
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in p-type polymeric electrode materials towards high-voltage 4.0 V-class organic lithium-ion batteries
Febri
Baskoro†
 *ab,
Santosh U.
Sharma†
a,
Andre Lammiduk
Lubis
a and
Hung-Ju
Yen
*ac
*ab,
Santosh U.
Sharma†
a,
Andre Lammiduk
Lubis
a and
Hung-Ju
Yen
*ac
aInstitute of Chemistry, Academia Sinica, Taipei 115, Taiwan. E-mail: febri_baskoro@yahoo.co.id; hjyen@gate.sinica.edu.tw
bMaterial Science and Engineering Research Group, Faculty of Mechanical and Aerospace Engineering, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia
cSustainable Chemical Science and Technology Program, Taiwan International Graduate Program (TIGP), Academia Sinica and National Taiwan University, Taipei 11529, Taiwan
First published on 12th November 2024
Abstract
Lithium-ion batteries stand at the forefront of energy storage technologies, facilitating the transition towards sustainable and electrified systems. However, to meet the increasing demands for energy density, safety, and longevity, the development of high-performance electrode materials is paramount. Although inorganic materials have been dominant in the current lithium-ion battery cathodes, the widely utilized inorganic cathode materials suffer from drawbacks, such as limited capacity, high energy consumption during their production, safety hazards associated with toxic metals (Li, Co, Mn, and Ni), and high raw material costs, due to their limited or localized resource distributions. Alternatively, polymeric materials have emerged as promising candidates to replace conventional inorganic materials due to their advantages such as abundance, environmentally friendly resources, structural diversity, ease of functionalization, fabrication, recycling, high capacity and rate capability, and excellent flexibility. This review article explores the strategic design principles underlying the synthesis and optimization of p-type polymeric electrode materials for next-generation 4.0 V-class batteries. Through a comprehensive analysis of recent advancements, morphology control, and interface engineering, this review elucidates the key strategies employed to achieve high-energy-density electrodes. Additionally, this review discusses the fundamental mechanisms governing the electrochemical performances of p-type polymeric electrodes and highlights the emerging trends and future directions in this field. By integrating insights from materials science, electrochemistry, and engineering, this review provides a roadmap for the rational design and development of p-type polymeric electrode materials towards the realization of high-performance 4.0 V-class lithium-ion batteries.
 Febri Baskoro | Dr Febri Baskoro obtained his PhD in Sustainable Chemical Science and Technology from National Yang Ming Chiao Tung University, Taiwan, in 2021, a joint PhD program between the Institute of Chemistry, Academia Sinica, and National Yang Ming Chiao Tung University, Taiwan. He earned his BS from Sebelas Maret University, Indonesia, and MS from Chang Gung University, Taiwan. Currently, he works as an Academia Sinica Postdoctoral Research Fellow in the Institute of Chemistry, Academia Sinica, Taiwan, under the guidance of Dr Hung-Ju Yen. His research interests include the development of novel functional electrode materials including MOFs, nanographenes, organic molecules, and polymers for high-performance and sustainable batteries. |
 Santosh U. Sharma | Dr Santosh U. Sharma completed his PhD in polymeric material chemistry for energy storage applications, such as lithium-ion batteries and supercapacitors, under the guidance of Prof. Jyh-Tsung Lee at National Sun Yat-sen University, Taiwan. He holds an MS in Industrial Polymer Chemistry and a BS in Chemistry, both from the University of Mumbai, India. Currently, he is a Postdoctoral Researcher at the Institute of Chemistry, Academia Sinica, Taiwan, working in Dr Hung-Ju Yen's group. His research interest includes the development and synthesis of redox-active molecules, including small molecules, polymers, and porous materials, for batteries and supercapacitors. |
 Andre Lammiduk Lubis | Andre Lammiduk Lubis obtained his Master's Degree in Chemistry from National Chung Hsing University, Taiwan, in 2021 under the supervision Prof. Kuan-Jiuh Lin. He was a Research Assistant in Dr Hung-Ju Yen's group in the Institute of Chemistry, Academia Sinica, Taiwan, in 2021–2024. His works mainly focus on the testing of polymer-based electrode materials for lithium-ion batteries. |
 Hung-Ju Yen | Dr Hung-Ju Yen is an Assistant Research Fellow in the Institute of Chemistry at Academia Sinica. He earned his BS (2006) and MS (2007) from National Chi Nan University and then completed his PhD with the 1st prize among all PhD graduates from National Taiwan University (NTU) in 2011. He joined Los Alamos National Laboratory as a J. Robert Oppenheimer Fellow (2013–2017) after his postdoctoral training at NTU (2011, 2012–2013). Dr Yen's main research interest lies in the organic synthesis of functional nanographenes and electroactive polymers for optoelectronic and energy applications. |
1. Introduction
In today's fast-paced and interconnected world, lithium-ion batteries (LIBs) have emerged as the linchpin of modern society, powering a vast array of devices that have become essential to our daily lives. From smartphones and laptops to electric vehicles (EVs) and renewable energy storage grids, LIBs serve as the backbone of our digital economy and the catalyst for the transition towards a sustainable energy future.1–3 As the world's dependency on LIBs continues to deepen, driven by advancements in technology, evolving consumer preferences, and global efforts to combat climate change, it becomes increasingly imperative to understand their significance, challenges, and prospects. However, the reliance on LIBs also presents challenges, including the need for improved safety, environmental impact of raw material extraction, and development of effective recycling strategies to manage end-of-life batteries.4 These challenges must be addressed to ensure the sustainable growth of LIB technology. In this regards, innovations in battery chemistry, design, and manufacturing processes are expected to further improve the energy density, safety, and lifecycle of LIBs, contributing to their continued importance in a low-carbon future.5Generally, a conventional LIB cell consists of a positive electrode (cathode), negative electrode (anode), and non-aqueous electrolyte system as well as a separator to prevent physical contact between these two electrodes (Fig. 1a). In principle, when a battery is being charged, the Li+ ions move from the cathode to anode through the electrolyte, whereas during discharge, the Li+ ions will move back from the anode to the cathode, releasing electrical current. As cathode materials, typical Li-intercalated materials, such as LiCoO2 (a lithium metal oxide with layered structure), have been widely used as lithium-ion sources. Meanwhile, graphite has been widely utilized as the anode material to store Li+ in its layers via the intercalation process. Notably, both the cathode and anode materials should be able to reversibly insert and remove Li+ from their respective structures.7 Furthermore, given that the cell voltage is established by the energy difference between the anode and cathode, the cathode energy should lie as low as possible, and the anode must lie as high as possible. This implies that cathode materials would require the stabilization of their higher oxidative states with a lower-lying energy band, while the anode materials would require the stabilization of their lower oxidative states with a higher-lying energy band.6,8,9 The electrolyte is the third component of LIBs, which serves as a medium to transport the Li+ ions involved in charging/discharging process of the LIBs.10 Although the role of the electrolytes is often considered trivial, the choice of electrolyte system is crucial depending on its compatibility with both electrodes.11 Besides the importance and technological advancement in the LIB components, the development of cathode materials has attracted research interest given that they play an important role in defining the working potential of LIB cells, thus having a significant impact on the energy density of the battery.
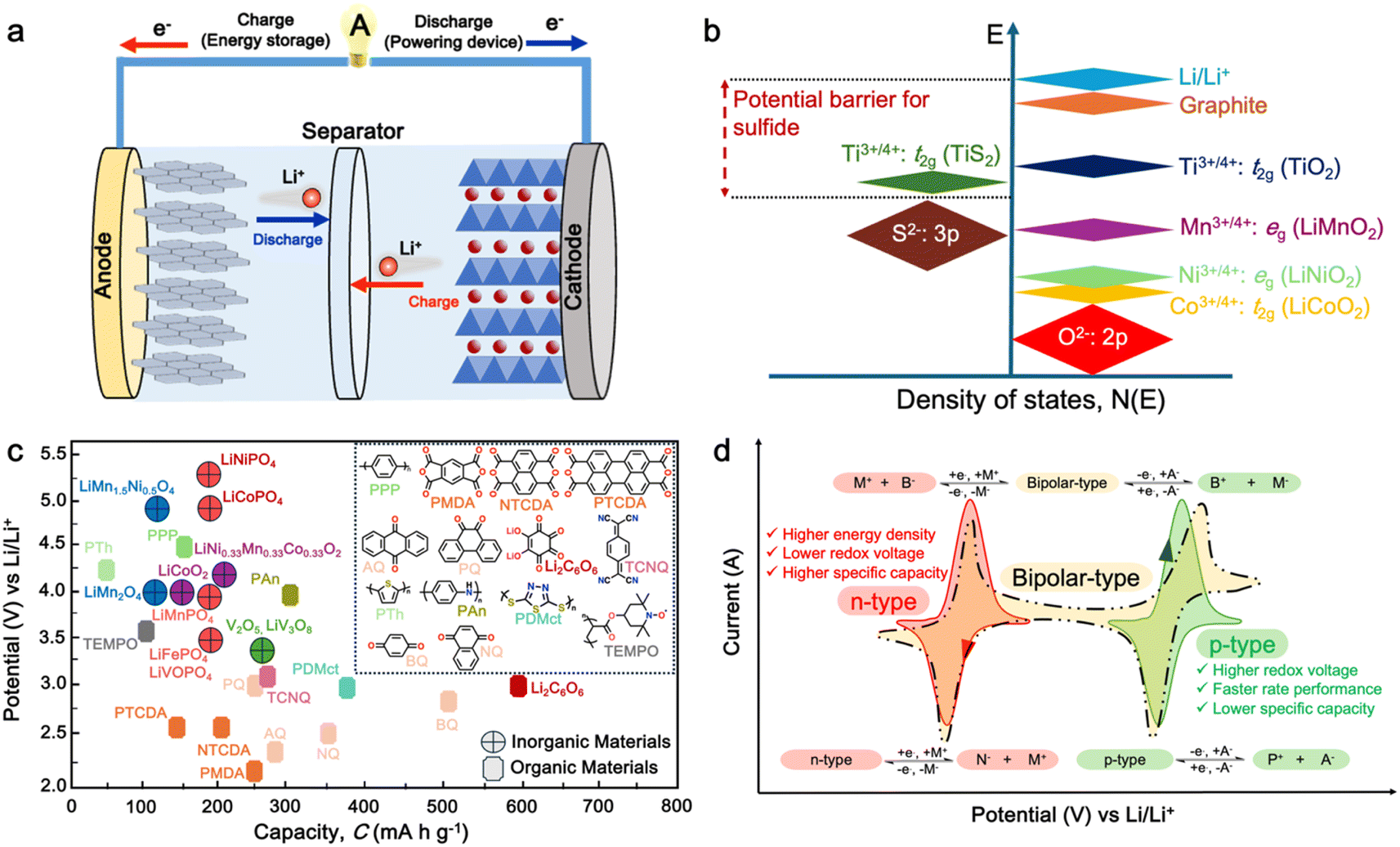 | ||
| Fig. 1 (a) Schematic of a full cell Li-ion battery. (b) Positions of the redox energies relative to the top of the anion: p bands. The top of the S2−: 3p band lying at a higher energy limits the cell voltage to <2.5 V with a sulfide cathode. In contrast, the top of the O2−: 2p band lying at a lower energy enables access to lower-lying energy bands with higher oxidation states and increases the cell voltage substantially to ∼4 V. Reproduced from ref. 6. (c) Recent development of cathode materials for LIBs. (d) Redox behavior of organic molecules in electrochemical processes. | ||
1.1 Current-status of cathode materials for LIBs
Given that the cathode materials are key in determining the cell voltage and, consequently, its energy density, the high-energy-density batteries require high working potential cathodes, paired with anodes that can provide a high specific capacity.2 In early study of LIB cathodes, Goodenough employed the basic understanding that the top of the S2−: 3p band lies at a higher energy than the top of the O2−: 2p band to design oxide cathodes (Fig. 1b). This means that the access to lower-lying energy bands with higher oxidation states such as Co3+/4+, and hence a higher cell voltage will be limited by the top of the S2−: 3p band, and attempts to lower the cathode redox energy by accessing higher oxidation states in a sulfide will result in the oxidation of the S2− ions to molecular disulfide ions (S2)2−. In contrast, in an oxide, the cathode redox energy can be significantly lowered by accessing lower-lying energy bands such as Co3+/4+, and hence the cell voltage can be increased to as high as 4 V given that the top of the O2−: 2p band lies at a lower energy compared to the top of the S2−: 3p band. This basic idea led to the discovery of oxide cathodes materials such as layered oxide, spinel oxide, and polyanion cathodes.6,8,9In general, the most common cathode materials used in commercial LIBs are based on transition metal oxides (Fig. 1c), such as lithium cobalt oxide (LiCoO2),12 lithium manganese oxide (LiMn2O4),13 lithium nickel manganese cobalt oxide (LiNiMnCoO2 or NMC),14 and lithium iron phosphate (LiFePO4 or LFP).15,16 However, they suffer from gradual capacity fading during cycling, which is attributed to the dissolution of the transition metal ions in the electrolyte, leading to their accumulation on the electrode, and thus resulting in a high resistive solid electrolyte interphase (SEI) layer on the cathode surface.17,18 Furthermore, there are several persisting challenges associated with the development of inorganic-based cathode materials.19–22 Firstly, with the increasing demand for high energy density batteries, there is a limitation in the theoretical capacity and working potential of current inorganic-cathode materials.23 Secondly, the high cost of raw materials due to their scarcity and environmental burden.6 Furthermore, complex and high-temperature synthesis processes are required, thus further increasing the material cost.24 Thirdly, the safety issue associated with unstable cathode materials such as LiCoO2, which can pose thermal runaway risks at high voltages, potentially leading to fires or explosions.25
Alternatively, organic cathode materials have emerged as promising contenders in the pursuit of next-generation energy storage solutions (Fig. 1c). Organic materials composed of elements such as C, O, N, and S have been recognized as a promising alternative to inorganic materials for battery electrodes.26 These materials have several advantages, including abundance, light weight, and environmental benignity.3,27,28 They can be used in various types of batteries, including metal-ion,29 dual-ion,30,31 molecular-ion,32 and anion-shuttle batteries,33 without being limited by the choice of counterions. Additionally, the flexible intermolecular packing of these materials has the potential to provide a higher rate capability than that of inorganic materials.3,27 Most importantly, the physicochemical and electrochemical properties of these materials can be easily tailored through elaborate molecular design.34
In general, electroactive organic materials can be broadly classified into three categories based on their charge states during redox reactions, namely p-type, n-type, and bipolar-type (Fig. 1d). As shown in Fig. 1d, p-type materials undergo oxidation from their neutral state, resulting in a positively charged state, while n-type materials accept an electron and become negatively charged.35 Bipolar-type materials usually contain both p- and n-type moieties, which can utilize both positively and negatively charged states. The specific moiety or redox center in the organic molecule governs the electrochemical response and defines the material type. Comparing the organic-based cathodes, p-type organic electrodes generally have higher redox potentials than that of n-type materials due to their lower electron energy level, thus resulting in higher redox potentials.36 Notably, the type of redox material should be carefully considered in understanding its electrochemical mechanism, given that it is sensitive to the nature of the anion and cation dissolved in the electrolyte. For example, the electrochemical activity of p-type materials is affected by the type of anions in the electrolyte. The anions in the electrolyte can coordinate with the redox centers of p-type materials, influencing their electrochemical behavior.36 Meanwhile, in n-type materials, the positively charged cations (Li+, K+, Na+, etc.) determine their electrochemical behavior such as voltage, specific capacity and cyclability.29,37–39
However, although organic materials offer many benefits as alternative electrode materials in LIBs, they still face some challenges that need to be addressed.27 One major hurdle is their typically low electronic conductivity, which can limit the rate capability and overall performance of the battery.40,41 Stability and cyclability are also critical considerations for organic cathode materials. Some organic compounds may suffer from poor stability over repeated charge–discharge cycles, leading to capacity fading and a reduced battery life.42 Additionally, solubility in the electrolyte poses a challenge for certain organic cathode materials. The dissolution of the active molecules in the electrolyte can result in capacity loss and electrolyte degradation over time.43,44 In brief, both inorganic and organic materials play distinct roles as cathode materials for LIBs. Although inorganic cathodes are currently dominant in commercial applications, ongoing research into organic cathodes promises advancements that can lead to more sustainable and cost-effective energy storage solutions.
1.2 Definition of polymeric electrodes
To improve the stability and cyclability of organic-based electrode materials, several approaches have been explored such as polymerization,45–47 use of high-concentration electrolytes,48,49 nanostructures,50–53 and composite formation with advanced carbon materials.54–57 Among them, the polymerization process is considered a significant strategy that can solve the dissolution problem and improve the material stability of organic-based electrodes in the electrolyte.58–63 Since then, the definition of polymeric electrodes in LIBs have emerged as a type of electrode material made from polymer-based materials. Polymer-based materials are known for their flexibility, conductivity, and chemical stability, making them suitable for use in various electrochemical devices such as batteries and capacitors.28 Furthermore, their design can be easily customized at the molecular level to meet specific energy storage requirements. Benefiting from organic-based cathodes and their excellent physical properties, polymeric electrodes have emerged as of new class of electrode materials to replace traditional inorganic electrodes in LIBs. Furthermore, polymeric materials have been proven to be the active component, which can serve as either the anode, cathode or both electrodes in an LIB setup.58,62–65 Unlike small-organic molecules, which have a major concern in terms of their inherent solubility in organic electrolytes and thus limits their widespread use and development, the higher molecular weight of polymer electrode materials often reduces their solubility in organic electrolytes, making them more promising candidates for the next generation of LIBs.66 One of unique properties of polymer-based electrodes is their flexibility, given that polymeric electrodes can be made into flexible films, thus opening a way for the fabrication of flexible batteries. Additionally, polymeric materials are generally less expensive than inorganic materials, making them a cost-effective alternative.67 Furthermore, polymeric materials are more environmentally friendly than inorganic materials, given that they do not require the mining and processing of heavy metals. This distinction underscores the potential of these polymers as a favorable trend in advancing LIB technologies. However, despite these advantages, polymeric electrodes also face some limitations, which need to be addressed. One major limitation is their lower electrical conductivity compared to inorganic materials. This can result in a lower charge transfer efficiency and reduced battery performance.27 Additionally, polymeric materials generally have lower lithium-ion conductivity than inorganic materials, which can lead to slower ion transport and reduced battery performance.3Earlier studies have shown that conducting polymers (CPs) such as polythiophene (PTh), polypyrrole (PPy), and polyaniline (PANI) can be oxidized by accepting anions, which significantly increases their electrical conductivity, and thus applied in rechargeable batteries.68–71 However, their use is limited due to their low specific capacity and sloppy voltage profiles. Furthermore, nitroxide radical polymers, which can undergo bipolar-type redox reactions, have also been proposed as alternative organic cathode materials.72 These polymers can accept an anion or cation, which allows them to undergo single-electron oxidation (p-type) or single-electron reduction (n-type). However, their low electrical conductivity and high solubility made them impractical.73,74 Since then, many researchers have focused on n-type organic electrode materials75–78 such as benzoquinone,79 anthraquinone,80 terephthalate,81 and imide53,82 derivatives. However, the redox voltage of these n-type organic materials is typically limited to below 3.0 V vs. Li/Li+, which is inferior to that of conventional cathode materials due to their redox mechanism (Fig. 1c).
Alternatively, high redox voltage p-type organic materials such as phenazine,83,84 carbazole,85,86 and phenylamine87,88 derivatives are gaining attention for their application in LIBs. These materials undergo reversible oxidations to form cationic species, which can lead to a faster rate performance. For example, phenoxazine (PXZ)-based cathodes exhibited a specific capacity of 112 mA h g−1 at 1C with an average discharge voltage of 3.7 V vs. Li/Li+ and retained 73% capacity at a high rate of 20C.89 A benzo[b]phenazine-based polymer (p-DPBPZ) displayed an initial discharge specific capacity of 151 mA h g−1, with energy and power densities reaching 537 W h kg−1 and 1965 W kg−1, respectively.90 Furthermore, indole[3,2-b]carbazole (DEICZ) showed stable plateaus at high discharge potentials of 3.44 V and 4.09 V vs. Li/Li+.91 Moreover, materials based on the extended π–π conjugation of dioxin have also been reported to surpass the redox potential limit of organic cathode materials, achieving high voltages of over 4.1 V.92 Additionally, based on the structural design of polymer electrodes, we successfully combined the triphenylamine (p-type) and naphthalimide (n-type) moieties in a polymer backbone, resulting in a significant increase in specific capacity and working voltage up to ∼202 mA h g−1 and 4.5 V vs. Li/Li+, respectively.62 Furthermore, our studies also found that conformational structures (isomerism) and bridge functionalization on the imide units impact the electrochemical performance of polymer electrodes by regulating their intrinsic properties such as charge storage behavior, ion diffusivity, and activation energy.62–64 These examples underscore the potential of polymer electrode materials and the importance of structural design in the polymer structure to achieve a high energy density and fast charge–discharge rates as well as high-voltage ability, making them promising candidates for advanced LIBs. Herein, we delve into the cutting-edge advancements in designing high-voltage p-type organic cathode materials for LIBs. We explore a variety of p-type organic materials that have emerged over the last decade, each based on distinct redox-active centers. Our discussion encompasses their electrochemical attributes, such as redox potentials and kinetics, and how these properties influence the overall performance of LIBs. By providing a comprehensive overview of the progress in p-type organic materials, we aim to highlight their potential to revolutionize the design of high-voltage polymer-based cathodes for LIB applications through a comparison of the molecular design strategies employed.
2. Requirement, properties, and challenges of polymer electrodes
2.1 General requirements of LIB cathode materials
The cathode plays a pivotal role in LIBs given that it serves as both the source and recipient of lithium ions during the charge and discharge cycles, and also define the working potential of LIB cells. To ensure the optimum performance, several aspects need to be considered in the design of next-generation cathode materials as, follows:93–95(i) Redox-active ion: the presence of a redox-active ion is crucial given that it enables the material to undergo reversible oxidation and reduction processes during the charge and discharge cycles. This redox activity allows the storage and release of lithium ions, contributing to the overall capacity of the battery. In general, materials with well-defined redox chemistry can exhibit high energy density and excellent cycling stability, making them ideal candidates for high-performance battery electrodes.
(ii) Reversible lithium reaction: the ability of the material to undergo reversible reactions with lithium ions is essential for maintaining the integrity of the host structure throughout multiple charge and discharge cycles. This reversible process ensures that the active material can efficiently accommodate and release lithium ions without significant structural degradation. Furthermore, a stable host structure not only prolongs the cycle life of the battery but also helps maintain its energy storage capacity over time.
(iii) High free energy of reaction: a high free energy reaction with lithium ions is desirable given that it directly correlates with the capacity of the battery electrode material. Materials with a high capacity can store a greater number of lithium ions per unit mass, leading to enhanced energy storage capabilities. Achieving a voltage of around 4 V, while considering the stability of the electrolyte is crucial for maximizing the energy density of LIBs and meeting the demands of high-energy storage applications.
(iv) Ionic electrical conductivity: high-power density in LIBs relies on the rapid kinetics of lithium-ion insertion/extraction and fast electronic conductivity within the electrode material. Fast lithium diffusion rates and low activation energies enable swift ion transport, ensuring efficient electrochemical reactions. This facilitates rapid energy transfer, which is ideal for applications such as electric vehicles and portable electronics. Additionally, high electrical conductivity can minimize the resistance and voltage losses during the electrochemical process, thus further enhancing the overall energy density and efficiency.
(v) Structural stability: maintaining structural stability is crucial to prevent mechanical deformation, pulverization, or structural collapse of the electrode material during prolonged cycling. Structural stability ensures that the active material retains its integrity and functionality, contributing to the long-term performance and cycle life of the battery. Materials with robust structures exhibit excellent mechanical strength and resistance to degradation, even under extreme operating conditions, enhancing the reliability and safety of LIB systems.
(vi) Cost-effectiveness and environmental challenges: cost-effectiveness and environmental considerations play pivotal roles in the development of LIB materials. Economically feasible materials not only enable widespread adoption by reducing manufacturing costs but also contribute to the competitiveness of LIBs in various markets, fostering innovation and advancement in battery technology. Addressing environmental concerns by utilizing non-toxic, recyclable, and sustainably sourced materials helps minimize the ecological footprint of batteries, aligning with global efforts towards a cleaner and more sustainable future. Furthermore, promoting the use of environmentally benign materials encourages the development of greener battery technologies, supporting the transition to renewable energy systems and reducing dependence on finite resources.
2.2 Properties of p-type polymer electrode
The redox potential of a material is a critical factor in determining its suitability for use in energy storage devices. The redox potential is influenced by the molecular structure of the material and the nature of the electrolyte used.96 In the case of p-type materials, their redox potential is higher than that of n-type materials, making them more suitable for use as cathodes in energy storage devices (Fig. 2a). At the core of their functionality, their conjugated molecular structure, featuring alternating single and double bonds along the polymer backbone, enables the delocalization of electrons, and thus facilitates the movement of charge carriers through the material.97,98 Furthermore, p-type polymers offer tunable energy levels through chemical modifications, which allows precise engineering of their electronic properties to match specific device requirements.99,100 Moreover, p-type polymers have also been recognized to provide good thermal and structural stability, which is essential for withstanding the processing and operational conditions of electronic devices.101 Some p-type polymers can be designed to be air-stable, thus eliminating the need for stringent inert atmospheres during the device fabrication.102,103 Additionally, their high charge carrier mobility, compatibility with other materials used in device architectures, and relatively low bandgap further contribute to their versatility and suitability for a wide range of applications.104–106 | ||
| Fig. 2 (a) Different p-type organic materials along with their average redox voltages. (b) Frontier molecular orbitals (FMOs) relevant to the redox reactions of redox-active organic materials. (c) Schematic showing a solvated anion and cation within the electrolyte. Reproduced from ref. 36. | ||
Furthermore, in p-type organic cathode materials, their performance is significantly affected by factors such as redox voltage and energy levels, as well as the impact of counter ions.36,89 The redox potential of p-type organic cathode materials is crucial for their electrochemical performance, which is determined by their highest occupied molecular orbital (HOMO) energy level (Fig. 2b).36 Therefore, adjusting this HOMO level of p-type organic cathode materials through heteroatom doping and functionalization can further optimize their performance in energy storage applications.107 Meanwhile, the counter ions, such as BF4−, PF6−, FSI−, and TFSI−, also significantly impact the performance of p-type organic cathode materials including their charge transport properties, ionic conductivity, and ultimately their storage capacity and power density (Fig. 2c).108 Overall, p-type polymers offer a combination of properties that make them promising candidates for cathode materials in LIBs.36,109
2.3 Challenges and limitations
P-type polymers possess unique properties, which make them attractive for various applications, including energy storage such as LIBs. These polymers are characterized by their ability to accept and transport charge ions within their structure. This property stems from the presence of electron-deficient units or dopants within the polymer backbone. Apart from the outstanding properties that have been elaborated in the previous section, the development of p-type polymer electrodes faces certain challenges, as follows:36,66,110–116(i) Susceptibility to degradation: given that p-type polymer cathodes offer a high voltage operation, and thus maintaining redox reversibility at higher potential becomes critical during the battery operation. This high voltage operation coupled with electrolyte incompatibility can induce irreversible reactions, structural changes or degradation during charge and discharge cycles, which can result in capacity fading and reduced cycling stability. Therefore, enhancing stability and electrolyte compatibility of materials is required to ensure the long operation of p-type polymer cathodes in LIBs.
(ii) Low specific capacity: many p-type polymers exhibit lower specific capacities than traditional inorganic cathode materials due to their lower number of redox active sites. This lower specific capacity limits the energy density of LIBs utilizing p-type polymers as cathodes, impacting their overall performance and suitability for certain applications. Additionally, due to the fact that p-type polymers operate by incorporating anions from the electrolyte during the electrochemical process, the corresponding anions play an important role in defining their specific capacity. Furthermore, this anion involvement during electrochemical processes together with the instability of organic solvents in the electrolyte at high-voltage (>4.0 V) can result in the degradation and consumption of the electrolyte upon cycling, leading to a lower energy density. Consequently, providing more redox active sites in the polymer backbone through material design and electrolyte optimization is essential to enhance the storage capacity.
(iii) Minimum rate capability: the rate capability of p-type polymers refers to their ability to deliver and accept mobile ions at high charge and discharge rates. Some p-type polymers may exhibit a limited rate capability as a result of the slow ionic diffusion kinetics due to their poor electronic conductivity. Hence, enhancing the electronic conductivity of p-type polymer cathodes is crucial to provide rapid ion mobility during electrochemical processes.
(iv) Interfacial issues: the interface between the p-type polymer cathode and electrolyte plays a crucial role in ion transport and the overall battery performance. In batteries, poor electrolyte wetting properties, resistive electrode–electrolyte interphase, or unwanted interface reactions significantly hinder ion diffusion, and consequently decrease their efficiency. Therefore, addressing these interfacial issues is essential to improve the ion transport kinetics and enhance the performance of batteries.
(v) Electrode preparation issues: although polymer-based cathodes have minimum solubility in electrolytes, they often suffer from aggregation during electrode fabrication, thus reducing the effective surface area for ion transport and electrochemical reactions. This problem is worse in high-mass-loading electrodes, leading to an uneven current distribution, increased resistance, and reduced capacity. In large-scale preparation, their low electrical conductivity further hinder their performances. Therefore, the appropriate solvent selection and structural design are crucial for improving processability. Furthermore, the introduction of conductive additives such as carbon black, nanotubes, and graphene can enhance the electrical and ionic conductivity of polymer electrodes.
(vi) Cost and scalability: the production cost and scalability of p-type polymers are important considerations for their practical application in large-scale energy storage systems. Polymers with rare or expensive dopants, complex synthesis, and low processability may face challenges in cost-effective production and scalability, hindering their practical application and commercial viability. Therefore, developing cost-effective synthesis methods and utilizing abundant and sustainable raw materials are essential for realizing the widespread adoption of p-type polymer cathodes in LIBs and other energy storage devices.
3. Current progress in p-type polymer cathode materials
3.1 Polytriphenylamine
As a p-type compound, the structural specificity of the polytriphenylamine (PTPA) is recognized to originate from its radical nitrogen redox centers, which are stabilized by phenyl groups, facilitating radical redox reactions and charge-transporting processes. This structural feature allows high power capability and high energy density upon prolonged cycling. Additionally, its porous polymer structure with abundant interconnected holes due to its rigid phenyl groups provides sufficient surface area and ionic channels for rapid ion mobility.117 Additionally, as a promising organic cathode material for LIBs, the redox activity of PTPA is attributed to the doping/de-doping process of anionic species on the nitrogen radical, providing a specific capacity of ∼109 mA h g−1.62,88,118,119 However, some issues related to the low theoretical capacity, conductivity and processability of PTPA-based polymers still hinder their practical application. In this regard, Feng and co-workers reported simple PTPA (Fig. 3a) as an organic cathode material for LIBs.120 The charge–discharge curves of the PTPA electrode exhibited an approximately linear relationship between voltage and capacity in the range of 4.2–3.6 V, resembling an electrochemical doping/de-doping process (Fig. 3b). At a moderate discharge rate of 0.5C, the discharge capacity reached 103 mA h g−1, which is approximately 94% of the theoretical capacity of 109 mA h g−1, but much lower than that of conventional electrodes, as shown in Fig. 3c.120 Additionally, to enhance the specific capacity, the combination of different redox centers can facilitate greater electron transfer, and thus extend the storage capability. Similarly, Chen and colleagues successfully synthesized a novel micro-/mesoporous polymer, PTDAPTz, containing triazine units.121 As shown in Fig. 3d, the energy gap between the lowest unoccupied molecular orbital (LUMO) and HOMO of TDAPTz was found to be stronger than that of TPA due to the stronger electron-withdrawing effect of the triazine unit, resulting in a lower band gap. This characteristic reflected in their electrochemical response as the redox peaks of the triphenylamine units in PTDAPTz shifted towards a higher potential than that of PTPAn (Fig. 3e) due to the electron-withdrawing effect of the triazine units, thus decreasing the electron cloud density nearby the nitrogen atom of triphenylamine and making it difficult to gain and lose the radical electron. This unique feature led to an improved cell performance, as shown in Fig. 3f. Furthermore, due to the high free radical density of the PTPA organic cathode, PTDATA, was synthesized to enhance the specific capacity via multiple electron transfer.119 As shown in Fig. 4a, PTDATA exhibited multiple redox peaks compared to the PTPA cathode. These multiple peaks of PTDATA can be ascribed to the four free radical center structures in the TDATA unit of PTDATA, which undergo a four-electron transfer reaction during the charge–discharge process. These multi electron transfer successfully improved the cycling performance of the PTDATA cathode with the initial capacity of ∼131 mA h g−1, which was maintained at 98 mA h g−1 after 100 cycles at a current density of 20 mA g−1 (Fig. 4b).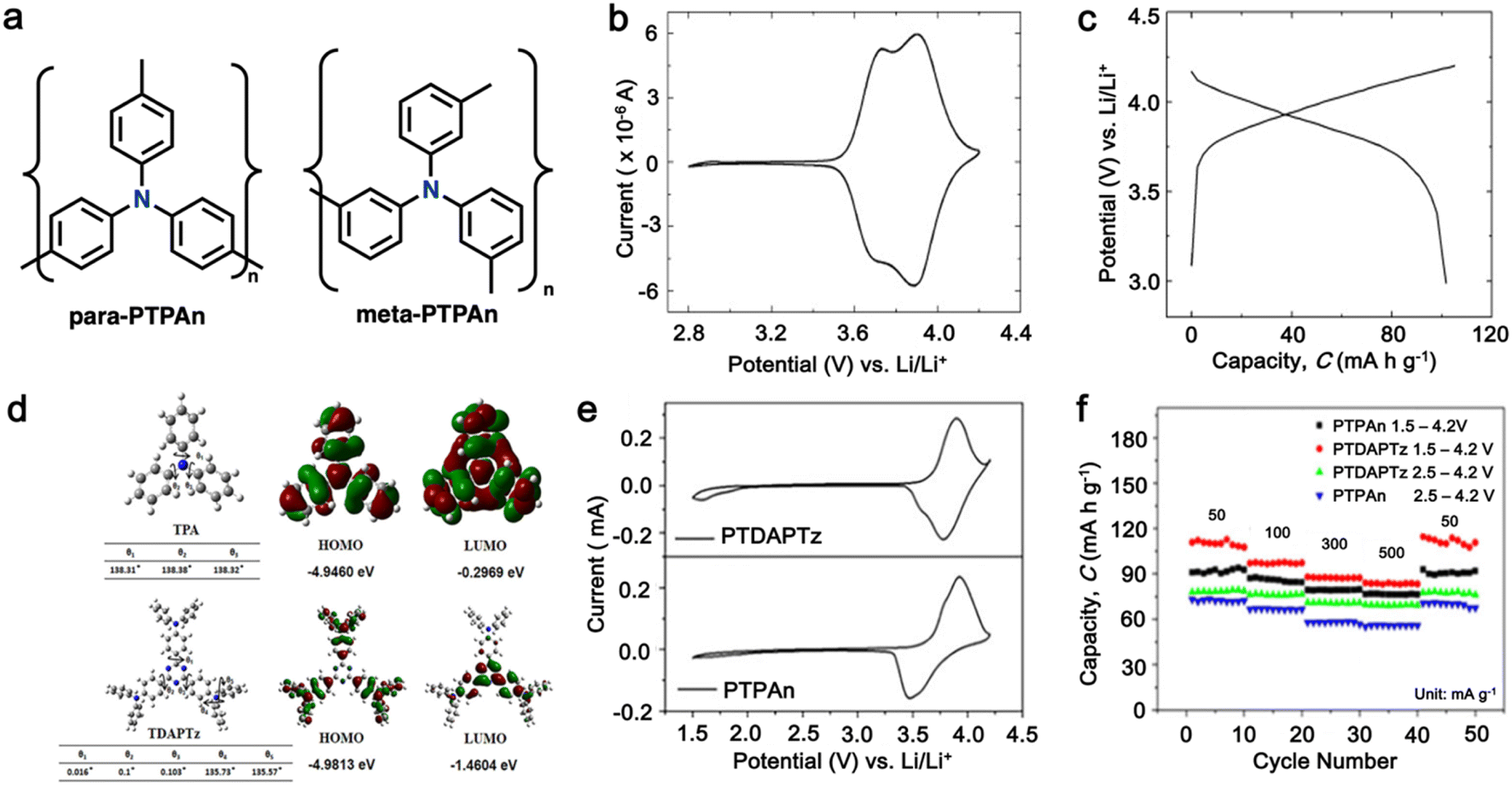 | ||
Fig. 3 (a) Two possible isomers of PTPAn, (b) cyclic voltammetry (CV) curve of PTPAn in 1 M LiPF6 EC/DMC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) measured at a scan rate 1 mV s−1, and (c) charge–discharge curves of Li-PTPAn test cells at a constant current of 50 mA g−1. Reproduced with permission from ref. 120 Copyright 2008, Elsevier. (d) Molecular orbital (HOMO and LUMO) and geometric structure of TPA and TDAPTz monomers calculated theoretically using DFT calculation at the B3LYP/6-31G level performed using Gaussian 09. (e) CVs of PTDAPTz and PTPAn in 1 M LiPF6 EC/DMC (v/v = 1:1) measured at 1 mV s−1 in the range of 1.5 of 4.2 V. (f) Rate performances of the PTDAPTz and PTPAn electrodes at various current rates. Reproduced with permission from ref. 121 Copyright 2018, John Wiley and Sons. :1 v/v) measured at a scan rate 1 mV s−1, and (c) charge–discharge curves of Li-PTPAn test cells at a constant current of 50 mA g−1. Reproduced with permission from ref. 120 Copyright 2008, Elsevier. (d) Molecular orbital (HOMO and LUMO) and geometric structure of TPA and TDAPTz monomers calculated theoretically using DFT calculation at the B3LYP/6-31G level performed using Gaussian 09. (e) CVs of PTDAPTz and PTPAn in 1 M LiPF6 EC/DMC (v/v = 1:1) measured at 1 mV s−1 in the range of 1.5 of 4.2 V. (f) Rate performances of the PTDAPTz and PTPAn electrodes at various current rates. Reproduced with permission from ref. 121 Copyright 2018, John Wiley and Sons. | ||
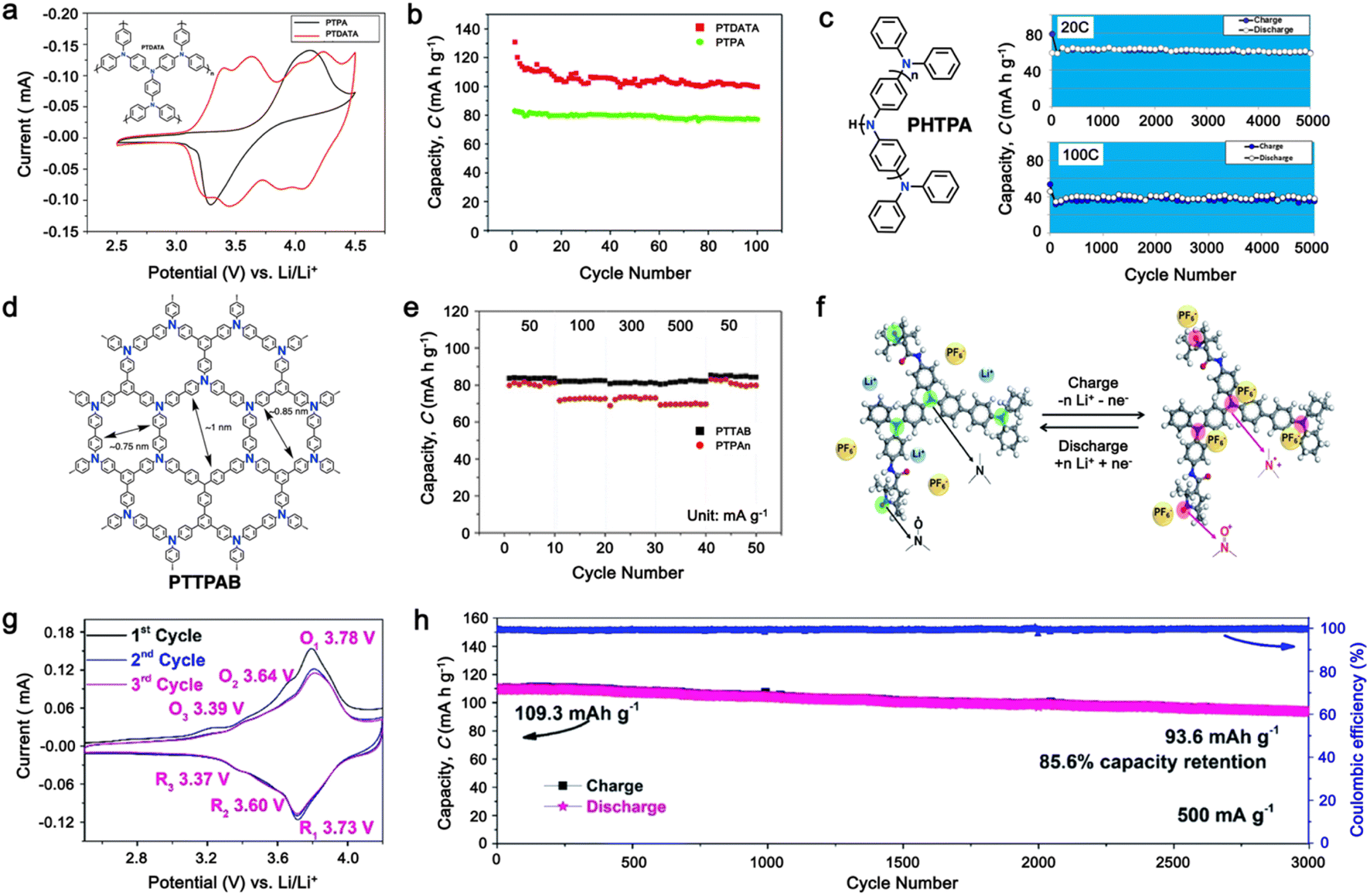 | ||
| Fig. 4 (a) and (b) CV at a scan rate of 1 mV s−1 and cycling performance under a current density of 20 mA g−1 for the PTDATA cathode, respectively. Reproduced with permission from ref. 119 Copyright 2017, The Royal Society of Chemistry. (c) Chemical structure and cycling performance of hyperbranched PHTPA cathode at a high current rate. Reproduced with permission from ref. 122 Copyright 2018, the American Chemical Society. (d) and (e) Chemical structure and rate performance of microporous PTTPAB cathode. Reproduced with permission from ref. 123 Copyright 2018, Elsevier. (f) Schematic of electrochemical reaction mechanism of P(TPA-co-DDP-PROXYL). (g) and (h) CV curve at a scan rate of 0.5 mV s−1 and long cycling performance of P(TPA-co-DDP-PROXYL) at 500 mA g−1. Reproduced with permission from ref. 124 Copyright 2022, The Royal Society of Chemistry. | ||
Moreover, different strategies such as the formation of the hyperbranched and microporous PTPA polymer have also been applied to further improve the processability and structural stability of the electrode materials.122,123 Yamamoto, et al. reported that hyperbranched phenylamine-based (PHTPA), prepared by Buchwald–Hartwig reaction (C–N coupling), exhibited high processability and delivered an ultrafast charge–discharge process (Fig. 4c).122 The high processability of PHTPA was proven by the fact that it could easily dissolve in most of organic solvents such as N-methyl-2-pyrrolidone (NMP), THF, chloroform, and toluene although the polymer has all aromatic structures. Meanwhile, it remained insoluble in the electrolyte solution of 1 M LiPF6 EC:DEC (30:70), indicating that PHTPA can be applied to LIBs. Furthermore, the PHTPA cathode delivered a stable specific capacity of ∼60 and ∼40 mA h g−1 at C rates of 20 and 100, respectively, up to 5000 cycles (Fig. 4c), demonstrating its ultralong stability and ultrafast charge–discharge ability due to the formation of the microsphere morphology of the hyperbranched polymer.122 Additionally, a star-shaped triphenylamine-based monomer with a benzene core was prepared to synthesize the conjugated microporous polymer poly[1,3,5-tris(4-diphenylamino-phenyl)benzene] (PTTPAB) via chemical oxidative polymerization (Fig. 4d).123 This study found that the microporous structure is beneficial for providing fast ion transport, thus significantly improving the rate capability. As depicted in Fig. 4e, the PTTPAB cathode delivered a specific capacity of ∼80 mA h g−1 without a significant capacity drop as the current density increased up to 500 mA g−1.
In addition, copolymer formation has also been introduced in the PTPA organic cathode to improve its specific capacity. For instance, a novel conjugated radical copolymer, poly(triphenylamine-co-N,N′-bis(4-carbamoyl-2,2,5,5-tetramethyl-pyrrolin-1-oxyl)-N,N′ diphenyl-1,4-phenylenediamine) (P(TPA-co-DDP-PROXYL)), was synthesized through chemical oxidative polymerization and utilized as a cathode material.124 This polymer incorporated both a crosslinking conjugated backbone and PROXYL nitroxide radical side chains. As shown in Fig. 4f, the presence of triphenylamine and nitroxide radicals within the polymeric backbone is expected to provide numerous active sites to improve the specific capacity. In this regard, a series of redox couples could be monitored in the CV profile of P(TPA-co-DDP-PROXYL) in the potential window of 2.5–4.2 V vs. Li/Li+ (Fig. 4g). The two pairs of reversible redox peaks at 3.73/3.78 V (R1/O1) and 3.6/3.64 V (R2/O2) correspond to the para-substitution and meta-substitution redox reactions of the triphenylamine unit, respectively, while the R3/O3 (3.37/3.39 V) redox couple is associated with the transition from the nitroxide radicals to the oxoammonium cations.124 The resulting P(TPA-co-DDP-PROXYL) cathode material exhibited a superb electrochemical performance and ultralong cycle life with 72% capacity retention at 2000 mA g−1 over 3000 cycles (Fig. 4h).124 This ultralong stability can be attributed to its rigid molecular structure and exceptional resistance when immersed in the electrolyte for extended periods. Moreover, Table 1 summarizes the recent development of phenylamine-based cathodes for organic LIBs. Overall, phenylamine-based electrodes have been explored and demonstrated superior high-rate capability, cycling stability, and energy density, making them promising cathode materials for next-generation organic LIBs operated at high voltage.
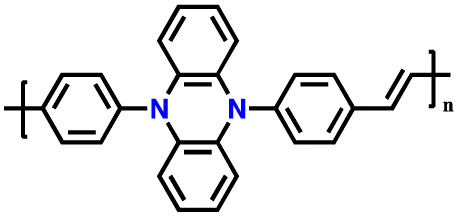
| No. | Polymer material | Chemical structure | Electrolyte | Voltage (V) vs. Li/Li+ | Capacity (mA h g−1) | Current applied | Ref. |
|---|---|---|---|---|---|---|---|
| Phenylamine-based cathodes | |||||||
| 1 | PDPA |

