thermodynamic considerations. At elevated temperatures, equilibrium shifts towards the desorption direction aligning with the reversible release of CO2 upon heating. The absorption capacity of CO2 gradually increases as the pressure increases, aligning with conventional wisdom. Elevated pressure facilitates the transportation of more CO2 molecules to the reaction site during crystal formation, thereby promoting nucleophilic attack reactions. When the molar ratio of the absorbent is adjusted to 1:2 or 2:1 (DBU:Py), the absorption reaction can still proceed; however, the resulting product no longer exhibits a white, solid appearance but rather transforms into a yellow, translucent, and thick solid. This alteration in physical properties arises due to the necessity for a 1:1 mole ratio within the crystal's structural unit, with an excess of DBU or Py leading to disruption of this crystalline structure.
Table 1 Adsorption data under different conditionsa
| Absorbent (molar ratio) |
Temperature (K) |
Pressure (MPa) |
Absorbing ability (molar ratio) |
Absorbing ability (wt%) |
Time (min) |
Energy intake (kJ mol−1 CO2) |
Status |
| (Note: The pressure of 0.1 MPa open indicates that the reaction is conducted in an open system, where the non-pressurized vessel (flask) and rubber hose are exposed to the surrounding atmospheric conditions. The reaction is conducted in the pressure range of 0.1–0.5 MPa using a stainless steel reactor and airway, with the utilization of a pressure-reducing valve to achieve the desired pressure level within the reactor). |
| DBU:Py = 1:1 |
293 |
0.1(open) |
0.65 |
12.8 |
30 min |
0.077 |
White slurry solid |
| DBU:Py = 1:1 |
293 |
0.1 |
0.71 |
14.0 |
30 min |
0.070 |
| DBU:Py = 1:1 |
293 |
0.2 |
0.77 |
15.2 |
30 min |
0.065 |
White hard solid |
| DBU:Py = 1:1 |
293 |
0.3 |
0.81 |
16.0 |
30 min |
0.062 |
| DBU:Py = 1:1 |
293 |
0.4 |
0.83 |
16.3 |
30 min |
0.060 |
| DBU:Py = 1:1 |
293 |
0.5 |
0.84 |
16.5 |
30 min |
0.059 |
| DBU:Py = 1:1 |
303 |
0.5 |
0.86 |
16.9 |
30 min |
0.174 |
| DBU:Py = 1:1 |
313 |
0.5 |
0.87 |
17.1 |
30 min |
0.172 |
| DBU:Py = 1:1 |
323 |
0.5 |
0.85 |
16.7 |
30 min |
0.176 |
| DBU:Py = 1:1 |
333 |
0.5 |
0.78 |
14.3 |
30 min |
0.192 |
| DBU:Py = 2:1 |
293 |
0.5 |
0.94 |
18.5 |
30 min |
0.053 |
Yellow translucent solid |
| DBU:Py = 1:2 |
293 |
0.5 |
0.86 |
16.9 |
30 min |
0.058 |
Yellow translucent solid |
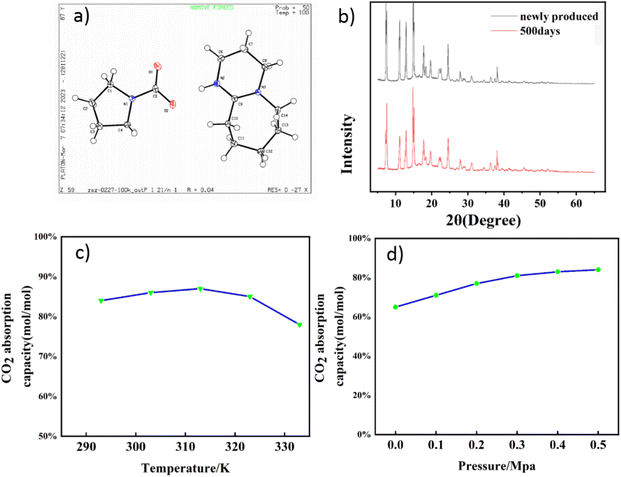
Both 13C NMR and FT-IR confirm the process of CO2 absorption. As depicted in Fig. 4a, the absorbent's spectrum exhibits a high chemical shift peak for No. 6 C prior to the reaction, indicating its connection to the N atom through a double bond. Following the reaction, a distinctive signal peak emerges at 164 ppm corresponding to No. 17 C (Fig. 4b), which is characteristic of carbonyl carbon formation.51 As a result of the transformation of DBU into the [DBUH] form following the reaction, the significant electrostatic interaction led to an increase in the chemical shift of No. 6 C from 160 ppm to 167 ppm. The presence of carbonyl carbon within the system signifies that N–H captures CO2 subsequent to nucleophilic attack. The absorption peaks of various hydrogen atoms in the absorbent after CO2 capture are individually assigned as shown in Fig. 4c. Given the evident H2O signal observed in Fig. 4c, it is plausible to infer that the presence of the [DBUH][HCO3] form within the crystal structure cannot be entirely excluded, which may also be one of the reasons for the thermal instability of the crystal. Fig. 4d shows the HMBC spectrum of the sample; two carbon signals with high chemical shifts can be observed, and the carbon signal with low chemical shifts in them is associated with two remote hydrogen signals. These two hydrogen signals are coming from Py 13, 16 and 14, 15, where 14 and 15 correspond to the hydrogen signals of the four bonds away from 17 and 13. 