
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabricating lignocellulosic films as potential biobased plastics†
Antonella Rozaria Nefeli
Pontillo
 ab,
Sirui
Chen
b,
Diego
Freire Ordóñez
a,
Niall
Mac Dowell
c,
Koon-Yang
Lee
a and
Tom
Welton
*b
ab,
Sirui
Chen
b,
Diego
Freire Ordóñez
a,
Niall
Mac Dowell
c,
Koon-Yang
Lee
a and
Tom
Welton
*b
aDepartment of Aeronautics, Imperial College London, London, UK
bDepartment of Chemistry, Imperial College London, London, UK. E-mail: t.welton@imperial.ac.uk
cCentre for Environmental Policy, Imperial College London, London, SW7 2AZ, UK
First published on 1st April 2025
Abstract
Replacing petroleum based plastic packaging with an environmentally sustainable and economically viable process is an important step towards the goal of displacing oil as a petrochemical feedstock. Here we report the preparation of thin films consisting only of cellulose and lignin, using a recyclable ionic liquid. A co-solvent, DMSO, was used to decrease the viscosity of the dope solutions and facilitate the dissolution of the lignin. The films exhibit high mechanical properties with the tensile strength ranging between 65.44 and 93.15 MPa, comparable to those of commercial counterparts, while the presence of lignin increases the viscosity of the dope solutions, adds UV blockage to the films and decreases the wettability of the films with the water contact angle increasing up to 48.6%. The films are stable in various solvents and when immersed in aqueous solutions can swell and double in mass. The research also confirms that the ionic liquid can be retrieved and reused for at least five cycles without hindering its chemical composition and thermal stability.
Sustainability spotlightThe growing demand for sustainable materials necessitates alternatives to petroleum-based plastics and non-recyclable materials, which contribute to pollution and climate change. This study presents a methodology to create films solely from abundant biomass, often regarded as waste, resulting in biodegradable, non-toxic materials with excellent properties. The use of harsh chemicals is avoided by using a non-volatile and recyclable ionic liquid, a non-hazardous co-solvent and water as the antisolvent, aligning with the principle of waste prevention and circular economy. This work aims to reduce plastic pollution, lower environmental impact, and promote a sustainable manufacturing process, thus supporting UN SDGs 9 (industry, innovation, and infrastructure), 12 (responsible consumption and production), 13 (climate action) and 14 (life below water). |
Introduction
Undeniably, plastic remains an integral part of our lives. Official data indicates that 400 Mt of plastic are produced annually, with a significant portion of 39% dedicated to the packaging industry in Europe.1 Alarmingly, plastic production is not decreasing, even though guidelines such as the European Directives on the reduction of the impact of certain plastic products on the environment2 and on packaging and packaging waste3 aim to reduce it. Instead, production is rising at an annual rate of approximately 3% and each citizen in the European Union generates an average of 34.5 kg of plastic waste per year,4 while plastic films made up 34% of total packaging waste in the UK in 2014.5 This growing reliance on plastic has exacerbated environmental pollution, with far-reaching chemical, geophysical, and biological consequences that are expected to worsen.5 Plastic packaging films pose a particularly acute problem, as only a small fraction can be recycled. Effective recycling requires these materials to be free of organic matter and other contaminants, an especially challenging task for food packaging.5,6 Transitioning towards sustainable alternatives and a circular economy is imperative in order to mitigate the environmental impact of plastic pollution, aligning with global initiatives towards environmental responsibility.7,8Cellulose has been a promising candidate as a packaging material for many years – this abundant polymer of natural origin can be transformed into transparent, colourless films with excellent mechanical and thermal properties.9,10 One major challenge in processing cellulose is its inherent insolubility in common organic solvents due to the strong intermolecular hydrogen bonding of the hydroxyl groups within the polymer. Although cellulosic films, such as cellophane, have been produced since 1924,11 only a few molecular solvents such as sodium hydroxide and carbon disulfide, N-methylmorpholine-N-oxide (NMMO)12 and LiCl-N,N-dimethylacetamide (DMAc),13 are effective for this polymer. Recent advances in sustainable chemistry have led to the use of ionic liquids to dissolve cellulose, with the 1-ethyl-3-methylimidazolium acetate ([C2C1im][OAc]) emerging as one of the most effective solvents, as it has demonstrated the ability to dissolve cellulose at concentrations up to 27%.14 The mechanism of the dissolution of the polymer has been identified – the anion breaks the intermolecular hydrogen bonds of the polymer, forming new bonds, while the cation forms hydrophobic interactions.15 Then, for the regeneration of the cellulosic material, an antisolvent, such as water, is added which interrupts the IL/polymer hydrogen bonds.
Cellulose is compatible with a wide range of materials, enabling the enhancement of the final material's properties and broadening its potential applications. Lignin can serve as an excellent additive. Abundantly available and often regarded as waste, lignin is a complex aromatic polymer that can provide antimicrobial, antioxidant and UV-blocking activity, properties that could be useful for many applications, such as food packaging.16–19 The combination of these two polymers could lead to the formation of non-toxic and biodegradable films.
This study explores the dissolution of cellulose and lignin in an [C2C1im][OAc]/DMSO solution and follows the process of fabricating films with different ratios of the two polymers while the main properties of the films are monitored. The recycling potential of the ionic liquid is also explored, aiming to develop a sustainable process for the fabrication of bio-plastic films.
Results and discussion
Preparation of the dope solutions
During the preparation of the dope solutions, a major challenge encountered using pure [C2C1im][OAc] was the rapid and significant increase of the samples' viscosity, due to the formation of hydrogen bonds between the polymers and the solvent,20–22 which impeded the dissolution of the polymers in high concentrations. On the other hand, preliminary experiments indicated that films fabricated from dope solutions with low concentration of cellulose were excessively thin and fragile, rendering their use very difficult.To address this challenge and to increase the concentration of the polymers that can be dissolved, an aprotic solvent, DMSO, was added as co-solvent.23 Although DMSO cannot dissolve cellulose, it effectively reduced the viscosity of the sample. Furthermore, it has been found that when DMSO is added as a cosolvent to ionic liquids, it successfully solvates the anions and cations of the [C2C1im][OAc],15,24 facilitating further the dispersion and dissolution of the polymer. An additional advantage of using DMSO as the co-solvent is that it dissolves lignin at relatively high concentrations (Fig. 1a). Therefore, the lignin was pre-dissolved in DMSO before being mixed with the ionic liquid, allowing homogenous solubilisation in the dope solutions.
