
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransforming waste fish bones into nanoparticles with ultrasound and aqueous organic acids†
Sarah
Boudreau
a,
Sabahudin
Hrapovic
b,
Emma
McIsaac
a,
Edmond
Lam
 b,
Fabrice
Berrué
*c and
Francesca M.
Kerton
*a
b,
Fabrice
Berrué
*c and
Francesca M.
Kerton
*a
aDepartment of Chemistry, Memorial University of Newfoundland, St. John's, NL, Canada. E-mail: fkerton@mun.ca
bAquatic and Crop Resource Development, National Research Council Canada, Montreal, QC, Canada
cAquatic and Crop Resource Development, National Research Council Canada, Halifax, NS, Canada. E-mail: fabrice.berrue@nrc-cnrc.gc.ca
First published on 18th March 2025
Abstract
Synthesizing materials from biomass has gained significant attention as a step towards achieving a circular economy. Seafood processing by-products (e.g., heads, fins, bones, and viscera) are currently disposed of using unsustainable practices including disposal in landfills and/or at sea. However, fish bones are made of 60% hydroxyapatite and therefore could be utilized as a sustainable feedstock for calcium phosphate materials. In this research, nano-hydroxyapatite particles were prepared from Atlantic salmon (Salmo salar) bones using ultrasound in combination with heat, ball milling, and aqueous acid treatment. The size of the synthesized hydroxyapatite nanoparticles can be tailored depending on the chosen conditions. The smallest particles (d = 29 nm) were produced using aqueous propanoic acid and 15 min ultrasound exposure, whereas heat pre-treatment and ultrasound treatment for 60 min led to more well-defined but larger particles (d = 69 nm). The presence of calcium propanoate on the surface of nanoparticles prepared with propanoic acid was detected by IR spectroscopy and X-ray diffraction. A simplified gate-to-gate life cycle assessment was used to demonstrate that this ultrasound process results in a 97% reduction in CO2 emissions compared to other methods reported in the literature to date.
Sustainability spotlightThe aquaculture and seafood processing industries are critical to meeting protein demands that are on the rise (SDG2); however, they produce significant amounts of biological waste. This can affect life at sea (SDG14) when disposed of in the ocean and also have further detrimental effects when landfills are used due to resulting greenhouse gas emissions (SDG13 and SDG15). Sustainable processes to utilize fish bones as a second product stream will increase the economic viability of fish processing facilities (SDG8, SDG9, and SDG12). Preparing materials that could potentially be used for higher-value applications is a step towards achieving a circular economy. A life cycle assessment demonstrates that our method for generating hydroxyapatite nanoparticles from waste bones results in a 97% reduction in CO2 emissions compared to other published methods. |
Introduction
As climate change continues to be driven by CO2 emissions, biomass feedstocks have gained significant attention to replace fossil fuels and assist in the achievement of net-zero goals.1 Biomass by-products from several industries, such as pulp and paper,2 and agriculture,3 are often wasted. However, these by-products have the potential to be useful for other applications and help in the journey towards a circular economy.4 For example, there has been much research into repurposing lignin5–7 and cellulose8–10 from plant waste, as well as smaller molecules derived from them.11,12 While most research in the literature has focused on these organic wastes, our group has looked at accessing inorganic materials and minerals from biomass, specifically seafood processing by-products. For example, we have isolated calcium carbonate from waste blue mussel (Mytilus edulis) shells13 and subsequently transformed the shells into a biogenic sponge-like material for the absorption of crude oil and dyes.14 Recently, our group has optimized an enzymatic method to isolate hydroxyapatite (HAP) from Atlantic salmon (Salmo salar) bones15 and has since been exploring potential applications for this material.Over the past century, nanomaterials16,17 have become highly important for biomedicine,18,19 food processing and packaging,20 energy storage,21 environmental remediation,22,23 agriculture,24 catalysis25 and more.26 As the number of potential applications continues to increase, there is a need to develop sustainable processes to synthesize nanoparticles. Current methods to synthesize nanoparticles typically involve bottom-up processes, such as sol–gel reactions, chemical vapour deposition, or chemical reduction.27 These processes often rely on mined or petroleum-derived feedstocks, using hazardous chemicals and generating waste. That being said, the principles of green chemistry can be applied to overcome these obstacles.28 An emerging green and sustainable technique to synthesize nanoparticles is mechanochemistry.29,30 For example, ball milling has been used for the synthesis of gold31 and zinc oxide nanoparticles,32 processes that typically rely on solution methods.33,34
Mechanochemistry has also been explored as a top-down method to yield nanoparticles from biomass.35 For example, Jin et al. have synthesized chitin and chitosan nanocrystals from green crab (Carcinus maenas) shells and soft wood pulp, respectively, using a combination of mechanochemistry and aging.36 Douard et al. prepared colloidal and stable nanocellulose materials by ball milling cotton fibres in deep eutectic solvents.37 Other organic bio-derived nanomaterials that have been investigated include starch, pectin, gum, and alginate.38 However, there remains a lack of research regarding the transformation of inorganic biomass into nanoparticles using methods based on green chemistry. While nano-hydroxyapatite (nHAP) has been prepared previously by Sharifianjazi et al. from pigeon (Columba livia) bones, the described process relies on high temperatures such as 850 °C before milling.39 In this study, we have successfully demonstrated that nHAP can be synthesized from Atlantic salmon bones without using significant amounts of energy. Two mechanochemical methods, ball milling and sonication,40 were used in combination to yield nHAP with tailored size and particle morphology.