|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2.5–4.5 | 116 | 0.1 A g−1 | 125 |
| 2 | PTPA |
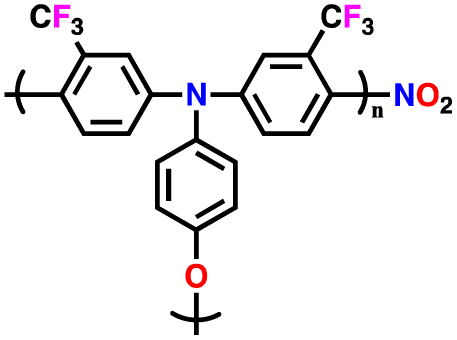
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2.5–4.2 | 69.7 | 0.05 A g−1 | 126 |
| 4 | PDPA-AQ |

|
1 M LiPF6 in EC/DMC (1:1, v/v) |
1.5–4.5 | 159 | 0.1C | 127 |
| 5 | PTPAn |
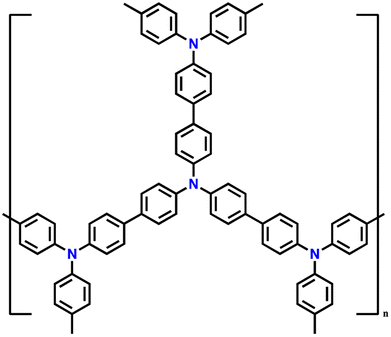
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2.5–4.2 | 100.4 | 0.02 A g−1 | 128 |
| PTPA-CN |
|
85.4 | |||||
| 6 | Poly(4-cyano)triphenylamine |
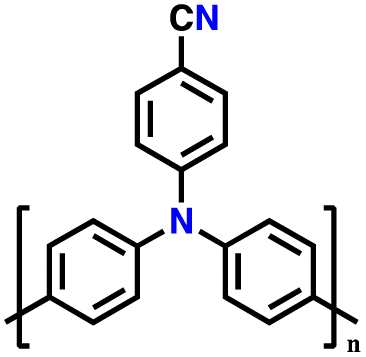
|
1 M LiPF6 in EC/DMC/EMC (1:1:1, v/v/v) |
3.0–4.2 | 75 | 0.08 A g−1 | 129 |
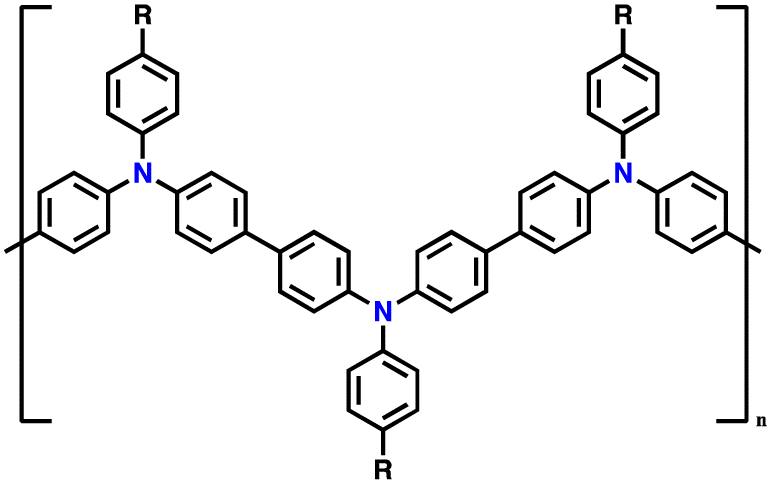
| 7 | PDDP |

|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2.5–4.2 | 110.6 | 0.02 A g−1 | 88 |
| 8 | PTPA |
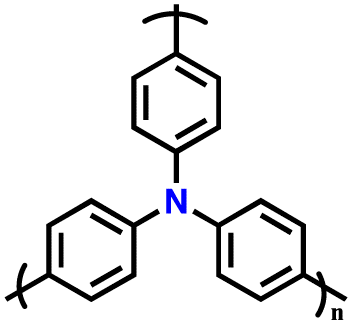
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2.5–4.2 | 80 | 0.02 A g−1 | 88 |
| 9 | PFTP |