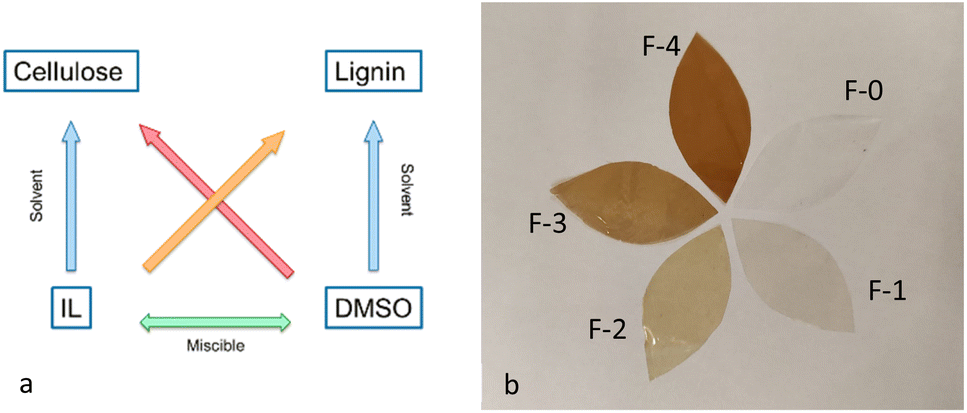 | ||
| Fig. 1 (a) Schematic representation of the dissolution system of the polymers (b) picture of the five films prepared. | ||
The lignin used in this study, is an organosolv, sulphur-free pine lignin. 31P NMR (Fig. S1†) revealed that it contains high concentration of aliphatic and phenolic hydroxyl groups (1.88 mmol g−1).25
Another critical factor impeding cellulose dissolution is moisture content, as water acts as an anti-solvent for the polymer. To mitigate this issue, all reagents were dried before use. In particular, the hygroscopic ionic liquid was dried on a Schlenk line at 45 °C until its relative water content was less than 1% w/w, measured by Karl Fischer titration.
Thus, to ensure successful sample preparation for film fabrication, the concentration of the polymers must be sufficiently high while the relative water content of all the reagents must be very low. Finally, immediately after adding the cellulose and lignin solution in DMSO to the ionic liquid, the mixtures were mixed by vortex for 2 minutes to achieve homogeneity and then heated to 70 °C until complete dissolution.
Rheological properties of the dope solutions
Creep and recovery testing was employed to calculate the viscosity of the ionic liquid and the mixture of the ionic liquid with DMSO (xDMSO = 0.4) at 70 °C (Fig. S2†). The viscosity of the solvent decreased from 17.8 mPa s to 9.2 mPa s when DMSO was added (Table 1).| Sample | η (Pa s) | η sp (×104 Pa s) | G′ = G′′ | f (Hz) at G′ = G′′ | η 0 (Pa s) |
|---|---|---|---|---|---|
| DS-0 | 780.6 | 8.5 | 467.6 | 0.8348 | 853.5 |
| DS-1 | 1626.0 | 17.7 | 226.1 | 0.3837 | 1393.4 |
| DS-2 | 1707.9 | 18.6 | 494.2 | 0.3885 | 1837.9 |
| DS-3 | 1818.3 | 19.8 | 293.3 | 0.2193 | 2050.7 |
| DS-4 | 2246.9 | 24.4 | 409.1 | 0.2297 | 2430.8 |
Then, the viscosity of five dope solutions (DS) was measured. The dope solutions were composed of a different ratio of cellulose and lignin dissolved in the [C2C1im][OAc]/DMSO mixture. The mass of cellulose is constant for all samples, while the concentration of lignin ranges between 0 and 3.40% w/w. All dope solutions exhibited viscoelastic behaviour leading to an elastic recovery upon removal of the stress, but the deformation was not fully reversible. The viscosity of the samples ranged from 780.6 Pa s for the non-lignin-containing dope solution (DS-0) to 2246.9 Pa s for DS-4 with the highest concentration of lignin.
The calculated specific viscosity (ηsp), describes the contribution of the polymers in the total viscosity, excluding the solvent. The results show that, although cellulose is the main contributor,26 the presence of lignin also has an effect on the viscosity. Hart27 has also reported that at high concentrations of lignin the sample reached a critical viscosity, limiting its further dissolution. Therefore, lignin can be seen as a thickening agent.
Next, the samples were subjected to oscillatory measurements at frequencies from 0.1–10 Hz to calculate the storage modulus (G′) representing the elastic, solid-like behaviour of the sample, the loss modulus (G′′) representing the viscous, liquid-like behaviour of the sample and their crossover point (G′ = G′′).
Above the crossover point, G′ is lower than G′′ and the material behaves more like a liquid, while below this point, the sample behaves more like a solid as G′ > G′′. Within the frequency range tested, all five dope solutions exhibited an elastic-dominant behaviour, with the crossover point occurring at low but decreasing frequencies, between 0.8 and 0.2 Hz, as the concentration of the lignin increases (Table 1 and Fig. S3a†), attributed to increased relaxation times being required.
The solvent exhibited an almost constant viscosity regardless of the shear rate applied, confirming a Newtonian fluid (Fig. S3b and c†). On the contrary, the dope solutions exhibited a non-Newtonian fluid profile, and the results fitted well the cross model28–30 (R2 > 0.993). The zero-shear viscosity (η0) was calculated and is presented on Table 1. This value confirms that the higher concentration of lignin leads to a higher entanglement of the polymeric chains.
Film preparation and characterisation
For the preparation of the films, firstly we aimed to determine the minimum amount of water and the number of coagulation baths required to completely remove the ionic liquid and obtain films composed only of cellulose and lignin. To do so, casted samples of the dope solutions were immersed in different amounts of water. After each coagulation bath, the water was collected and analysed with 1H NMR to detect any remaining ionic liquid. Our findings indicated that washing with a water-to-ionic liquid ratio of 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in four consecutive coagulation baths were required. The regenerated films produced under these conditions were thin and transparent, as illustrated in Fig. 1b. The concentration of the two polymers present in each film is detailed in Table 2.