16 is the bond away from 17, while 14 and 15 signals are slightly stronger. In summary, this shows that among the two carbon signals with a high chemical shift, the one with a low chemical shift is carbonyl carbon. After your prompt, we have confirmed this fact and corrected the error in the manuscript.
 |
| | Fig. 4 (a) 13C NMR of absorbent DBU:Py = 1:1, (b) 13C NMR of absorbent DBU:Py = 1:1 after CO2 capture, (c) 1H NMR of absorbent DBU:Py = 1:1 after CO2 capture, and the HMBC spectrum of the sample (d). | |
In the FT-IR spectrum (Fig. 5), the prominent feature is observed as a peak at 1627 cm−1 corresponding to the DBU–Py–CO2 signal, which serves as a characteristic indication of carbonyl carbon and confirms the capture of CO2. The broad peak at 3367 cm−1 can be attributed to N–H stretching vibration. Signals in proximity to 1550 cm−1 are categorized as N–H bending vibrations. Due to the constrained nature of N–H caused by the five-membered ring structure of Py, the N–H signal from Py is not distinctly evident.
 |
| | Fig. 5 FT-IR spectra of DBU, Py, and DBU–Py–CO2. | |
The three-dimensional structure of the crystal was created using Mercury software (Fig. 6a). The adsorption and desorption process for captured CO2 was conducted under atmospheric pressure (approximately 0 MPa) for a total of 5 cycles, during which the mass variation of the absorbent and its absorption capacity were recorded. The specific details of each cycle are documented in Table 2. Upon completing all 5 cycles, the absorbent retained 93.5% of its initial mass and exhibited an absorption capacity of 0.61 mol per mol of absorbent (Fig. 6b). Notably, the main source of loss in this process is attributed to operational consumption, as the temperature employed during adsorption and desorption is significantly lower than the decomposition temperature of DBU and Py compounds. If measures can be taken to minimize both volatilization and operational losses, this scheme would hold greater practical value.
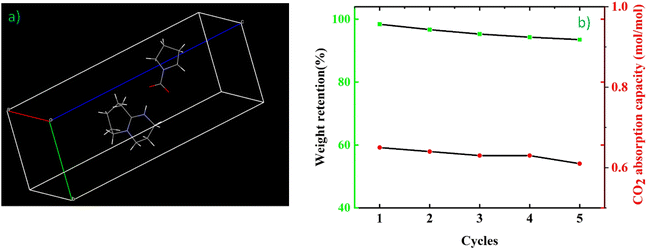 |
| | Fig. 6 (a) Intracellular three-dimensional structure of the DBU–Py–CO2 crystal drawn using Mercury software and (b) mass retention and absorption capacity of the absorbent during adsorption and desorption. | |
Table 2 Adsorption resolution cycle
| Recycling times |
Absorbent (molar ratio) |
Absorbent quality retention |
Absorbing ability (molar ratio) |
Absorbing ability (wt%) |
| 1 |
DBU:Py = 1:1 |
98.4% |
0.65 |
12.8 |
| 2 |
DBU:Py = 1:1 |
96.7% |
0.64 |
12.6 |
| 3 |
DBU:Py = 1:1 |
95.3% |
0.63 |
12.4 |
| 4 |
DBU:Py = 1:1 |
94.3% |
0.63 |
12.4 |
| 5 |
DBU:Py = 1:1 |
93.5% |
0.61 |
12.0 |
Experimental
Materials
All chemicals were purchased and used without further purification, purchased from ACMEC, Shanghai, China. Tetrahydropyrrole (Py), dioxane, and ethyl acetate were purchased from Sinopharm Chemical Reagents Co. LTD. (Shanghai, China). A 50 ml reactor was purchased from Parr, USA. CO2, analytically pure, was purchased from Anhui Hefei Hengxing Industrial Gas Co., Ltd. (Hefei, China), and stored in cylinders.