|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2.5–4.2 | 74.2 | 0.02 A g−1 | 130 |
| 10 | PTPA-PO |
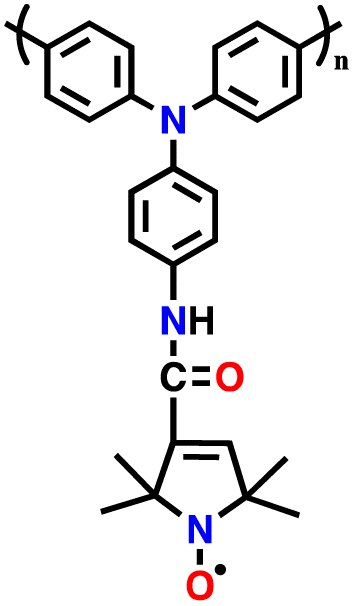
|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2.0–4.2 | 134.5 | 0.02 A g−1 | 131 |
| 11 | PTPA-CNT |
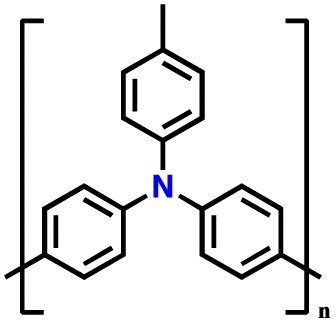
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2–4.2 | 103.1 | 0.02 A g−1 | 132 |
| PTPO-CNT |
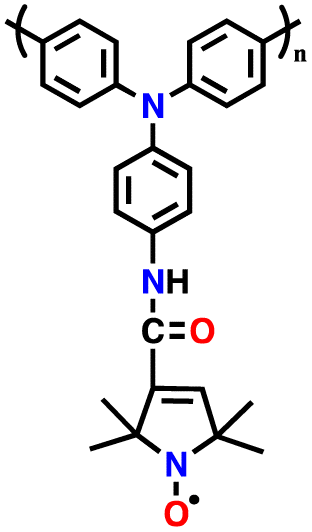
|
128 | |||||
| 12 | PTTPAB |
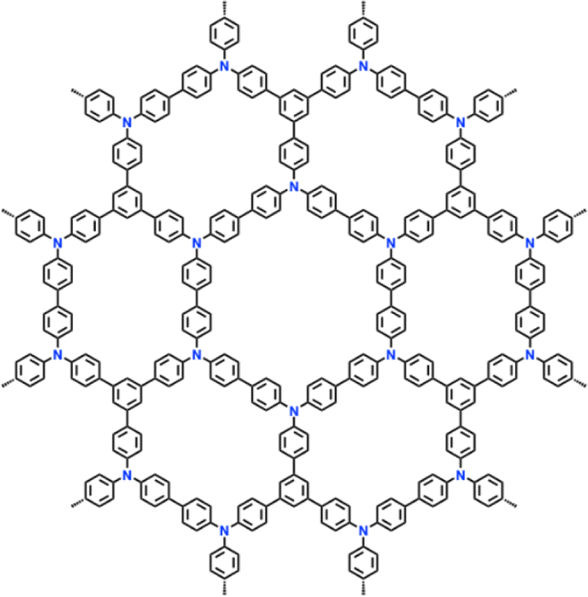
|
1 M LiPF6 in EC/DMC/DEC (1:1:1, v/v/v) |
2.5–4.2 | 86.5 | 0.02 A g−1 | 123 |
| 13 | PHTPA |
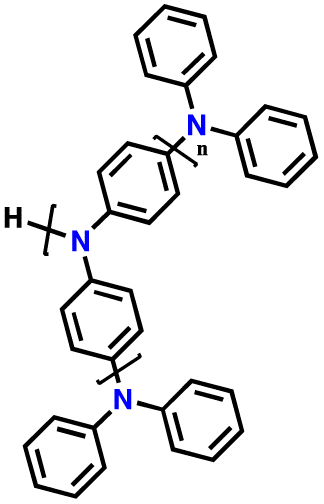
|
1 M LiPF6 in EC/DEC (3:7, v/v) |
2.8–4.0 | 60 | 20C (1C = 0.06 A g−1) | 122 |
| 14 | PTDAPTz |
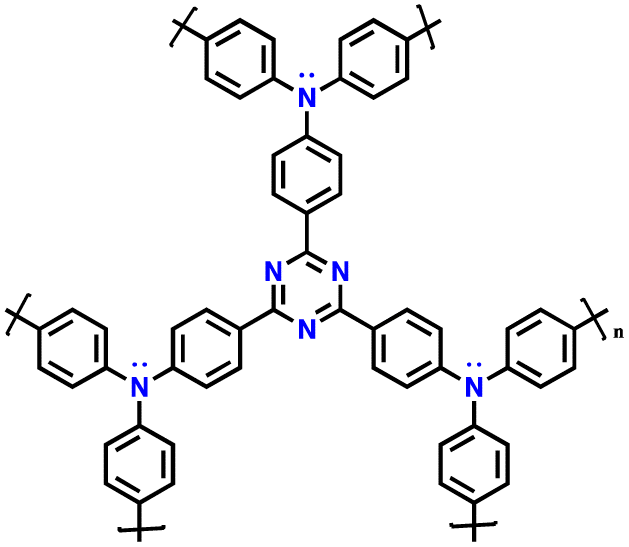
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
1.5–4.2 | 123 | 0.02 A g−1 | 121 |
| 15 | YPTPA |
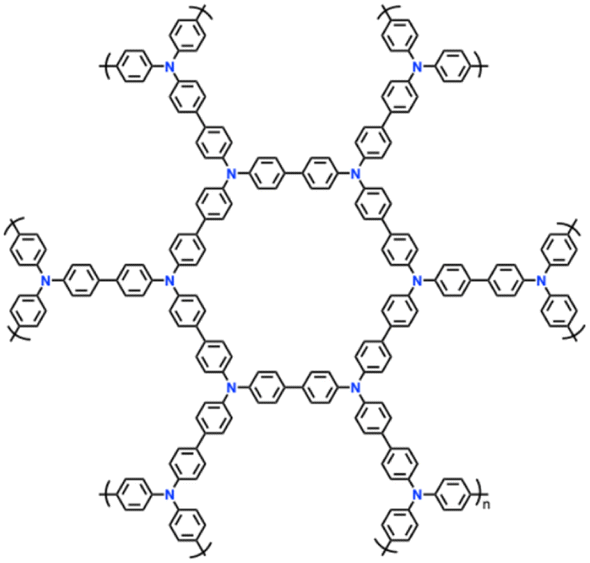
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
2.5–4.2 | 100 | 0.1 A g−1 | 87 |
3.2 Thioether polymers
Organosulfur compounds have been recognized as potential cathode materials for LIBs due to their high theoretical capacity (1675 mA h g−1) and energy density (2600 W h kg−1) via the reversible formation/deformation of S–S bond formation.28 However, despite their abundance and low cost, challenges such as their non-conductivity and the polysulfide shuttle effect limit their practical use.133,134 Although organosulfur cathodes with diverse macromolecular structures can offer an improvement in stability and kinetics, some issues associated with S–S bond recovery have been encountered, thus impacting their cycling stability.135–137 Alternatively, thioether polymers, which are distinct from organosulfur compounds containing single S–S bonds, offer a different redox mechanism, involving the loss of electrons and formation of stable oxidation states.138 This characteristic makes them potential candidates for cathodes with high voltage outputs, corresponding to the intercalation of anions (ClO4−, PF6−, BF4−, and TFSI−).139,140 Additionally, the charge storage mechanism of thioether polymers differs from organosulfur polymers, given that it does not involve chain breaking, potentially leading to higher cyclability.141 Over the past few decades, numerous polymeric organosulfide materials have been developed, which can be categorized into four types including main-chain-type, side-chain-type, polysulfides with –Sx groups where x > 2, and thioether-type organosulfur polymers.134 Ren et al. synthesized two polyacetylene derivatives, P1 and P2, containing pendant thianthrene groups through polymerization using the [(2,5-norbornadiene)RhCl]2/Et3N catalyst with thianthrene-1-ylmethyl 4-ethynylbenzoate (M1) or bis(thianthrene-1-ylmethyl)-4-ethynylphthalate (M2), as shown in Fig. 5a.144 Both P1 and P2 exhibited reversible redox activity with a high oxidation potential of 4.16 V vs. Li/Li+ due to the presence of p-type thioethers groups.144 Notably, P2 showed a higher specific capacity of 100 mA h g−1 compared to P1 (33 mA h g−1) (Fig. 5b and c), which is attributed to the more active units within the P2 molecule. Moreover, adopting the same redox behavior, Vaid et al. reported a polythianthrene cathode with an impressive working potential of up to 4.1 V vs. Li/Li+, which is comparable that of inorganic cathodes.145 | ||
| Fig. 5 (a) Chemical structure of polyacetylene derivatives containing thioether-based cathode, namely, P1 and P2. (b) and (c) Charge–discharge profiles of thioether-based cathodes P1 and P2. Reproduced with permission from ref. 142 Copyright 2020, Elsevier. (d) and (e) Charge–discharge profile and chemical structure of tetrathiafulvalene (TTF)-based cathode 3a and 3d, respectively. (f) Possible redox reaction of TTF-based cathode. Reproduced with permission from ref. 143 Copyright 2019, John Wiley and Sons. (g) Examples of representative p-type organosulfur polymers. Reproduced with permission from ref. 138. | ||
Additionally, Misaki et al. demonstrated tris-fused tetrathiafulvalene (TTF) analogues, including unsubstituted- and bis(ethylenedioxy)-derivatives as cathode LIBs (Fig. 5d and e).146 As shown in Fig. 5d and e, these compounds delivered a voltage window of up to 3.6 V with a high capacity of 192 and 160 mA h g−1. These distinct electrochemical performances were associated with the typical p-type reaction mechanism and six-electron electron transfer, as illustrated in Fig. 5f. However, these materials suffered from rapid capacity decay in the initial cycles, indicating the need to address their poor cycling stability (Fig. 5d and e). Furthermore, inspired by polymers, as presented in Fig. 5g(i and ii), polyphenyls with dithiolane moieties, as shown in Fig. 5g(iii), were prepared, showing promising stability and a stable capacity of up to 300 mA h g−1.147,148 However, these materials often exhibit large polarization due to their low conductivity, thus limiting their stability. Moreover, the incorporation of two sulfur atoms into a six-membered ring, as seen in thianthrene, results in exceptional stability.149 Thianthrene-based polymers can lose two electrons alongside the intercalation of anions, leading to a high discharge plateau at around 4.0 V without a significant overpotential.30 The Fig. 5g(iv) polymer exhibited highly reversible charge–discharge curves at 4.05 V for charging and 3.95 V for discharging, with a capacity of 105 mA h g−1 after the first cycle and 81% capacity retention after 250 cycles.30 Employing a similar strategy, Schubert et al. utilized 1,3-dithiane in a five-membered ring polymer, which resulted in the discharge potential of about 3.2 V, where the charge storage mechanism involved anion intercalation (Fig. 5g (v)).150 These findings highlight the potential of p-type thioether-based cathodes for high-potential LIBs, offering improved capacity, voltage, and cycle stability. Furthermore, Table 2 provides the recent trends in thioether-based cathodes for organic LIBs.
| No. | Polymer material | Chemical structure | Electrolyte | Voltage (V) vs. Li/Li+ | Capacity (mA h g−1) | Current applied | Ref. |
|---|---|---|---|---|---|---|---|
| Thioether-based polymers | |||||||
| 1 | PPPS-14 |

|
1 M LiTFSI in DOL/DME (1:1 v/v) |
1.8–3.0 | 382.5 | 1C (1C = 0.622 A g−1) | 151 |
| 2 | Crosslinked polybenzenehexasulfide |
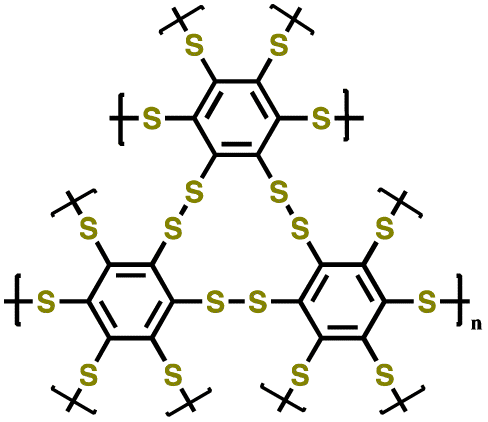
|
1 M LiTFSI in DOL/DME (1:1 v/v) |
0.75–3.2 | 150 | 0.1C | 152 |
| 3 | PBDTD |
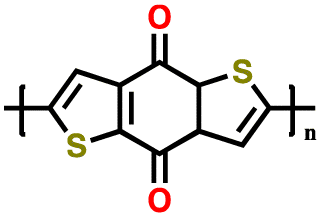
|
1 M LiClO4 in DOL/DME (1:1 v/v) |
1.9–3.2 | 180 | 5C (1C = 0.214 A g−1) | 153 |
| 4 | PVBDT |
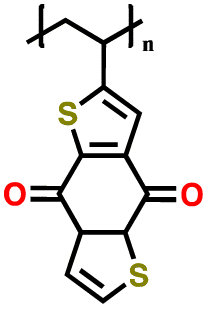
|
1 M LiClO4 in EC/DMC (1:1 v/v) |
1.5–3.25 | 116 | 1C | 154 |
| 5 | PPTS |
|
1 M LiTFSI in DOL/DME (1:1 v/v) with 0.2 M LiNO3 |
1.8–3.0 | 633 | 1C (1C = 0.778 A g−1) | 155 |
| 6 | PBTTS |
|
1 M LiTFSI in DOL/DME (1:1 v/v) with 0.2 MLiNO3 |
1.8–3.0 | 616.6 | 0.1C (1C = 0.901 A g−1) | 156 |
| 7 | PEHS |
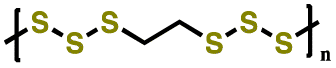
|
1 M LiTFSI in DOL/DME (1:1 v/v) with 0.2 MLiNO3 |
1.8–3.0 | 774 | 1C (1C = 1.217 A g−1) | 157 |
| 8 | S-BOP |
|
1 M LiTFSI in DIOX/TEGDME (0.67:0.33 v/v) with 0.2 M LiNO3 |
1.7–2.7 | 630 | 1C (1C = 0.72 A g−1) | 158 |
| 9 | S-TTCA |
|
1 M LiTFSI in DIOX/TEGDME (0.67:0.33 v/v) with 0.2 M LiNO3 |
1.7–2.7 | 813 | 0.2C (1C = 1.675 A g−1) | 159 |
| 10 | CP(S3BT) |
|
1 M LiTFSI in DOL/DME (1:1 v/v) with 0.1 M LiNO3 |
1.5–3.0 | 682 | 1C | 160 |
3.3 Dihydrophenazine-based polymers
In the preceding section, we explored how organic electrodes derived from triphenylamine-based polymers exhibit notably high redox potentials, though their specific capacities are typically around 100 mA h g−1. This restriction arises from the fact that the triphenylamine unit tends to undergo a single-electron redox reaction, depending solely on a single nitrogen heteroatomic redox center on their polymer backbone. Thus, to overcome this limitation, recent studies have focused on developing p-type organic electrodes that are capable of multi-electron redox reactions to achieve higher capacities. In this regard, dihydrophenazine and its derivatives have been explored as new alternative p-type polymers with multi-electron redox reactions. These organic electrodes feature two heteroatomic redox centers (N, S, O) in their core six-membered ring, fused by two benzene rings, allowing two single-electron redox reactions and a significantly higher theoretical specific capacity (∼270 mA h g−1) than that of triphenylamine-based electrodes. The redox potentials of these phenazines are in the range of 3.1 to 4.2 V vs. Li/Li+ based on the electron-donating/withdrawing strength of the heteroatoms.Among the phenazines, N,N′-substituted phenazine derivatives have captured interest due to their ability to undergo two successive one-electron transfer reactions, categorizing them as p-type molecules with higher redox potentials.161 As shown in Fig. 6a, the reversibility of these electron transfer reactions depends on the substituted groups on the two N atoms. A significant challenge in applying polymeric materials lies in their insufficient electrical and/or ionic conductivity, especially for those employing the anion-exchange mechanism.22,165–167
 | ||
| Fig. 6 (a) Chemical structure and redox mechanism of N,N′-substituted phenazine derivatives. Reproduced with permission from ref. 84 Copyright 2019, Elsevier. (b) Schematic of enhancing the power density by decreasing the rigidity of the polymer chains. (c) Proposed polymers with variable chain rigidity based on DPPZ. (d) CV profiles of p-DPPZR1, p-DPPZR2, and p-DPPZR3 cells at sweeping rates of 0.2, 0.4, 0.6, 0.8, and 1 mV s−1. (e) Typical charge/discharge profiles of p-DPPZR1, p-DPPZR2, and p-DPPZR3 at 1C rate. Reproduced with permission from ref. 162 Copyright 2019, The Royal Society of Chemistry. (f) CV profiles of poly(Ph-PZ) at 0.25 mV s−1 in Li metal half cells. (g) Rate capability of poly(Ph-PZ)-based cathode. Reproduced with permission from ref. 163 Copyright 2021, The Royal Society of Chemistry. (h) Redox mechanism and DFT calculation of TPZB cathode. Reproduced with permission from ref. 164 Copyright 2020, the American Chemical Society. | ||
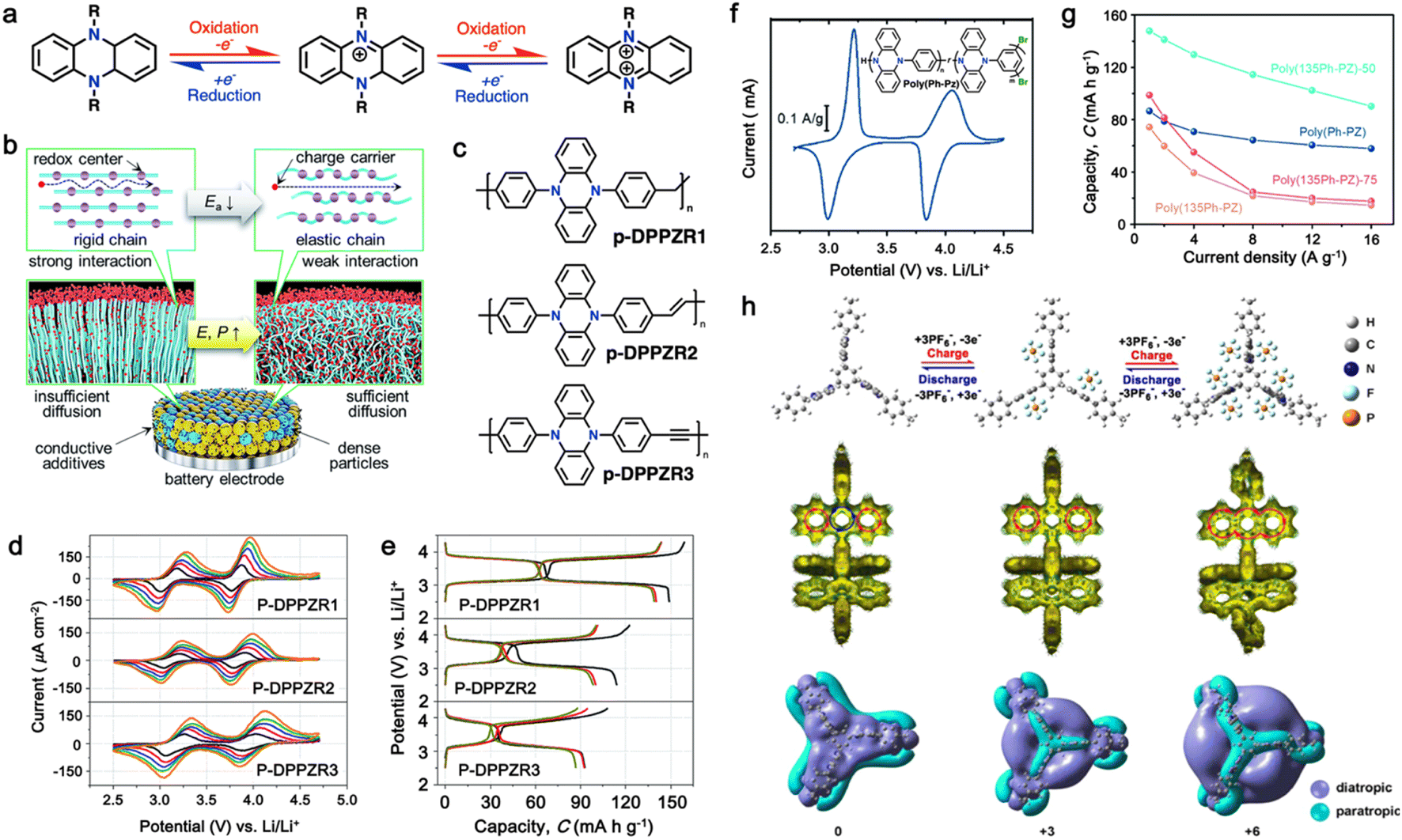
In this regard, Niu and colleagues explored manipulating the rigidity of polymer chains to alter the ion diffusion behavior in polymeric materials.162 Their study revealed that introducing a suitable twisted group in phenazine-based polymers significantly enhances their ionic diffusion coefficient, leading to improved power densities despite their lower surface area and electrical conductivity.168 This approach emphasized the different structural packing of the polymer chains, where the rigid chains often hinder ion diffusion through the polymer chain due to the increase in activation energy, while more elastic chains could accelerate the ion diffusion via the formation of an amorphous phase (Fig. 6b).169,170 To validate this approach, three polymers based on redox-active N,N′-diphenyl-5,10-dihydrophenazine (DPPZ) were synthesized with varying chain rigidities, namely, p-DPPZR1 (–CH2– bridging), p-DPPZR2 (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– trans-isomer), and p-DPPZR3 (–C
CH– trans-isomer), and p-DPPZR3 (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) (Fig. 6c).162 As shown Fig. 6d, two pairs of redox peaks can be observed, corresponding to the two successive two-steps of one-electron redox reactions of DPPZ. In terms of specific capacity, p-DPPZR1 delivered a specific capacity of approximately 145.9 mA h g−1 (Fig. 6e), surpassing that of p-DPPZR2 (112.1 mA h g−1) and p-DPPZR3 (93.7 mA h g−1).162 Additionally, to address the limitations posed by the strong π–π interactions narrowing the internal free volume, Gannett et al. developed a network polymer of PZ by incorporating a phenylene linker (Ph) with three points of connectivity, as shown in Fig. 6f.163 This modification aimed to enhance the rate performance of the polymer system. Among the copolymers, poly(135Ph-PZ)-50 demonstrated the highest performance due to its optimal characteristics, as shown by the redox reactions in cyclic voltammetry (Fig. 6f). The polymer electrode achieved a specific capacity of up to 135 mA h g−1 at 1.0 A g−1, retaining 65% of its low-rate capacity at a high rate of 16 A g−1 (Fig. 6g).163 Furthermore, the optimized geometries of poly(1,3,5-tris(10-(4-vinylphenyl)phenazin-5(10H)-yl)benzene) (TPZB) at the 0, +3, and +6 valence states were investigated, revealing the formation of a twist angle by the dihydrophenazine group with a centered benzene ring, creating storage space for PF6− ions resembling piers (Fig. 6h).164 This configuration facilitated their diffusion and intercalation/deintercalation. The calculation of iso-chemical shielding surfaces (ICSSs) further explored the aromaticity of the redox intermediates and their impact on the electrode performance (Fig. 6h).164
C–) (Fig. 6c).162 As shown Fig. 6d, two pairs of redox peaks can be observed, corresponding to the two successive two-steps of one-electron redox reactions of DPPZ. In terms of specific capacity, p-DPPZR1 delivered a specific capacity of approximately 145.9 mA h g−1 (Fig. 6e), surpassing that of p-DPPZR2 (112.1 mA h g−1) and p-DPPZR3 (93.7 mA h g−1).162 Additionally, to address the limitations posed by the strong π–π interactions narrowing the internal free volume, Gannett et al. developed a network polymer of PZ by incorporating a phenylene linker (Ph) with three points of connectivity, as shown in Fig. 6f.163 This modification aimed to enhance the rate performance of the polymer system. Among the copolymers, poly(135Ph-PZ)-50 demonstrated the highest performance due to its optimal characteristics, as shown by the redox reactions in cyclic voltammetry (Fig. 6f). The polymer electrode achieved a specific capacity of up to 135 mA h g−1 at 1.0 A g−1, retaining 65% of its low-rate capacity at a high rate of 16 A g−1 (Fig. 6g).163 Furthermore, the optimized geometries of poly(1,3,5-tris(10-(4-vinylphenyl)phenazin-5(10H)-yl)benzene) (TPZB) at the 0, +3, and +6 valence states were investigated, revealing the formation of a twist angle by the dihydrophenazine group with a centered benzene ring, creating storage space for PF6− ions resembling piers (Fig. 6h).164 This configuration facilitated their diffusion and intercalation/deintercalation. The calculation of iso-chemical shielding surfaces (ICSSs) further explored the aromaticity of the redox intermediates and their impact on the electrode performance (Fig. 6h).164
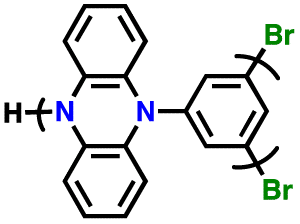
Additionally, substituting one of the N atoms with an S or O atom in the phenazine ring resulted in two further types of redox active compounds known as phenothiazine and phenoxazine, respectively. This substitution has been reported increase in the redox potential phenazine-based cathodes due to its weaker electron-donating strength. For instance, Kolek et al. introduced a phenothiazine (PTZ)-based polymer cathode, PVMPT, where N-methylated PTZ (MPT) units were attached to a vinyl backbone as pendant groups.171 In this study, each MPT unit underwent two single-electron oxidations sequentially at 3.44 V (from neutral state A to radical cation state C) and 4.18 V vs. Li/Li+ (to the di-cationic state D), respectively (Fig. 7a). Interestingly, as shown in Fig. 7b, the oxidation of PVMPT resulted in strong π–π interactions between neighboring MPT dimers, leading to the formation of the intermediate oxidation state at potentials of 3.44 and 4.18 V vs. Li/Li+. Upon oxidation, the PTZ units in PVMPT could associate intra- or inter-molecularly, stabilizing oxidized states B and C, as depicted in Fig. 7b. These interactions became evident in the CV curves measured at a slow scan rate (20 mV s−1), where the second cathodic peak, corresponding to the reduction of the radical cation to the neutral species (C → A), split into two peaks separated by 95 mV.171
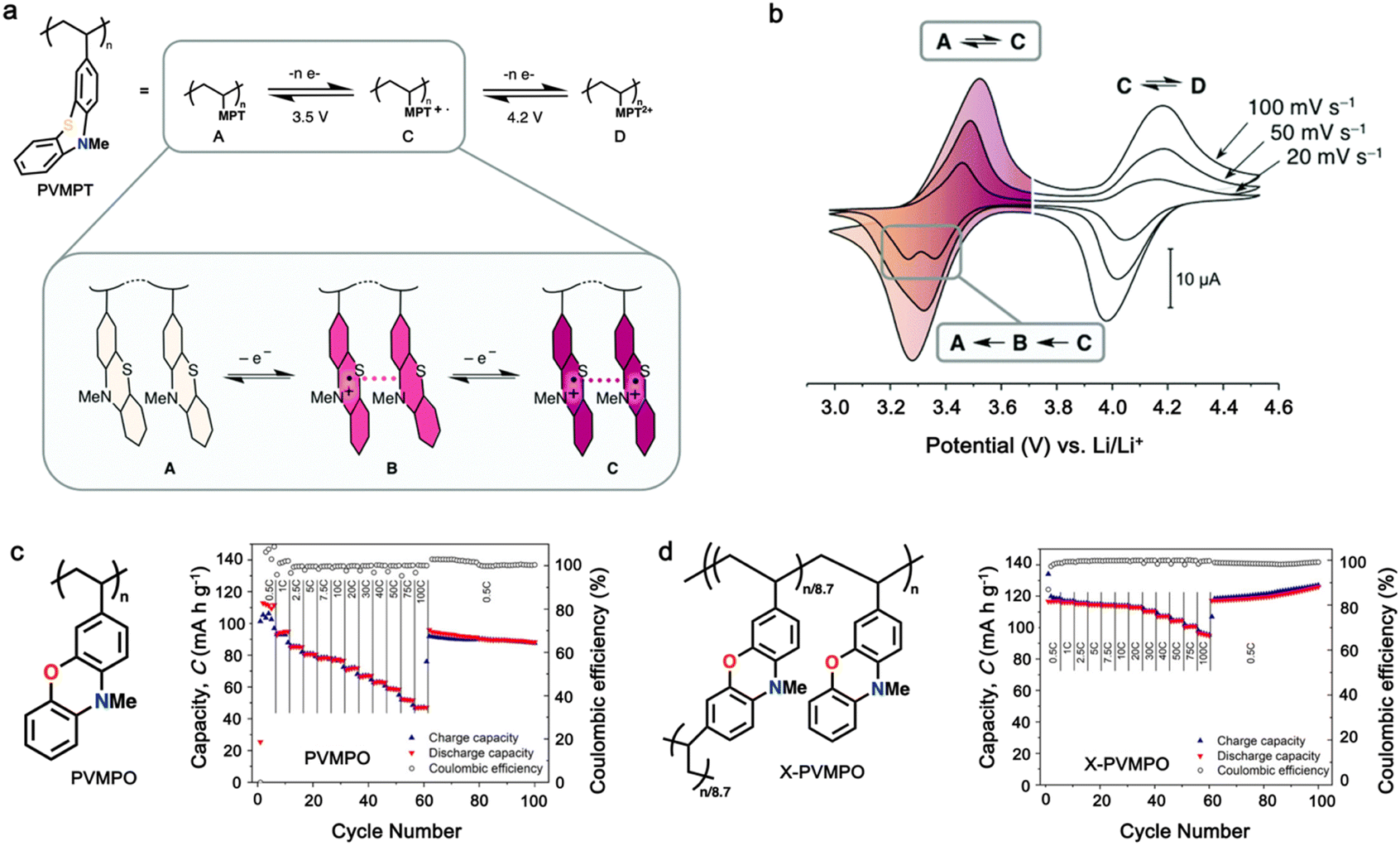 | ||
| Fig. 7 (a) Schematic of the redox processes in PVMPT cathode. (b) Cyclic voltammograms of PVMPT in solution (1 mM in CH2Cl2, 0.1 M n-Bu4NPF6, glassy carbon working electrode). Reproduced with permission from ref. 171 Copyright 2017, The Royal Society of Chemistry. (c) Chemical structure and rate performance of PVMPO cathode. (d) Chemical structure and rate performance of X-PVMPO cathode. Reproduced with permission from ref. 172 Copyright 2020, The American Chemical Society. | ||
Furthermore, phenoxazine (PXZ), a phenazine derivative, features an oxygen/nitrogen pair in its core ring and has been explored for cathode LIBs.173 Otteny et al. replaced the MPT unit in the PVMPT polymer with N-methylphenoxazine (MPO) to create a poly(3-vinyl-N-methylphenoxazine) (PVMPO) polymer (Fig. 7c).172 Showing similar electrochemical behavior to that of PVMPT, the PVMPO electrode exhibited two single-electron oxidation reactions at 3.35 and 4.22 V vs. Li/Li+, although the second reaction was not entirely reversible. Notably, PVMPO displayed inferior cycle stability due to the lack of π–π interactions between the MPO dimers compared to that of PVMPT. Additionally, the cross-linked formation of PVMPO to form the X-PVMPO polymer (Fig. 7d) effectively enhanced the cycle stability by its preventing dissolution.171,174 This strategy has significantly improved the cycling and rate capability performance of PXZ-based polymers. In addition, Table 3 illustrates the recent development of phenazine-based cathodes for organic LIBs.
| No. | Polymer material | Chemical structure | Electrolyte | Voltage (V) vs. Li/Li+ | Capacity (mA h g−1) | Current applied | Ref. |
|---|---|---|---|---|---|---|---|
| Dihydrophenazine-based polymer | |||||||
| 1 | p-DPPZ |