:1 in four consecutive coagulation baths were required. The regenerated films produced under these conditions were thin and transparent, as illustrated in Fig. 1b. The concentration of the two polymers present in each film is detailed in Table 2.
| Sample | Ratio of polymers (wt%) | Thickness (μm) | WCA (°) | WVP (×106 g per (m day Pa)) | σ max (MPa) | E (GPa) | ε failure (%) | Crystallinity (%) | T onset (°C) | T d (°C) | Mass residue (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cellulose | Lignin | |||||||||||
| F-0 | 100 | 0 | 19.4 ± 4 | 45.63 ± 1.7 | 8.01 ± 1.4 | 80.03 ± 9.9 | 7.06 ± 1.2 | 2.80 ± 0.7 | 68 | 298 | 353.7 | 17.7 |
| F-1 | 96 | 4 | 24.4 ± 3 | 62.48 ± 0.8 | 12.33 ± 0.4 | 65.44 ± 8.4 | 6.32 ± 1.0 | 2.73 ± 0.8 | 58 | 296 | 353.8 | 16.3 |
| F-2 | 92 | 8 | 24.4 ± 5 | 65.36 ± 7.0 | 10.55 ± 1.7 | 93.15 ± 6.6 | 8.54 ± 0.6 | 3.07 ± 0.7 | 59 | 302 | 355.7 | 19.7 |
| F-3 | 80 | 20 | 25.6 ± 3 | 67.78 ± 8.1 | 11.62 ± 1.2 | 76.75 ± 5.5 | 6.55 ± 0.5 | 2.88 ± 0.4 | 51 | 286 | 305.8 | 23.9 |
| 354.1 | ||||||||||||
| F-4 | 71 | 29 | 28.5 ± 3 | 65.00 ± 10.2 | 12.17 ± 2.2 | 72.31 ± 4.3 | 5.83 ± 0.3 | 3.18 ± 0.5 | 55 | 280 | 304.4 | 25.3 |
| 349.0 | ||||||||||||
The film F-0, which consisted only of cellulose, is a transparent and colourless thin film. As the concentration of lignin gradually increases, the films become progressively darker while still retaining their transparency. The water contact angle (WCA) is a critical property of films for applications such as food packaging. It measures surface wettability and indicates the hydrophobicity of the tested surfaces, which is vital for prolonging the shelf life of packaged goods.31 Cellulose films are typically hydrophilic, with film F-0 exhibiting a WCA of approximately 45°. The addition of lignin increases the WCA, and the film F-1 reaches a WCA of 62° increasing to 65° for film F-2. Beyond this concentration, further increases in lignin do not significantly affect the WCA, which plateaus at around 65°. This indicates that while lignin enhances the hydrophobicity of the films, it is not inherently a strongly hydrophobic polymer due to its hydrophilic hydroxyl groups (Fig. S1†).
Cellulose films naturally exhibit high water vapour permeability (WVP) values, which is a critical property for food packaging.19 The WVP increased from 8.00 × 10−6 g per m per day per Pa to 12.17 × 10−6 g per m per day per Pa with the addition of lignin, resulting in a more permeable film. This change is attributed to the disruption of cellulose–cellulose intermolecular interactions with cellulose–lignin interactions which lead to a more open structure.
All the films are brittle and break at relatively low strains (Fig. S4†). The maximum tensile stress (σmax) required to fracture the films ranges between 65–93 MPa while the Young's modulus (E) is between 5.8 and 8.5 GPa (Table 2).
These values are comparable to the values reported in literature for commercial cellophane, a widely used cellulose film that contains plasticisers and other additives, with a σmax between 40 and 125 MPa and the E ranging from 0.9 to 3 GPa.32,33
The maximum tensile stress and the Young's modulus exhibited fluctuations with the increase of the lignin concentration. A trend is observed in the elongation values, indicating a correlation between the increase of the lignin concentration and the elongation at break.
The tensile properties of cellulose films are influenced by various factors including the type of cellulose, the polymer concentration, the final composition, and preparation process. Su34 reported that microcrystalline cellulose dissolved in cryogenic aqueous phosphoric acid exhibited a tensile strength of 707 MPa and elongation of 4.12%. For cellulose films regenerated from ionic liquids, tensile strength ranged from 62 to 160 MPa, with elongation between 3% and 20%. Yang35 compared the tensile properties of lignocellulosic films prepared with a DMAc/LiCl solvent system and found that both the concentration and the type of lignin had an impact on the mechanical properties of the films. They reported a tensile strength of 78.5 MPa for the cellulose film but with the addition of lignin, tensile strength values ranged from 74.3 to 79.2 MPa for 2% lignin and from 74.0 MPa to 94.5 MPa for 4% loading while the elongation at break decreased, although no clear trend was observed in relation to the lignin concentration. This effect was attributed to the chemical composition of the different lignin samples: higher concentration of hydroxyl groups led to the formation of additional hydrogen bonds between the two polymers, restricting their movement. The overall conclusion though was that adding lignin into the films, hinders the flexibility but not the tensile properties.
A commercial cellophane was also tested at the same conditions. It was found that the mechanical properties of the lignocellulosic films are comparable to the commercial counterpart (Fig. S5†). Interestingly, the lignocellulosic films require higher maximum strength to break than cellophane, while the Young's modulus is comparable. The significantly higher elongation before break that cellophane exhibits is attributed to the presence of plasticiser in its composition.36
In this study, the tensile strength and the Young's modulus obtained experimentally are very high but the elongation at break is very low. The rigidity of the films is attributed to the low degree of polymerisation and the high concentration of amorphous cellulose.
To assess how the films interact with incident light, the light transmittance through the films and the haze were measured (Fig. 2a and S6†). The F-0 film, consisted only of cellulose, exhibited a high light transmittance in the visible spectrum (400–700 nm), ranging from 87% to 91% and very low haze, less than 6%, confirming that the light can pass through the film without scattering.