Adsorption and desorption
1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and tetrahydropyrrole (Py) were magnetically stirred in the reactor while being heated by using an oil bath exposed to 0.1–0.5 MPa CO2 gas. A phase transition occurred upon a significant pressure drop, indicating completion of the reaction within the predetermined time. The reaction time is 30 min. During the desorption process, all solid products are heated in a 50 ml three-necked glass flask at a temperature of 80 °C. Desorption is considered complete when there is no further reduction in mass within a period of 2 minutes. The remaining liquid in the flask should be utilized for the subsequent cycle, while the balance should be used to weigh the mass and calculate absorption. In this study, the energy demand for CO2 capture ranged from a minimum of 0.053 kJ mol−1 CO2 to a maximum of 0.192 kJ mol−1 CO2. When compared with other recently developed capture methods,52–56 this represents relatively low energy consumption. The primary reason for this efficiency is the reduced time required for CO2 capture. At 293 K, only the stirring power (100 W) of the device was utilized, rather than the heating power (300 W). Overall, the low energy consumption associated with CO2 absorption under mild conditions constitutes one of the key advantages of this study.
Characterization
Fourier transform infrared (FT-IR) spectra were recorded on a Bruker vector-2 spectrophotometer in the 400–4000 cm−1 range. XRD analysis was performed on a Philips X′ Pert PRO SUPER X-ray diffractometer with Cu Ka radiation (l = 1.54056 Å). Nuclear magnetic resonance (NMR) studies were carried out on a Bruker NMR (400 M) spectrometer with DMSO-d as solvent. A four-circle single crystal diffractometer, manufactured by Oxford Diffraction Company, Gemini S Ultra was used.
Conclusions
In this study, we discovered that DBU and Py serve as absorbents for CO2 capture and crystal formation. The spatial arrangement of the crystals is determined through single crystal diffraction analysis. Additionally, we investigate the factors influencing the adsorption process and reversible release process. These findings demonstrate the feasibility of converting CO2 into crystals, although practical application remains a distant goal at this stage. Nevertheless, it underscores the immense potential for future advancements in crystal design and applications.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
S. Zhang carried out the experiments and wrote the original manuscript and L. Yan designed, supported, and wrote the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2023YFA1507500), the National Natural Science Foundation of China (No. 52373159), and the Fundamental Research Funds for the Central Universities (No. YD2060002015).
Notes and references
- X. Guo, B. Xiao and L. Song, J. Cleaner Prod., 2020, 271, 122691 CrossRef CAS.
- B. Lin and B. Xu, Sci. Total Environ., 2020, 719, 137503 CrossRef CAS PubMed.
- B. J. Wang, J. L. Zhao, Y. F. Wu, C. Q. Zhu, Y. N. He and Y. X. Wei, Environ. Sci. Pollut. Res. Int., 2019, 26, 17950–17964 CrossRef CAS PubMed.
- S. Solomon, G. K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- M. A. Abdelkareem, K. Elsaid, T. Wilberforce, M. Kamil, E. T. Sayed and A. Olabi, Sci. Total Environ., 2021, 752, 141803 CrossRef CAS PubMed.
- L. Li, N. Zhao, W. Wei and Y. Sun, Fuel, 2013, 108, 112–130 CrossRef CAS.