|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2.5–4.5 | 150 | 0.25C (1C = 0.209 A g−1) | 84 |
| 2 | p-DPPZS |
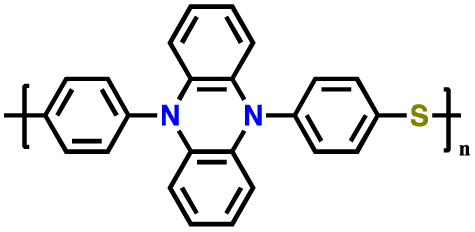
|
1 M LiPF6 EC/DEC | 2.9–4.3 | 133 | 5C (1C = 0.147 A g−1) | 175 |
| 3 | p-DPPZR-1 |
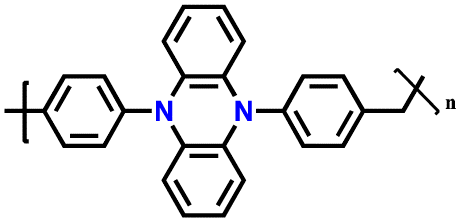
|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2.5–4.3 | 140 | 1C (1C = 0.153 A g−1) | 162 |
| p-DPPZR-2 |
|
102 | 1C (1C = 0.148 A g−1) | ||||
| p-DPPZR-3 |

|
88 | 1C (1C = 0.149 A g−1) | ||||
| 4 | Poly(135Ph-PZ) |
|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2.6–4.5 | 158 | 1 A g−1 | 163 |
| Poly(135Ph-PZ)-X |

|
180 | |||||
| 5 | Poly(Ph-PZ) |

|
1 M LiPF6 in EC/DEC (1:1, v/v) |
1.5–4.3 | 209 | 5C | 176 |
| 6 | PBEMP |
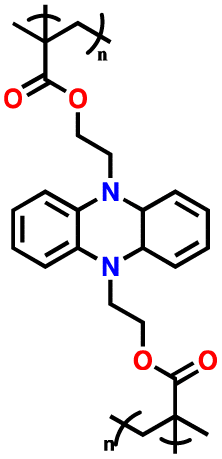
|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2–4.5 | 101 | 1 A g−1 | 177 |
| 7 | p-TPPZ |
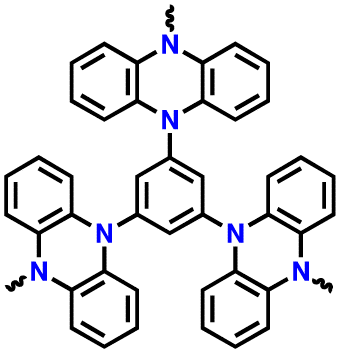
|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2.5–4.5 | 171.9 | 0.5C (1C = 0.233 A g−1) | 178 |
| p-DPPZ |
|
169.7 | |||||
| 8 | PDPAPZ |
|
1 M LiPF6 in EC/DMC | 2.5–4.2 | 107 | 0.1 A g−1 | 179 |
| PPTZPZ |
|
83 | |||||
| 9 | p-TPZB |
|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2.5–4.5 | 145 | 2C | 164 |
| 10 | CPP |
|
1 M LiPF6 in EC/DEC (1:1, v/v) |
2.5–4.2 | 184 | 0.2 A g−1 | 180 |
| NCPP |
|
149 | |||||
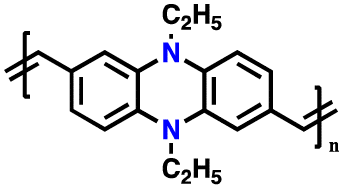
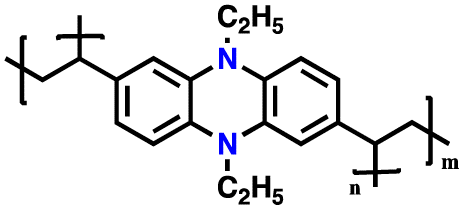
| 11 | R = Me, PMPPZ |
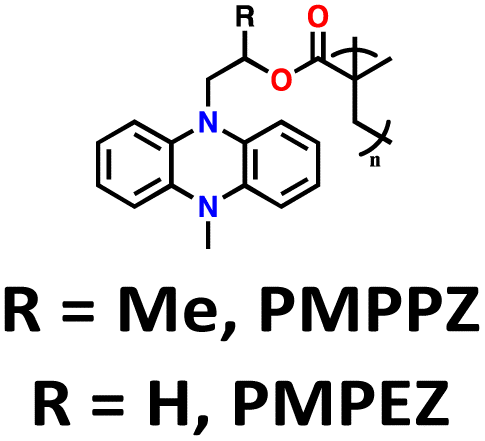
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
1.5–4.2 | 99 | 0.2 A g−1 | 181 |
| R = H, PMPEZ | 130 | ||||||
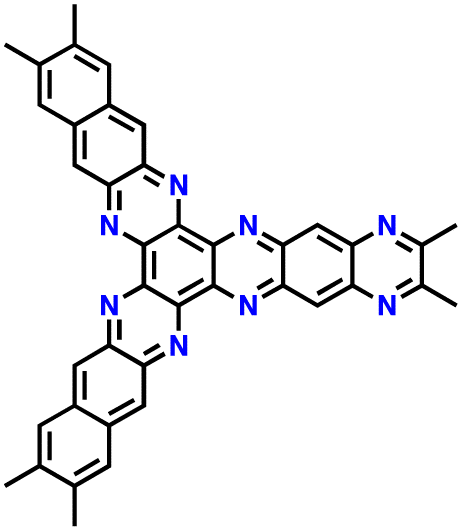
| 12 | HAT |

|
1 M LiPF6 in EC/DMC (1:1, v/v) |
1.0–3.5 | 99 | 10 A g−1 | 182 |
| 13 | N2-HATN |
|
LiCF3SO3 in G4 | 1.2–4.0 | 183 | 0.5 A g−1 | 183 |
3.4 Thianthrene and dibenzodioxin
Sharing a similar foundational structure with phenazine-based cathodes, thianthrene (TT) and dibenzodioxin (DD) have been considered next-generation high-voltage cathodes for LIBs by replacing nitrogen atoms with a sulfur atom (TT) and oxygen atom (DD), resulting in redox reactions at a higher voltage of ∼4.1 V. These heteroatoms play a vital role by providing multiple redox-active sites, enabling the essential multi-electron transfer reactions necessary for efficient charge and discharge processes in batteries. This characteristic is fundamental for achieving high specific capacities and ensuring a stable cycling performance, making both compounds highly attractive candidates for high-energy-density LIBs. A notable difference between TT and DD is the specific heteroatoms incorporated into their molecular structures. As shown in Fig. 8a, the redox mechanism of TT- and DD-based molecules demonstrates how each unit accommodates a single-electron transfer reaction. Recently, Speer and colleagues reported the preparation of three norbornene polymers, namely P1–P3, featuring one or two TT pendant groups as organic cathodes (Fig. 8b–d), respectively.149 During the CV measurement, the first oxidation of the polymers showed a high redox potential (∼4.1 V vs. Li/Li+), but the second one was irreversible for the P2 polymer (Fig. 8b). Given that only one electron was utilized during the electrochemical process, their theoretical specific capacity was limited to 73 mA h g−1 and they practically delivered a specific capacity of ∼66 mA h g−1, accounting for almost 90% of the total specific capacity. However, rapid decay over 100 cycles was observed due to irreversible anion intercalation (Fig. 8c). Importantly, the P1 electrode still delivered a specific energy of 274 W h kg−1, highlighting its promise for the preparation of high potential and high energy materials. Recently, Wild his group developed an all-organic battery consisting of a poly(2-vinylthianthrene) cathode and poly(2-methacrylamide-TCAQ) anode.185 | ||
| Fig. 8 (a) Redox mechanism of thianthrene (TT) and dibenzodioxin (DD). Chemical structure and cyclic voltammogram of TT-substituted polynorbornenes (b) P1, (c) P2, and (d) P3, respectively. Reproduced with permission from ref. 149 Copyright 2015, The Royal Society of Chemistry. (e) Schematic of the synthesis and (f) crystallographically determined solid-state structure of hyperjunction 3D-DD cathode. (g) Cyclic voltammograms and the design scheme of DMPZ, TT, and DD. (h) Charge–discharge profiles of 3D-DD. Inset figure shows MEP maps of 3D-DD and [3D-DD]+ showing the charge delocalization over the three-blade components. Reproduced with permission from ref. 92 Copyright 2023, The Royal Society of Chemistry. (i) Rate capability of DD–TCNQ cathode. (j) Redox mechanism of DD–TCNQ cathode. Reproduced with permission from ref. 184 Copyright 2019, Elsevier. | ||
The cathode material of poly(2-vinylthianthrene) displayed a discharging plateau at 3.95 V vs. Li/Li+ and a discharge capacity of 105 mA h g−1, corresponding to a specific energy of about 415 mW h g−1.
Despite their remarkable cell potential, DD-based cells suffered from severe capacity loss, reaching almost 50% during the first 10 cycles, due to the high solubility of DD molecules in the carbonate-based electrolytes system. In the early investigation of DD-based cathodes, Lee and group devised a modification strategy for a low-solubility DD analogue using a rigid iptycene scaffold to maximize the intermolecular interactions and form a 3D-DD design incorporating three DD(CN)2 units supported by the iptycene scaffold via facile nucleophilic aromatic substitution reactions (Fig. 8e).92 The methyl-substituted quaternary carbon centers at the bridgehead positions prevented C–H activation reactions during the redox cycle. The stacked 3D-DD structures were reported to form a complex structure by accommodating large lattice deformation through sliding motions of neighboring π-faces (Fig. 8f). Fig. 8g shows the voltage distribution of phenazine derivatives. As depicted in Fig. 8g, the DD electrode (blue line) displayed a redox peak at around 1.1 V vs. Ag/Ag+, which is much higher than that of PNZ (orange line) or thianthrene (TT) (green line).83,185 Notably, an increase in the redox potential of the DD molecules was achieved without the addition of any redox-inactive functional groups, which is generally accompanied by a reduction in the specific capacity.186,187 Consequently, when oxygen atoms were introduced in the core structure, it showed a remarkable increase in cell potential (4.1 V) against lithium, consequently delivering a discharge capacity of 90 mA h g−1 at 50 mA g−1 (Fig. 8h).92 Besides the formation of a complex structure, different approaches have also been employed by incorporating conductive molecules. In this regard, Lee et al. synthesized a charge-transfer complex by combining DD with tetracyanoquinodimethane (TCNQ).184 This DD–TCNQ complex demonstrated decreased solubility compared to the pristine DD and TCNQ molecules, which is attributed to the robust π–π interactions and coulombic attraction between their layers. Consequently, the DD–TCNQ electrode achieved a specific capacity of approximately 170 mA h g−1 at 50 mA g−1 within the voltage range of 2.6–4.2 V (Fig. 8i). The spectroscopic analysis unveiled that DD undergoes a single-electron redox reaction above the 4 V region, while TCNQ undergoes a two-electron redox reaction below the 4 V region during battery cycling. Throughout the charge and discharge process, it was hypothesized that the initial DD–TCNQ state transitions into DD–LiTCNQ, DD–Li2TCNQ, and DD+–TCNQ (Fig. 8j). Their shared capability for multi-electron transfer reactions, coupled with the distinct advantages offered by their respective heteroatoms, underscores their potential for next-generation high-voltage cathodes. In brief, besides the TT and DD-based compounds capability of providing higher redox activity of > 4.0 V, addressing the related issue of high solubility due to solvent interaction and side reactions associated with C–S–C bond cleavage during cycling can pave the way for their practical application as LIB cathodes with enhanced energy density, extended cycle life, and improved overall performance. Moreover, Table 4 presents the recent advances in TT and DD-based cathodes for organic LIBs.
| No. | Polymer material | Chemical structure | Electrolyte | Voltage (V) vs. Li/Li+ | Capacity (mA h g−1) | Current applied | Ref. |
|---|---|---|---|---|---|---|---|
| Thianthrene-based polymers | |||||||
| 1 | P1 |
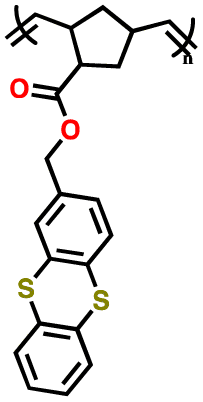
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
3.3–4.5 | 73 | 1C | 149 |
| P2 |