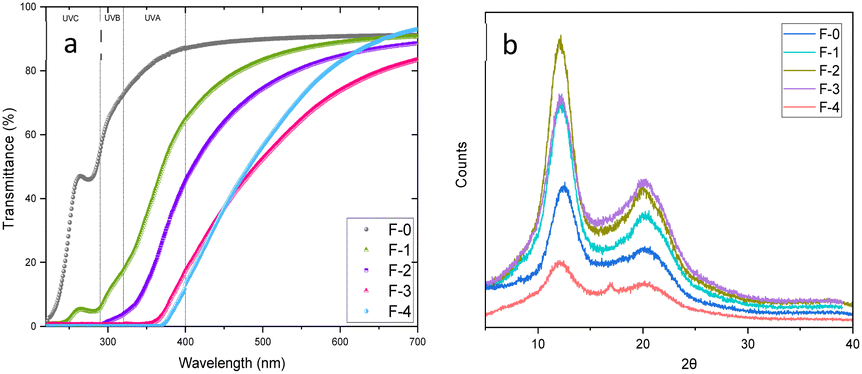 | ||
| Fig. 2 (a) Optical properties: light transmittance through the films (b) XRD diffractograms. | ||
Lignin-containing films also exhibit low haze but reduced light transmission, particularly in the UV spectrum. Specifically, films F-1, F-2, F-3 and F-4 almost completely block the transmittance of light in the UVC region (200–280 nm). For the F-1 film, with only 4% lignin, transmittance decreased to 5–15% for the UVB and 15–65% for the UVA range. At higher wavelengths, light transmittance decreases further as the lignin concentration increases. As the lignin concentration increases, light transmittance further decreases, and at the highest lignin concentrations shows no light transmittance in the UVC and UVB regions, and most of the UVA region (320–370 nm). This confirms that lignin acts as an effective UV-blocking agent, as it is rich in UV-absorbing aromatic functional groups, such as phenols (Fig. S1†).37,38
Sigmacell cellulose exhibits three diffraction peaks at 15°, 17°, 22° and 34° attributed to the (1 −1 0), (1 1 0), and (0 2 0) crystal planes of the cellulose Iβ allomorph (Fig. S7†). On the other hand, the XRD patterns of the films exhibited diffraction peaks at 2θ values of 12°, 20° and 22°, corresponding to the crystal planes (1 −1 0), (1 1 0) and (0 2 0) of cellulose II.
Analysis of the XRD results with Ritveld refinement confirmed that the regenerated cellulose consisted of cellulose II and amorphous cellulose. The dissolution of the cellulose in the ionic liquid along with the use of moderately elevated temperature,39 the presence of DMSO40 for the dissolution of the polymer and the use of water as the coagulation antisolvent facilitated the transition from cellulose Iβ to cellulose II. Rietveld refinement calculated that the crystallinity of the F-0 film is the highest at 68% while for the lignin-containing films the crystallinity ranged from 51–59% due to the presence of the amorphous lignin.
The same crystallinity (68%) was also reported by El-Wakil37 for the cellulose obtained from dissolving Egyptian bleached bagasse pulp fibres in N-methylmorpholine N-oxide (NMMO) and regeneration with water. In the study of Wang,41 regenerated cellulose from 1-ethyl-3-methylimidazolium diethylphosphate ([C2C1im][DEP]) with water exhibited a crystallinity index of 59.67%. On the other hand, Wang30 dissolved dissolving wood pulp in the superbase-derived ionic liquid 1,8-diazabicyclo[5.4.0]undec-7-enium ethoxyacetate and the crystallinity index of the regenerated cellulose ranged from 36.2% to 38.8% depending on the antisolvent used. The discrepancy between the reported values of the crystallinity of regenerated cellulose can be attributed to various influencing factors. For instance, Rana42 demonstrated that the duration of cellulose treatment with an ionic liquid significantly impacts crystallinity. Additionally, the crystallinity of the native cellulose also affects the final result.43 Therefore, while there are many variables to be considered, the transformation of cellulose I to cellulose II is confirmed.
The main results from the thermogravimetric analysis (TGA) of the samples are summarised in Table 2. All samples exhibited a sharp mass drop within the temperature range 200 and 400 °C (Fig. S8†). For the films F-0, F-1 and F-2 a single step drop was observed at around 355 °C, while for the films F-3 and F-4, it appears that there is another step at 305 °C. This is attributed to the presence of the lignin, as it constitutes the 20% or 29% of the film respectively.
There are some differences observed between the TGA curves of the films and the lignin and cellulose. The degradation temperature of the cellulose is found to be 350.3 °C, which is almost the same as the Td of the films. However, the mass residue after the thermal decomposition of the polymer is less than 10% of the initial mass, which is significantly lower than the remaining mass of F-0 film. This is attributed to the different crystallinity of the two cellulosic samples. The regenerated cellulose (F-0) is constituted manly of the cellulose II allomorph, which is thermodynamically more stable than cellulose Iβ.44
A clear trend exists between the lignin concentration in the films and the mass residue at 700 °C. The remaining ash from the thermal treatment of the lignin at the same temperature is approximately 36%, while for the regenerated cellulose, as discussed above, it is significantly lower. Therefore, as the concentration of the lignin increases, so does the remaining mass.
The FTIR spectra of the films exhibited the characteristic peaks of cellulose II (Fig. S9†). For films F-3 and F-4 two more small peaks were present, one at 1511 cm−1 and one at 1269 cm−1 both of which are attributed to C–C stretch (in-ring) of aromatic compounds of lignin. The lack of peaks at around 1560 cm−1, 1376 cm−1 and 1178 cm−1 confirms the absence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch, C–H bending and CO stretch,45 and as a result it confirms that the ionic liquid has been washed out of the films. Similarly, no peaks characteristic of DMSO are present in the films. The complete wash-out of the ionic liquid from the films was also confirmed via elemental analysis, with the absence of N in the films (Fig. S9†).
N stretch, C–H bending and CO stretch,45 and as a result it confirms that the ionic liquid has been washed out of the films. Similarly, no peaks characteristic of DMSO are present in the films. The complete wash-out of the ionic liquid from the films was also confirmed via elemental analysis, with the absence of N in the films (Fig. S9†).
The swelling behaviour of the films in a range of solvents was examined to assess the interaction with different chemical environments (Fig. S10†). The solvents tested comprised of four aqueous solutions, namely water, a 5 wt% acetic acid solution to simulate vinegar, a 1 wt% NaOH solution to evaluate the effect of a strongly basic medium, and brine with a salt concentration of 3.5%, commonly found in food.