- L. Desport and S. Selosse, Resour., Conserv. Recycl., 2022, 180, 106150 CrossRef CAS.
- A. Parekh, G. Chaturvedi and A. Dutta, Sustain. Energy Technol. Assessments, 2023, 55, 102942 CrossRef.
- I. Sreedhar, T. Nahar, A. Venugopal and B. Srinivas, Renewable Sustainable Energy Rev., 2017, 76, 1080–1107 CrossRef CAS.
- S. Krishnamurthy, A. Lind, A. Bouzga, J. Pierchala and R. Blom, Chem. Eng. J., 2021, 406, 127121 CrossRef CAS.
- I. P. Koronaki, L. Prentza and V. Papaefthimiou, Renewable Sustainable Energy Rev., 2015, 50, 547–566 CrossRef CAS.
- C. Chao, Y. Deng, R. Dewil, J. Baeyens and X. Fan, Renewable Sustainable Energy Rev., 2021, 138, 110490 CrossRef CAS.
- K. Xie, Q. Fu, J. Kim, H. Lu, Y. He, Q. Zhao, J. Scofield, P. A. Webley and G. G. Qiao, J. Membr. Sci., 2017, 535, 350–356 CrossRef CAS.
- Y. Xu, C. Luo, H. Sang, B. Lu, F. Wu, X. Li and L. Zhang, Chem. Eng. J., 2022, 435, 134960 CrossRef CAS.
- C. Song, Q. Liu, S. Deng, H. Li and Y. Kitamura, Renewable Sustainable Energy Rev., 2019, 101, 265–278 CrossRef CAS.
- Y. Xu, B. Lu, C. Luo, F. Wu, X. Li and L. Zhang, Chem. Eng. J., 2022, 435, 134852 CrossRef CAS.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- Z. W. Ma, P. Zhang, H. S. Bao and S. Deng, Renewable Sustainable Energy Rev., 2016, 53, 1273–1302 CrossRef CAS.
- K. O. Yoro, M. O. Daramola, P. T. Sekoai, E. K. Armah and U. N. Wilson, Renewable Sustainable Energy Rev., 2021, 147, 111241 CrossRef CAS.
- H. F. Svendsen, E. T. Hessen and T. Mejdell, Chem. Eng. J., 2011, 171, 718–724 CrossRef CAS.
- H. C. Mantripragada, H. Zhai and E. S. Rubin, Int. J. Greenhouse Gas Control, 2019, 82, 59–68 CrossRef.
- M. T. Mota-Martinez, P. Brandl, J. P. Hallett and N. Mac Dowell, Mol. Syst. Des. Eng., 2018, 3, 560–571 RSC.
- X. Tan, X. Sun and B. Han, Natl. Sci. Rev., 2022, 9, nwab022 CrossRef PubMed.
- J. Wolfs, R. Nickisch, L. Wanner and M. A. R. Meier, J. Am. Chem. Soc., 2021, 143, 18693–18702 CrossRef CAS PubMed.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustain. Chem. Eng., 2018, 6, 8826–8835 CrossRef CAS.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- H. Fu, Y. Hou, H. Sang, T. Mu, X. Lin, Z. Peng, P. Li and J. Liu, AIChE J., 2021, 67, 7 Search PubMed.
- L. Veltri, R. Amuso, P. Vitale, M. A. Chiacchio, C. Benincasa and B. Gabriele, J. CO2 Util., 2021, 52, 101695 CrossRef CAS.
- Q. Gu, J. Fang, Z. Xu, W. Ni, K. Kong and Z. Hou, New J. Chem., 2018, 42, 13054–13064 RSC.
- N. Fanjul-Mosteirín, C. Jehanno, F. Ruipérez, H. Sardon and A. P. Dove, ACS Sustain. Chem. Eng., 2019, 7, 10633–10640 CrossRef.
- X.-C. Chen, K.-C. Zhao, Y.-Q. Yao, Y. Lu and Y. Liu, Catal. Sci. Technol., 2021, 11, 7072–7082 RSC.
- Z.-Z. Yang, L.-N. He, Y.-N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971 RSC.
- C. Wang, H. Luo, D.-e. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CrossRef CAS PubMed.