|
||||||
| P3 |
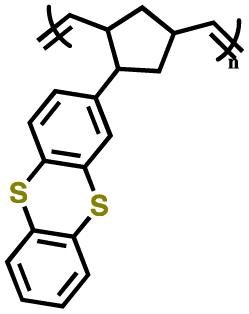
|
||||||
| 2 | Poly(2-vinylthianthrene) |
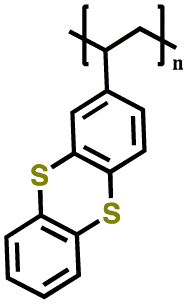
|
1 M LiClO4 in EC/DMC (3:7 v/v) |
3.2–4.2 | 105 | 1C | 185 |
| 3 | P1 |
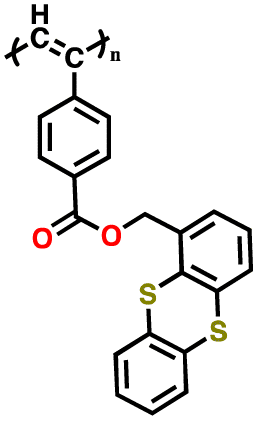
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
3.3–4.4 | 35 | 0.05 mA cm−2 | 144 |
| P2 |
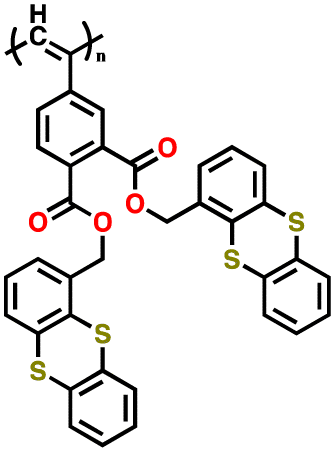
|
100 | |||||
|
|||||||
| Dibenzodioxin-based polymers | |||||||
| 4 | PIM 1 |
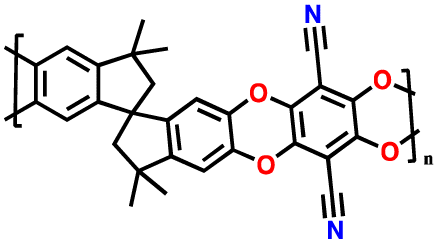
|
Dried PEO with LiTFSI (0.08 g) in 8 mL CH3CN | 1.6–2.7 | 1181 | 0.5C | 188 |
|
|||||||
| Polyviologen-based polymers | |||||||
| 5 | PVPTOCl2 |
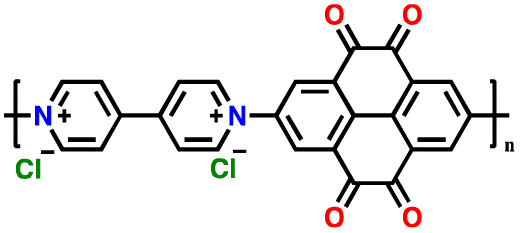
|
1 M LiTFSI in (DOL:DME, 1:1 (v/v)) |
1.5–3.8 | 235 | 0.2 A g−1 | 189 |
| PVAQCl2 |
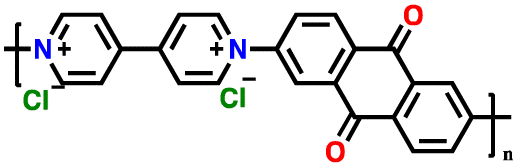
|
113 | |||||
| 6 | PBV-Cl2 |
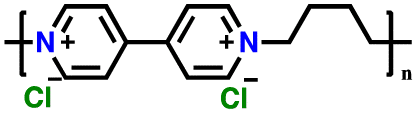
|
1 M LiPF6 in EC/DEC, (1:1, v/v) |
1.4–3.1 | 177 | 0.2 A g−1 | 190 |
| 7 | PVBVEtX2 |
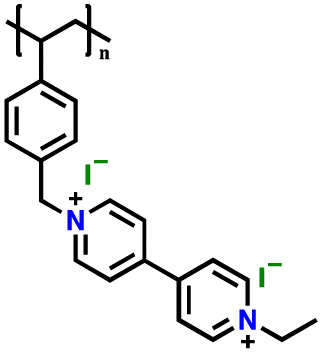
|
2 M LiClO4 in TEGDME | 1.2–3.8 | 192 | 0.1 A g−1 | 191 |
| 8 | P-MV |
|
1 M LiTFSI in DME | 1.8–3.2 | 60 | 0.33C | 192 |
| 9 | PXVCl2 |
|
0.5 M tributylmethylammonium chloride in propylene carbonate | 1.5–3.2 | 140 | 0.01 A g−1 | 193 |
| 10 | EV(ClO4)2 |
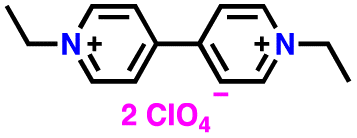
|
2 M LiClO4 in TEGDME | 1.6–3.0 | 176 | 0.5C | 194 |
| EVI2 |
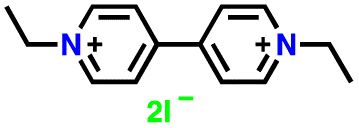
|
227 | |||||
| 11 | POF |
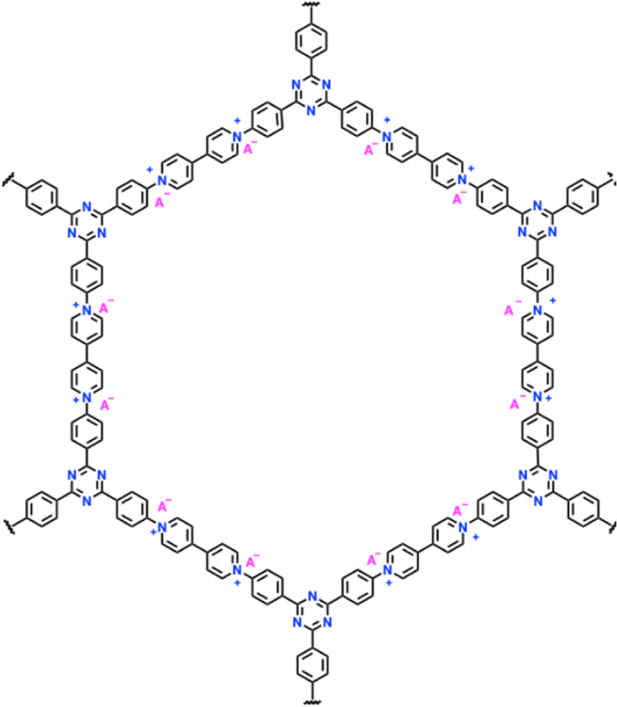
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
1.5–4.5 | 130 | 3 A g−1 | 195 |
3.5 Polyviologens
Polyviologens, also known as poly(4,4′-bipyridinium) compounds, are a class of organic materials that have gained significant attention for potential application in high-performance LIBs.196–198 These materials as cathode LIBs are characterized by unique redox-active viologen units, which consist of an anion doping/de-doping process on two nitrogen-containing pyridinium rings connected by a conjugated bridge, leading to the formation of stable radical cations and dications.190–194,199–201 Additionally, polyviologen-based cathodes offer several desirable properties for use in LIBs, including high electrochemical stability, good conductivity, and excellent cycling performance.202 To further explore the potential of viologens, in 2015, Yao and colleagues introduced the first solid-type electrode based on viologen, presenting the viologen polymer poly(1,1′-pentyl-4,4′-bipyridinium dihexafluorophosphate (PBPy)), as shown in Fig. 9a (inset). The PBPy electrode exhibited two voltage plateaus at 2.6 and 2.1 V vs. Li/Li+, delivering a specific capacity of 79 mA h g−1 in the initial cycle.203 However, the low degree of polymerization led to rapid capacity degradation to 36 mA h g−1 after 20 cycles. Nonetheless, when utilized as an anode, PBPy enabled the fabrication of an all-organic full-cell with poly(N-vinylcarbazole) (PVK) as the cathode, achieving a specific capacity of 100 mA h g−1 at a voltage of 1.8 V, relying on the PF6− anion charge carrier (Fig. 9a).203 Furthermore, to improve the specific capacity and cycling performance, an anion insertion approach was reported to effectively increase the performance of viologen-based cathodes. For instance, Wang et al. reported the first Cl−-insertable p-type polymer, namely poly(butyl viologen dichloride) (PBV-Cl2), as a cathode for LIBs (Fig. 9b).190 As shown in Fig. 9c, PBV-Cl2 demonstrated a significant capacity improvement of up to 200 mA h g−1 in the initial cycles compared to that of PVB(PF6)2 at a current density of 50 mA g−1. The improved specific capacity is associated with the presence of an inserted Cl− ion in the structure, which contributed to the ion storage mechanism during the charge/discharge process. However, rapid capacity decay was observed during cycling in the first 50 cycles due to the dissolution of the PBV-A2 polymer in the carbonate-ether-based electrolyte and the inevitable anion exchange during the discharge–charge process (for PBV-Br2 and PBV-Cl2).190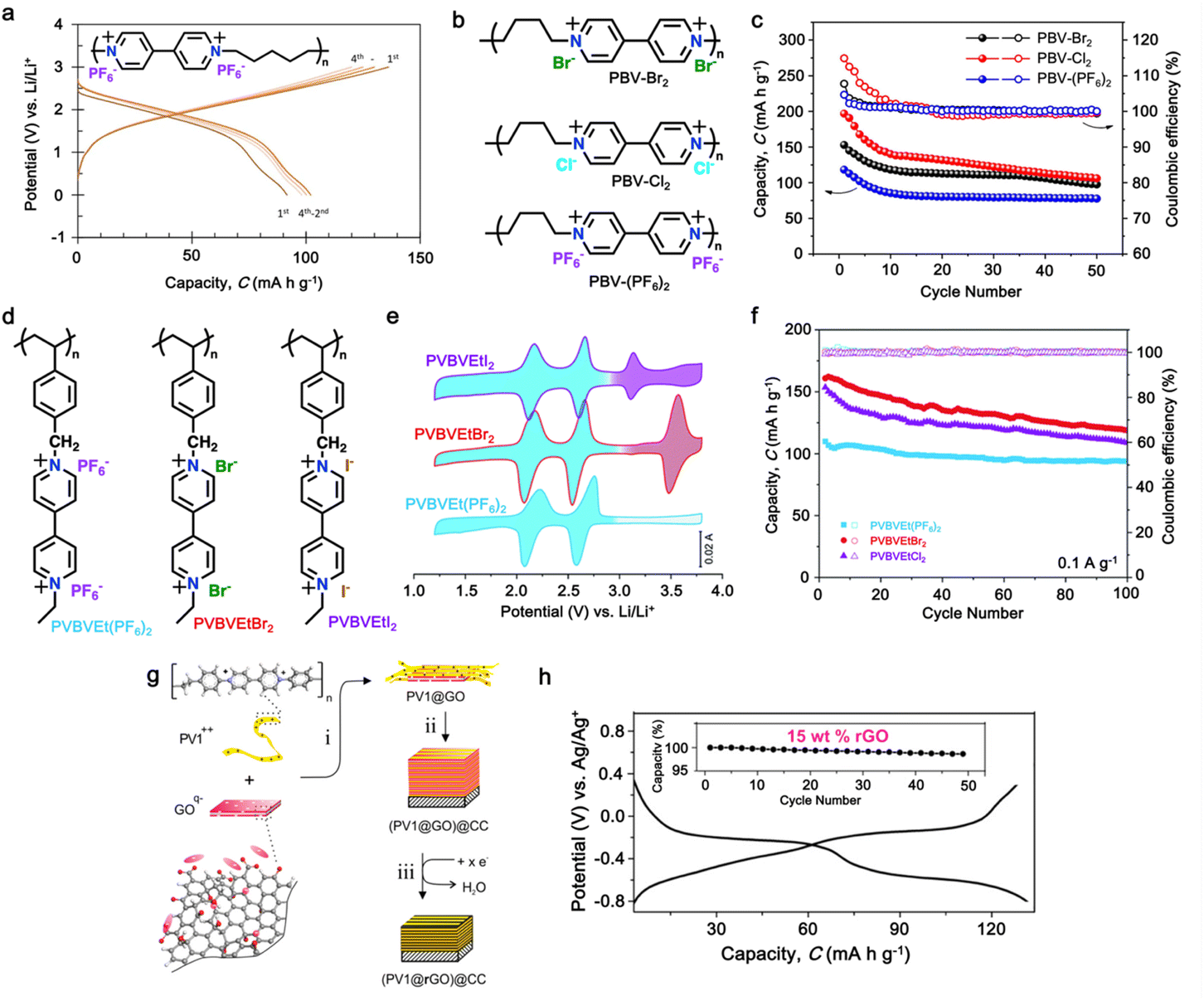 | ||
| Fig. 9 (a) Galvanostatic charge–discharge profile of full-cell PBPy. Reproduced with permission from ref. 203 Copyright 2015, Springer Nature. (b) Chemical structure and (c) cycling performance under 50 mA g−1 in 1 M LiPF6 EC:DEC electrolyte (voltage window of 1.4–3.1 V) of poly(butyl viologen)-based cathode. Reproduced with permission from ref. 190 Copyright 2022, Elsevier. (d) Chemical structures of viologen-based ionic polymers PVBVEtX2 (X = PF6−, Br− or I−). (e) and (f) CV curves at 0.1 mV s−1 scan rate and cycling performance under 0.1 A g−1 current density of PVBVEt(PF6)2, PVBVEtBr2, and PVBVEtI2 electrodes. Reproduced with permission from ref. 204 Copyright 2022, The Royal Society of Chemistry. (g) Overview of the preparation of poly-viologen/rGO composite electrode, (PV1@rGO)@CC. (h) Chronopotentiometric measurements of (PV1@rGO)@GC in 3 M KCl/H2O at cutoff voltages in the range of 0.3–0.8 V, at a rate of 5 A g−1. Reproduced with permission from ref. 199 Copyright 2017, The American Chemical Society. | ||
Similarly, a series of viologen-based cathodes was prepared to investigate the impact of the counter anion in the polymer structure.191 As shown in Fig. 9d, poly(viologen halide)-based cationic polymers with Br− and I− as counter anions were prepared and nominated as PVBVEtBr2 and PVBVEtI2, respectively. Notably, an additional redox peak appeared when the counter anions of Br− and I− were introduced in the viologen-based cathode. As shown in Fig. 9e, a strong and reversible redox peak was present at 3.53 V for PVBVEtBr2, which can be assigned to the one-step reaction of the Br−/Br3− redox couple; while two redox peaks centered at 3.12 and 3.66 V were observed for PVBVEtI2, corresponding to the oxidation of I− to I3− (3.12 V), and then to I2 (3.66 V). This involvement of the counter anion significantly improved the specific capacity of the viologen-based cathodes in the initial cycles, before gradually decaying up to 100 cycles (Fig. 9f). The capacity loss observed for the PVBVEtBr2 and PVBVEtI2 electrodes was mostly caused by the slight diffusion of Br3−, I3− and I2 species from the electrodes during the charge/discharge process.191 In addition, it was reported that the formation of a composite electrode could improve the cycling stability of viologen-based polymers during the electrochemical process (Fig. 9g).199 As shown Fig. 9h, the formation of a viologen-based polymer composite with 15 wt% reduced graphene oxide (rGO) significantly enhanced the cycling performance, given that negligible capacity loss was observed up to 50 cycles. Overall, the unique electrochemical properties and tunable structures of polyviologens make them promising candidates for the fabrication of high-performance LIBs and other energy storage technologies, with ongoing research aimed at further optimizing their performance and expanding their applications in the future. Moreover, Table 4 shows the recent development of viologen-based cathodes for organic LIBs.
3.6 Nitroxide radical polymers
During the past decade, radical organic compounds have garnered significant attention in the realm of polymer-based batteries.72,205–207 They feature polymers with pendant stable organic radicals, characterized by an unpaired electron in their uncharged state. These polymers offer superior redox chemistry with favorable kinetics, facilitated by redox reactions involving the singly occupied molecular orbital, enabling rapid electron transfer and high-rate performance capability. Considering the nonconjugated backbones and stable open-shell pendant groups of nitroxide radical polymers, they offer charge transport through a series of redox reactions between the pendant open-shell sites. Therefore, stable radicals such as nitroxyl, phenoxyl,205,208 nitroxylbenzene,209 nitronylnitroxyl,210 and hydrazyl211 groups, with structures such as 2,2,6,6-tetramethylpiperidine-N-oxy (TEMPO), have been explored as LIB cathodes.207,212,213 Among the radical polymers, poly(2,2,6,6-tetramethylpiperidinyloxy-4-vinylmethacrylate) (PTMA)54,166,214,215 and 2,2,6,6-tetramethyl-1-piperidinyloxy (PTVE)213,216 have been investigated and showed high redox potentials of ∼3.6 V vs. Li/Li+. In this regard, a nitroxide radical polymer based on an ethylene oxide backbone, poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl) (PTEO), was designed and reported (Fig. 10a). As shown in Fig. 10b, a two-step redox reaction of PTEO was observed in the CV curves, which is associated with the formation of the aminoxy anion at 2.75/3.20 V and oxoammonium cation at 3.48/3.60 V.217 Due to the super high electrical conductivity of PTEO due to the unique open-shell site of the monomer and the completed conservation of all of the radical sites in the monomer, this PTEO delivered a specific capacity of 154 mA h g−1 at a 1C rate after 150 cycles, corresponding to 91% capacity retention (Fig. 10c). Additionally, to improve the material stability and minimize the polymer solubility, Jin et al. grafted a PTMA-based polymer onto the surface of reduced graphene oxide (rGO) via in situ free radical polymerization (FRP), as shown in Fig. 10d. The electrochemical performance of rGO-g-PTMA showed that specific capacities ranging from 176 to 119 mA h g−1 can be obtained at a current rate of 1C by varying the PTMA loading from 38 to 54 wt% (Fig. 10e). Notably, the rGO-g-PTMA50 cathode exhibited the highest specific capacity. Moreover, CV measurement confirmed the electrochemical performance of rGO-g-PTMA, with the rGO-g-PTMA50 sample demonstrating a superior performance, as shown in Fig. 10f.218 Employing a similar approach, Zhang et al.219 investigated a PTMA-based copolymer cathode combined with nanostructured carbon-based electrodes through non-covalent interactions (Fig. 10g). Prior to the addition of conductive carbon such as rGO and CNTs, ultrafast single electron transfer-nitroxide radical coupling was applied to introduce pyrene groups in PTMA, forming P(TEMPO-co-PyMA) copolymers.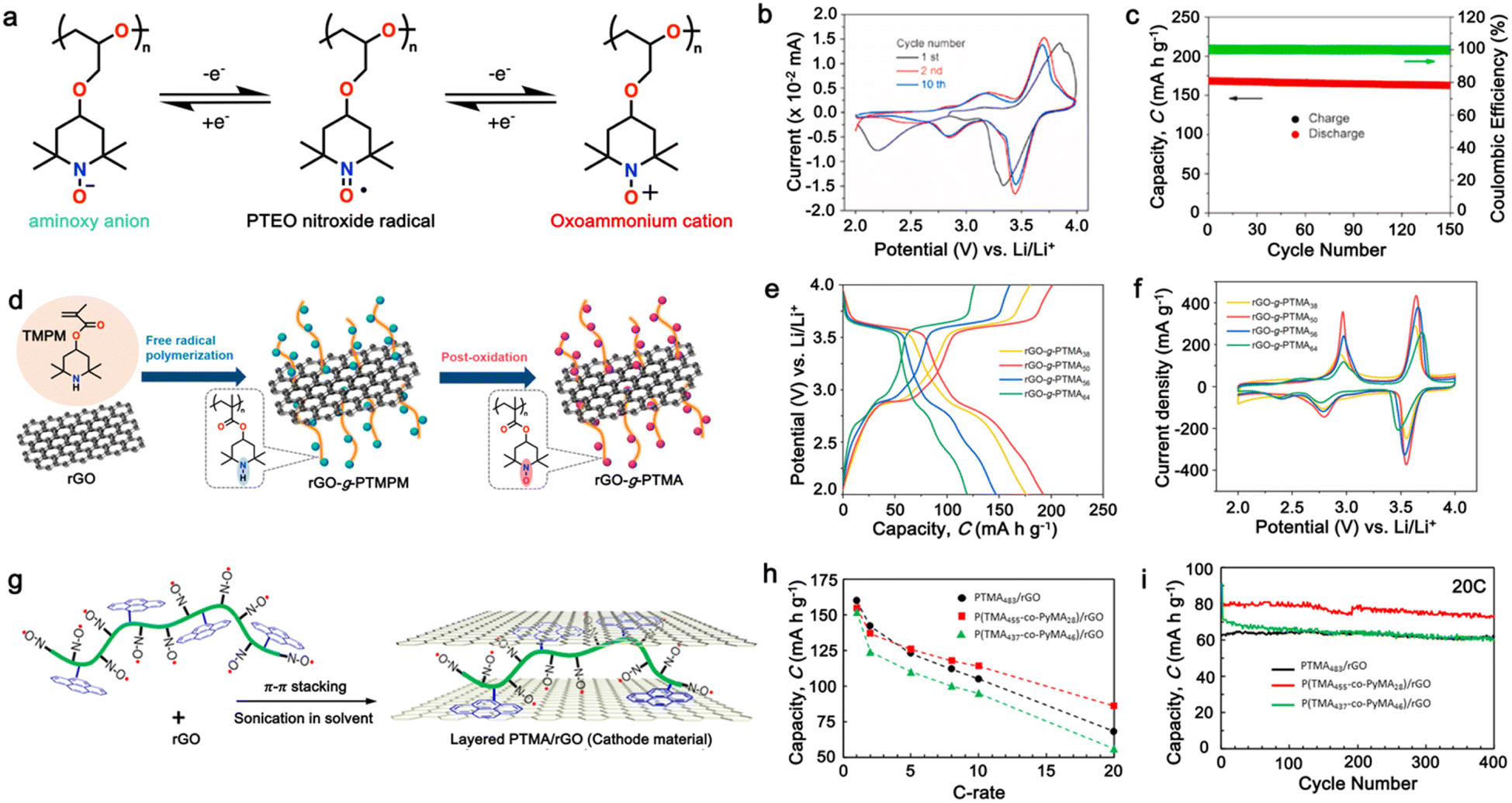 | ||
| Fig. 10 (a) Electrochemical mechanism of PTEO nitroxide radical during charge/discharge cycles. (b) and (c) CV curves at a scan rate of 0.5 mV s−1 and cycling performance under 1C rate (1C = 240 mA g−1) of PTEO nitroxide radical, respectively. Reproduced with permission from ref. 217 Copyright 2020, Elsevier. (d) Schematic of the preparation of PTMA-grafted graphene sheets via free radical polymerization. (e) Galvanostatic charge–discharge curves (2nd cycle) under 1C and (f) CV curves of rGO-g-PTMA with different PTMA loadings. Reproduced with permission from ref. 218 Copyright 2021, Elsevier. (g) Formation of a layered PTMA/rGO composite through noncovalent π–π stacking between pyrene groups and rGO sheets. (h) and (i) Rate performance and cycling stability of layered PTMA/rGO composite, respectively. Reproduced with permission from ref. 219 Copyright 2017, the American Chemical Society. | ||
After that, the P(TEMPO-co-PyMA) copolymers were wrapped with rGO using ultrasonication and ultracentrifugation. Subsequently, these copolymers were bound to rGO flakes, forming electrode composites with a multilayered sandwich-like structure (Fig. 10g). This approach successfully enhanced the performance of the PTMA-based cathode, which delivered a high capacity of ∼150 mA h g−1 at a 1C rate (Fig. 10h) and exhibited outstanding stability under a high current rate of 20C (Fig. 10i).219Table 5 describes the current trends in developing nitroxyl radical-based cathodes for organic batteries.
| No. | Polymer material | Chemical structure | Electrolyte | Voltage (V) vs. Li/Li+ | Capacity (mA h g−1) | Current applied | Ref. |
|---|---|---|---|---|---|---|---|
| Nitroxide radical-based polymers | |||||||
| 1 | PNPP |
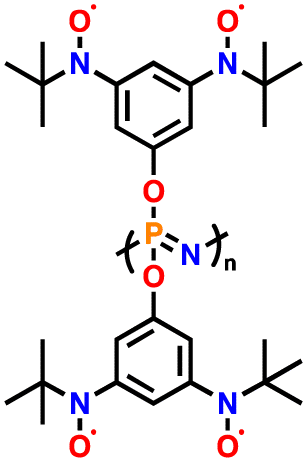
|
1 MLiPF6 in EC:DMC (1:1, v/v) |
2.6–4.0 | 100 | 0.5C | 220 |
| 2 | PTMA-filled NCNT |
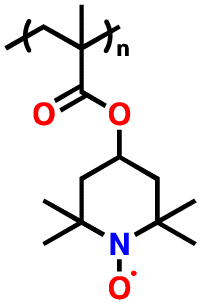
|
1.2 M LiPF6 in EC:DEC, (1:1, v/v) |
1.5–4.0 | 159.6 | 1C | 221 |
| 3 | MWNT-g-PTMA |

|
1 M LiPF6 in (DMC:EC:EMC, 1:1:1 v/v/v) |
2.0–4.0 | 243 | 1C (1C = 0.222 A g−1) | 222 |
| 4 | PTMA |

|
1 M LiPF6 in DMC | 2.8–4.0 | 67 | 1C | 223 |
| 5 | PTEO |

|
1 M LiPF6 in PC | 2.0–4.0 | 220 | 0.2C (1C = 0.24 A g−1) | 217 |
| 6 | PTMA |

|
1 M LiPF6 in EC:DMC, (1:1, v/v) |
1.5–4.0 | 219.8 (2e−) | 1C | 224 |
| 110.9 (1e−) | |||||||
| 7 | PETM |
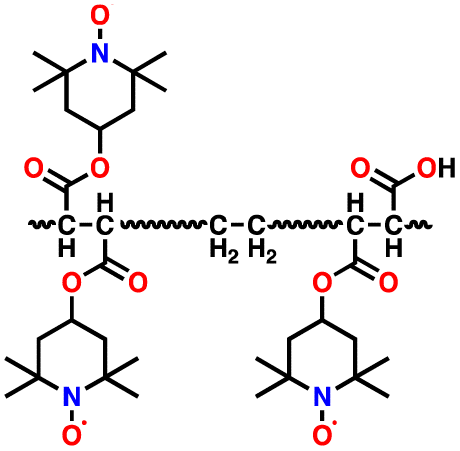
|
1 M LiPF6 in EC:DMC:EMC, (1:1:1 v/v/v) |
3.0–4.2 | 90 (binder free) | 10C | 225 |
| 99.5 (with PVDF) | |||||||
| 8 | PTMA |
|
1 M LiPF6 in EC:DEC, (1:1, v/v) |
3.0–4.2 | 32 | 0.025 mA | 226 |
| 9 | P(TMA-r-PyrM) |

|
1 M LiPF6 in EC:DEC, (1:1, v/v) |
3.0–4.2 | 105 | 0.5C | 227 |
4. Alternative design of p-type polymer electrodes
4.1 Bipolar-type polymers
Bipolar-type polymers have emerged as promising candidates for next-generation high-performance LIBs due to their unique redox properties involving both p-type and n-type charge carriers, enabling the optimum charge storage and transport within the electrode material.11 Due to the synergistic contribution of p- and n-type charge storage character, the nature of these polymers allows them to store and release both cations and anions during the charging and discharging processes, leading to enhanced energy storage capabilities.22 This feature is particularly advantageous for achieving high specific capacities and energy density in LIBs.228 Bipolar-type polymers have demonstrated impressive electrochemical performance metrics in LIBs, including high specific capacities, excellent rate capabilities, and long-term cycling stability.229 These polymers hold promise for a wide range of applications in portable electronics, electric vehicles, and grid energy storage systems, where high energy density, fast charging/discharging rates, and durability are critical requirements.230 Recently, a series of bipolar polymers has been reported to significantly improve the specific capacity of LIBs. For instance, Zhang et al. reported two bipolar-type polymers by combining arylamine-based (p-type) and carbonyl-based (n-type) moieties to form poly(arylamine-imide)s, namely poly(N,N,N′,N′-tetraphenyl-1,4-benzenediamine naphthalenediimide) (PDDP-NI) and poly(N,N,N′,N′-tetraphenyl-1,4-benzenediamine perylenediimide) (PDDP-PI), as shown in Fig. 11a and b.118 Bipolar activity was observed given that these polymers exhibited specific redox properties associated with their arylamine- and imide-units. As shown in Fig. 11a and b, both PDDP-NI and PDDP-PI exhibited three pairs redox peaks at ∼2.46/2.30, 3.56/3.44, and 3.98/3.87 V, respectively. The first pair redox peak is ascribed to the redox activity of the carbonyl group in the imide unit (n-type), while the other two redox couples are associated with the doping/de-doping process of the arylamine unit (p-type). Furthermore, this strategy successfully improved the cathode specific capacity up to 150.9 (PDDP-NI) and 119.4 (PDDP-PI) mA h g−1 after 70 cycles at 0.1C.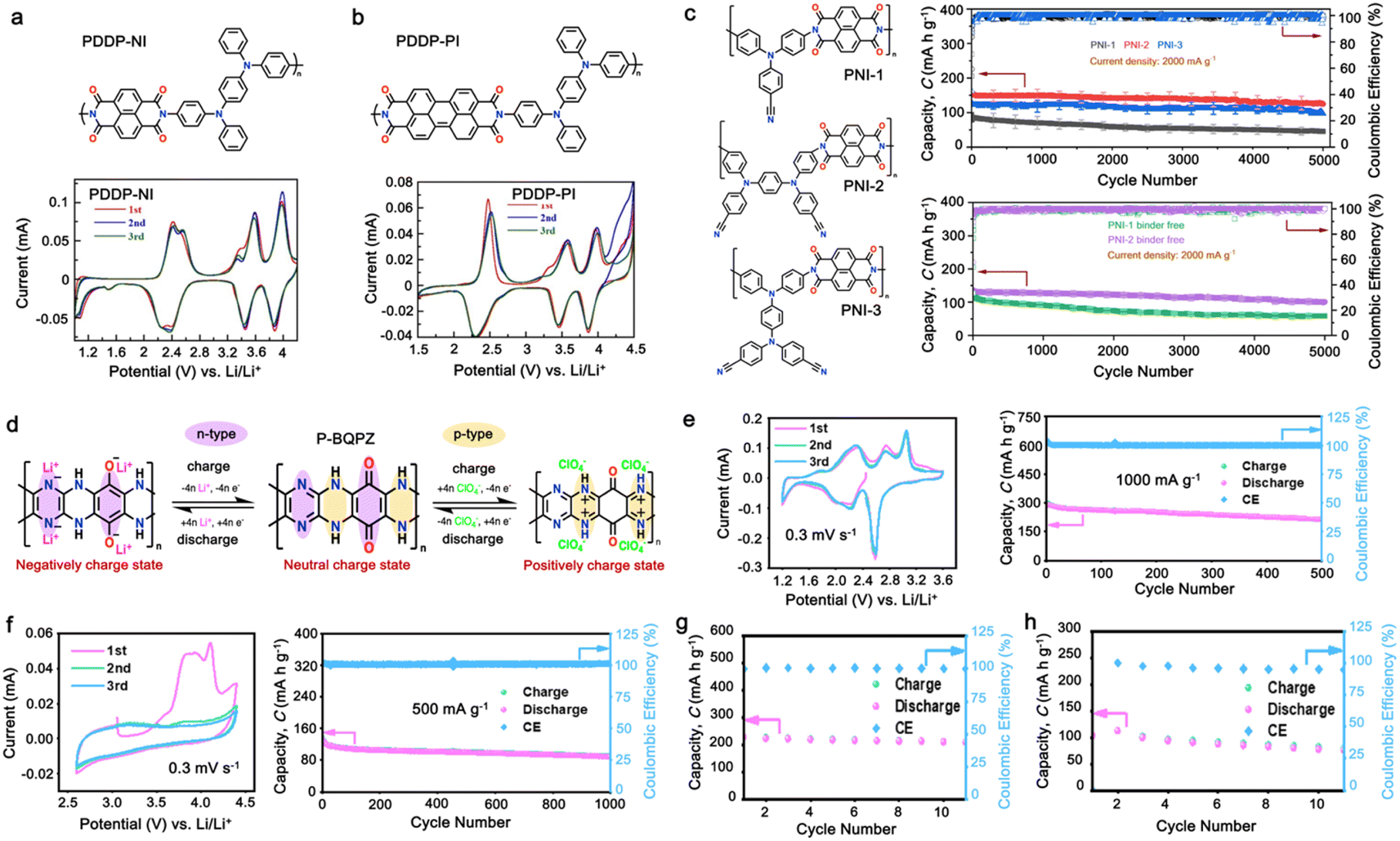 | ||
| Fig. 11 (a) and (b) Chemical structure and cyclic voltammogram of PDDP-NI and PDDP-PI bipolar polymer, respectively. Reproduced with permission from ref. 231 Copyright 2021, The Electrochemical Society. (c) Chemical structures and long cycling performance of redox-active polynaphthalimides (PNIs). Reproduced with permission from ref. 62 Copyright 2023, The Royal Society of Chemistry. (d) Chemical structure and redox reaction mechanism of the bipolar polymer P-BQPZ. (e) and (f) CV curves and long cycling performance of n-type P-BQPZ and p-type P-BQPZ, respectively. (g) Cycling performance of the full cell based on n-type redox reaction of P-BQPZ at a current of 500 mA g−1 between 0.1 and 2.7 V. (h) Cycling performance of the full cell based on p-type redox reaction of P-BQPZ at current of 50 mA g−1 between 0.1 and 3.4 V. Reproduced with permission from ref. 232 Copyright 2022, Elsevier. | ||
Employing a similar approach, a series of bipolar triphenylamine-based polynaphthalimides (TPA-PNIs) was reported not only as a cathode, but also as an anode and binder-free cathode in LIBs.62 These TPA-PNI cathodes delivered a high specific capacity of up to 195 mA h g−1 after 100 cycles at 50 mA g−1 due to the synergistic contribution from both the p- and n-type units. As shown in Fig. 11c, these polymers exhibited outstanding stability up to 5000 cycles under an extreme current density of 2000 mA g−1. Notably, no significant capacity drop was observed even as a binder-free cathode (Fig. 11c), suggesting the excellent material stability and dual-role ability as active materials and electrode binder simultaneously. In another study, Labasan et al. developed two polyimide (PI) derivatives, TPA-PMPI and TPA-NTCPI, as electrode materials for LIBs.58 These polymers exhibited excellent thermal stability and bipolar properties. The TPA-NTCPI cathode delivered a reversible specific capacity of 150 mA h g−1 at 0.1 A g−1 and showed stability up to 1000 cycles, while the TPA-PMPI anode achieved a high specific capacity of up to 1600 mA h g−1 at 0.1 A g−1 after 100 cycles.58 Furthermore, various polymerization methods have also been studied to integrate bipolar moieties. Wang and colleagues successfully polymerized amino-phenyl carbazole naphthalene diimide (APCNDI) using in situ electropolymerization to eliminate the dissolution problem.233 The electropolymerized cathode demonstrated an excellent electrochemical performance, stable cycling performance, and superior rate performance. In addition, employing a different strategy, Zhao and colleagues integrated a series of n- and p-type redox-active moieties into one stable polymer backbone to minimize the redox-inactive moieties (Fig. 11d).232 As shown in Fig. 11e, the CV curves of P-BQPZ demonstrate clear and reversible redox peaks of n-type redox reactions, which are attributed to the CO bond at 2.6, 2.7, and 3.0 V (vs. Li/Li+) and CN bond at 2.3 and 2.2 V (vs. Li/Li+). This redox activity contributed to the specific capacity of 213.3 mA h g−1 up to 500 cycles at a current density of 1000 mA g−1 (Fig. 11e). Additionally, the activity of the P-BQPZ p-type cathode is depicted in Fig. 11f, showing higher redox activity at 3.1 V (vs. Li/Li+) and contributing a capacity of ∼120 mA h g−1 up to 1000 cycles at 500 mA h g−1. Additionally, the cycling performance of the full cell based on the n-type and p-type redox reaction of P-BQPZ is presented in Fig. 11g and h, respectively, depicting the successful incorporation of P-BQPZ in the full cell LIB system. These findings provide a novel strategy for designing and fabricating high-performance cathode LIBs by combining two different electrochemical characters, thus offering potential solutions to improve the energy density. Continued research efforts are aimed at advancing the synthesis, characterization, and understanding of these polymers as well as their minimizing synthetic cost, which are essential for realizing their full potential in energy storage applications. Furthermore, Table 6 describes the current trends in developing alternative designed p-type cathodes for organic batteries.
| No. | Polymer material | Chemical structure | Electrolyte | Voltage (V) vs. Li/Li+ | Capacity (mA h g−1) | Current applied | Ref. |
|---|---|---|---|---|---|---|---|
| Bipolar-type polymers | |||||||
| 1 | PAQDPZ |
|
3 M LiFSI in TEGDME | 1.6–4.3 | 105 (symmetric organic battery) | 0.2 A g−1 | 234 |
| 1 M LiPF6 in EC/DEC | 1.5–4.2 | 208 (Li-ion full cell) | 0.2 A g−1 | ||||
| 3 M LiFSI in TEGDME | 1.6–4.3 | 222 (Li-ion half-cell) | 0.2 A g−1 | ||||
| 2 | Poly(CoL)n |