Experimental
Materials
HAP was isolated from Atlantic salmon (Salmo salar) by-products based on our previous work.15 Salmon frames were donated by local seafood markets in St. John's, NL (The Seafood Shop, Sis' Seafood, and The Fish Depot). Briefly, salmon frames (backbones) were manually cleaned with a spatula to remove excess meat, minced in a blender for 1 min, boiled in tap water for 1 h, and finally treated with 15 μL g−1 Neutrase and 7.5 μL g−1 Lipozyme CALB L for 6 h at 40 °C in tap water. The bones were dried overnight in air and then pulverized into a powder, termed sHAP, for various lengths of time using a SPEX Sample Prep 8000 M mixer mill and a stainless-steel vial with 10 × 2.5′′ stainless steel balls.Propanoic acid (CAS: 79-09-4), glacial acetic acid (CAS: 64-19-7), and oleic acid (CAS: 112-80-1) were obtained from Fisher Scientific. Propanoic/propionic and propanoate/propionate are used interchangeably herein.
Characterization
Fourier transform infrared (FTIR) spectroscopy in attenuated total reflectance (ATR) mode was performed with a Bruker INVENIO-R (Bruker, USA), equipped with a single-bounce diamond ATR platform. The spectra were obtained using a scan range from 400 to 4000 cm−1 with 32 scans and a resolution of 4 cm−1.Powder X-ray diffraction (XRD) was performed using a Rigaku Mini Flex 600 6 G (Rigaku, Japan) with Cu Kβ radiation (40 kV, 15 mA) in continuous scan mode. The scan speed was set to 2.000° min−1, the sample width to 0.020°, and the scan range of 2θ = 3.000 to 90.000°. The sample holder used for XRD analyses was a Rigaku Si510 10 mm × 0.2 mm well (906163).
Samples were imaged using scanning electron microscopy (SEM) on a FEI MLA 650 FED under high vacuum. The voltage was set to 10–15 kV and the current to 10 mA with a working distance of 10.5–14.4 mm. The SEM is equipped with a dual Bruker 5th generation XFlash SDD X-ray detector for energy dispersive X-ray (EDX) spectroscopy to study the chemical composition of materials' surface.
Thermogravimetric analysis (TGA) was performed using a Discovery TGA 550 (TA Instruments, USA). The samples were heated from 25 to 1000 °C at a rate of 10 °C min−1 in the presence of air (20 L min−1).
Negatively stained samples were analysed using a transmission electron microscope (TEM, HITACHI H-7500, Japan) in bottom-mounted contrast mode. TEM grids (copper 200 mesh, 12–25 nm carbon supported, Ted Pella Inc.) were freshly glow-discharged using an EMS GloQube-D, dual chamber glow discharge system (Electron Microscopy Sciences, PA) in negative mode with a plasma current of 25 mA for 45 s. These grids were floated on 10 μL sample aliquots on Parafilm for 2 min. The excess droplets were subsequently wicked away from the edge of the grid with filter paper strips (Whatman™ 541). The grid was then rinsed with droplets of double distilled water. Immediately after rinsing with water, the grid was exposed to 10 μL of Van Gieson's staining solution for 60 s and the stain was carefully removed using a fresh piece of filter paper. Finally, the grid was dried under ambient conditions for 2 h and used for TEM analysis.
Size distribution histograms were created using ImageJ software to measure the diameter of particles on a selected TEM image. The average particle size and standard error were calculated by tabulating measurements from 2–3 TEM images from a single sample in a spreadsheet.
Dynamic light scattering (DLS) was used to determine the average particle size (hydrodynamic radius) and the zeta potential of samples (1 mg mL−1) using a Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK). The DLS analyses were performed in triplicate and the average was reported.