In all the aqueous solutions, the films exhibited significant swelling. When immersed in water or the acetic acid solution, the films swelled over 200%. The swelling was even more notable in brine and NaOH, where the mass of the films more than doubled. For all the samples and all the solvents, the swelling occurred within the first 30 minutes of immersion. After that time, the mass remained stable for the duration of the test. Additionally, it was observed that the films with the lowest concentration of lignin exhibited the highest swelling as lignin is less hydrophilic than cellulose, which restricts water absorption and swelling.
Furthermore, three organic solvents were also examined: ethanol, hexane (a non-polar solvent mimicking oil-like behaviour), and dichloromethane (a polar aprotic solvent with poor plastic compatibility). For none of those solvents did the mass of the films change significantly. This suggests that the films do not interact substantially with these organic solvents, highlighting their hydrophilic nature and the limited affinity for non-polar or plastic-incompatible environments.
The films with varying lignin content were immersed in the solutions for 72 h, before recovery and drying to assess the stability in each solvent. The lignocellulose films showed excellent stability for the solvents except for the NaOH solution and the brine solution (Table S1†). In NaOH(aq), rapid discolouration of the films was observed (Fig. S12†). This is likely due to the leaching of the lignin, as supported by the significant mass loss roughly corresponding to the mass ratio of lignin in the films. This result aligns with expectations, as an alkali treatment is the industrial standard for the delignification process of wood. A weaker base such as sodium hydrogen carbonate (baking soda) was unable to bleach the lignin-containing films. For films soaked in brine, a mass increase was observed, likely due to the trapped salt crystals on the films.
Recycling of ionic liquid
After the regeneration of the F-2 film, the coagulation solution was collected for recovery of the ionic liquid and subsequent reuse for the preparation of new films with the same concentration of polymers. The solution primarily consisted of water, but also contained ionic liquid, DMSO, and small concentrations of the two polymers, resulting in a light brown solution. Following the purification step, the recovered ionic liquid was colourless and transparent, similar to the freshly synthesised ionic liquid.After each recycling cycle, no reduction in the ability of the ionic liquid to dissolve the two polymers was observed. Additionally, the yield of the conversion of the polymers into films was consistently high for all the cycles studied and ranged between 93–96% (Table 3). The yield of recycling of the ionic liquid was around 90% for each of the first two cycles. Thereafter, a decrease of the recycling yield was observed, which was found to be 86.4% for the third cycle and almost 84% for the fourth cycle. Although the yield lies within the reported values for recycling the ionic liquid via vacuum evaporation of the antisolvent,46 it is considered to be very low for industrial use as the cumulative yield of recovery after the 5th cycle is only 60.4%. The low solvent recovery yield is attributed primarily to the selected isolation method which may not be the optimal for this system, and mechanical losses inherent in the multistep process. The solvent losses may occur during the collection of the coagulation bath, the purification step as well as the recovery phase. Therefore, optimisation would be required as part of the scaling-up process.
| Cycle | Polymer conversion (%) | Recycling yield (%) | Cumulative recycling yield (%) | T onset (°C) | T d (°C) | Mass residue (%) |
|---|---|---|---|---|---|---|
| 0 | — | — | — | 207 | 243 | 0.8 |
| 1st | 96.0 ± 4 | 89.3 ± 6 | 89.3 | 219 | 248 | 0 |
| 2nd | 92.4 ± 2 | 93.3 ± 5 | 83.3 | 213 | 244 | 0.6 |
| 3rd | 94.9 ± 0 | 86.4 ± 11 | 72.0 | 221 | 247 | 0.3 |
| 4th | 95.8 ± 2 | 83.9 ± 1 | 60.4 | 209 | 245 | 2.5 |
Spectroscopically, it was confirmed that the ionic liquid remained structurally stable throughout the recycling process. The 1H NMR and 13C NMR spectra of the freshly synthesised ionic liquid (IL-0) and the recycled ionic liquids (IL-1, IL-2, IL-3, IL-4) revealed that the acid-to-base ratio remained the same, and that no major impurities nor either of the polymers accumulated in the ionic liquid (Fig. S13 and S14†). No significant differences in the chemical shift peaks were observed, with the exception of the peak of the proton in the 2-position of the imidazolium ring, attributed to the different concentration of the ionic liquid in the NMR solvent47 and not to structural changes of the sample occurring after recycling. The FTIR analysis verified that the structure of the ionic liquid remained stable through the recycling process (Fig. S15†).
These results suggest that the recycled ionic liquid could be reused for the fabrication of cellulose films for at least four cycles of recycling, indicating that recycling could be the key component for an economically and environmentally friendly process. The process developed in this study is depicted in Fig. 3.
 | ||
| Fig. 3 Simplified process flow diagram of the process developed in this study for the preparation of the lignocellulosic films and the recycling of the ionic liquid. | ||
To assess the thermal and structural stability of the ionic liquid throughout the recycling cycles, the TGA, 1H NMR, 13C NMR and FTIR was measured for all samples, details of which are provided in the ESI.†
TGA analysis confirmed that the thermal properties of the ionic liquid remained practically unchanged throughout the recycling process. For all samples measured, the decomposition temperature ranged between 243 and 248 °C, confirming the stability of the ionic liquid (Fig. S16†).