|
1 M LiPF6 in EC:DEC, (1:1, v/v) |
1.5–4.5 | 125.16 | 2 A g−1 | 235 |
| 3 | P-BQPZ |
|
1 M LiTFSI in DOL:DME (1:1, v/v) |
2.6–4.4 | 130 (p-type) | 0.5 A g−1 | 232 |
| 1 M LiClO4 in EC:DMC (1:1, v/v) |
1.2–3.6 | 298 (n-type) | 1 A g−1 | ||||
| 4 | PNI-1 |

|
1 M LiPF6 in EC:DEC (1:1, v/v) |
1.5–4.5 | 125 | 0.05 A g−1 | 62 |
| PNI-2 |
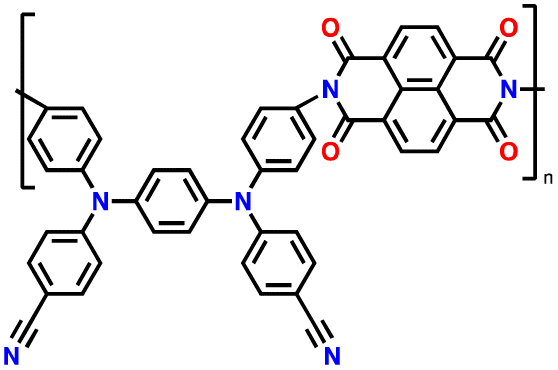
|
195 | |||||
| PNI-3 |
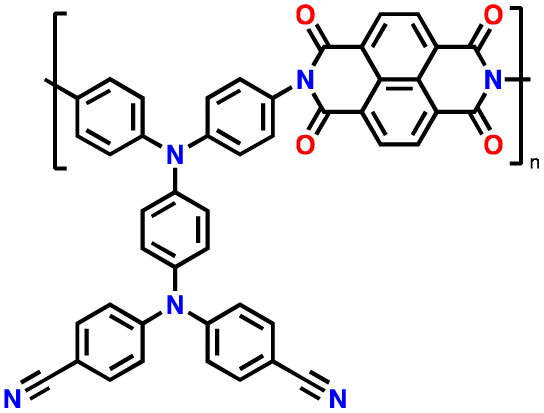
|
170 | |||||
| 5 | DAAQ |
|
1 M LiPF6 in EC:DMC (1:1, v/v) |
1.5–4.5 | 311 | 0.05 A g−1 | 236 |
| 6 | TPA-PMPI |
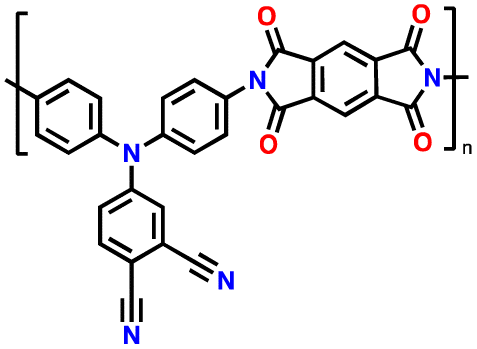
|
1 M LiTFSI in DOL:DME (1:1, v/v) |
1.5–3.5 (cathode) | 150 | 0.1 A g−1 | 58 |
| TPA-NTCPI |
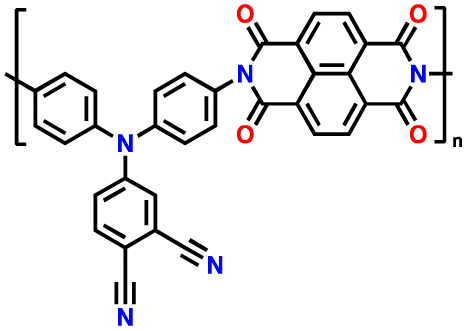
|
||||||
|
|||||||
| Hybrid organic–inorganic polymer | |||||||
| 7 | LiV3O8/polythiophene (LiV3O8/PTh) | 1 M LiPF6 in EC:DMC (1:1, v/v) |
1.8–4.0 | 213.3 | 1C (1C = 0.3 A g−1) | 237 | |
| 8 | LiV3O8/polydiphenylamine | 1 M LiPF6 in EC:DMC:EMC, (1:1:1, v/v/v) |
1.8–4.0 | 311 | 0.06 A g−1 | 238 | |
| 9 | pCPDT-Fc P1 |

|
1 M LiPF6 in EC:DMC (1:1, v/v) |
2.7–3.9 | 59.7 | 0.1C (1C = 0.066 A g−1) | 239 |
| pDTP-Fc P2 |

|
59.8 | |||||
| 10 | NMC/PPy | 1 M LiPF6 in EC:DMC:EMC, (1:1:1 (v/v/v) |
2.5–4.5 | 202.2 | 0.2C | 240 | |
| 11 | LMNC@Li3PO4&PANI | 1 M LiPF6 in EC:DMC:EMC, (1:1:1) (v/v/v) |
2.0–4.8 | 172.60 | 1C | 241 | |
| 12 | Li1.2Ni0.2Mn0.6O2/PEDOT:PSS | 1 M LiPF6 in EC:DMC (1:1, v/v) |
2.0–4.8 | 285 | 0.2C (1C = 0.3 A g−1) | 242 | |
|
|||||||
| Copolymer materials | |||||||
| 13 |
|
2 M LiTFSI in DOL/DME (1:1, v/v) |
1.5–3.0 | 127 | 0.1C | 243 | |
| 84 | 1C (1C = 0.172 A g−1) | ||||||
| 14 | Pyromellitic polyimide-b-PEO |
|
1 M LiTFSI/MeTHF | 1.0–3.4 | 225 | 1.1C (1C = 0.203 A g−1) | 244 |

|
160 | ||||||
| 15 | pBTC-TEMPO |
|
1 M LiPF6 in EC/DMC, (1:1, v/v) |
3.0–4.0 | 50 | 1C (1C = 0.06 A g−1) | 245 |
| 16 | Poly-PPDA-PYR |

|
1 M LiPF6 in EC/DMC, (1:1, v/v) |
2.0–4.2 | 75 | 0.5 A g−1 | 246 |
| 17 | PPh-PTO |
|
1 M LiTFSI in DOL/DME(1:1, v/v) |
1.5–3.8 | 235 | 0.1 A g−1 | 247 |
| 18 | PENDI |

|
1 M LiPF6 in EC/DMC, (1:1, v/v) |
1.0–4.0 | 110 | 0.1C (1C = 0.202 A g−1) | 248 |
| 19 | PTMA-co-GMA |

|
1 M LiPF6 in EC/DEC (1:1, v/v) |
3.0–4.0 | 104 | 0.1C | 249 |
| 20 | p(APT-T2) |
|
1 M LiPF6 in EC/DMC (1:1, v/v) |
3.2–4.2 | 68.5 | 1C | 250 |
| 21 | PTPA-HATN |

|
1 M LiPF6 in EC/DMC/EMC (1:1:1, v/v/v) |
1.5–4.2 | 120 | 0.025 A g−1 | 251 |
| PDTPA-HATN |
|
103.6 | |||||
| 22 | NSHATN |