- R. Shi, H. Cheng, H. Li, P. Wu, C. Zhang, M. Arai and F. Zhao, Green Energy Environ., 2022, 7, 477–484 CrossRef CAS.
- L. Zheng, G. Yang, J. Liu, X. Hu and Z. Zhang, Chem. Eng. J., 2021, 425, 131452 CrossRef CAS.
- V. R. Velpuri, S. Kumari and K. Muralidharan, J. Chem. Sci., 2023, 135, 24 CrossRef CAS.
- S. Jiang, H. Y. Cheng, R. H. Shi, P. X. Wu, W. W. Lin, C. Zhang, M. Arai and F. Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47413–47421 CrossRef CAS PubMed.
- H. Koizumi, K. Takeuchi, K. Matsumoto, N. Fukaya, K. Sato, M. Uchida, S. Matsumoto, S. Hamura and J.-C. Choi, ACS Sustain. Chem. Eng., 2022, 10, 5507–5516 CrossRef CAS.
- A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113 CrossRef CAS.
- Y. Qu, Y. Chen and J. Sun, J. CO2 Util., 2022, 56, 101840 CrossRef CAS.
- Y. Chai, Q. Chen, C. Huang, Q. Zheng, M. North and H. Xie, Green Chem., 2020, 22, 4871–4877 RSC.
- J. K. Mannisto, L. Pavlovic, T. Tiainen, M. Nieger, A. Sahari, K. H. Hopmann and T. Repo, Catal. Sci. Technol., 2021, 11, 6877–6886 RSC.
- J. Sun, W. Cheng, Z. Yang, J. Wang, T. Xu, J. Xin and S. Zhang, Green Chem., 2014, 16, 3071 RSC.
- T. Moore, A. J. Varni, S. H. Pang, S. A. Akhade, S. Li, D. T. Nguyen and J. K. Stolaroff, Energy Environ. Sci., 2023, 16, 484–490 RSC.
- T. Kitamura, Y. Inoue, T. Maeda and J. Oyamada, Synth. Commun., 2015, 46, 39–45 CrossRef.
- P. Rigby, Nano Today, 2007, 2, 9 Search PubMed.
- N. J. Williams, C. A. Seipp, F. M. Brethomé, Y.-Z. Ma, A. S. Ivanov, V. S. Bryantsev, M. K. Kidder, H. J. Martin, E. Holguin, K. A. Garrabrant and R. Custelcean, Chem, 2019, 5, 719–730 CAS.
- C. A. Seipp, N. J. Williams, M. K. Kidder and R. Custelcean, Angew Chem. Int. Ed. Engl., 2017, 56, 1042–1045 CrossRef CAS PubMed.
- T. Elliott, L. Charbonneau, E. Gazagnaire, I. Kilpeläinen, B. Kótai, G. Laczkó, I. Pápai and T. Repo, RSC Sustainability, 2024, 2, 1753–1760 RSC.
- H. Wu, S. Wen, X. Zhang, S. Zhang, X. Guo and Y. Wu, Chem. Eng. J., 2024, 488, 150771 CrossRef CAS.
- P. G. J. D. J. Heldebrant, C. A. E. C. A. Thomas and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef PubMed.
- J. W. Lee, H. Ahn, S. Kim and Y. T. Kang, J. Cleaner Prod., 2023, 390, 136141 CrossRef CAS.
- J. Zheng, Y. Zhang, L. Zhao, H. Li, R. Zhao, X. Nie, S. Deng and P. Linga, Energy Convers. Manage., 2024, 314, 118656 CrossRef CAS.
- X. Chen, Y. LiJin, Y. Chen, G. Jing, B. Lv, J. Dong and Z. Zhou, Sep. Purif. Technol., 2024, 340, 126751 CrossRef CAS.
- H. Yin, C. Ma, Y. Duan, S. Shi, Z. Zhang, S. Zeng, W. Han and X. Zhang, Chem. Eng. J., 2024, 496, 153867 CrossRef CAS.
- M. Li, Z. Liang, J. Sun, T. Xu, L. Xing, T. Qi, S. An, J. Li, F. Dong and L. Wang, Sep. Purif. Technol., 2025, 362, 131607 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Lifeng Yan
and
Lifeng Yan