Procedure
Waste Atlantic salmon bones were cleaned following our previously reported method.15 Briefly, salmon frames were manually cleaned of excess meat, blended for 1 min, boiled for 1 h in tap water, and enzymatically treated with 15 μL g−1 Neutrase and 7.5 μL g−1 Lipozyme CALB L for 6 h in water at 40 °C. The bones, referred to as sHAP herein, were allowed to dry in air overnight before being pulverized in a ball mill for 1–4 h with cooling breaks after every 20 min to prevent overheating (see the ESI†). A portion of salmon bones was put in an oven for 24 h at 200 °C prior to milling to study the effects of this heat on nanoparticle synthesis.Samples subjected to ultrasound were treated by dispersing 10 mg sHAP in 10 mL of selected media (e.g., water and aqueous organic acids), as shown in Fig. 1. The solution was sonicated for 15–60 min with a Misonix S-4000 sonicator employed with a circulating cooling bath set to 3 °C to prevent temperature fluctuations. The amplitude of the ultrasonic vibration was set to 50% and different energies were provided to each sample depending on the chosen solvent. This process was repeated under identical conditions for several samples to ensure reproducibility. Next, the mixture was centrifuged for 5–60 min at 6000 rpm. The supernatant was decanted and retained for TEM and DLS analyses while the residual pellet was discarded.
 | ||
| Fig. 1 Milling and ultrasound steps to transform Atlantic salmon bones into nHAP. | ||
Results and discussion
The time periods for ball milling and centrifuging were varied, and their impacts on the average size and distribution of nHAP particles were evaluated. For ball milling, fish bones were ground for 1, 2, 3, and 4 h, and 10 mg of the resulting sHAP powder was dispersed in 10 mL water. DLS data (Table S1†) and TEM imaging (Fig. S1†) confirmed that increased milling time did not have an impact on particle size, and therefore 1 h was chosen for further experiments. The centrifugation time was set to 15 min, as a longer time did not affect particle size and distribution (Table S2†).The choice of sonicating medium was the most important variable for the size and uniformity of nHAP particles. Deionized (DI) water, 5% (v/v) propanoic acid (PA), 5% (v/v) acetic acid (AA), and 5% (w/v) oleic acid (OA) were investigated as media by comparing the DLS data and TEM images of the resulting particles. The nHAP particles synthesized with OA had a significantly higher average particle size of 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 950 nm according to DLS (Table 1) and non-uniformity as observed by TEM (Fig. S2†), so they were not investigated further. We believe this to be the result of OA being more hydrophobic than other organic acids studied. OA has a reported logKow value of 7.64 (ref. 41), which is significantly higher than those of AA (−0.17)42 and PA (0.33).43 Therefore, OA is less efficient at permeating into sHAP aggregates and interacting with the surface –OH groups of HAP. A more hydrophilic acid with a shorter aliphatic chain is necessary to disassemble agglomerates of HAP within sHAP to form nHAP.
950 nm according to DLS (Table 1) and non-uniformity as observed by TEM (Fig. S2†), so they were not investigated further. We believe this to be the result of OA being more hydrophobic than other organic acids studied. OA has a reported logKow value of 7.64 (ref. 41), which is significantly higher than those of AA (−0.17)42 and PA (0.33).43 Therefore, OA is less efficient at permeating into sHAP aggregates and interacting with the surface –OH groups of HAP. A more hydrophilic acid with a shorter aliphatic chain is necessary to disassemble agglomerates of HAP within sHAP to form nHAP.
| Entry | Medium | Size (nm) | SDa | PDIa | SDa | Zeta (mV) | SDa |
|---|---|---|---|---|---|---|---|
| a Abbreviations: standard deviation, SD; polydispersity index, PDI; oleic acid, OA; acetic acid, AA; propanoic acid, PA. b Prior to analysis, the sample was centrifuged for 15 min at 6000 rpm with the supernatant decanted for analysis and the pellet discarded. | |||||||
| 1 | 5% OA | 19950 |
6181 | 0.354 | 0.247 | −16.3 | 5.98 |
| 2 | Water | 677.60 | 108.4 | 0.557 | 0.0840 | 5.62 | 0.755 |
| 3 | 5% AA | 345.70 | 3.889 | 0.546 | 0.885 | 16.3 | 0.985 |
| 4 | 5% PA | 252.60 | 38.70 | 0.545 | 0.043 | 15.2 | 0.686 |
Water, 5% PA, and 5% AA yielded spherical nanoparticles that were compared using size distribution histograms (Fig. 2) – throughout this communication size represents the diameter of particles from TEM data, which were analyzed using ImageJ software. While water produced the lowest average particle size (Fig. 2a and b), the histogram also had large error bars, especially in the 6–10 and >100 nm ranges, possibly from the tendency of water-based nHAP to agglomerate. We propose that agglomeration has a greater tendency to occur in water-based systems as the surface of nHAP has not been modified in any way. Under aqueous carboxylic acid conditions, nHAP will be modified, presenting carboxylate groups on the surface. TEM analysis shows that 5% AA resulted in lower agglomeration; however, most nanoparticles were >100 nm (Fig. 2c and d). 5% PA was chosen for further investigation because the resulting nHAP particles were colloidal and the smallest based on TEM (Fig. 2e and f) and DLS analyses (Table 1).
 | ||
| Fig. 2 Size distributions and TEM images of nHAP samples prepared by ultrasound (US) in water or 5% organic acid for 15 min. Samples a and b (entry 2, top, blue, scale bar: 200 nm, Fig. S3†) were sonicated in water, c and d (entry 3, middle, orange, scale bar: 600 nm, Fig. S4†) in 5% AA, and e and f (entry 4, bottom, green, scale bar: 500 nm, Fig. S5†) in 5% PA. Prior to analysis, the sample was centrifuged for 15 min at 6000 rpm with the supernatant decanted for analysis and the pellet discarded. Reported diameters are labelled as “length”. | ||
Next, the concentration of PA was increased from 5 to 10% (Fig. 3). nHAP particles sonicated in 10% PA for 15 min had more defined edges compared to those produced using 5% PA. We hypothesize that this results from residual collagen within sHAP being decomposed by the higher concentration of acid. Collagen does not conduct electrons and therefore causes a charge build-up during TEM analysis, distorting the image and making observed particles less resolved. The organic acid also plays a role in stabilizing the surface of nHAP particles, so a reasonable concentration is required for sufficient stabilization. The size distribution histogram of nHAP in 10% PA had a slightly lower average particle size and only 0.5% of particles were larger than 100 nm. Therefore, 10% PA was used herein for further experiments. Throughout these studies, experiments were repeated to ensure reproducibility in terms of particle size across a specific set of conditions.
 | ||
| Fig. 3 Size distribution histograms and TEM images of nHAP samples prepared by ultrasonication (US) in 10% propanoic acid. Samples a and b (top left, Fig. S6,† scale bar: 200 nm) and c and d (top right, Fig. S7,† scale bar: 500 nm) were sonicated for 15 min while samples e and f (bottom left, scale bar: 400 nm, Fig. S8†) and g and h (bottom right, scale bar: 200 nm, Fig. S9†) were sonicated for 60 min. Samples c and d (top right, scale bar: 500 nm) and g and h (bottom right, scale bar: 200 nm) were pre-treated by placing sHAP into an oven for 24 h at 200 °C before milling. Prior to analysis, the samples were centrifuged for 15 min at 6000 rpm with the supernatant decanted for analysis and the pellet discarded. | ||
Using 10% PA, the sonicating time was increased from 15 to 60 min. Interestingly, while the edges of particles were even more defined (Fig. 3f), the average particle size increased significantly from 28.7 ± 0.50 (Fig. 3a) to 43.4 ± 1.2 nm (Fig. 3e). Also, 7.7% of nHAP particles have diameters greater than 100 nm, but the reported PDI from DLS data improved from 0.443 to 0.389 (Table S3†). This could be caused by a longer ultrasound time leading to a greater proportion of the original sHAP disaggregating into nanoparticles and there is no longer sufficient PA available to stabilize the surfaces. The optimum conditions depend on the carboxylic acid (Kow), concentration, and ultrasound treatment time.
Increased diameters and PDIs were also obtained when sHAP was heated for 24 h at 200 °C before milling. For example, heated samples sonicated for 15 min had a larger average particle size (Fig. 3c) than unheated sHAP sonicated for 60 min (Fig. 3e). Also, sonicating heated sHAP for 60 min yielded particles with an average particle size of 102 ± 14 nm (Fig. 3g) with 45% of particles being greater than 100 nm and a PDI of 0.837 (Table S3†).
On the basis of other literature mechanisms regarding ultrasound action,44 we propose that the nanoparticles form through etching of semi-crystalline sHAP. It is observed by SEM imaging that once the original sHAP material is ball-milled for 1 h, microparticles are formed (Fig. 4a and b). These particles have cracks on their surface that are further deepened and break due to the added energy from the cavitation principle during ultrasound, thus producing nano-sized particles (Fig. 4c).
 | ||
| Fig. 4 SEM images of sHAP (precursor for nHAP) ball milled for 1 h (prior to ultrasound treatment) at 1 mm (a) and 10 μm (b) scales. Image (b) shows cracks and agglomerations of particles present on the surface of sHAP. Scheme (c) demonstrates the proposed method for synthesizing nHAP: the nHAP particles embedded in the protein matrix are released from sHAP through the energy supplied by ultrasound-induced cavitation. A portion of the sample remains embedded in the protein matrix that is separated by centrifugation. | ||
The nanoparticles prepared with 10% PA for 15 min were analyzed using FTIR, XRD, SEM-EDX, and TGA. Samples were prepared by allowing the nHAP suspensions to evaporate under ambient conditions. Peaks observed in the IR spectrum suggest that the nanoparticles do not only consist of HAP, but also calcium propanoate45 resulting from the ultrasound processing with PA (Fig. 5). Biogenic HAP is confirmed by its characteristic phosphate ion vibrations at 1076 and 546 cm−1, while collagen (1651, 1549, and 1242 cm−1)46,47 and carbonate (1468 and 1419 cm−1)46,47 signals are also present (Fig. S10†).15 Compared with sHAP, the peak at 1549 cm−1 for nHAP has higher intensity. This is thought to be caused by the formation of calcium propanoate, which has its strongest vibration at 1569 cm−1.45 Other peaks associated with the presence of calcium propanoate were observed at 2974, 2939, 2881, 1375, 1242, 852, and 814 cm−1 (Fig. S10†).45
 | ||
| Fig. 5 FTIR spectra of nHAP (top) and its precursor, sHAP (bottom). | ||
XRD analysis also indicated that calcium propanoate was formed in this process and possibly on the surface of nHAP during the ultrasound procedure (Fig. 6). Compared to sHAP,15 the nHAP sample is more crystalline, as indicated by the sharper peaks, and this may be due to removal of significant quantities of collagen present in the original sHAP starting material. Diffractions below 2θ = 30.6° in the nHAP sample are consistent with the presence of propanoate48 and those above 2θ = 34.3° are from nHAP (HAP and carbonates).14,15,49 Additionally, there are several peaks that overlap (e.g., calcium carbonate and calcium propanoate both have peaks around 2θ = 23.4 and 29.4°).14,48
 | ||
| Fig. 6 XRD diffractogram of nHAP composed of calcium propanoate (orange circles), calcium carbonate (green diamonds), and HAP (blue hearts). | ||
We analyzed the chemical composition of nanoparticles using SEM-EDX. As expected, the resolution of images was not high due to the sample's small particle size; however, we were still able to obtain sufficient EDX analysis (Fig. 7 and S11–17 and Table S6†). The average Ca/P ratio of nHAP was 2.50, significantly higher than the stoichiometric Ca/P ratio of 1.67. This is the result of biogenic HAP containing carbonate impurities necessary for biological function that often replace a phosphate group, thus lowering the overall P content. Furthermore, we suspect that the reported average carbon content of 6.41 wt% is from the calcium propanoate formed on the surface of the particles during the ultrasound process, thus leading to an increased Ca/P ratio, as phosphates or hydroxyl groups will have been replaced by propanoates. Compared to sHAP, the C content decreased significantly by 22.1 wt% (Fig. S18–S20 and Table S7†), suggesting that residual collagen was degraded during the ultrasound treatment. The O content also decreased by 16.1 wt%, which is reasonable because collagen has O-containing functional groups present.
 | ||
| Fig. 7 Selected EDX spectrum of nHAP prepared with 10% propanoic acid (15 min); C mass%: 9.35, O mass%: 30.7, P mass%: 14.0, Ca mass%: 45.9, and Ca/P: 2.54. | ||
Finally, the thermal stability of nHAP was studied with TGA up to 1000 °C (Fig. 8). Overall, there was a total mass loss of 45.3%. From 28 to 200 °C, there was a mass loss of 16.4% attributed to the evaporation of water. The mass loss of 25.3% from 200to 590 °C was the result of any residual proteins remaining from the bone matrix and primarily calcium propanoate on the surface being degraded, which is in agreement with the literature data.50 The final loss of 3.58% up to 1000 °C is explained by the degradation of calcium carbonate, producing carbon dioxide. 54.7% of the sample remains intact up to 1000 °C and can thus be explained by the presence of HAP. Therefore, the synthesized nHAP is relatively stable up to 200 °C and there is no further decomposition after 700 °C because HAP does not show any sign of degrading until 1360 °C.51 The TGA curve of nHAP is almost identical to that of sHAP,15 suggesting that the product is similar to the original bone material but is also consistent with the presence of calcium propanoate on the surface.
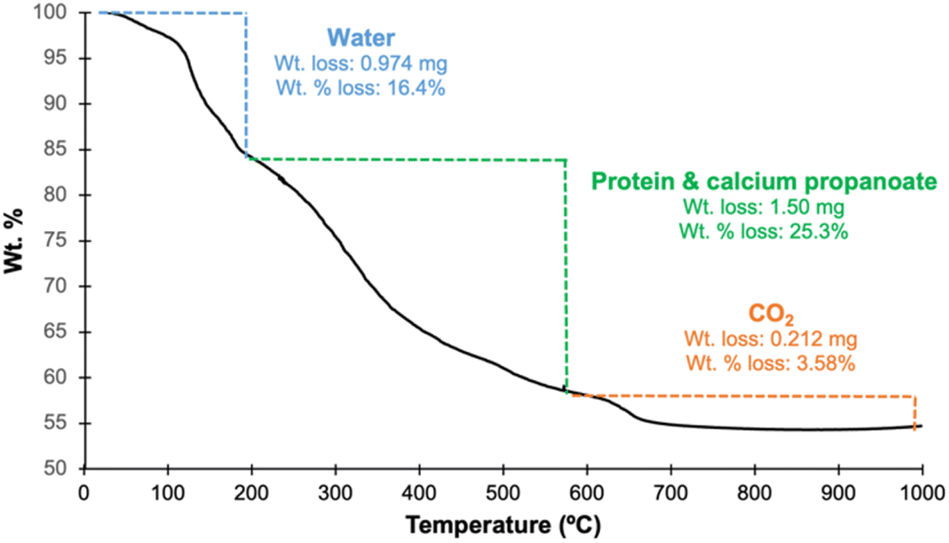 | ||
| Fig. 8 TGA curve of nHAP prepared with 10% propanoic acid (15 min) from 0 to 1000 °C; wt loss 16.4% (28.5–210 °C): water; wt loss 25.3% (210–588 °C): protein and calcium propanoate; wt loss 3.58% (588–998 °C): carbon dioxide. | ||
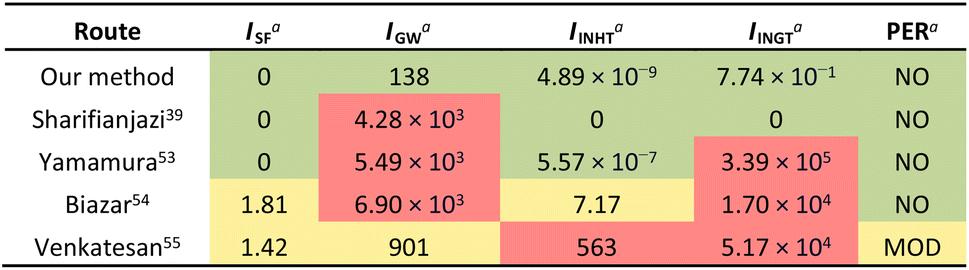
To validate the improved sustainability of our process compared to others reported in the literature, we have performed a simplified gate-to-gate life cycle assessment (LCA) that is shown in Table 2.52 This LCA is based on the transformation of fish waste into nHAP without considering the manufacture of additional chemicals used (e.g., PA). More details regarding LCA calculations can be found in the ESI.† Our method that relies on ultrasound was compared with processes that have used calcination and/or alkaline deproteinization. Yamamura et al., Biazar et al., and Venkatesan et al. used NaOH to dissolve the protein residues53–55 and therefore possess significant human ingestion potential. While the method reported by Sharifianjazi et al. is not hazardous for human exposure because of the lack of chemicals required, it relies on temperatures up to 850 °C to produce nHAP,39 resulting in a high global warming potential from increased CO2 emissions.
| a Abbreviations: Life Cycle Assessment, LCA; ISF, smog formation potential; IGW, global warming potential; IINHT, human inhalation toxicity potential; IINGT, human ingestion toxicity potential; PER, persistence potential (see the ESI for more information). b Values determined for each potential are coloured according to their impact: low is green, moderate is yellow and high is red. |
|---|
|
Conclusions
In conclusion, we have successfully transformed fish processing industry discards into nHAP particles using environmentally benign conditions. The size and definition of particles can be tailored by heating the bones before sonication, modifying the ultrasound medium and changing the time period of sonication. Higher concentrations of organic acid and heat lead to larger particle sizes, but more defined particles. Larger or smaller particles (30 nm vs. 100 nm) can therefore be prepared depending on their desired application. For example, nHAP particles have been widely used in biomedicine as drug carriers to inhibit the growth of tumor cells.56 nHAP has also been used for the environmental remediation of heavy metals57 and several industrial dyes.58 Furthermore, our LCA demonstrates a 97% reduction in CO2 emissions compared with traditional calcination routes.39 We are now performing ongoing studies to understand the nature of the sHAP remaining in the pellet formed upon centrifugation and uses for nHAP.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, F. M. K.; methodology, S. B., E. M., and F. M. K.; characterization, S. B. and S. H.; writing – original draft preparation, S. B.; writing – review and editing, S. B., S. H., E. L., F. M. K., and F. B.; supervision, F. M. K. and F. B.; funding acquisition, F. M. K. and F. B. All authors listed have agreed to the final version of this communication and have made significant contributions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NRC Ocean Program, OGEN (OCN-110-4), Ocean Frontier Institute, NSERC of Canada, Canada Foundation for Innovation, Dr Liqin Chen, and Memorial University of Newfoundland (MUN) for funding. The authors also thank Sachel Christian-Robinson (PhD Candidate, MUN) for TGA analysis and Dylan Goudie (CREAIT, MUN) for SEM-EDX analysis and advice.References
- United Nations, Climate Change, https://www.un.org/en/global-issues/climate-change, accessed February 20, 2025.
- A. Haile, G. G. Gelebo, T. Tesfaye, W. Mengie, M. A. Mebrate, A. Abuhay and D. Y. Limeneh, Bioresour. Bioprocess., 2021, 8, 35 Search PubMed.
- S. Babu, S. Singh Rathore, R. Singh, S. Kumar, V. K. Singh, S. K. Yadav, V. Yadav, R. Raj, D. Yadav, K. Shekhawat and O. Ali Wani, Bioresour. Technol., 2022, 360, 127566 CAS.
- J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC.
- B. L. Tardy, E. Lizundia, C. Guizani, M. Hakkarainen and M. H. Sipponen, Mater. Today, 2023, 65, 122–132 CrossRef CAS.
- M. J. Gan, Y. Q. Niu, X. J. Qu and C. H. Zhou, Green Chem., 2022, 24, 7705–7750 RSC.
- C. Zhang, X. Shen, Y. Jin, J. Cheng, C. Cai and F. Wang, Chem. Rev., 2023, 123, 4510–4601 CrossRef CAS PubMed.
- F. D'Acierno, C. A. Michal and M. J. MacLachlan, Chem. Rev., 2023, 123, 7295–7325 CrossRef PubMed.
- Y. Ai, L. Zhang, M. Cui, R. Huang, W. Qi, Z. He, J. J. Klemeš and R. Su, Green Chem., 2022, 24, 6406–6434 RSC.
- M. Gao, Y. Shang, B. Li and H. Du, Green Chem., 2022, 24, 9346–9372 RSC.
- R. Hu, J. Zhan, Y. Zhao, X. Xu, G. Luo, J. Fan, J. H. Clark and S. Zhang, Green Chem., 2023, 25, 8970–9000 CAS.
- A. Kumar, A. Singh Chauhan, R. Bains and P. Das, Green Chem., 2023, 25, 849–870 CAS.
- J. N. Murphy, K. Hawboldt and F. M. Kerton, Green Chem., 2018, 20, 2913–2920 CAS.
- J. N. Murphy, C. M. Schneider, K. Hawboldt and F. M. Kerton, Matter, 2020, 3, 2029–2041 Search PubMed.
- S. Boudreau, S. Hrapovic, Y. Liu, A. C. W. Leung, E. Lam and F. M. Kerton, RSC Sustainability, 2023, 1, 1554–1564 CAS.
- M. Österberg, K. A. Henn, M. Farooq and J. J. Valle-Delgado, Chem. Rev., 2023, 123, 2200–2241 CrossRef PubMed.
- D. Yan, Chem. – Eur. J., 2015, 21, 4880–4896 CAS.
- J. Zhao and M. H. Stenzel, Polym. Chem., 2018, 9, 259–272 CAS.
- F. Fang, M. Li, J. Zhang and C.-S. Lee, ACS Mater. Lett., 2020, 2, 531–549 CAS.
- J. Chen, Y. Guo, X. Zhang, J. Liu, P. Gong, Z. Su, L. Fan and G. Li, J. Agric. Food Chem., 2023, 71, 3564–3582 CrossRef CAS PubMed.
- Z. Gan, J. Yin, X. Xu, Y. Cheng and T. Yu, ACS Nano, 2022, 16, 5131–5152 CAS.
- H. Rafeeq, A. Hussain, A. Ambreen, Z.-e Huma, M. Waqas, M. Bilal and H. M. N. Iqbal, J. Nanostruct. Chem., 2022, 12, 1007–1031 CAS.
- R. Das, T. Lindström, P. R. Sharma, K. Chi and B. S. Hsiao, Chem. Rev., 2022, 122, 8936–9031 CrossRef CAS PubMed.
- R. Saberi Riseh, M. Hassanisaadi, M. Vatankhah, R. S. Varma and V. K. Thakur, Nano-Micro Lett., 2024, 16, 147 CrossRef CAS PubMed.
- A. Srivastava, H. Kaur, H. Pahuja, T. M. Rangarajan, R. S. Varma and S. Pasricha, Coord. Chem. Rev., 2024, 507, 215763 CrossRef CAS.
- B. Thomas, M. C. Raj, A. K. B, R. M. H, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625 CrossRef CAS PubMed.
- N. Baig, I. Kammakakam and W. Falath, Mater. Adv., 2021, 2, 1821–1871 RSC.
- M. Lancaster, Green Chemistry 3rd Edition: an Introductory Text, Royal Society of Chemistry, 2016 Search PubMed.
- B. G. Fiss, A. J. Richard, G. Douglas, M. Kojic, T. Friščić and A. Moores, Chem. Soc. Rev., 2021, 50, 8279–8318 RSC.
- D. Yan, R. Gao, M. Wei, S. Li, J. Lu, D. G. Evans and X. Duan, J. Mater. Chem. C, 2013, 1, 997–1004 RSC.
- A. J. Richard, M. Ferguson, B. G. Fiss, H. M. Titi, J. Valdez, N. Provatas, T. Friščić and A. Moores, Nanoscale Adv., 2023, 5, 2776–2784 RSC.
- S. Wirunchit, P. Gansa and W. Koetniyom, Mater. Today: Proc., 2021, 47, 3554–3559 CAS.
- P. Suchomel, L. Kvitek, R. Prucek, A. Panacek, A. Halder, S. Vajda and R. Zboril, Sci. Rep., 2018, 8, 4589 CrossRef PubMed.
- Th. A. Singh, J. Das and P. C. Sil, Adv. Colloid Interface Sci., 2020, 286, 102317 CrossRef CAS PubMed.
- F. Hajiali, T. Jin, G. Yang, M. Santos, E. Lam and A. Moores, ChemSusChem, 2022, 15, e202102535 CrossRef CAS PubMed.
- T. Jin, T. Liu, F. Hajiali, M. Santos, Y. Liu, D. Kurdyla, S. Régnier, S. Hrapovic, E. Lam and A. Moores, Angew. Chem., Int. Ed., 2022, 61, e202207206 CrossRef CAS PubMed.
- L. Douard, M. N. Belgacem and J. Bras, ACS Sustain. Chem. Eng., 2022, 10, 13017–13025 CrossRef CAS.
- M. Nasrollahzadeh, M. Sajjadi, S. Iravani and R. S. Varma, Carbohydr. Polym., 2021, 251, 116986 CrossRef CAS PubMed.
- F. Sharifianjazi, A. Esmaeilkhanian, M. Moradi, A. Pakseresht, M. S. Asl, H. Karimi-Maleh, H. W. Jang, M. Shokouhimehr and R. S. Varma, Mater. Sci. Eng., B, 2021, 264, 114950 CrossRef CAS.
- D. Yan, D.-K. Bučar, A. Delori, B. Patel, G. O. Lloyd, W. Jones and X. Duan, Chem. – Eur. J., 2013, 19, 8213–8219 CrossRef CAS PubMed.
- National Center for Biotechnology Information, Oleic Acid, https://pubchem.ncbi.nlm.nih.gov/compound/Oleic-Acid, accessed February 20, 2025.
- National Center for Biotechnology Information, Acetic Acid,https://pubchem.ncbi.nlm.nih.gov/compound/Acetic-Acid, accessed February 20, 2025.
- National Center for Biotechnology Information, Propionic Acid (Compound), https://pubchem.ncbi.nlm.nih.gov/compound/Propionic-Acid, accessed February 20, 2025.
- A. Gedanken, Ultrason. Sonochem., 2004, 11, 47–55 CrossRef CAS PubMed.
- NIST Chemistry WebBook, SRD 69, Sodium propionate, 2025, https://webbook.nist.gov/cgi/cbook.cgi?ID=B6010184&Units=SI&Mask=80, accessed February 3 Search PubMed.
- P. Surya, A. Nithin, A. Sundaramanickam and M. Sathish, J. Mech. Behav. Biomed. Mater., 2021, 119, 104501 CrossRef CAS PubMed.
- O. A. Osuchukwu, A. Salihi, I. Abdullahi and D. O. Obada, Mater. Today: Proc., 2022, 62, 4182–4187 CAS.
- C. A. O'Connell and D. Dollimore, Thermochim. Acta, 2000, 357–358, 79–87 CrossRef.
- B. El, M. Hicham, A. Mohamed, R. El, M. Benanni-Ziatni, R. El Hamri and T. Abderrahim, J. Mater. Environ. Sci., 2016, 7, 4049–4063 Search PubMed.
- S. Zaidi, D. Sanchez-Rodriguez, J. Farjas, D. Mohamed and P. Roura-Grabulosa, J. Therm. Anal. Calorim., 2023, 148, 13039–13049 CrossRef CAS.
- C.-J. Liao, F.-H. Lin, K.-S. Chen and J.-S. Sun, Biomater., 1999, 20, 1807–1813 CrossRef CAS PubMed.
- S. M. Mercer, J. Andraos and P. G. Jessop, J. Chem. Educ., 2012, 89, 215–220 CrossRef CAS.
- H. Yamamura, V. H. P. da Silva, P. L. M. Ruiz, V. Ussui, D. R. R. Lazar, A. C. M. Renno and D. A. Ribeiro, J. Mech. Behav. Biomed. Mater., 2018, 80, 137–142 CrossRef CAS PubMed.
- E. Biazar, M. Daliri J, S. Heidari K, D. Navayee A, M. Kamalvand, M. Sahebalzamani, F. Royanian, M. Shabankhah and F. Farajpour L, J. Bioeng. Res., 2020, 2, 10–19 Search PubMed.
- J. Venkatesan, B. Lowe, P. Manivasagan, K.-H. Kang, E. P. Chalisserry, S. Anil, D. G. Kim and S.-K. Kim, Mater., 2015, 8, 5426–5439 CAS.
- X. Gui, W. Peng, X. Xu, Z. Su, G. Liu, Z. Zhou, M. Liu, Z. Li, G. Song, C. Zhou and Q. Kong, Nanotechnol. Rev., 2022, 11, 2154–2168 CAS.
- A. Nayak and B. Bhushan, Mater. Today: Proc., 2021, 46, 11029–11034 CAS.
- S. Pai, M. S. Kini and R. Selvaraj, Environ. Sci. Pollut. Res., 2021, 28, 11835–11849 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, ultrasound procedure, characterization information, TEM images, DLS, and LCA data. See DOI: https://doi.org/10.1039/d5su00164a |
| This journal is © The Royal Society of Chemistry 2025 |