|
LiCF3SO3 in G4 | 1.5–4.0 | 337 | 0.05 A g−1 | 252 |
| 23 | HATCNOC-poly |
|
1 M LiTFSI in TEGDME | 1.0–2.8 | 158.6 | 0.05 A g−1 | 253 |
4.2 Hybrid organic–inorganic polymers
Hybrid organic–inorganic polymers have emerged as promising alternatives to improve the electrochemical performance and stability of polymer-based electrodes. This synergistic combination of both organic and inorganic character offers opportunities to overcome the limitations associated with conventional electrode materials and develop high-performance LIBs with enhanced energy storage capabilities. One common approach in designing organic–inorganic hybrid electrodes involves incorporating a polymer matrix into the structure of inorganic materials. For example, organic molecules such as conducting polymers, viologens, and redox-active organic compounds can be integrated with inorganic materials such as metal oxides, sulfides, phosphides, and carbon-based materials to form hybrid electrode composites.254 These composites exhibit improved electrochemical properties, including high capacity, cycling stability, and rate capability, compared to their individual components. Another strategy involves the synthesis of nanostructured hybrid materials, where organic and inorganic components are intimately intertwined at the nanoscale. This approach enables precise control of the morphology, surface area, and interfacial properties of the electrode materials, leading to enhanced ion diffusion kinetics and electrochemical performance.255 Nanostructured hybrid materials can be fabricated using techniques such as sol–gel synthesis, hydrothermal synthesis, chemical vapor deposition, and electrodeposition.256Several types of organic–inorganic hybrid materials have been investigated for LIB electrodes, including metal–organic frameworks (MOFs),255,257,258 covalent organic frameworks (COFs),53,65,259–261 conductive polymers/carbon composites,262,263 and redox-active organic–inorganic composites.264,265 In particular, Zhu et al. developed a composite cathode material by introducing polydiphenylamine (PDPA) in the lithium trivanadate (LiV3O8, LVO) using an in situ oxidative polymerization method, leading to significant improvements in electrochemical properties and the inhibition of adverse reactions, as shown in Fig. 12a.238 The TEM analysis revealed that the surfaces of LiV3O8 were coated with a layer of PDPA, with an average polymer thickness of around 20 nm (Fig. 12b). The 10 wt% LiV3O8/PDPA composite exhibited a high initial specific discharge capacity of 311 mA h g−1, which decreased to 272 mA h g−1 after 50 cycles at a current density of 60 mA g−1. Furthermore, the composite displayed a remarkable improvement in rate capability, with its discharge capacities at various current densities outperforming that of the pure LVO electrodes (Fig. 12c). This increased performance of the LVO-based cathode could be ascribed to its high conductive coating of PDPA. With a similar aim, LiFePO4/poly(3,4-ethylenedioxythiophene) (PEDOT) composites were prepared.267 Furthermore, poly(aniline) was also explored to coat an LiFePO4 cathode, resulting in a hybrid polymer–inorganic composite. It was demonstrated that the polymer not only served as a conductive matrix and binder but also as an additional host for lithium-ion intercalation. At a 0.2C rate, it achieved a capacity of 165 mA h g−1, which decreased by 25% to 123 mA h g−1 at a 10C rate. Despite this, the discharging curves remained flat, suggesting its good cycling stability.268
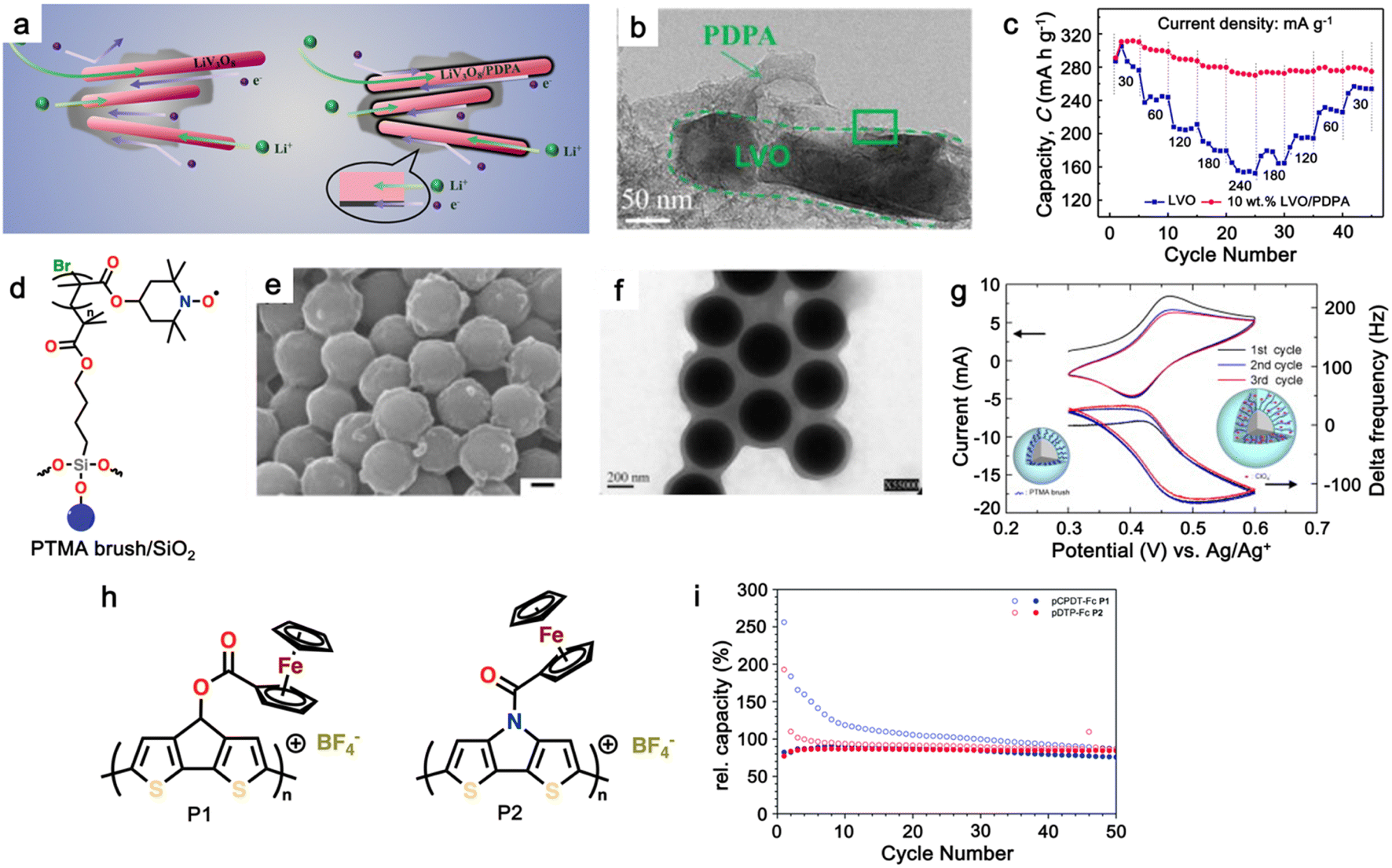 | ||
| Fig. 12 (a) Schematic depicting the Li+ and electron-transfer pathway for LVO and LVO/PDPA composites. (b) TEM micrographs showing the morphology of 10 wt% LVO/PDPA composite. (c) Comparison of the rate capabilities between LVO and 10 wt% LVO/PDPA composites at various current densities. Reproduced with permission from ref. 238 Copyright 2018, the American Chemical Society. (d) Chemical structure of PTMA brush/SiO2 hybrid materials. (e) and (f) SEM and TEM images of PTMA brush/SiO2, respectively. (g) Cyclic voltammogram of PTMA brush/SiO2 on the Au electrode and its corresponding resonance frequency change in EQCM sensor. The electrolyte was 0.1 M (C4H9)4NClO4 in acetonitrile. Reproduced with permission from ref. 266 Copyright 2011, Elsevier. (h) Chemical structures of ferrocene-functionalized polyheteroacenes, namely P1 and P2. (i) Cycling performances of P1 and P2 ferrocene-functionalized polyheteroacenes under 0.1C. Reproduced with permission from ref. 239 Copyright 2018, The Royal Society of Chemistry. | ||
In addition, a triphenylamine-based MOF, Cu-TCA (H3TCA = tricarboxytriphenyl amine), was reported as a LIB cathode active material.269 In this framework architecture, the redox activity of Cu-TCA is associated with the redox activity of both the metal clusters (Cu+/Cu2+) and organic ligand radicals (N/N+) with separated voltage plateaus and a high working potential of up to 4.3 V (vs. Li/Li+). Due to its abundant redox-active constituents and highly stable organic ligands, Cu-TCA has a theoretical capacity of 145 mA h g−1, which is comparable to that of commercial materials such as LiFePO4 (170 mA h g−1). Moreover, Cu-TCA achieved long cycling stability over 200 cycles at a 2C rate, with an average coulombic efficiency of 96.5%, suggesting that Cu-TCA is capable of delivering a high recharge rate with high-capacity retention. Inspired by these findings, Lin et al. explored the potential of nitroxide radical-based PTMA as a cathode material (Fig. 12d) by polymerizing a polymer brush over silica nanoparticles.266 The nitroxide polymer brushes, grafted onto silica nanoparticles via surface-initiated atom transfer radical polymerization, prevented the dissolution of the polymer in organic electrolytes. The SEM and TEM images confirmed that the nitroxide polymer brushes were successfully grafted onto the silica nanoparticles (Fig. 12e and f). Further, the electrochemical quartz crystal microbalance results indicated that the non-crosslinking nitroxide polymer brushes prevented the polymer from dissolving in organic electrolytes (Fig. 12g). These electrodes showed high discharge capacities and excellent cycle-life performance, demonstrating the potential of organic–inorganic hybrid systems for energy storage applications. Similarly, Schwartz et al. synthesized conjugated polymers based on ferrocene-functionalized cyclopentadithiophene and poly(dithieno[3,2-b:2′,3′-d]pyrrole) backbones (Fig. 12h).239 These polymers exhibited high reversible capacities and excellent capacity retention over multiple cycles, showcasing their suitability for battery applications (Fig. 12i). Together, these studies highlight the potential of organic–inorganic hybrid materials as electrodes for high-performance LIBs.
4.3 Copolymer materials
Recently, copolymerization has emerged as an alternative approach to enhance the electrochemical performance of polymer-based electrodes. These materials typically consist of two or more different polymerized monomer units, offering synergistic benefits compared to individual polymers.270,271 One notable advantage of copolymer materials is their ability to combine the desirable properties of different polymer components. For example, copolymers can incorporate both electron-rich and electron-deficient monomers, enabling improved charge transfer kinetics and stability during cycling. Additionally, the incorporation of various functional groups within copolymer structures can be introduced to facilitate rapid lithium-ion diffusion and enhance the number of active sites.271 Moreover, copolymer materials often offer enhanced mechanical strength and flexibility compared to individual polymer or traditional inorganic electrodes, making them suitable for use in flexible and lightweight battery applications.272 Furthermore, copolymer-based electrodes can be processed using scalable solution-based techniques, enabling cost-effective and large-scale manufacturing.Recent research efforts have focused on designing copolymer materials with tailored properties for specific battery applications. For instance, copolymers containing conjugated backbones, such as polythiophenes and polypyrrole, have been investigated due to their high electrical conductivity and lithium-ion storage capacity.92,273 Additionally, copolymers incorporating redox-active moieties, such as viologen and anthraquinone, have shown promise for achieving high capacity and long-term cycling stability.274 Copolymers such as that derived from pyrrole and dopamine (PPy-DA) offer distinct advantages in LIB applications by combining the charge storage properties of different monomers.275 The copolymer presented a porous NF morphology, which was different from the plate-like structure of PDA and the aggregated nanospheres of PPy (Fig. 13a–c). PPy-DA was produced via the copolymerization of dopamine (n-type) and PPy (p-type) monomer, leading to a copolymer with a shorter polaron delocalization length. This reduction in delocalization was effective in enhancing its redox potential (around 3–3.5 V) and specific capacity (∼160 mA h g−1) (Fig. 13d). Furthermore, two novel copolymers, namely poly(dihydrophenazine-co-diphenylamine) (PDPAPZ) and poly(dihydrophenazine-co-phenothiazine) (PPTZPZ), were synthesized and evaluated as cathode materials (Fig. 13e and f), respectively.179 These copolymers exhibited a high average discharge potential (3.5–3.6 V) in lithium cells. Remarkably, the PDPAPZ/Li cells demonstrated a steady specific capacity of ∼101 mA h g−1 at a current density of 0.1 A g−1 up to 100 cycles (Fig. 13e). Alternatively, PPTZPZ exhibited a lower capacity of 80 mA h g−1 and later demonstrated a gradual decrease in capacity up to 100 cycles (Fig. 13f) due to the inactive sulfur species. Similarly, Yao and colleagues synthesized a new conjugated copolymer for use as an organic cathode material by incorporating both conducting aniline and pyrene units (Fig. 13g).246 This poly-PPDA-PYR achieved a reversible specific capacity of 113 mA h g−1 at a current density of 20 mA g−1 with a high voltage output of 3.2 V and impressive capacity retention of 75.2% after 180 cycles (Fig. 13g). Additionally, the copolymer exhibited an excellent rate performance of up to 1500 mA g−1, and the highest specific capacity ∼100 mA h g−1 could be recovered once the current density switched back to 20 mA g−1 (Fig. 13g).
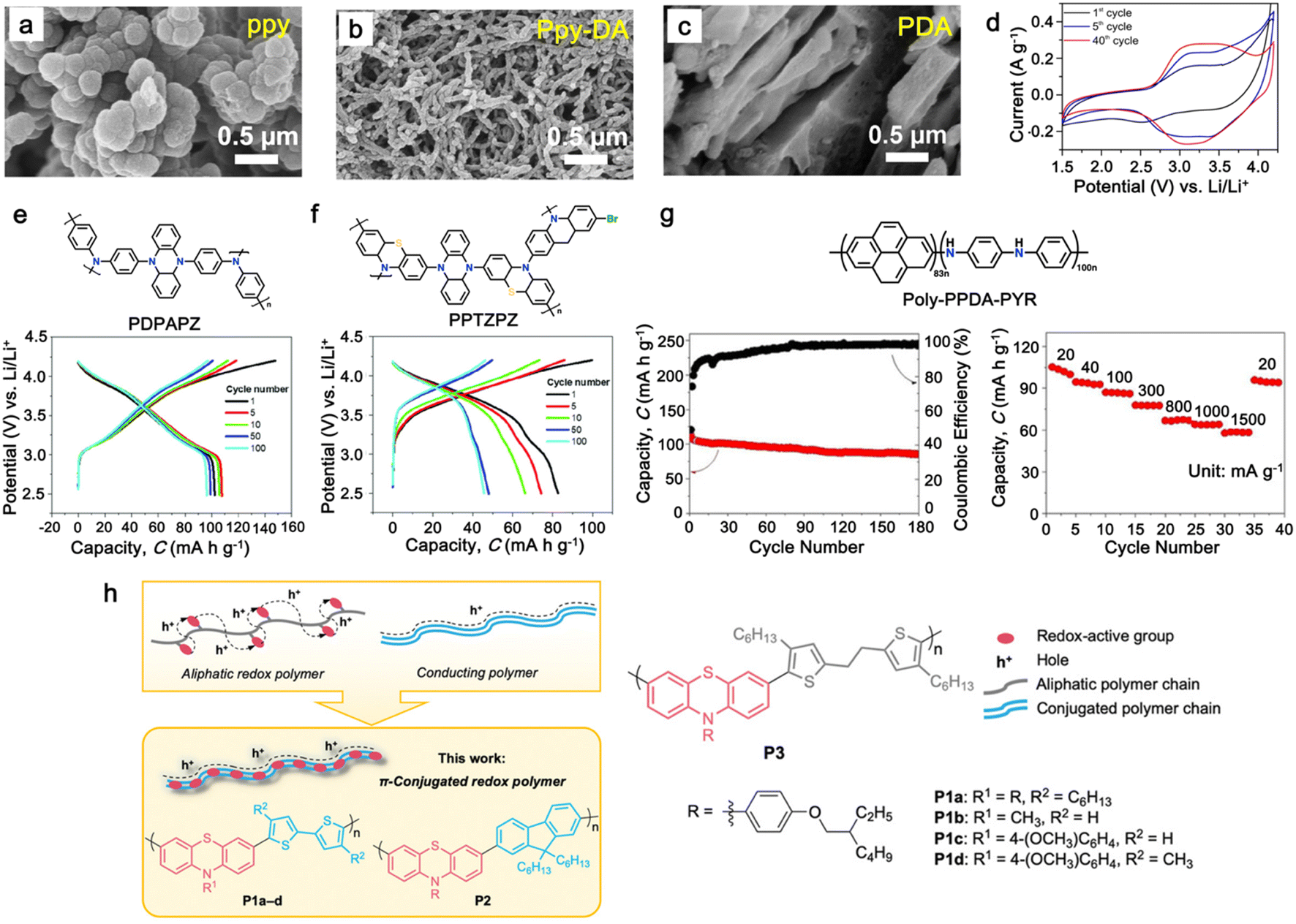 | ||
| Fig. 13 SEM images of (a) PPy, (b) PPy-DA, and (c) PDA. (d) CV curves of PPy-DA at a scan rate of 1 mV s−1. Reproduced with permission from ref. 275 Copyright 2018, Elsevier. (e) and (f) Chemical structure and galvanostatic charge–discharge of PDPAPZ and PPTZPZ copolymers, respectively. Reproduced with permission from ref. 179 Copyright 2020, John Wiley and Sons. (g) Cycling performance at a current density of 20 mA g−1 and rate performance of poly-PPDA-PYR copolymer. Reproduced with permission from ref. 246 Copyright 2019, John Wiley and Sons. (h) Schematic of the π-conjugated redox polymer. Reproduced with permission from ref. 276 Copyright 2019, John Wiley and Sons. | ||
In addition to the copolymerization of redox-active components, the incorporation of inactive components into copolymers has also been extensively investigated due to their impact on the electrochemical performance. These inactive components serve various functions such as enhancing the cycle stability, electrical conductivity, ionic conductivity, cohesiveness, and flexibility; however, a reduced theoretical capacity is often observed.276 For instance, Hernández et al. combined a redox-active polyimide (PI) with ion-conductive polyether blocks (PEO), where PEO acted as both a binder and conductive agent.244 The resulting binder-free and conductive agent-free copolymer electrode exhibited an excellent discharge capacity and cycling life. Similarly, Zhang et al. synthesized a copolymer based on poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PTMA) with controllable pyrene side groups, which enhanced the electron transfer rates and resulted in improved specific capacity and rate capability when uniformly dispersed in a composite with reduced graphene oxide (rGO).219 Additionally, an ultra-high-rate capability and long stability were demonstrated using phenothiazine copolymer.276 The design of this copolymer cathode was based on the combination of the high oxidation of phenothiazine at 3.6 V (vs. Li/Li+) and good hole conductivity of the bithiophene and fluorene comonomer (Fig. 13h). An ultra-high-rate capability and long cycling stability were demonstrated given that the π-conjugated copolymer could be operated for up to 30000 cycles at an extreme current rate of 100C with >97% capacity retention. These studies emphasized the importance of the copolymerization strategy to enhance the electrochemical performance of the polymer cathode.
5. Conclusions and perspective
Polymeric electrode materials have emerged as promising alternative cathode materials towards high-performance and high-energy-density 4.0 V-class organic LIBs. By tailoring their chemical structure through functionalization, doping, and incorporating both redox-active and inactive components, researchers can optimize the key parameters of batteries such as specific capacity, cycling stability, and rate capability. This innovative approach addresses many of the limitations associated with traditional electrode materials, paving the way for more efficient, durable, and high-performing LIBs. As the demand for higher energy storage solutions continues to grow, the advancements in polymeric electrode design are poised to play a crucial role in meeting the energy needs of the future. Owing to their flexibility in design, scalability of synthesis, and compatibility with high-energy-density applications compared to the conventional inorganic cathode materials, a series of high-voltage polymers such as arylamine-based, thioether, phenazine, viologen and radical polymers has been explored and discussed in detail. Additionally, alternative approaches such as the formation of bipolar polymers and hybrid organic–inorganic polymers and copolymerization strategies have also been shown to be promising to enhance the electrochemical performance and stability of polymer cathodes. By harnessing the unique properties of each polymer, such as tunable redox chemistry, mechanical flexibility, and chemical stability, researchers have unlocked new possibilities for achieving a superior electrochemical performance in LIBs. Moving forward, continued research efforts in this field will be essential to further optimize the design and synthesis of polymeric-based electrode materials, paving the way for the next generation of high-performance LIBs.Looking ahead, the future of polymeric electrode materials for high-energy-density LIBs holds exciting prospects and challenges. Firstly, it is necessary to gain a deeper understanding of the fundamental electrochemical processes occurring within polymer electrodes, including the ion transport mechanisms, charge storage mechanisms, and degradation pathways. In this case, advanced characterization techniques, coupled with computational modeling, will play a crucial role in elucidating these complex phenomena and guiding the rational design of next-generation polymer electrodes. Furthermore, efforts should be directed towards the development of scalable synthesis methods for producing polymer materials with well-defined structures and tailored properties. The integration of advanced manufacturing techniques, such as additive manufacturing and roll-to-roll processing, will enable the fabrication of large-area, high-performance polymer electrodes for practical applications. Additionally, research should focus on exploring novel polymer chemistries, functionalization strategies, and electrode architectures to further enhance the energy density, safety, and lifespan of 4.0 V-class organic LIBs. Additionally, collaboration among academia, industry, and government institutions will be crucial for accelerating the translation of research findings into commercial products. Investment in infrastructure, pilot-scale production facilities, and collaborative research initiatives will facilitate the transition of polymeric electrode materials from the laboratory to the marketplace. Ultimately, the continued advancement of polymeric electrode materials holds immense potential to drive innovation in energy storage technology and address the growing demand for high-performance LIBs in a wide range of applications.
Data availability
The data supporting this article have been included as Notes and references.Author contributions
Conceptualization and methodology: F. B. and H. J. Y. Investigation: F. B., S. U. S. and A. L. L. Writing-original draft: F. B. and S. U. S. Writing – review & editing: F. B., S. U. S., and H. J. Y. Visualization: F. B. Supervision: H. J. Y. All authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. B. acknowledges the postdoctoral fellowship program supported by Academia Sinica (AS-PD-11201-M04). H.-J. Yen acknowledges the financial support by Ministry of Science and Technology in Taiwan (MOST 110-2124-M-001-001) and National Science and Technology Council (NSTC 112-2113-M-001-012).Notes and references
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- J. Xie and Y.-C. Lu, Nat. Commun., 2020, 11, 2499 CrossRef CAS.
- C. N. Gannett, L. Melecio-Zambrano, M. J. Theibault, B. M. Peterson, B. P. Fors and H. D. Abruña, Mater. Rep.: Energy, 2021, 1, 100008 CAS.
- T. Wulandari, D. Fawcett, S. B. Majumder and G. E. J. Poinern, Battery Energy, 2023, 2, 20230030 CrossRef.
- C. P. Grey and D. S. Hall, Nat. Commun., 2020, 11, 6279 CrossRef CAS PubMed.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- C. Liu, Z. G. Neale and G. Cao, Mater. Today, 2016, 19, 109–123 CrossRef CAS.
- A. Saxena, N. Gnanaseelan, S. K. Kamaraj and F. Caballero-Briones, in Rechargeable Lithium-Ion Batteries, CRC Press, 2020, pp. 260–288 Search PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. Li, C. Lin, M. Weng, Y. Qiu, P. Chen, K. Yang, W. Huang, Y. Hong, J. Li, M. Zhang, C. Dong, W. Zhao, Z. Xu, X. Wang, K. Xu, J. Sun and F. Pan, Nat. Nanotechnol., 2021, 16, 599–605 CrossRef CAS PubMed.
- B. Tao, I. J. McPherson, E. Daviddi, C. L. Bentley and P. R. Unwin, ACS Sustainable Chem. Eng., 2023, 11, 1459–1471 CrossRef CAS.
- D. D. MacNeil, Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1332 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- S. Gantenbein, M. Schönleber, M. Weiss and E. Ivers-Tiffée, Sustainability, 2019, 11(23), 6697 CrossRef CAS.
- C. Zeng, F. Fan, R. Zheng, X. Wang, G. Tian, S. Liu, P. Liu, C. Wang, S. Wang and C. Shu, ACS Appl. Mater. Interfaces, 2024, 16, 11377–11388 CrossRef CAS PubMed.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef CAS.
- S. Zhao, K. Yan, J. Zhang, B. Sun and G. Wang, Angew. Chem., Int. Ed., 2021, 60, 2208–2220 CrossRef CAS PubMed.
- K. Amin, L. Mao and Z. Wei, Macromol. Rapid Commun., 2019, 40, 1800565 CrossRef PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- H. Ji, J. Wu, Z. Cai, J. Liu, D.-H. Kwon, H. Kim, A. Urban, J. K. Papp, E. Foley, Y. Tian, M. Balasubramanian, H. Kim, R. J. Clément, B. D. McCloskey, W. Yang and G. Ceder, Nat. Energy, 2020, 5, 213–221 CrossRef CAS.
- M. Malik, K. H. Chan and G. Azimi, Mater. Today Energy, 2022, 28, 101066 CrossRef CAS.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS.
- W. Du, X. Du, M. Ma, S. Huang, X. Sun and L. Xiong, Adv. Funct. Mater., 2022, 32, 2110871 CrossRef CAS.
- S. Lee, J. E. Kwon, J. Hong, S. Y. Park and K. Kang, J. Mater. Chem. A, 2019, 7, 11438–11443 RSC.
- A. Wild, M. Strumpf, B. Häupler, M. D. Hager and U. S. Schubert, Adv. Energy Mater., 2017, 7, 1601415 CrossRef.
- L. M. Zhu, A. W. Lei, Y. L. Cao, X. P. Ai and H. X. Yang, Chem. Commun., 2013, 49, 567–569 RSC.
- K. Hatakeyama-Sato, T. Tezuka, R. Ichinoi, S. Matsumono, K. Sadakuni and K. Oyaizu, ChemSusChem, 2020, 13, 2443–2448 CrossRef.
- É. Deunf, P. Moreau, É. Quarez, D. Guyomard, F. Dolhem and P. Poizot, J. Mater. Chem. A, 2016, 4, 6131–6139 RSC.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- M. E. Baumert, V. Le, P.-H. Su, Y. Akae, D. Bresser, P. Théato and M. M. Hansmann, J. Am. Chem. Soc., 2023, 145, 23334–23345 CrossRef CAS PubMed.
- H. Kye, Y. Kang, D. Jang, J. E. Kwon and B.-G. Kim, Adv. Energy Sustainability Res., 2022, 3, 2200030 CrossRef CAS.
- D. J. Min, K. Lee, S. Y. Park and J. E. Kwon, ChemSusChem, 2020, 13, 2303–2311 CrossRef CAS.
- X. Liu and Z. Ye, Adv. Energy Mater., 2021, 11, 2003281 CrossRef CAS.
- Y. Hu, Y. Gao, L. Fan, Y. Zhang, B. Wang, Z. Qin, J. Zhou and B. Lu, Adv. Energy Mater., 2020, 10, 2002780 CrossRef CAS.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S. K. Jung, H. D. Lim and K. Kang, Adv. Mater., 2018, 30, 1704682 CrossRef.
- X. Feng, X. Chen, B. Ren, X. Wu, X. Huang, R. Ding, X. Sun, S. Tan, E. Liu and P. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 7178–7187 CrossRef CAS PubMed.
- R. Chen, D. Bresser, M. Saraf, P. Gerlach, A. Balducci, S. Kunz, D. Schröder, S. Passerini and J. Chen, ChemSusChem, 2020, 13, 2205–2219 CrossRef CAS.
- J. Heiska, M. Nisula and M. Karppinen, J. Mater. Chem. A, 2019, 7, 18735–18758 RSC.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS.
- T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae and H. Tsuyama, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Haupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- J. Yang, Z. Wang, Y. Shi, P. Sun and Y. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 7179–7185 CrossRef CAS.
- T. Cai, Y. Han, Q. Lan, F. Wang, J. Chu, H. Zhan and Z. Song, Energy Storage Mater., 2020, 31, 318–327 CrossRef.
- F. Wan, X.-L. Wu, J.-Z. Guo, J.-Y. Li, J.-P. Zhang, L. Niu and R.-S. Wang, Nano Energy, 2015, 13, 450–457 CrossRef CAS.
- Y. Wang, Y. Ding, L. Pan, Y. Shi, Z. Yue, Y. Shi and G. Yu, Nano Lett., 2016, 16, 3329–3334 CrossRef CAS PubMed.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861–868 CrossRef CAS.
- Q. Dong, T. Naren, L. Zhang, W. Jiang, M. Xue, X. Wang, L. Chen, C.-S. Lee and Q. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202405426 CrossRef CAS PubMed.
- W. Guo, Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Energy Environ. Sci., 2012, 5, 5221–5225 RSC.
- Z. Song, T. Xu, M. L. Gordin, Y.-B. Jiang, I.-T. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205–2211 CrossRef CAS PubMed.
- C. Luo, Y. Zhu, Y. Xu, Y. Liu, T. Gao, J. Wang and C. Wang, J. Power Sources, 2014, 250, 372–378 CrossRef CAS.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
- K. B. Labasan, H.-J. Lin, F. Baskoro, J. J. H. Togonon, H. Q. Wong, C.-W. Chang, S. D. Arco and H.-J. Yen, ACS Appl. Mater. Interfaces, 2021, 13, 17467–17477 CrossRef CAS.
- J. Wang, H. Liu, C. Du, X. Zhang, Y. Liu, H. Yao, Z. Sun and S. Guan, Chem. Eng. J., 2022, 444, 136598 CrossRef CAS.
- X.-Y. Han, C.-X. Chang, L.-J. Yuan, T.-L. Sun and J. Sun, Adv. Mater., 2007, 19, 1616–1621 CrossRef CAS.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS.
- F. Baskoro, A. L. Lubis, H. Q. Wong, G.-S. Liou and H.-J. Yen, J. Mater. Chem. A, 2023, 11, 11210–11221 RSC.
- F. Baskoro, H.-J. Lin, C.-W. Chang, C.-L. Wang, A. L. Lubis and H.-J. Yen, J. Mater. Chem. A, 2023, 11, 569–578 RSC.