Comparison with other bioplastics
The growing environmental impact of petroleum-based plastics has become a critical concern. The official data are alarming: the annual production of plastic in Europe alone is nearly 26 million tonnes48 while, according to calculations, around 36% of the plastic produced worldwide is used in packaging.49A potential solution could be the adoption of more sustainable plastics, such as the use of bioplastics, which are biodegradable and compostable. However, bioplastics currently represent a mere 0.5% of total plastic production1 – a figure that must increase significantly to make a meaningful difference. If current trends persist, research shows that the rate of plastic waste production could triple, reaching 155 to 265 Mt per year.50 This increase will only worsen the already critical issue of microplastics, which harm ecosystems and pose risks to human health.51
The most commonly used bioplastic is cellophane, which was discovered about 100 years ago11 and is primarily composed of cellulose.50 Despite its potential as a sustainable alternative, cellophane has several significant drawbacks related to its production and use. One major issue is the harmful chemicals involved in its manufacturing process – a mixture of strong alkali, carbon disulfide, a bath of sulfuric acid and sodium sulfate.11,52 Some of those chemicals are considered to be hazardous,53 with risks including flammability, acute toxicity, reproductive toxicity, specific organ toxicity from repeated exposure, and skin corrosion, while harmful by-products may also occur, posing additional risks to human health and the environment. Another disadvantage is that the composition of cellophane does not consist only of cellulose, but always contains additives and plasticisers at a ratio of approximately 10%, used to enhance properties such as elasticity and stability. However, these additives make recycling more challenging, reducing the overall sustainability of the material. Additionally, cellophane films are often coated with other materials to further enhance their properties. These coatings frequently include non-biodegradable plastics, which severely compromise the biodegradability of the final product.54
Even when considering the second strongest alternative bioplastic – polylactic acid (PLA) – there are two significant drawbacks that hinder its broader production and adoption. First, while PLA's ability to be produced biologically is an important factor in its sustainability, its production relies heavily on large quantities of edible crops such as corn and sugarcane, competing with the global demand for food. Second, despite being derived from biological sources, PLA is not truly biodegradable under natural conditions. It requires industrial composting facilities to break down effectively – facilities that are often unavailable. Without access to such infrastructure, PLA degrades very slowly in the environment, undermining its sustainability benefits.10
Therefore, the process and the films presented in this work constitute a highly promising approach to addressing environmental concerns associated with plastics. The process involves the use of 1-ethyl-3-methylimidazolium acetate an ionic liquid that is widely known and used, commercially available, and can also be synthesised from relatively safe raw materials. This ionic liquid can lead to skin irritation (H315) and an allergic skin reaction (H317) but no other hazards have been reported, while like all ionic liquids, it has negligible vapour pressure and high stability, minimising its environmental impact. A key advantage of this ionic liquid is its recyclability, which allows it to be reused multiple times, significantly reducing both the environmental footprint and the overall cost of the process. Furthermore, the final product is composed solely of cellulose and lignin; two biocompatible, inherently non-toxic, biodegradable, and compostable polymers of natural origin. Therefore, their use would require nothing more than their disposal in the organic waste bin, making the end-of-life phase simple and sustainable.54,55
In terms of performance, the non-lignin containing cellulose films exhibit high transparency with low haze. The addition of lignin results in darker films that allow less light to pass through, with the UV protection increasing as lignin concentration increases, without significantly affecting the haze. Complete UV protection is achieved when the films are consisted of at least 29% lignin, although some UV protection is provided even when the concentration of lignin is as low as 4%. The tensile strength of the lignocellulosic films measured ranged between 65 and 93 MPa with an elongation at break of around 3%. Furthermore, cellulose films have a WVP of approximately 8 × 10−6 g per m per day per Pa, which increased to 10.5–12.3 × 10−6 g per m per day per Pa when lignin is added. Finally, cellulose is a hydrophilic polymer. Therefore, the cellulose films exhibited water contact angle of around 46° and with the addition of lignin the value raised to 62°–65°.
A comparison of the properties of the lignocellulosic films prepared in this study with the data available in the literature regarding other biogenic films and conventional films reveals that these films exhibit excellent performance, that is comparable and often surpassing available alternatives (Table S2†). For instance, the tensile strength of lignin/cellulose films is higher than PLA's (TS = 20–69.1 MPa, E = 2.24 GPa) or chitosan's (TS = 6.64–64.3 MPa, E = 0.445–3.0 GPa) and significantly higher than starch (TS = 3 MPa). Biopolymers exhibit significant variations in their properties. Many factors can affect the final properties of films, such as the polymers molecular weight, or its purity or the fabrication method employed.
Non-biodegradable films from HDPE and LDPE also exhibit lower tensile strength of around 23–30 and 10–40 MPa respectively. Interestingly PVA, PVC, PBAT, PP and PET also present lower tensile strength and Young's modulus. Additionally, the tested films exhibit good water vapour permeability, comparable to PLA (3.45 × 10−6 g d−1 m−1 Pa−1) and chitosan films (4.74 ± 0.05 × 10−6 g d−1 m−1 Pa−1). In contrast, PVA and PCL films are slightly less permeable (60.4 × 10−6 g m−1 d−1 Pa−1 and 1.54 ± 0.04 × 10−6 g m−1 d−1 Pa−1 respectively), while cellophane films are reported to be slightly more permeable (14 × 10−6 g d−1 m−1 Pa−1). PET remains significantly less permeable (9.5 ± 0.8 × 10−8 g d−1 m−1 Pa−1).
The primary drawback of the process lies in its energy-intensive stage, the isolation of the ionic liquid from the washings, as substantial amount of water is required to fully remove the solvent from the film. However, this challenge is amenable to optimisation during scale-up, which fell out the scope of the current study. Importantly, all solvents and antisolvents used in the production of lignocellulosic films can be recycled and reused without compromising their effectiveness, further enhancing the sustainability of the process. The resulting films exhibit excellent properties comparable to those of other bioplastics, yet the manufacturing process is safer and greener. Additionally, the final product is free of undesirable components, aligning it closely with the principles of sustainability.
Experimental
Materials and methods
:23 w/w, equivalent to a molar ratio of 0.6:0.4. For each film, in 1.3 g of the aforementioned mixture, 0.12 g of cellulose and 0, 0.005, 0.01, 0.03 or 0.05 g of lignin were added to form five different dope solutions (DS). The samples left at 70 °C overnight, to ensure complete polymer dissolution.
The resulting dope solutions were casted into films using a 250 μm casting knife and an Elcometer 4340 Automatic film applicator. Each film was promptly immersed into a water coagulation bath to regenerate the cellulose. After 15 minutes, the film was transferred to a fresh coagulation bath. This process was repeated two more times to thoroughly remove the solvent. The films were secured on a glass surface and left to air dry at 22 ± 2 °C for 24 hours.
The collected water from the coagulation baths was purified via overnight stirring with activated charcoal, followed by filtration and evaporation to retrieve the ionic liquid.
Characterisation of lignin
:1 v/v ratio and transferred in a Shigemi NMR microtube. 50 μL of an internal standard and a relaxation reagent solution were added (20.5 mg mL−1 of cyclohexanol and 5.6 mg mL−1 of chromium(III) acetylacetonate solution in anhydrous pyridine/deuterated chloroform respectively). Finally, 50 μL of chromium(III) 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) were added and the sample was filled up to 300 μL. The NMR experiments were held at 298 K, for 1000 scans and an inverse gated decoupling pulse with a relaxation delay of 11 s was used between 30° pulses.
Characterisation of dope solutions
![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) ). The specific viscosity (ηsp) of each dope solution was calculated with regards to the solvent (ηs) according to eqn (1):
). The specific viscosity (ηsp) of each dope solution was calculated with regards to the solvent (ηs) according to eqn (1): | (1) |
Linear viscoelastic moduli (G′ and G′′) as a function of frequency between 0.1 to 100 Hz at a 0.01 strain (γ) were calculated via oscillatory measurements. Finally, the solvent and the dope solutions were subjected to a rotational stress with increasing shear rate and the viscosity was monitored to assess the rheological profile of the samples.58,59
Characterisation of films
The water vapour permeability (WVP) of the films was measured according to the wet cup method as described in the ASTM D1653 method. More specifically, the permeability cups with a surface area of 10 cm2 were filled with 10 mL of water and the film was used to seal the opening. The test set up was then placed in a conditioning chamber with temperature of 22 ± 2 °C and relative humidity of 50 ± 2%. The evaporation rate was measured by regularly recording the weight of the samples over a period of two days. The samples were measured in triplicate for both analyses.
For the stability test, the films were immersed in the solvents for 72 hours and then were dried on the Schlenk line. Films tested with aquaous NaOH or brine were thoroughly rinsed with distilled water to remove any residual salts. Once dried, the films were exposed to open air to reach equilibrium, and their final masses were recorded. Infrared spectroscopy (IR) confirmed the absence of any residual solvent in the films.
Conclusions
Here we report the preparation of lignocellulosic films regenerated from a 1-ethyl-3-methylimidazolium acetate and DMSO mixtures with excellent mechanical and optical properties. The rheological measurements of the dope solutions revealed that the lignin is a thickening agent, as is the cellulose, and thus, increasing their concentration would make the dissolution process and the handling of the dope solutions challenging.The cellulosic films prepared are very robust, exhibiting a high tensile strength and Young's modulus. The addition of lignin did not drastically alter the mechanical properties, but it did improve the optical and barrier properties. The addition of even the smallest amount of lignin blocks the UV light to pass through the film. Furthermore, the wettability of the films was improved with the addition of the second polymer while the swelling ratio when immersed in various aqueous solvents was decreased.
The use of an ionic liquid as the medium for the dissolution of cellulose, ensures the recyclability of the process. It was confirmed that the entire process could be repeated five times, without and thermal or structural impact on the [C2C1im][OAc] which retained the capability to dissolve cellulose at high concentrations.
Future research should focus on optimising ionic liquid recycling to enhance the sustainability of the production process. This method constitutes a proof of concept that the lignocellulosic films exhibit excellent properties and that the ionic liquid can be recovered and reused multiple times. As the global effort to combat plastic pollution intensifies, the use of lignocellulosic films offers a viable and environmentally responsible solution. This process represents a proposal grounded in sustainable practices and holds great potential for reducing the environmental impact of plastics.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, writing – review & editing: A. R. N. P., S. C., D. F. O., N. M. D., K. Y. L., T. W, Funding acquisition, project administration, supervision: N. M. D., K. Y. L, T. W., investigation: A. R. N. P., S. C., methodology, formal analysis, visualization, validation, writing – original draft: A. R. N. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank TNO for providing the lignin. We also greatly acknowledge the funding provided by the Imperial College President's Excellence Fund for Frontier Research for funding A. R. N. P. and D. F. O.Notes and references
- Plastics Europe, Plastics - the fastFacts 2023, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2023/.
- Directive (EU) 2019/904 of the European Parliament and of the Council of 5 June 2019 on the reduction of the impact of certain plastic products on the environment.
- Directive 94/62/EC of 20 December 1994 on packaging and packaging waste.
- C. H. Bullock, N. Thorball, C. Somlai, J. Gallagher and Environmental Protection Agency, Packaging waste statistics, producer motivations and consumer behaviour, Environmental Protection Agency, 2021, Report No. 426 Search PubMed.
- O. Horodytska, F. J. Valdés and A. Fullana, Waste Manage., 2018, 77, 413–425 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- P. Ghisellini, C. Cialani and S. Ulgiati, J. Cleaner Prod., 2016, 114, 11–32 Search PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- Q. Qin, S. Zeng, G. Duan, Y. Liu, X. Han, R. Yu, Y. Huang, C. Zhang, J. Han and S. Jiang, Chem. Soc. Rev., 2024, 53, 9306–9343 CAS.
- R. Mori, RSC Sustainability, 2023, 1, 179–212 Search PubMed.
- G. C. Inskeep and P. V. Horn, Ind. Eng. Chem., 1952, 44, 2511–2524 Search PubMed.
- H.-P. Fink, P. Weigel, H. Purz and J. Ganster, Prog. Polym. Sci., 2001, 26, 1473–1524 CAS.
- B.-x. Zhang, J.-i. Azuma and H. Uyama, RSC Adv., 2015, 5, 2900–2907 Search PubMed.
- C. S. Lovell, A. Walker, R. A. Damion, A. Radhi, S. F. Tanner, T. Budtova and M. E. Ries, Biomacromolecules, 2010, 11, 2927–2935 Search PubMed.
- K. M. Gupta and J. Jiang, Chem. Eng. Sci., 2015, 121, 180–189 CAS.
- A. Alzagameem, S. E. Klein, M. Bergs, X. T. Do, I. Korte, S. Dohlen, C. Hüwe, J. Kreyenschmidt, B. Kamm and M. Larkins, Polymers, 2019, 11, 670 CrossRef CAS PubMed.
- H.-M. Wang, T.-Q. Yuan, G.-Y. Song and R.-C. Sun, Green Chem., 2021, 23, 3790–3817 CAS.
- J. Ruwoldt, F. H. Blindheim and G. Chinga-Carrasco, RSC Adv., 2023, 13, 12529–12553 Search PubMed.
- Y. He, H.-C. Ye, T.-T. You and F. Xu, Food Hydrocolloids, 2023, 137, 108355 CAS.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325–327 CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- H. Cruz, M. Fanselow, J. D. Holbrey and K. R. Seddon, Chem. Commun., 2012, 48, 5620–5622 RSC.
- Y. Lv, J. Wu, J. Zhang, Y. Niu, C.-Y. Liu, J. He and J. Zhang, Polymer, 2012, 53, 2524–2531 CrossRef CAS.
- Y. Zhao, X. Liu, J. Wang and S. Zhang, J Phys. Chem. B, 2013, 117, 9042–9049 CrossRef CAS PubMed.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- R.-L. Wu, X.-L. Wang, F. Li, H.-Z. Li and Y.-Z. Wang, Bioresour. Technol., 2009, 100, 2569–2574 CrossRef CAS PubMed.
- W. E. Hart, J. B. Harper and L. Aldous, Green Chem., 2015, 17, 214–218 CAS.
- K. S. Lefroy, B. S. Murray and M. E. Ries, J. Phys. Chem. B, 2021, 125, 8205–8218 CAS.
- X. Chen, Y. Zhang, L. Cheng and H. Wang, J. Polym. Environ., 2009, 17, 273–279 CAS.
- X. Wang, W. Zheng, Z. Guo, H. Nawaz, T. You, X. Li and F. Xu, Green Chem., 2023, 25, 1597–1610 CAS.
- N. Asim, M. Badiei and M. Mohammad, Emergent Mater., 2021, 1–16 Search PubMed.
- E. González Carmona, I. Schlapp-Hackl, S. Jääskeläinen, M. Järvinen, K. Nieminen, D. Sawada, M. Hummel and H. Sixta, Cellulose, 2023, 30, 11633–11648 Search PubMed.
- I. S. Makarov, L. K. Golova, G. N. Bondarenko, T. S. Anokhina, E. S. Dmitrieva, I. S. Levin, V. E. Makhatova, N. Z. Galimova and G. K. Shambilova, Membranes, 2022, 12, 297 CAS.
- H. Su, B. Wang, Z. Sun, S. Wang, X. Feng, Z. Mao and X. Sui, Carbohydr. Polym., 2022, 277, 118878 CAS.
- F. Yang, L. Xu, G. Dai, L. Luo, K. Yang, C. Huang, D. Tian and F. Shen, Molecules, 2022, 27, 1637 CAS.
- B. A. Morris, The Science and Technology of Flexible Packaging: Multilayer Films from Resin and Process to End Use, William Andrew, 2022 Search PubMed.
- H. Sadeghifar, R. Venditti, J. Jur, R. E. Gorga and J. J. Pawlak, ACS Sustain. Chem. Eng., 2017, 5, 625–631 CAS.
- B. Cui, L. Liu, S. Li, W. Wang, L. Tan, C. Liu and W. Wang, Mater. Chem. Front., 2023, 7, 897–905 CAS.
- J.-M. Andanson, E. Bordes, J. Devémy, F. Leroux, A. A. Pádua and M. F. C. Gomes, Green Chem., 2014, 16, 2528–2538 CAS.
- E. N. Durmaz and P. Z. Çulfaz-Emecen, Chem. Eng. Sci., 2018, 178, 93–103 CAS.
- N. El-Wakil, M. Taha, R. Abouzeid and A. Dufresne, Biomass Convers. Biorefin., 2024, 14, 5399–5410 CAS.
- M. M. Rana and H. De la Hoz Siegler, J. Mater. Res., 2023, 38, 328–336 CAS.
- G. Jiang, W. Huang, L. Li, X. Wang, F. Pang, Y. Zhang and H. Wang, Carbohydr. Polym., 2012, 87, 2012–2018 CAS.
- L. Szabó, R. Milotskyi, G. Sharma and K. Takahashi, Green Chem., 2023, 25, 5338–5389 Search PubMed.
- M. L. Williams, S. P. Holahan, M. E. McCorkill, J. S. Dickmann and E. Kiran, Thermochim. Acta, 2018, 669, 126–139 Search PubMed.
- J. Zhang, D. Zou, S. Singh and G. Cheng, Sustainable Energy Fuels, 2021, 5, 1655–1667 RSC.
- M. X. Du, L. X. Han, S. R. Wang, K. J. Xu, W. R. Zhu, X. Qiao and C. Y. Liu, ChemPhysChem, 2023, 24, e202300292 CrossRef CAS PubMed.
- https://environment.ec.europa.eu/topics/plastics_en, accessed 7 May 2024.
- U. E. programme, https://www.unep.org/interactives/beat-plastic-pollution/, 2024, accessed 15/09/2024.
- P. Rai, S. Mehrotra, S. Priya, E. Gnansounou and S. K. Sharma, Bioresour. Technol., 2021, 325, 124739 CrossRef CAS PubMed.
- M. Sendra, P. Pereiro, A. Figueras and B. Novoa, J. Hazard. Mater., 2021, 409, 124975 CrossRef CAS PubMed.
- B. Morris, The Science and Technology of Flexible Packaging, 2017, 69–119 Search PubMed.
- M. Davidson and M. Feinleib, Am. Heart J., 1972, 83, 100–114 CrossRef CAS PubMed.
- P. Benyathiar, S. Selke and R. Auras, J. Polym. Environ., 2015, 23, 449–458 CrossRef CAS.
- S. RameshKumar, P. Shaiju and K. E. O'Connor, Curr. Opin. Green Sustainable Chem., 2020, 21, 75–81 CrossRef CAS.
- S. Koutsoukos, J. Becker, A. Dobre, Z. Fan, F. Othman, F. Philippi, G. J. Smith and T. Welton, Nat. Rev. Methods Primers, 2022, 2, 49 CrossRef CAS.
- A. Brandt, L. Chen, B. E. van Dongen, T. Welton and J. P. Hallett, Green Chem., 2015, 17, 5019–5034 RSC.
- A. Simões, M. Miranda, C. Cardoso, F. Veiga and C. Vitorino, Pharmaceutics, 2020, 12, 820 CrossRef PubMed.
- C. E. Owens, J. Du and P. B. Sánchez, Biomacromolecules, 2022, 23, 1958–1969 Search PubMed.
- İ. Uzun, J. Polym. Res., 2023, 30, 394 CrossRef.
- A. D. French, Cellulose, 2014, 21, 885–896 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00197h |
| This journal is © The Royal Society of Chemistry 2025 |