- A. L. Lubis, F. Baskoro, T.-H. Lin, H. Q. Wong, G.-S. Liou and H.-J. Yen, ACS Appl. Mater. Interfaces, 2024, 16, 48722–48735 CrossRef CAS.
- J. Sun, Y. Xu, Y. Lv, Q. Zhang and X. Zhou, CCS Chem., 2023, 5, 1259–1276 CrossRef CAS.
- M. Majumder, A. K. Thakur, A. S. Patole and S. P. Patole, in Organic Electrodes: Fundamental to Advanced Emerging Applications, ed. R. K. Gupta, Springer International Publishing, Cham, 2022, pp. 171–188 Search PubMed.
- Z. Yang, F. Wang, P. Meng, J. Luo and C. Fu, Energy Storage Mater., 2022, 51, 63–79 CrossRef.
- F. Goto, K. Abe, K. Ikabayashi, T. Yoshida and H. Morimoto, J. Power Sources, 1987, 20, 243–248 CrossRef CAS.
- M. Kobayashi, N. Colaneri, M. Boysel, F. Wudl and A. J. Heeger, J. Chem. Phys., 1985, 82, 5717–5723 CrossRef CAS.
- S. Taguchi and T. Tanaka, J. Power Sources, 1987, 20, 249–252 CrossRef CAS.
- L. W. Shacklette, R. L. Elsenbaumer, R. R. Chance, J. M. Sowa, D. M. Ivory, G. G. Miller and R. H. Baughman, J. Chem. Soc., Chem. Commun., 1982, 361–362 RSC.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839–19843 CrossRef CAS PubMed.
- T. Suga, M. Sakata, K. Aoki and H. Nishide, ACS Macro Lett., 2014, 3, 703–707 CrossRef CAS PubMed.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., 2015, 127, 14153–14157 CrossRef.
- K. Zhang, C. Guo, Q. Zhao, Z. Niu and J. Chen, Adv. Sci., 2015, 2, 1500018 CrossRef PubMed.
- J. E. Kwon, C.-S. Hyun, Y. J. Ryu, J. Lee, D. J. Min, M. J. Park, B.-K. An and S. Y. Park, J. Mater. Chem. A, 2018, 6, 3134–3140 RSC.
- W. Huang, Z. Zhu, L. Wang, S. Wang, H. Li, Z. Tao, J. Shi, L. Guan and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 9162–9166 CrossRef CAS.
- H. Senoh, M. Yao, H. Sakaebe, K. Yasuda and Z. Siroma, Electrochim. Acta, 2011, 56, 10145–10150 CrossRef CAS.
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600–605 CrossRef CAS.
- Q. Zhao, J. Wang, C. Chen, T. Ma and J. Chen, Nano Res., 2017, 10, 4245–4255 CrossRef CAS.
- Y. Shi, H. Tang, S. Jiang, L. V. Kayser, M. Li, F. Liu, F. Ji, D. J. Lipomi, S. P. Ong and Z. Chen, Chem. Mater., 2018, 30, 3508–3517 CrossRef CAS.
- M. Lee, J. Hong, B. Lee, K. Ku, S. Lee, C. B. Park and K. Kang, Green Chem., 2017, 19, 2980–2985 RSC.
- G. Dai, X. Wang, Y. Qian, Z. Niu, X. Zhu, J. Ye, Y. Zhao and X. Zhang, Energy Storage Mater., 2019, 16, 236–242 CrossRef.
- J. Kim, H.-S. Park, T.-H. Kim, S. Y. Kim and H.-K. Song, Phys. Chem. Chem. Phys., 2014, 16, 5295–5300 RSC.
- M. Yao, H. Senoh, T. Sakai and T. Kiyobayashi, J. Power Sources, 2012, 202, 364–368 CrossRef CAS.
- C. Zhang, X. Yang, W. Ren, Y. Wang, F. Su and J.-X. Jiang, J. Power Sources, 2016, 317, 49–56 CrossRef CAS.
- C. Su, F. Yang, L. Ji, L. Xu and C. Zhang, J. Mater. Chem. A, 2014, 2, 20083–20088 RSC.
- K. Lee, I. E. Serdiuk, G. Kwon, D. J. Min, K. Kang, S. Y. Park and J. E. Kwon, Energy Environ. Sci., 2020, 13, 4142–4156 RSC.
- M. Li, R. Wang, T. Wu, Y. Liu, Y. Chen, W. He, S. Feng, X. Zhang, G. Dai and Y. Zhao, ACS Appl. Energy Mater., 2023, 6, 6834–6841 CrossRef CAS.
- H. Park, H. Kye, J.-S. Lee, Y.-C. Joo, D. J. Min, B.-G. Kim, S. Y. Park and J. E. Kwon, Energy Environ. Mater., 2024, 7, e12694 CrossRef CAS.
- S. Lee, G. Kwon, T. Kang, J. Kim, B. Lee, C. Kim, C. Lee, Y. Kim, J. Noh, Y.-S. Yu, D. Lee and K. Kang, J. Mater. Chem., 2023, 11, 22441–22448 RSC.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- J. Xu, X. Cai, S. Cai, Y. Shao, C. Hu, S. Lu and S. Ding, Energy Environ. Mater., 2023, 6, e12450 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2014, 114, 11414–11443 CrossRef CAS PubMed.
- S. V. Venkatesan, A. Nandy, K. Karan, S. R. Larter and V. Thangadurai, Electrochem. Energy Rev., 2022, 5, 16 CrossRef CAS.
- R. Hildner, A. Köhler, P. Müller-Buschbaum, F. Panzer and M. Thelakkat, Adv. Energy Mater., 2017, 7, 1700314 CrossRef.
- S. Wang, G. Zuo, J. Kim and H. Sirringhaus, Prog. Polym. Sci., 2022, 129, 101548 CrossRef CAS.
- H. Sun, F. Chen and Z.-K. Chen, Mater. Today, 2019, 24, 94–118 Search PubMed.
- J.-H. Dou, Z.-A. Yu, J. Zhang, Y.-Q. Zheng, Z.-F. Yao, Z. Tu, X. Wang, S. Huang, C. Liu, J. Sun, Y. Yi, X. Cao, Y. Gao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2019, 141, 6561–6568 CrossRef CAS.
- C. Bian, S. Wang, Y. Liu and X. Jing, RSC Adv., 2016, 6, 55007–55016 Search PubMed.
- A. Onwubiko, W. Yue, C. Jellett, M. Xiao, H.-Y. Chen, M. K. Ravva, D. A. Hanifi, A.-C. Knall, B. Purushothaman, M. Nikolka, J.-C. Flores, A. Salleo, J.-L. Bredas, H. Sirringhaus, P. Hayoz and I. McCulloch, Nat. Commun., 2018, 9, 416 CrossRef PubMed.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed.
- S. Prodhan, J. Qiu, M. Ricci, O. M. Roscioni, L. Wang and D. Beljonne, J. Phys. Chem. Lett., 2020, 11, 6519–6525 CrossRef CAS.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 Search PubMed.
- S. Bitton and N. Tessler, Adv. Electron. Mater., 2024, 2300766 CrossRef CAS.
- K. D. Fong, J. Self, K. M. Diederichsen, B. M. Wood, B. D. McCloskey and K. A. Persson, ACS Cent. Sci., 2019, 5, 1250–1260 CrossRef CAS PubMed.
- T. Junkers, J. Vandenbergh, P. Adriaensens, L. Lutsen and D. Vanderzande, Polym. Chem., 2012, 3, 275–285 RSC.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- T. L. D. Tam, M. Lin, S. W. Chien and J. Xu, ACS Macro Lett., 2021, 11, 110–115 CrossRef PubMed.
- A. Dai, A. Wan, C. Magee, Y. Zhang, S. Barlow, S. R. Marder and A. Kahn, Org. Electron., 2015, 23, 151–157 Search PubMed.
- I. M. Ward and J. Sweeney, Mechanical Properties of Solid Polymers, John Wiley & Sons, 2012 Search PubMed.
- H.-Y. Wu, J.-D. Huang, S. Y. Jeong, T. Liu, Z. Wu, T. van der Pol, Q. Wang, M.-A. Stoeckel, Q. Li and M. Fahlman, Mater. Horiz., 2023, 10, 4213–4223 RSC.
- L. Bondi, C. Marzuoli, E. Gutiérrez-Fernández, G. Tullii, J. Martín, B. Fraboni, D. Mecerreyes, M. R. Antognazza and T. Cramer, Adv. Electron. Mater., 2023, 9, 2300146 Search PubMed.
- I. K. Yakushchenko, M. G. Kaplunov, O. N. Efimov, M. Y. Belov and S. N. Shamaev, Phys. Chem. Chem. Phys., 1999, 1, 1783–1785 RSC.
- C. Zhang, S. Chen, G. Zhou, Q. Hou, S. Luo, Y. Wang and G. Shi, J. Electrochem. Soc., 2021, 168, 050548 CrossRef CAS.
- C. Su, H. He, L. Xu, K. Zhao, C. Zheng and C. Zhang, J. Mater. Chem. A, 2017, 5, 2701–2709 RSC.
- J. K. Feng, Y. L. Cao, X. P. Ai and H. X. Yang, J. Power Sources, 2008, 177, 199–204 CrossRef CAS.
- Z. Chen, C. Su, X. Zhu, R. Xu, L. Xu and C. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2574–2583 CrossRef CAS.
- K. Yamamoto, D. Suemasa, K. Masuda, K. Aita and T. Endo, ACS Appl. Mater. Interfaces, 2018, 10, 6346–6353 CrossRef CAS.
- Z. Chen, W. Li, Y. Dai, N. Xu, C. Su, J. Liu and C. Zhang, Electrochim. Acta, 2018, 286, 187–194 CrossRef CAS.
- Y. Ou, Y. Xiong, Z. Hu, Y. Zhang and L. Dong, J. Mater. Chem. A, 2022, 10, 10373–10382 RSC.
- F. A. Obrezkov, A. F. Shestakov, S. G. Vasil'ev, K. J. Stevenson and P. A. Troshin, J. Mater. Chem. A, 2021, 9, 2864–2871 RSC.
- I. Kang, T. Lee, Y. R. Yoon, J. W. Kim, B.-K. Kim, J. Lee, J. H. Lee and S. Y. Kim, Mater, 2021, 14, 7885 CrossRef CAS.
- W. Huang, T. Jia, G. Zhou, S. Chen, Q. Hou, Y. Wang, S. Luo, G. Shi and B. Xu, Electrochim. Acta, 2018, 283, 1284–1290 CrossRef CAS.
- C. Su, X. Zhu, L. Xu, N. Zhou, H. He and C. Zhang, Electrochim. Acta, 2016, 196, 440–449 CrossRef CAS.
- L. Zhu and X. Cao, Mater. Lett., 2015, 150, 16–19 CrossRef CAS.
- C. Su, L. Ji, L. Xu, N. Zhou, G. Wang and C. Zhang, RSC Adv., 2016, 6, 22989–22995 RSC.
- J. Xiong, Z. Wei, T. Xu, Y. Zhang, C. Xiong and L. Dong, Polymer, 2017, 130, 135–142 CrossRef CAS.
- T. Xu, J. Xiong, X. Du, Y. Zhang, S. Song, C. Xiong and L. Dong, J. Phys. Chem. C, 2018, 122, 20057–20063 CrossRef CAS.
- X. Zhao, C. Wang, Z. Li, X. Hu, A. A. Razzaq and Z. Deng, J. Mater. Chem. A, 2021, 9, 19282–19297 RSC.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- P. Sang, Q. Chen, D.-Y. Wang, W. Guo and Y. Fu, Chem. Rev., 2023, 123, 1262–1326 Search PubMed.
- X. Zhang, W. Guo and Y. Fu, Acc. Mater. Res., 2024, 5, 316–328 Search PubMed.
- M. A. Weret, C.-F. J. Kuo, W.-N. Su, T. S. Zeleke, C.-J. Huang, N. A. Sahalie, T. A. Zegeye, Z. T. Wondimkun, F. W. Fenta, B. A. Jote, M.-C. Tsai and B. J. Hwang, J. Power Sources, 2022, 541, 231693 Search PubMed.
- Y. Chen, S. Zhuo, Z. Li and C. Wang, EnergyChem, 2020, 2, 100030 Search PubMed.
- D.-Y. Wang, W. Guo and Y. Fu, Acc. Chem. Res., 2019, 52, 2290–2300 CrossRef CAS.
- W. Guo, D. Y. Wang, Q. Chen and Y. Fu, Adv. Sci., 2022, 9, 2103989 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- J. Ren, X. Wang, H. Liu, Y. Hu, X. Zhang and T. Masuda, React. Funct. Polym., 2020, 146, 104365 CrossRef CAS.
- D. Ogi, Y. Fujita, M. Kato, T. Yamauchi, T. Shirahata, M. Yao and Y. Misaki, Eur. J. Org Chem., 2019, 2019, 2725–2728 CrossRef CAS.
- J. Ren, X. Wang, H. Liu, Y. Hu, X. Zhang and T. Masuda, React. Funct. Polym., 2020, 146, 104365 CrossRef CAS.
- T. P. Vaid, M. E. Easton and R. D. Rogers, Synth. Met., 2017, 231, 44–50 CrossRef CAS.
- D. Ogi, Y. Fujita, M. Kato, T. Yamauchi, T. Shirahata, M. Yao and Y. Misaki, Eur. J. Org Chem., 2019, 2019, 2725–2728 CrossRef CAS.
- J. Tang, L. Kong, J. Zhang, L. Zhan, H. Zhan, Y. Zhou and C. Zhan, React. Funct. Polym., 2008, 68, 1408–1413 CrossRef CAS.
- L. Zhan, Z. Song, N. Shan, J. Zhang, J. Tang, H. Zhan, Y. Zhou, Z. Li and C. Zhan, J. Power Sources, 2009, 193, 859–863 CrossRef CAS.
- M. E. Speer, M. Kolek, J. J. Jassoy, J. Heine, M. Winter, P. M. Bieker and B. Esser, Chem. Commun., 2015, 51, 15261–15264 RSC.
- B. Häupler, R. Burges, C. Friebe, T. Janoschka, D. Schmidt, A. Wild and U. S. Schubert, Macromol. Rapid Commun., 2014, 35, 1367–1371 CrossRef PubMed.
- P. Sang, Y. Si and Y. Fu, Chem. Commun., 2019, 55, 4857–4860 RSC.
- M. B. Preefer, B. Oschmann, C. J. Hawker, R. Seshadri and F. Wudl, Angew. Chem., Int. Ed., 2017, 56, 15118–15122 CrossRef CAS PubMed.
- Y. Jing, Y. Liang, S. Gheytani and Y. Yao, Nano Energy, 2017, 37, 46–52 CrossRef CAS.
- B. Häupler, T. Hagemann, C. Friebe, A. Wild and U. S. Schubert, ACS Appl. Mater. Interfaces, 2015, 7, 3473–3479 CrossRef PubMed.
- A. Bhargav, M. E. Bell, Y. Cui and Y. Fu, ACS Appl. Energy Mater., 2018, 1, 5859–5864 CrossRef CAS.
- P. Sang, J. Song, W. Guo and Y. Fu, Chem. Eng. J., 2021, 415, 129043 CrossRef CAS.
- A. Bhargav, C.-H. Chang, Y. Fu and A. Manthiram, ACS Appl. Mater. Interfaces, 2019, 11, 6136–6142 CrossRef CAS PubMed.
- S. H. Je, T. H. Hwang, S. N. Talapaneni, O. Buyukcakir, H. J. Kim, J.-S. Yu, S.-G. Woo, M. C. Jang, B. K. Son, A. Coskun and J. W. Choi, ACS Energy Lett., 2016, 1, 566–572 CrossRef CAS.
- H. Kim, J. Lee, H. Ahn, O. Kim and M. J. Park, Nat. Commun., 2015, 6, 7278 CrossRef CAS.
- S. Zeng, L. Li, D. Zhao, J. Liu, W. Niu, N. Wang and S. Chen, J. Phys. Chem. C, 2017, 121, 2495–2503 CrossRef CAS.
- R. F. Nelson, D. W. Leedy, E. T. Seo and R. N. Adams, Fresenius' Z. Anal. Chem., 1966, 224, 184–196 CrossRef.
- Z. Niu, H. Wu, L. Liu, G. Dai, S. Xiong, Y. Zhao and X. Zhang, J. Mater. Chem. A, 2019, 7, 10581–10588 RSC.
- C. N. Gannett, B. M. Peterson, L. Melecio-Zambrano, C. Q. Trainor, B. P. Fors and H. D. Abruña, J. Mater. Chem. A, 2021, 9, 5657–5663 RSC.
- L. Huang, Y. Chen, Y. Liu, T. Wu, H. Li, J. Ye, G. Dai, X. Zhang and Y. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 17868–17875 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639–4647 CrossRef.
- D. Golodnitsky, E. Strauss, E. Peled and S. Greenbaum, J. Electrochem. Soc., 2015, 162, A2551 CrossRef CAS.
- E. B. Trigg, T. W. Gaines, M. Maréchal, D. E. Moed, P. Rannou, K. B. Wagener, M. J. Stevens and K. I. Winey, Nat. Mater., 2018, 17, 725–731 CrossRef CAS PubMed.
- M. Kolek, F. Otteny, P. Schmidt, C. Mück-Lichtenfeld, C. Einholz, J. Becking, E. Schleicher, M. Winter, P. Bieker and B. Esser, Energy Environ. Sci., 2017, 10, 2334–2341 RSC.
- F. Otteny, V. Perner, D. Wassy, M. Kolek, P. Bieker, M. Winter and B. Esser, ACS Sustainable Chem. Eng., 2020, 8, 238–247 CrossRef CAS.
- H. Kye, Y. Kang, D. Jang, J. E. Kwon and B.-G. Kim, Adv. Energy Sustainability Res., 2022, 3, 2200030 CrossRef CAS.
- M. Kolek, F. Otteny, J. Becking, M. Winter, B. Esser and P. Bieker, Chem. Mater., 2018, 30, 6307–6317 CrossRef CAS.
- G. Dai, Y. Liu, Z. Niu, P. He, Y. Zhao, X. Zhang and H. Zhou, Matter, 2019, 1, 945–958 CrossRef.
- C. N. Gannett, B. M. Peterson, L. Shen, J. Seok, B. P. Fors and H. D. Abruña, ChemSusChem, 2020, 13, 2428–2435 CrossRef CAS PubMed.
- Y. Wang, Y. Huang, Y. Hua, Y. Du and H. Yang, New J. Chem., 2022, 46, 14314–14317 RSC.
- S. Xu, H. Dai, S. Zhu, Y. Wu, M. Sun, Y. Chen, K. Fan, C. Zhang, C. Wang and W. Hu, eScience, 2021, 1, 60–68 CrossRef.
- F. A. Obrezkov, A. I. Somova, E. S. Fedina, S. G. Vasil'ev, K. J. Stevenson and P. A. Troshin, Energy Technol., 2021, 9, 2000772 CrossRef CAS.
- X. Zhao, X. Qiu, H. Xue, S. Liu, D. Liang, C. Yan, W. Chen, Y. Wang and G. Zhou, Angew. Chem., 2023, 135, e202216713 CrossRef.
- Q. He, S. Lv, Y. Huang, J. Guo, X. Peng, Y. Du and H. Yang, RSC Adv., 2023, 13, 12464–12468 RSC.
- R. R. Kapaev, I. S. Zhidkov, E. Z. Kurmaev, K. J. Stevenson and P. A. Troshin, J. Mater. Chem. A, 2019, 7, 22596–22603 RSC.
- J. Wang, J. C. Z. En, S. N. Riduan and Y. Zhang, Chem.–Eur. J., 2020, 26, 2581–2585 CrossRef CAS PubMed.
- S. Lee, J. Hong, S.-K. Jung, K. Ku, G. Kwon, W. M. Seong, H. Kim, G. Yoon, I. Kang, K. Hong, H. W. Jang and K. Kang, Energy Storage Mater., 2019, 20, 462–469 CrossRef.
- A. Wild, M. Strumpf, B. Häupler, M. D. Hager and U. S. Schubert, Adv. Energy Mater., 2017, 7, 1601415 CrossRef.
- M. Lee, J. Hong, D.-H. Seo, D. H. Nam, K. T. Nam, K. Kang and C. B. Park, Angew. Chem., Int. Ed., 2013, 52, 8322–8328 CrossRef CAS PubMed.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- Y. Ji, K. Yang, M. Liu, S. Chen, X. Liu, B. Yang, Z. Wang, W. Huang, Z. Song and S. Xue, Adv. Funct. Mater., 2021, 31, 2104830 CrossRef CAS.
- A. Yu, C. Li, X. Chen, C. Zhang, S. Mei and C. J. Yao, ChemSusChem, 2024, 17, e202301809 CrossRef CAS.
- F. Wang, J. Wang, G. Li, Z. Guo, J. Chu, X. Ai and Z. Song, Energy Storage Mater., 2022, 50, 658–667 CrossRef.
- Z. Wang, A. Duan, W. Jin, X. Huang and Y. Li, J. Mater. Chem. A, 2022, 10, 10026–10032 RSC.
- M. Stolar, C. Reus and T. Baumgartner, Adv. Energy Mater., 2016, 6, 1600944 CrossRef.
- T. Xia, T. Zhu, Y. Miao and X. Zhao, ACS Appl. Energy Mater., 2022, 5, 6980–6985 CrossRef CAS.
- T. Ma, L. Liu, J. Wang, Y. Lu and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 11533–11539 CrossRef CAS PubMed.
- W. Sun, C. Zhou, Y. Fan, Y. He, H. Zhang, Z. Quan, H. Kong, F. Fu, J. Qin and Y. Shen, Angew. Chem., 2023, 135, e202300158 CrossRef.
- T. Škorjanc, D. Shetty, M. A. Olson and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 6705–6716 CrossRef.
- K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K.-C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC.
- M. Kathiresan, B. Ambrose, N. Angulakshmi, D. E. Mathew, D. Sujatha and A. M. Stephan, J. Mater. Chem. A, 2021, 9, 27215–27233 RSC.
- S. M. Beladi-Mousavi, S. Sadaf, A. M. Mahmood and L. Walder, ACS Nano, 2017, 11, 8730–8740 CrossRef CAS PubMed.
- V. Kolivoška, M. Gál, L. Pospíšil, M. Valášek and M. Hromadová, Phys. Chem. Chem. Phys., 2011, 13, 11422–11429 RSC.
- W. Haiss, H. van Zalinge, H. Höbenreich, D. Bethell, D. J. Schiffrin, S. J. Higgins and R. J. Nichols, Langmuir, 2004, 20, 7694–7702 CrossRef CAS PubMed.
- M. J. Lacey, J. T. Frith and J. R. Owen, Electrochem. Commun., 2013, 26, 74–76 CrossRef CAS.
- M. Yao, H. Sano, H. Ando and T. Kiyobayashi, Sci. Rep., 2015, 5, 10962 CrossRef CAS.
- Z. Wang, A. Duan, W. Jin, X. Huang and Y. Li, J. Mater. Chem. A, 2022, 10, 10026–10032 RSC.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed.
- J. Qu, T. Katsumata, M. Satoh, J. Wada, J. Igarashi, K. Mizoguchi and T. Masuda, Chem.–Eur. J., 2007, 13, 7965–7973 CrossRef CAS.
- H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- Y. Xie, K. Zhang, Y. Yamauchi, K. Oyaizu and Z. Jia, Mater. Horiz., 2021, 8, 803–829 RSC.
- T. Suga, Y.-J. Pu, S. Kasatori and H. Nishide, Macromolecules, 2007, 40, 3167–3173 CrossRef CAS.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 6, 751–754 CrossRef.
- J. C. Barbosa, A. Fidalgo-Marijuan, J. C. Dias, R. Gonçalves, M. Salado, C. M. Costa and S. Lanceros-Méndez, Energy Storage Mater., 2023, 60, 102841 CrossRef.
- H. Nishide and T. Suga, Electrochem. Soc. Interface, 2005, 14, 32 CrossRef CAS.
- M. Suguro, S. Iwasa, Y. Kusachi, Y. Morioka and K. Nakahara, Macromol. Rapid Commun., 2007, 28, 1929–1933 CrossRef CAS.
- K. Nakahara, J. Iriyama, S. Iwasa, M. Suguro, M. Satoh and E. J. Cairns, J. Power Sources, 2007, 165, 398–402 CrossRef CAS.
- J.-K. Kim, Y. Kim, S. Park, H. Ko and Y. Kim, Energy Environ. Sci., 2016, 9, 1264–1269 RSC.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Macromol. Chem. Phys., 2009, 210, 1989–1995 CrossRef CAS.
- W. Deng, W. Shi, Q. Liu, J. Jiang, Q. Wang and C. Guo, J. Power Sources, 2020, 479, 228796 CrossRef CAS.
- W. Jin, T. Zhou, Z. Wang, W. Xue, C. Feng, F. Zhang, X. Huang, D. Yang, P. Théato and Y. Li, J. Power Sources, 2021, 511, 230363 CrossRef CAS.
- K. Zhang, Y. Hu, L. Wang, M. J. Monteiro and Z. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 34900–34908 CrossRef CAS.
- S. Yeşilot, F. Hacıvelioğlu, S. Küçükköylü, E. Demir, K. B. Celik and R. Demir-Cakan, Polym. Adv. Technol., 2019, 30, 2977–2982 CrossRef.
- H. Byeon, B. Gu, H.-J. Kim, J. H. Lee, I. Seo, J. Kim, J. W. Yang and J.-K. Kim, Chem. Eng. J., 2021, 413, 127402 CrossRef CAS.
- T. Zhou, W. Jin, W. Xue, B. Dai, C. Feng, X. Huang, P. Théato and Y. Li, J. Power Sources, 2021, 483, 229136 CrossRef CAS.
- K. Zhang, Y. Hu, L. Wang, J. Fan, M. J. Monteiro and Z. Jia, Polym. Chem., 2017, 8, 1815–1823 RSC.
- J.-K. Kim, J. Power Sources, 2020, 477, 228670 CrossRef CAS.
- Y. Chen, Y. Zhang, X. Liu, X. Fan, B. Bai, K. Yang, Z. Liang, Z. Zhang and K. Mai, Macromol. Rapid Commun., 2018, 39, 1800195 CrossRef.
- A. Gopinath and A. S. Nasar, Polymer, 2019, 178, 121601 CrossRef CAS.
- N. Hergué, B. Ernould, A. Minoia, R. Lazzaroni, J.-F. Gohy, P. Dubois and O. Coulembier, Batteries Supercaps, 2018, 1, 102–109 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS.
- B. Dunn, J.-M. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- C. Zhang, S. Chen, G. Zhou, Q. Hou, S. Luo, Y. Wang and G. Shi, J. Electrochem. Soc., 2021, 168, 050548 CrossRef CAS.
- Y. Zhao, M. Wu, H. Zhang, Z. Ge, C. Li, Y. Ma and Y. Chen, Energy Storage Mater., 2022, 47, 141–148 CrossRef.
- W. Wang, C. Zhao, J. Yang, P. Xiong, H. Su and Y. Xu, Sci. China Mater., 2021, 64, 2938–2948 CrossRef CAS.
- B. Wei, Y. Hong, W. Tang, M. Guo, X. He, C. Tang, J. Hu and C. Fan, Chem. Eng. J., 2023, 451, 138773 CrossRef CAS.
- C.-X. Zhang, X.-H. Chen, W.-S. Zhang, Y. Wang, S.-L. Mei, Y.-W. Zhong and C.-J. Yao, Chem. Eng. J., 2024, 483, 149198 CrossRef CAS.
- T. Liu, K. C. Kim, B. Lee, S. Jin, M. J. Lee, M. Li, S. Noda, S. S. Jang and S. W. Lee, ACS Appl. Energy Mater., 2020, 3, 3728–3735 CrossRef CAS.
- H. Guo, L. Liu, H. Shu, X. Yang, Z. Yang, M. Zhou, J. Tan, Z. Yan, H. Hu and X. Wang, J. Power Sources, 2014, 247, 117–126 CrossRef CAS.
- L. Zhu, L. Xie and X. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 10909–10917 CrossRef CAS PubMed.
- P.-O. Schwartz, S. Förtsch, E. Mena-Osteritz, D. Weirather-Köstner, M. Wachtler and P. Bäuerle, RSC Adv., 2018, 8, 14193–14200 RSC.
- L. Zhu, L. Xie, C. Bao, X. Yan and X. Cao, Int. J. Energy Res., 2020, 44, 298–308 CrossRef CAS.
- X. Li, X. Tang, K. Ouyang, P. Deng, L. Huang and W. Dang, Ionics, 2021, 27, 4649–4661 CrossRef CAS.
- F. Wu, J. Liu, L. Li, X. Zhang, R. Luo, Y. Ye and R. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 23095–23104 CrossRef CAS.
- J. Yang, Y. Shi, M. Li, P. Sun and Y. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 32666–32672 CrossRef CAS.
- G. Hernández, N. Casado, R. Coste, D. Shanmukaraj, L. Rubatat, M. Armand and D. Mecerreyes, RSC Adv., 2015, 5, 17096–17103 RSC.
- L. Assumma, Y. Kervella, J. M. Mouesca, M. Mendez, V. Maurel, L. Dubois, T. Gutel and S. Sadki, ChemSusChem, 2020, 13, 2419–2427 CrossRef CAS.
- C.-J. Yao, J. Xie, Z. Wu, Z. J. Xu, S. Zhang and Q. Zhang, Chem.–Asian J., 2019, 14, 2210–2214 CrossRef CAS.
- S. Zheng, L. Miao, T. Sun, L. Li, T. Ma, J. Bao, Z. Tao and J. Chen, J. Mater. Chem. A, 2021, 9, 2700–2705 RSC.
- H. Zhang, Y. Xie, X. Chen, T. Jia, W. Huang, S. Luo, Q. Hou, R. Zeng and Z. Sun, J. Electrochem. Soc., 2017, 164, A290 CrossRef CAS.
- S. Wang, A. M. G. Park, P. Flouda, A. D. Easley, F. Li, T. Ma, G. D. Fuchs and J. L. Lutkenhaus, ChemSusChem, 2020, 13, 2371–2378 CrossRef CAS PubMed.
- P. Acker, J. S. Wössner, G. Desmaizieres and B. Esser, ACS Sustainable Chem. Eng., 2022, 10, 3236–3244 CrossRef CAS.
- Z. Chen, S. Mei, W. Li, N. Xu, Y. Dong, Y. Jin, M. Ouyang and C. Zhang, J. Mater. Chem. A, 2021, 9, 27010–27018 RSC.
- J. Wang, Y. Lee, K. Tee, S. N. Riduan and Y. Zhang, Chem. Commun., 2018, 54, 7681–7684 RSC.
- R. Akiyoshi, M. Fujiwara, Y. Kamakura, T. Shimizu, R. Inoue, Y. Morisaki, A. Saeki, H. Yoshikawa and D. Tanaka, ACS Appl. Energy Mater., 2022, 5, 12760–12767 CrossRef CAS.
- N. Cheng, L. Ren, X. Xu, Y. Du and S. X. Dou, Mater. Today Phys., 2020, 15, 100289 CrossRef.
- T. Mehtab, G. Yasin, M. Arif, M. Shakeel, R. M. Korai, M. Nadeem, N. Muhammad and X. Lu, J. Energy Storage, 2019, 21, 632–646 CrossRef.
- G. Zhang, C. Xie, P. You and S. Li, in Introduction to Organic Electronic Devices, ed. G. Zhang, C. Xie, P. You and S. Li, Springer Nature Singapore, Singapore, 2022, pp. 283–307 Search PubMed.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- G. Xu, P. Nie, H. Dou, B. Ding, L. Li and X. Zhang, Mater. Today, 2017, 20, 191–209 CrossRef CAS.
- X. Chen, W. Sun and Y. Wang, ChemElectroChem, 2020, 7, 3905–3926 CrossRef CAS.
- Y. Deng, Y. Wang, X. Xiao, B. J. Saucedo, Z. Zhu, M. Xie, X. Xu, K. Yao, Y. Zhai and Z. Zhang, Small, 2022, 18, 2202928 CrossRef CAS PubMed.
- H. Kong, Y. Guan, J. Wang, W. Sun, L. Chen, J. Ou, L. Xie, F. Fu, H. Zhang and H. Chen, J. Mater. Chem. A, 2022, 10, 20866–20873 RSC.
- X. Du, Z. Zhang, W. Liu and Y. Deng, Nano Energy, 2017, 35, 299–320 CrossRef CAS.
- X. Yao and Y. Zhao, Chem, 2017, 2, 171–200 CAS.
- P. Sengodu and A. D. Deshmukh, RSC Adv., 2015, 5, 42109–42130 RSC.
- S. Maiti, A. Pramanik, T. Dhawa and S. Mahanty, Mater. Lett., 2017, 209, 613–617 CrossRef CAS.
- H.-C. Lin, C.-C. Li and J.-T. Lee, J. Power Sources, 2011, 196, 8098–8103 CrossRef CAS.
- J.-Y. Shi, C.-W. Yi and K. Kim, Bull. Korean Chem. Soc., 2010, 31, 2698–2700 CrossRef CAS.
- W.-M. Chen, L. Qie, L.-X. Yuan, S.-A. Xia, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Electrochim. Acta, 2011, 56, 2689–2695 CrossRef CAS.
- Z. Peng, X. Yi, Z. Liu, J. Shang and D. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 14578–14585 CrossRef CAS.
- J. Wang, W. Zhang, H. Wei, X. Zhai, F. Wang, Y. Zhou, F. Tao, P. Zhai, W. Liu and Y. Liu, Sustainable Energy Fuels, 2022, 6, 2901–2923 RSC.
- S. Lei, Y. Dong, Y. Dou, X. Zhang, Q. Zhang and Y. Yang, Mater. Adv., 2021, 2, 5785–5790 RSC.
- P. Das and B. C. Thompson, Polym. J., 2023, 55, 317–341 CrossRef CAS.
- K.-L. Wang, K.-T. Chen, Y.-H. Yi, Y.-H. Hung, H.-Y. Tuan and M. Horie, ACS Sustainable Chem. Eng., 2020, 8, 1043–1049 CrossRef CAS.
- V. V. Kondratiev and R. Holze, Chem. Pap., 2021, 75, 4981–5007 CrossRef CAS.
- C. Liedel, X. Wang and M. Antonietti, Nano Energy, 2018, 53, 536–543 CrossRef CAS.
- P. Acker, L. Rzesny, C. F. N. Marchiori, C. M. Araujo and B. Esser, Adv. Funct. Mater., 2019, 29, 1906436 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |