
Pore engineering of porous framework materials for efficient SF6 capture
Xiaoxuan
Sun
,
Liqin
Zhou
,
Jianmin
Chen
,
Zhaowei
Jia
,
Zhongxing
Zhao
and
Zhenxia
Zhao
 *
*
Key Laboratory of New Low-carbon Green Chemical Technology, Education Department of Guangxi Zhuang Autonomous Region, School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China. E-mail: zhaozhenxia@gxu.edu.cn
First published on 17th October 2024
Abstract
Sulfur hexafluoride (SF6) is an artificial inert gas widely used in the power and semiconductor industries and is known to be a significant contributor to the greenhouse effect due to its high global warming potential. Porous framework materials (PFMs), including metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and porous aromatic frameworks (PAFs), are well-established SF6 adsorbents characterized by highly ordered pore structures that can efficiently adsorb SF6via various electrostatic interactions. Recent advancements have focused on optimizing electrostatic interactions with SF6 molecules to enhance their SF6 adsorption performance in industrial settings. However, a comprehensive review on precise control of the structural and chemical properties through pore engineering strategies for PFMs has not been reported yet. This review systematically outlines the structure–activity relationship and the dominant mechanisms of host–guest interaction modes for effective SF6 capture and discusses pore engineering strategies to achieve efficient SF6 capture. Specifically, adjusting the pore structure to align with SF6 molecules, modifying the pore surface to strengthen electrostatic interactions and designing hierarchical pore structures to enhance SF6 adsorption kinetics are involved. Lastly, the potential of PFMs for SF6 capture is discussed from perspectives of AI-driven creation of high-performance PFMs, construction of PFMs with enhanced water- and acid-resistance under specialized conditions and shaping methods, aiming to guide the development of advanced SF6 adsorbents based on PFMs.
1. Introduction
With the development of economy and the increasing electricity demand, the electric power industry is progressively moving towards high voltage and ultra-high voltage.1,2 Insulating materials play a crucial role in ensuring the safe and stable operation of power systems.3,4 Gas insulation materials are extensively utilized in high-voltage and ultra-high-voltage power equipment due to their advantages of relatively light weight, blast resistance and especially their self-healing ability after insulation damage.5–8Sulfur hexafluoride (SF6) is an artificial insulating gas synthesized by two French chemists, Moissan and Lebeau, in 1900 and has enjoyed a history of over a hundred years.9 SF6 is composed of six fluorine atoms and a central sulfur atom with a kinetic diameter of 5.2 Å. Its octahedral molecular structure exhibits short bond distance and high bond energy, imparting exceptional chemical stability.10 SF6 is an electronegative gas that readily attracts free electrons to diminish the ionization process after electric breakdown, making it an excellent insulator and arc extinguisher.11 Consequently, SF6 has been the predominant choice in power facilities since 1947,12 including gas insulated switchgear (GIS),13 generator circuit breaker (GCB),14 gas insulated transmission line (GIL)15 and gas insulated transformer (GIT).16 Additionally, SF6 serves as a plasma etchant in the semiconductor industry for silicon-based chip etching.17,18
SF6 poses significant environmental concerns despite its numerous advantages, since SF6 has a global warming potential 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 times that of CO2 and an atmospheric lifetime of up to 3200 years due to its extremely high chemical stability.19 As an important contributor to the greenhouse effect, the increase of SF6 concentration would significantly warm up the Earth and cause other environmental problems, which has triggered global efforts to reduce the concentration of SF6 in the atmosphere.20 The Kyoto Protocol stipulates SF6 as one of the six major greenhouse gases and advocates for using the SF6/N2 mixture to mitigate the environmental impact without compromising its insulating properties.5,21 However, the emission of SF6 from aging pipelines and improper storage has still caused it to increase by threefold of the global mean concentration up to 10.5 ppt (t for trillion) in the past 25 years.10,22 Therefore, an efficient method for selectively capturing SF6 is needed to avoid its environmental hazards and facilitate its reuse.9,23,24 The adsorption separation technology with the advantages of easy operation and without SF6 phase change can capture and recover valuable fluorinated gases from industrial waste gases. Considering the low environmental concentration of SF6, non-thermal adsorption technology is the most promising and energy-efficient separation technique.20,25,26
900 times that of CO2 and an atmospheric lifetime of up to 3200 years due to its extremely high chemical stability.19 As an important contributor to the greenhouse effect, the increase of SF6 concentration would significantly warm up the Earth and cause other environmental problems, which has triggered global efforts to reduce the concentration of SF6 in the atmosphere.20 The Kyoto Protocol stipulates SF6 as one of the six major greenhouse gases and advocates for using the SF6/N2 mixture to mitigate the environmental impact without compromising its insulating properties.5,21 However, the emission of SF6 from aging pipelines and improper storage has still caused it to increase by threefold of the global mean concentration up to 10.5 ppt (t for trillion) in the past 25 years.10,22 Therefore, an efficient method for selectively capturing SF6 is needed to avoid its environmental hazards and facilitate its reuse.9,23,24 The adsorption separation technology with the advantages of easy operation and without SF6 phase change can capture and recover valuable fluorinated gases from industrial waste gases. Considering the low environmental concentration of SF6, non-thermal adsorption technology is the most promising and energy-efficient separation technique.20,25,26
Various porous materials, such as zeolites,27–29 porous polymer and organic cages,30–32 activated carbon,33,34 and porous framework materials (PFMs), have been investigated for the selective capture of SF6. Among them, PFMs, including metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and porous aromatic frameworks (PAFs), are reported as famous SF6 adsorbents.35–40 Their high surface area and tunable pore structure endow high adsorption uptake and selectivity for SF6via high affinity from electrostatic interaction sites.41–45 Effective pore engineering promises enhanced SF6 adsorption performance of PFMs by controlling their characteristics, including pore sizes, surface chemistry and flexibility.46–49 The burgeoning research demonstrated promising SF6 separation capabilities of PFMs over the past decade. However, a comprehensive overview focusing on the correlation between the PFM structure and their SF6 adsorption performance has not been reported till now, which is pivotal for guiding the design and fabrication of PFMs for efficient SF6 capture.
This review summarized the dominant mechanisms of SF6 adsorption on PFMs and delved into the latest advancements in PFMs as SF6 adsorbents, based on pore engineering strategies to enhance their adsorption performance. To begin with, the standards for evaluating the performance of adsorbents and the host–guest interaction modes for effective SF6 capture were introduced. Subsequently, modulation strategies for pore structure and surface chemistry were discussed for achieving enhanced capture of SF6. Finally, future challenges and prospects of PFMs as SF6 adsorbents in wider applications were addressed, aiming to improve their SF6 adsorption performance in industrial settings.
2. Properties and mechanisms of SF6 adsorption on PFMs
2.1. Properties
In order to effectively evaluate the performance of PFMs, the following aspects during the adsorption process should be considered.2.2. Adsorption mechanisms
2.2.1.1 Lewis acid–base interaction. SF6 with highly electronegative F atoms can act as a Lewis base and interact with Lewis acid sites through coordination. Typical Lewis acid sites include open metal sites (OMS) and hydrogen bonds (H-bonds).61,62
(i) Open metal sites: according to the Lewis acid–base theory, OMS are a typical kind of Lewis acid sites with unfilled bond orbitals and tend to match paired electrons in SF6 through the interaction of σ bonds and π bonds.63 For instance, Chowdhury and co-workers found that HKUST-1 (Cu3(1,3,5-benzenetricarboxylate)2) contained a certain amount of unsaturated CuII sites, which allowed HKUST-1 to adsorb SF6via strong electrostatic attraction between electropositive CuII and electron-rich SF6.64 Experimental results showed that HKUST-1 had a much higher SF6 adsorption capacity of up to 2.23 mmol g−1 than UiO-66 (0.82 mmol g−1) with similar pore sizes at 0.1 bar.65 Subsequently, OMS have been confirmed to enhance the interaction between the adsorbate and adsorbent and may further promote the adsorption through the interaction between the adsorbate and adsorbate.
(ii) H-bonds: H atoms in hydrogen-containing functional groups tend to show partial positive charge and become Lewis acid sites to form strong polar H-bonds with F atoms in SF6.66 For example, the electron cloud in the –NH2 group of UiO-66-NH2 deflected towards N atoms with higher electronegativity, while the H atoms were almost in a proton state, capable of forming N–H⋯F bonds with SF6.67 As a result, UiO-66-NH2 showed an increased Henry's constant for SF6 up to 38.2 mmol g−1 bar−1 compared with that of UiO-66 (22.2), indicating the improvement of the intrinsic interaction between the framework and SF6.
2.2.1.2. F–π interaction. The F–π interactions are electrostatically driven non-covalent interactions governed by the high electronegativity F atoms.68,69 The large electron cloud in aromatic molecules of the delocalized π-bonding system can form multiple F–π interactions with F atoms in SF6, which act as strong π-electron donors.70,71 The stability of the F–π interaction can be enhanced by increasing the electron deficiency in the aromatic system, thus correlating with the electrostatic surface potential (ESP) of the aromatic system. For example, SBMOF-1 with a pocket-like pore formed by four benzene rings attracted SF6 through F–π interactions.72 This is because the surface potential of the aromatic ring of the SDB ligands (4,4′-sulfonyldibenzoic acid) in SBMOF-1 was positive, while the surface potential of SF6 was negative.
2.2.1.3. Induced dipole moment effect. The electric field of polar functional groups can cause the deformation of the electron cloud and generate induced dipole moments to polarize SF6 molecules.73 The S–F bond in SF6 molecules would be polarized by the induced electric field gradient created by polar functional groups in the framework, generating electron transfer between PFMs and gas molecules. For instance, the entry of strongly electronegative F atoms in fluorinated PAFs (PAF-4F and PAF-8F) caused localized charge separation on the pore surface, making benzene rings become electropositive sites and intensify the adsorption affinity towards electronegative SF6.74
On the other hand, PFMs with mesopores can reduce the mass transfer resistance of SF6 to improve the absorption performance and reduce the time required for SF6 absorption to reach equilibrium. For example, Chuah et al. synthesized HKUST-1c nanocrystals with a hierarchical pore structure, which exhibited higher SF6 uptake (4.98 mmol g−1vs. 3.56 mmol g−1 at 25 °C and 1 bar) and better SF6/N2 selectivity (70 vs. 50 at 25 °C) compared to bulk HKUST-1 crystals.76 This result may indicate that the mesopores in HKUST-1c not only shortened the diffusion length and promoted the transport of SF6 molecules to active sites but also increased the number of accessible active sites for SF6 molecules.
3. Pore engineering strategies for enhancing the SF6 adsorption performance of PFMs
In the process of SF6 adsorption, SF6 molecules first diffuse through porous channels to the adsorption sites and then are anchored via various electrostatic interactions. Therefore, the adsorption performance of PFMs would be influenced by pore sizes, pore shape and surface properties.26 Based on the properties and mechanisms of PFMs mentioned before, it can be concluded that PFMs capable of capturing SF6 efficiently in their pores have a strong tendency to form various electrostatic interactions with SF6 molecules. The metal node composition and functionality present in ligands are fundamentally important structural parameters that determine the affinity of PFMs to adsorb SF6. In addition, such PFMs must exhibit appropriate pore apertures and pore shapes matching the kinetic diameters of SF6. In this section, several useful strategies and approaches from the perspective of pore engineering were given to enhance the interaction between PFMs and SF6 and achieve effective SF6 capture.3.1. Metal–organic frameworks
Metal–organic frameworks (MOFs) are known as a class of relatively new porous materials with a periodic network composed of metal ions/clusters and organic ligands linked via coordination bonds.77–79 Different from other traditional porous materials such as zeolites and activated carbons, MOFs possess clearly defined and adjustable crystal structures that are more suitable for on-demand targeted design.80 Evidently, gaining a better understanding of the structure–property relationship offers great potential for the enhancement of SF6 adsorption performance on MOFs by structural adjustments via isoreticular chemistry or by the design and construction of new MOF structures via the practice of reticular chemistry.81,82 This section focused on enhancing pore-matching effects and multiple interactions between the framework and gas molecules to improve the SF6/N2 separation of MOFs, and Table 1 summarizes recent advances in MOFs for SF6 adsorption.| Sample name | BET surface area (m2 g−1) | Pore size (Å) | SF6 uptake at 0.1 bar (mmol g−1) | SF6 uptake at 1.0 bar (mmol g−1) | SF6/N2 selectivity (1:9) |
SF6 isosteric heat of adsorption, Qst (kJ mol−1) | Ref. |
|---|---|---|---|---|---|---|---|
| UiO-66 | 1074 | 9 | 0.82 | 1.45 | 140.0 | 32.0 | 64 |
| UiO-66-Br2 | 616 | 9 | 0.78 | 220 | 45.0 | ||
| SBMOF-1 | 169 | 5.5 | 1.14 | 92.6 | 34.2 | 66 | |
| V-TBAPy | 1370 | 6.40 | 1.32 | 3.28 | 360.7 | 30.48 | 78 |
| Ga-TBAPy | 1484 | 5.90 | 1.33 | 3.50 | 418.5 | 30.44 | |
| V-TCPB | 1107 | 5.38 | 2.29 | 3.07 | 65.6 | 28.08 | |
| Ga-TCPB | 1144 | 7.85 | 2.25 | 2.95 | 55.4 | 27.18 | |
| Cu(peba)2 | 814 | 12.0 | 0.15 | 2.36 | 18.2 | 14.4 | 79 |
| Ni(pba)2 | 807 | 8.2 | 1.70 | 3.50 | 200.6 | 24.0 | |
| Ni(ina)2 | 470 | 6.0 | 2.39 | 2.84 | 375.1 | 33.4 | |
| ZIF-8 | 1540 | 3.4 | 0.15 | 1.7 | 80 | ||
| ZIF-70.26-80.74 | 948 | 7.2 | 2.08 | 40 | |||
| SNNU-202 | 1319 | 9.0 | 4.94 | 9 | 20.0 | 81 | |
| SNNU-203 | 1522 | 7.8 | 5.43 | 27 | 20.8 | ||
| SNNU-204 | 2170 | 7.6 | 6.06 | 49 | 21.0 | ||
| Mg-MOF-74 | 1631 | 11.0 | 2.10 | 6.42 | 18.0 | 32.0 | 82 |
| Co-MOF-74 | 1219 | 11.0 | 2.00 | 5.34 | 34.0 | 40.0 | |
| Zn-MOF-74 | 992 | 11.0 | 1.40 | 3.90 | 46.0 | 26.0 | |
| Ni(3-mbpa)2 | 835 | 11.9 | 1.79 | 2.83 | 221.0 | 30.2 | 83 |
| Co(3-mbpa)2 | 626 | 12.5 | 1.77 | 3.25 | 161.0 | 26.8 | |
| Ni(adc)(dabco)0.5 | 744 | 5.5 | 2.23 | 2.38 | 919.4 | 47.6 | 84 |
| Cu-MOF-NH2 | 2145 | 6 | 3.39 | 7.88 | 266 | 55.2 | 85 |
The coordination pattern of metal ions profoundly influences the topological structure of MOFs, leading to tunable gas adsorption capability. Åhlén et al. designed a series of MOFs based on TBAPy4− (1,3,6,8-tetrakis(4-carboxyphenyl)pyrene) to investigate the impact of metal species on pore size.86 Yb- and Tm-TBAPy exhibited similar diamond-shaped channels with a pore size distribution of approximately 6.5 Å, while Hf-TBAPy featured hexagonal channels spanning about 7.2 Å in diameter. Smaller pores allowed for greater potential overlap with SF6 molecules, thereby enhancing their SF6 adsorption and resulting in higher adsorption capacities for Yb- and Tm-TBAPy. Experimental results showed that the SF6 uptake of Yb- and Tm-TBAPy reached 2.33 mmol g−1 and 1.83 mmol g−1 at 1 bar, surpassing that of Hf-TBAPy (1.38 mmol g−1).
Subsequently, they designed a series of rod MOFs constructed by high valence metal ions (Ga(III) and V(IV)) with suitable pore size and uniform one-dimensional channels for efficient capture of SF6 (Fig. 1a and b).83 The rod secondary building units (SBUs) of MOFs coordinated with carboxylic acid groups of the ligands to form one-dimensional rhombohedral channels. The benzene ring of the linkage contracted along the channel direction with pore sizes of about 5.5 Å, which increased the affinity toward SF6 molecules, resulting in steep adsorption isotherms at low pressure (Fig. 1c). Experimental results showed the SF6 uptake of V-TCPB and Ga-TCPB (TCPB = 1,2,4,5-tetrakis(4-carboxlatephenyl)benzene) up to 3.0 mmol g−1 with excellent SF6/N2 selectivity above 350.
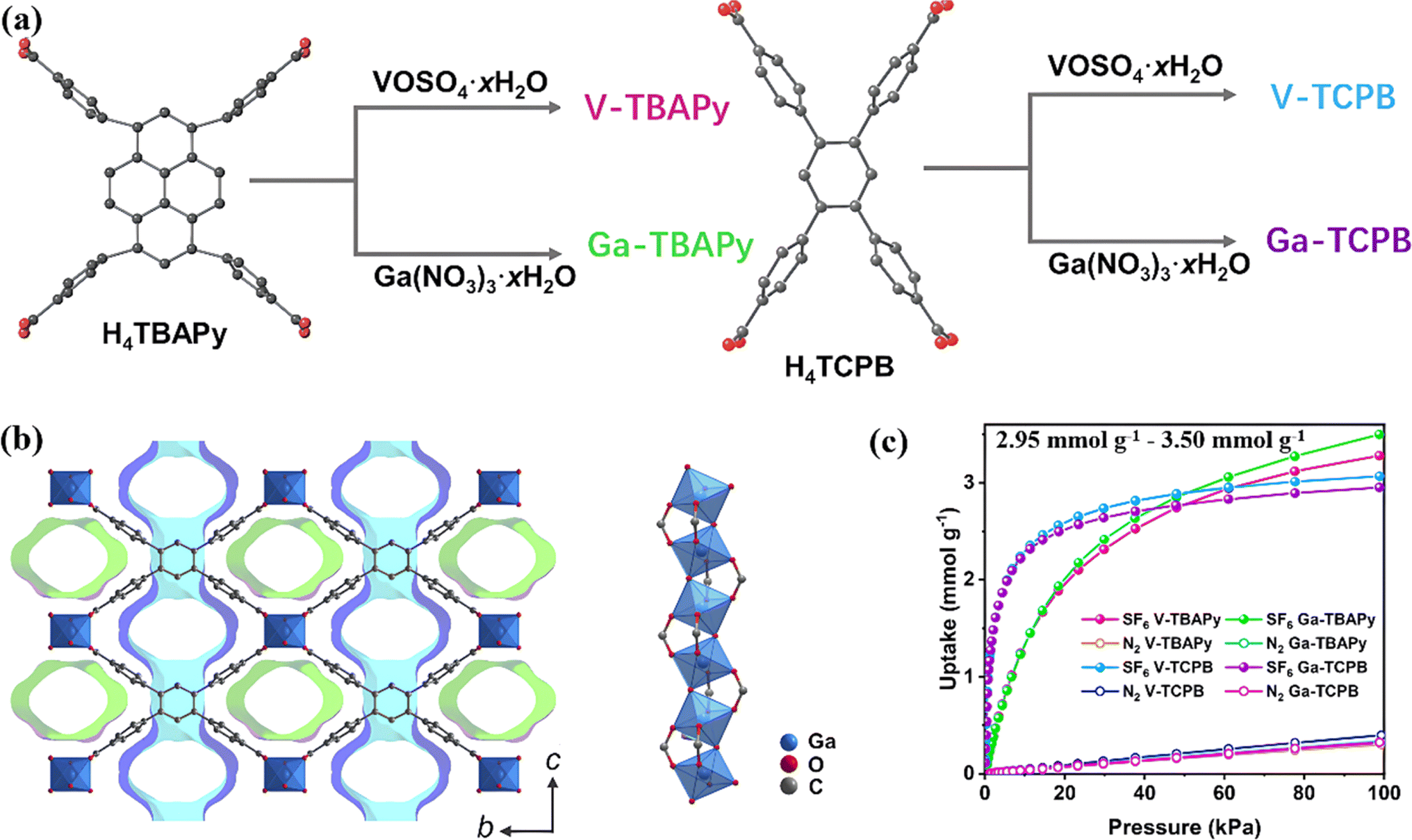 | ||
| Fig. 1 (a) Synthesis route of Ga- and V-based MOFs with the respective tetratopic H4TBAPy and H4TCPB ligands. (b) Structure of Ga-TCPB as viewed along the a-axis showing the presence of two unique pores (colored in green and blue) and the rod SBU running along the a-axis in the structure. (c) SF6 and N2 adsorption isotherms recorded at 293 K. Reproduced with permission from ref. 78. Copyright 2023 Royal Society of Chemistry. | ||
The pore size of MOFs can also be easily modified by linearly adjusting the length of the ligand within the existing parent structure, offering viable approaches to strengthen the host–guest interaction and enhance their SF6 adsorption properties. For example, Wang et al. used a family of bifunctional pyridylcarboxylate ligands with different lengths to create ultra-microporous MOFs for SF6 selective adsorption at low pressure.87 The pore size decreased from 12 to 6 Å with the reduction of the ligand length (Fig. 2a). Notably, the SF6 adsorption isotherm of Ni(ina)2 reached a plateau at very low pressure (>0.1 bar), indicating strong interactions between SF6 molecules and the framework (Fig. 2b). This was attributed to the perfect match between the pore and SF6 molecules (6 Å vs. 5.2 Å), which allowed the framework to generate multipoint van der Waals interactions with SF6 through several F–H bonds. As a result, Ni(ina)2 exhibited an unprecedented SF6/N2 selectivity up to 375.1 (Fig. 2c).
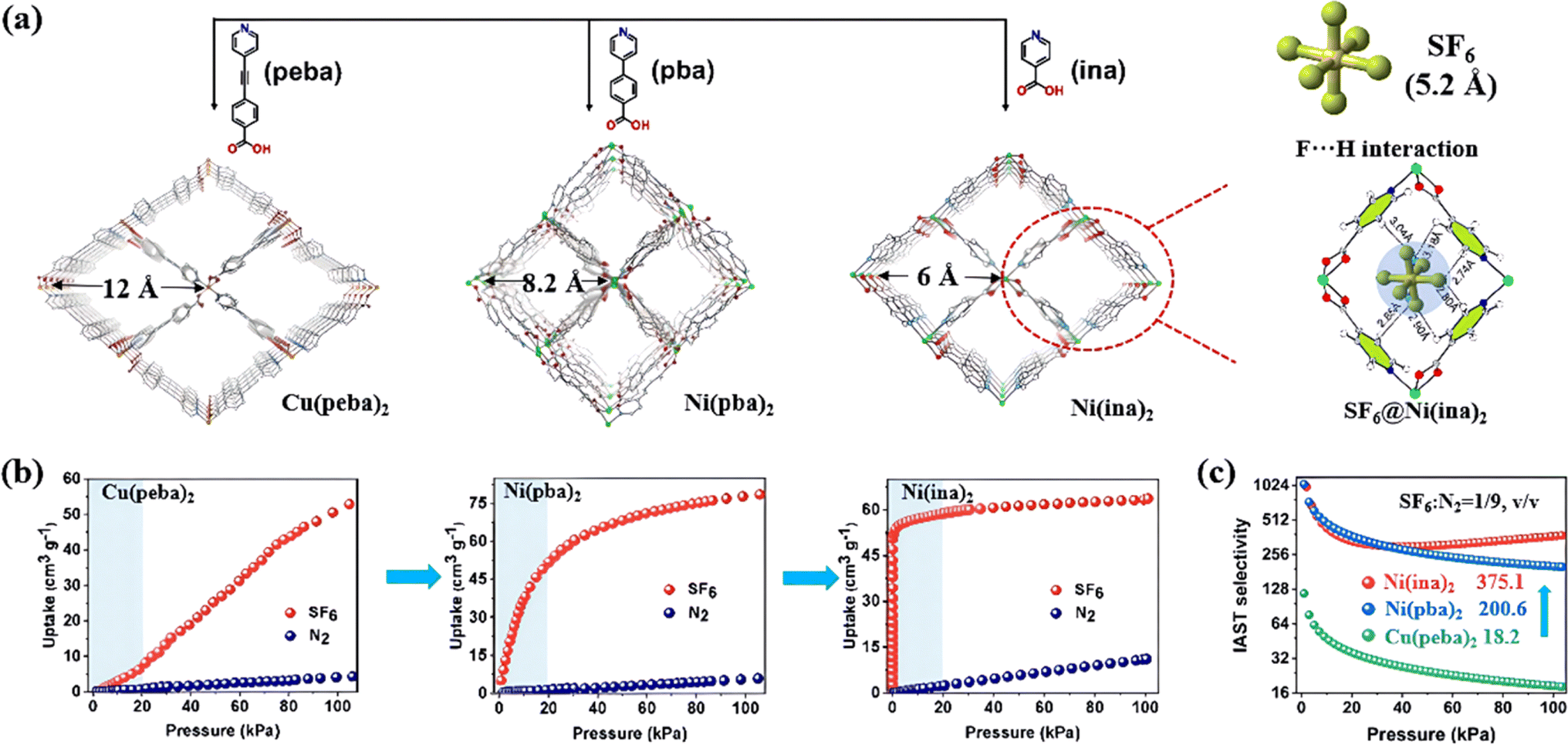 | ||
| Fig. 2 (a) The pore structures of Cu(peba)2, Ni(pba)2 and Ni(ina)2 and the calculated adsorption binding sites of SF6 in Ni(ina)2. (b) SF6 and N2 adsorption isotherms of Cu(peba)2, Ni(pba)2 and Ni(ina)2 at 298 K. (c) SF6/N2 selectivity of the three MOFs calculated at 298 K (SF6/N2, 1/9, v/v) using IAST. Reproduced with permission from ref. 79. Copyright 2022 Wiley-VCH. | ||
Introducing multiple ligands into the same framework can adjust the structure of MOFs from the coordination sites and redistribute their pore sizes, thereby enhancing their affinity with SF6 molecules. For instance, Åhlén and colleagues developed a series of mixed-linker ZIF-7-8s by substituting the 2-methylimidazolate (mIm) ligand in ZIF-8 with a larger benzimidazolate (bIm) ligand to reduce its pore size and enhance the selective adsorption for SF6.88 The inner cage sizes of ZIF-8 were reduced from 12.5 Å to 7.3 Å by adjusting the ratio between bIm and mIm ligands, thereby enhancing the van der Waals interaction between SF6 molecules and the framework. Among them, ZIF-70.2-80.8 with a bIm ligand concentration of 20% exhibited notable improvements compared with the original ZIF-8, surging the adsorption capacity from 0.15 mmol g−1 to 2.08 mmol g−1 and SF6/N2 selectivity from 1.39 to 40.04.
Subsequently, Li et al. achieved pore-window size control in Cu-MOFs via a mixed-linker strategy.84 Pore window partitions were realized by connecting adjacent square metal nodes through ditopic linear linkages, allowing for stepwise modification of pore sizes from 9.0 to 7.6 Å (SNNU-202-204) (SNNU-202 = Cu6(H2O)3(NTB)4(PYZ)1.5, SNNU-203 = Cu12(H2O)3(NTB)8(PYZ)3(BPY)1.5 and SNNU-204 = Cu6(NTB)4(DABCO)1.5(BPY)1.5, H3NTB = nitrilotribenzoic acid, PYZ = pyrazine, BPY = 4,4′-bipyridine and DABCO = 1,4-diazabicyclo[2.2.2]octane) (Fig. 3). The ultra-microporous structure of SNNU-204 formed strong interaction with SF6 molecules, leading to a significant increase of SF6/N2 selectivity from 9 to 49.
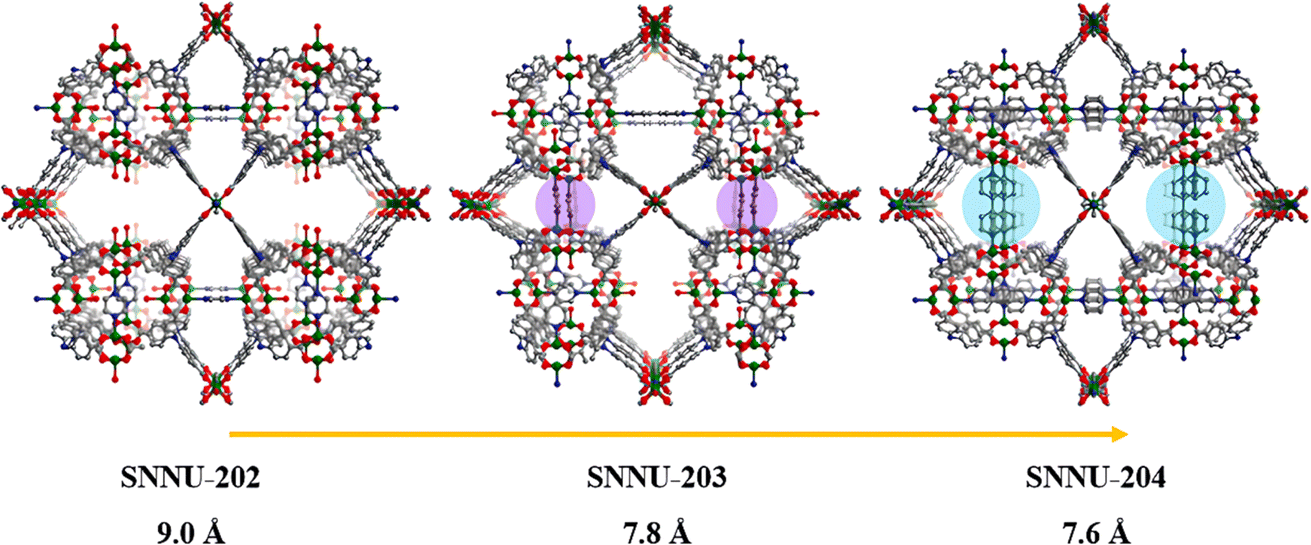 | ||
| Fig. 3 Progressive process of stepwise pore window partitions based on different partitional linkers. Reproduced with permission from ref. 81. Copyright 2024 Elsevier. | ||
3.1.2.1. Open metal sites. As reported earlier, the species and quantity of OMS in MOFs can greatly affect their adsorption properties towards SF6via electrostatic attraction. Kim et al. proposed adjusting OMS species to change their SF6 adsorption properties.85 Primarily, in this work, a series of M-MOF-74s (M = Mg, Co, and Zn) with highly dense OMS have been constructed and used to capture SF6 under low pressure (Fig. 4a). Experimental results showed that the SF6 adsorption capacity of these three MOFs exhibited the order of Co-MOF-74 > Mg-MOF-74 > Zn-MOF-74. Among them, Co-MOF-74 featuring unsaturated CoII sites had the highest adsorption uptake and adsorption heat for SF6 (Fig. 4b and c). This was ascribed to the strongest polarization of CoII sites, which generated very strong electrostatic interaction with S–F bonds of SF6. Meanwhile, Co-MOF-74 was proved to possess about 1.5 times higher density of CoII sites compared to CuII sites in HKUST-1,89 achieving a SF6 adsorption capacity of up to 5.34 mmol g−1 at 1 bar.
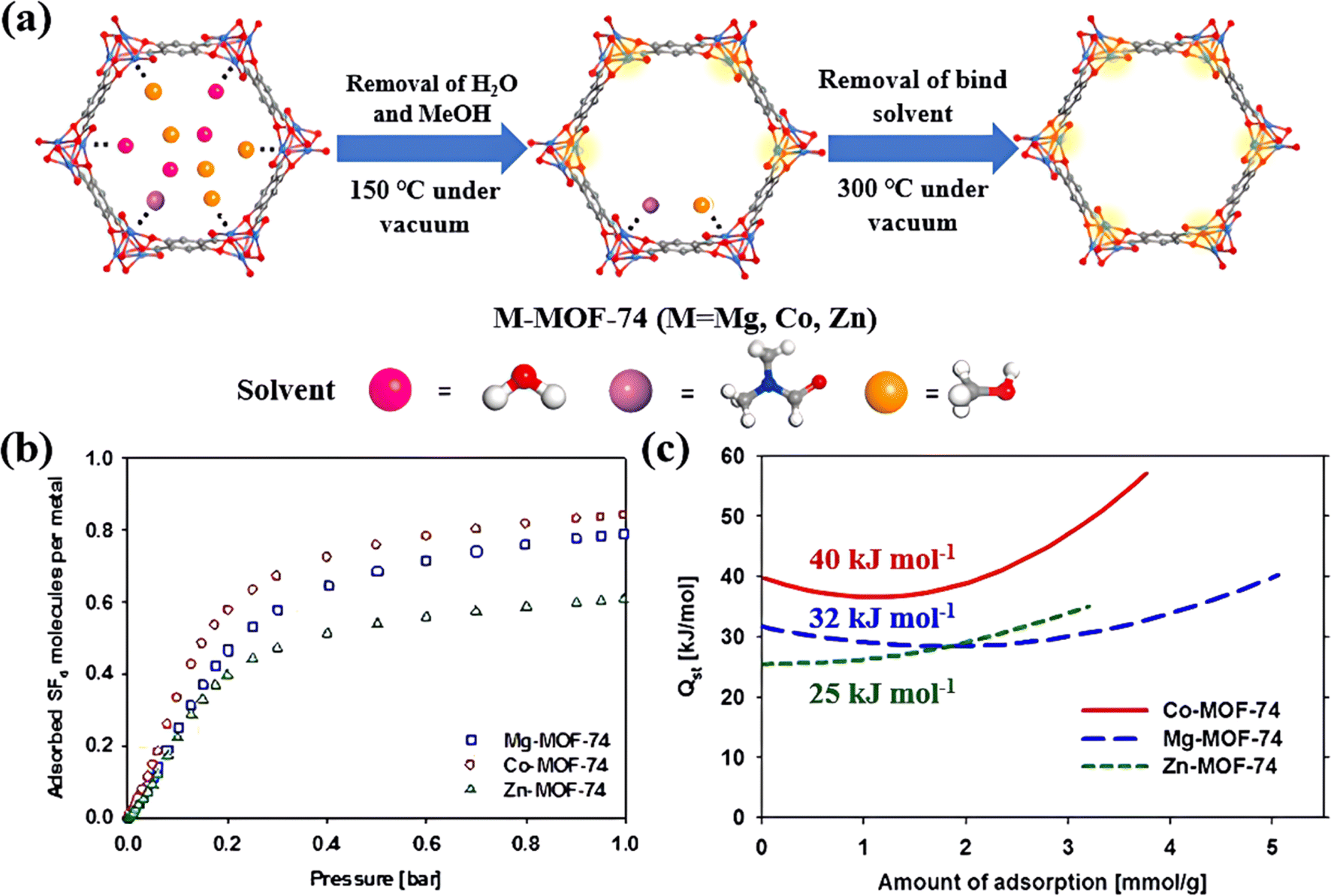 | ||
| Fig. 4 (a) The effective removal of guest molecules from Co-MOF-74 during the activation process. Reproduced with permission from Liang et al.89 Copyright 2021 Elsevier. (b) Adsorbed SF6 molecules per metal site at 298 K. (c) Isosteric heat of adsorption (Qst) for SF6 adsorption in three MOFs. Reproduced with permission from ref. 82. Copyright 2014 Elsevier. | ||
3.1.2.2. Functional groups of MOF ligands. Functional groups in MOF ligands also play a crucial role in SF6 adsorption performance through influencing the formation of H-bonds and pore polarizability in MOFs. For instance, introducing functional groups containing H atoms, such as methyl (–CH3) and amino (–NH2) groups, into ligands can create more affinity adsorption sites towards SF6 in MOFs. Zheng et al. implanted a methyl group (–CH3) into 4-(4-pyridyl)benzoate (pba) ligands to synthesize Ni(3-mpba)2 (3-mpba = 3-methyl-4-(4-pyridyl)benzoate) (Fig. 5a).90 In this system, methyl group in 3-mpba efficiently formed H-bonds with SF6, which strengthened its SF6 adsorption via multiple site bindings (Fig. 5b). This increased affinity could also be expressed by the change of their isosteric heat of adsorption (Qst) and SF6/N2 selectivity. Methyl-modified Ni(3-mpba)2 exhibited 6.2 kJ mol−1 higher enthalpy value of SF6 compared with its pristine Ni(pba)2, and its SF6/N2 selectivity increased from 160 to 221 (Fig. 5c).
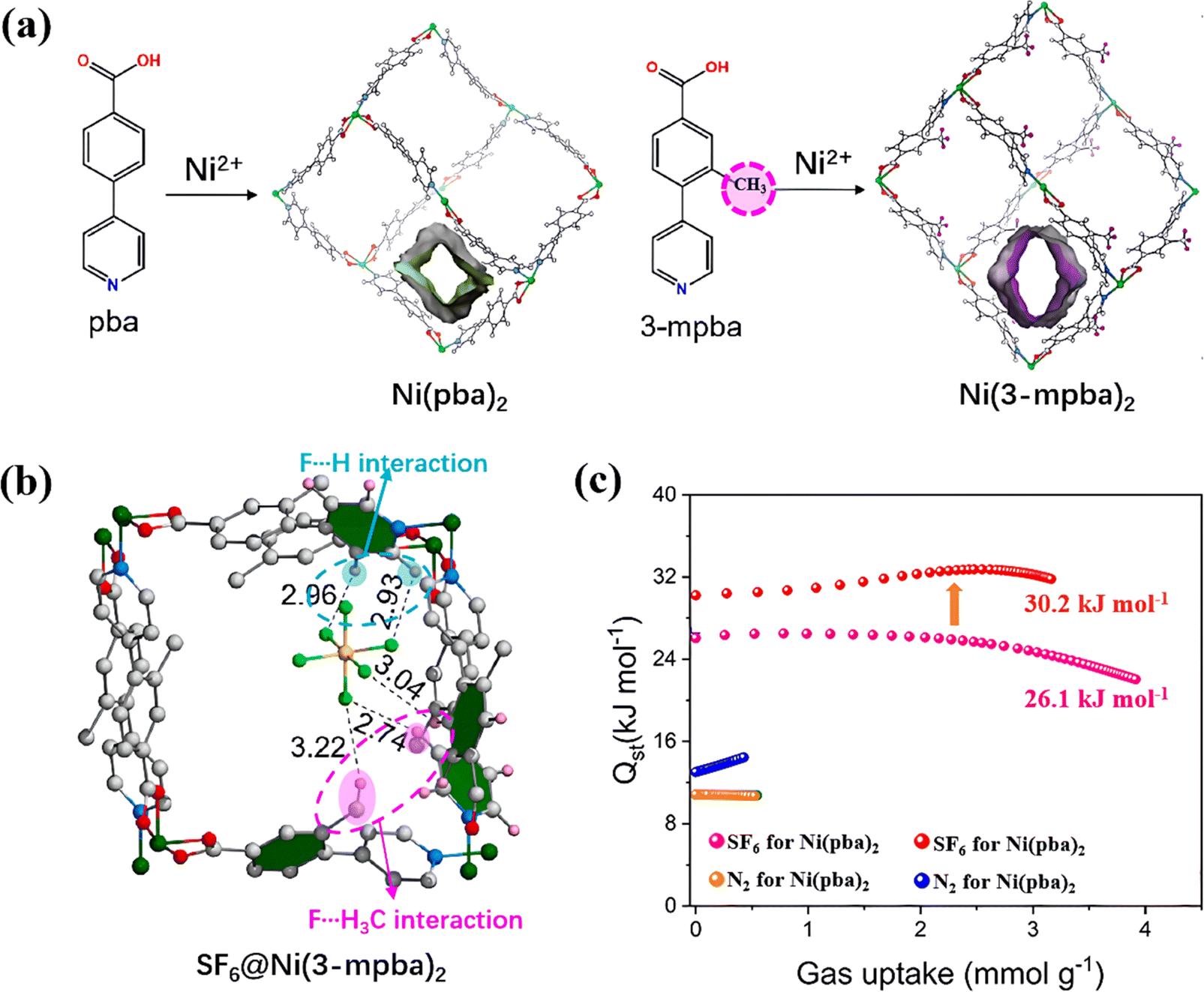 | ||
| Fig. 5 (a) The coordination geometry and pore structures of Ni(pba)2 and Ni(3-mpba)2. (b) Simulation of SF6 binding sites in Ni(3-mpba)2. (c) Isosteric heat of adsorption (Qst) for SF6 and N2 adsorption in Ni(pba)2 and Ni(3-mpba)2. Reproduced with permission from ref. 83. Copyright 2023 Elsevier. | ||
Additionally, Kim et al. introduced a series of halogen groups (Cl, Br, and I) in UiO-66 to explore the polarizability effect of the adsorbent surface on the adsorption properties of SF6.67 Their adsorption capacity of SF6 under low pressure is given as follows: UiO-66-Br2 > UiO-66-I > UiO-66-Br > UiO-66-Cl > UiO-66. It was found that their adsorption affinity increased with the atomic weight of these halogen atoms, which facilitated the van der Waals interaction towards SF6. Moreover, the introduction of bromine atoms partially blocked its pores and narrowed the pore size from 12 to 9 Å, thereby improving the SF6/N2 selectivity from 140 to 220. Chen et al. introduced F atoms into tetrazole ligands to synthesize extensively fluorinated MOFF-5.91 However, MOFF-5 only exhibited a modest SF6 adsorption capacity of 0.08 mmol g−1 at 0.1 bar, which was due to the mismatch between the highly polarized environment inside fluorinated pores and SF6 with low polarity, resulting in the absence of strong binding sites.
3.1.2.3. Topological structure. Topological features of MOFs could affect SF6 capture through forming multiple sites with the aid of the pore confinement effect.
Wu et al. constructed a benzene encircled SBMOF-1 with four benzene rings that formed a 5.5–8.5 Å pocket-shaped micropore to accommodate SF6 (5.2 Å) (Fig. 6a).72 High amounts of C–H groups in aromatic rings formed multiple van der Waals interactions with the F atoms of SF6 molecules and efficiently increased synergistic interactions (Fig. 6b). As a result, SBMOF-1 exhibited SF6 selectivity reaching 727 at 300 ppm.
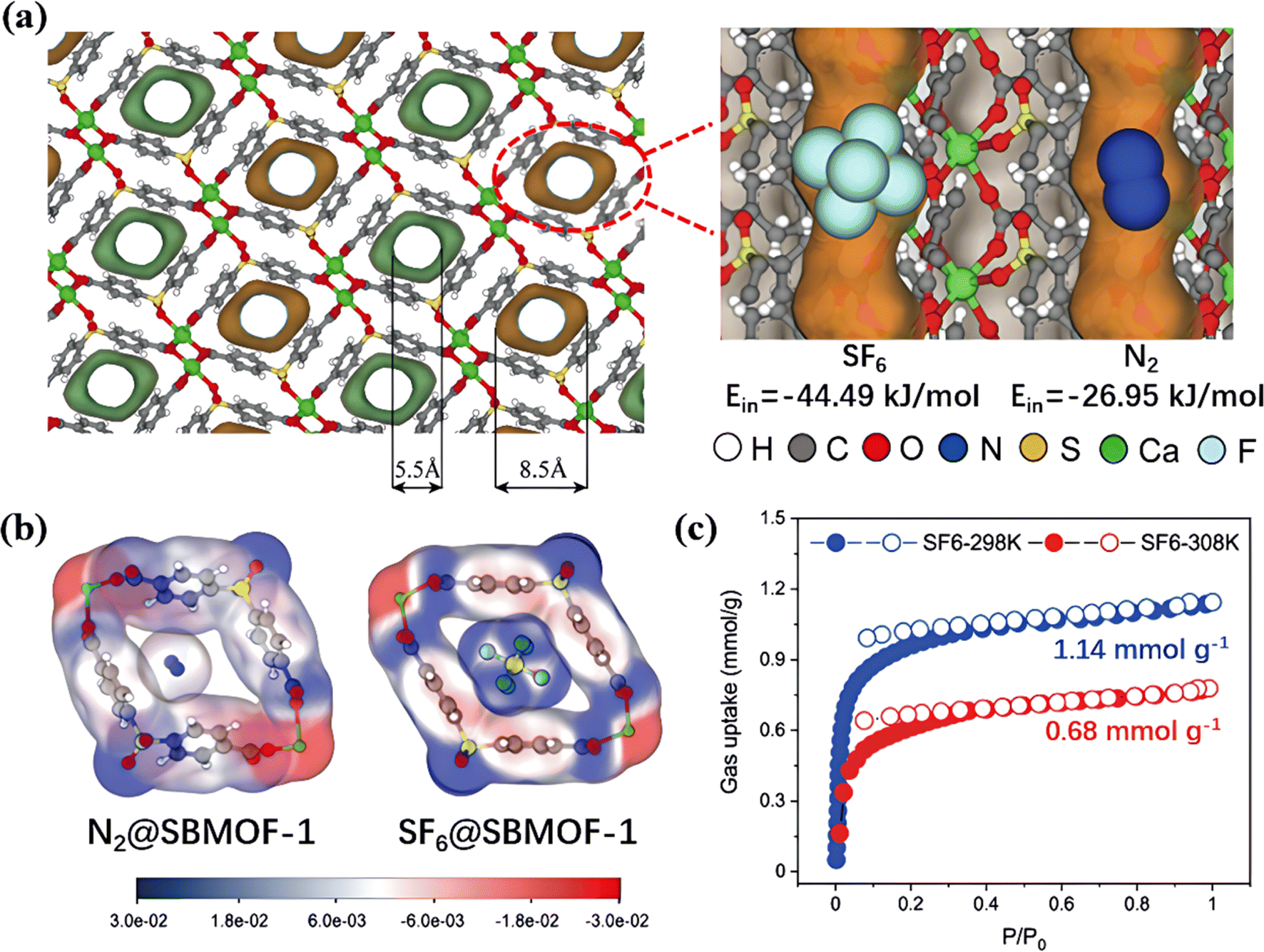 | ||
| Fig. 6 (a) Pore structure of SBMOF-1 from the front view. (b) Electrostatic potential of N2 and SF6 inside the pore channel of SBMOF-1. (c) SF6 and N2 adsorption isotherms of SBMOF-1 at 298 K. Reproduced with permission from ref. 66. Copyright 2022 Elsevier. | ||
Yang and co-workers used naphthalene rings as ligands to construct a splint-like pore in Ni(NDC)(TED)0.5 (H2NDC = 1,4-naphthalenedicarboxylic acid, TED = triethylenediamine), which could form a stronger potential overlap with SF6.92 The F–π interactions between naphthalene rings and SF6 were proved to be stronger than H-bonds. Therefore, Ni(NDC)(TED)0.5 exhibited a SF6 selectivity of up to 750 and a SF6 uptake of up to 2.76 mmol g−1 at 0.1 bar.
In addition, Chang et al. designed a single-molecule SF6 trap based on a pillar-layered MOF (Ni(adc)(dabco)0.5) (H2adc = 9,10-anthracenedicarboxylic acid, dabco = 1,4-diazabicyclo[2.2.2]octane).93 The parallel anthracene rings created 5.5 Å splint-like pores with a giant potential overlap with SF6, thereby confining gas molecules within the pore (Fig. 7a). Therefore, this single-molecule trap attracted SF6 through multiple affinity sites and a fluorophilic microenvironment, achieving unprecedented separation selectivity (Fig. 7b). As a result, Ni(adc)(dabco)0.5 exhibited an adsorption heat of up to 47.6 kJ mol−1 with ultra-high SF6/N2 selectivity (919.4) (Fig. 7c).
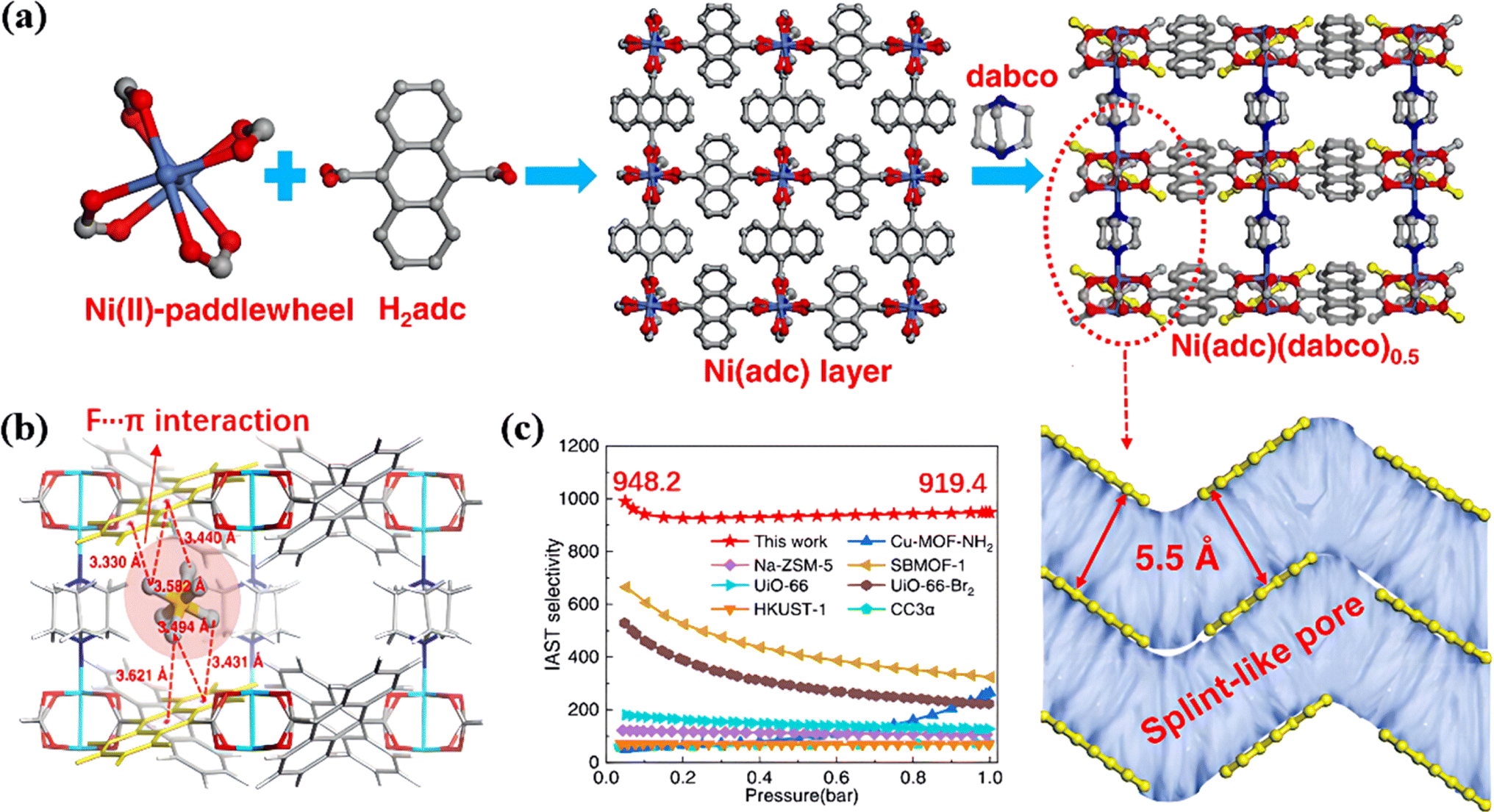 | ||
| Fig. 7 (a) Structure description of Ni(adc)(dabco)0.5. (b) Periodic DFT-optimized adsorption configuration of the SF6 molecule in the splint-like pore. Atom colors: Ni, cyan; O, red; N, blue; C, gray; H, white. (c) Comparison of the IAST-predicted selectivity of Ni(adc)(dabco)0.5 with some selected benchmark materials reported in the literature. The molar composition of the bulk mixture: SF6/N2 = 10/90; operation conditions: 298 K. Reproduced with permission from ref. 84. Copyright 2022 American Chemical Society. | ||
Ren et al. found that the MOF (Cu-MOF-NH2) with calix-shaped pores surrounded by benzene rings and multiple annularly pendant amino groups shows excellent SF6 capture (Fig. 8a).94 Such an internal environment endowed the framework with a multi-dimensional confinement towards SF6via strong H-bonds and van der Waals-type interactions. As a result, Cu-MOF-NH2 showed a SF6 adsorption capacity of 3.39 mmol g−1 at 0.1 bar and a SF6/N2 selectivity of up to about 266 (Fig. 8b and c).
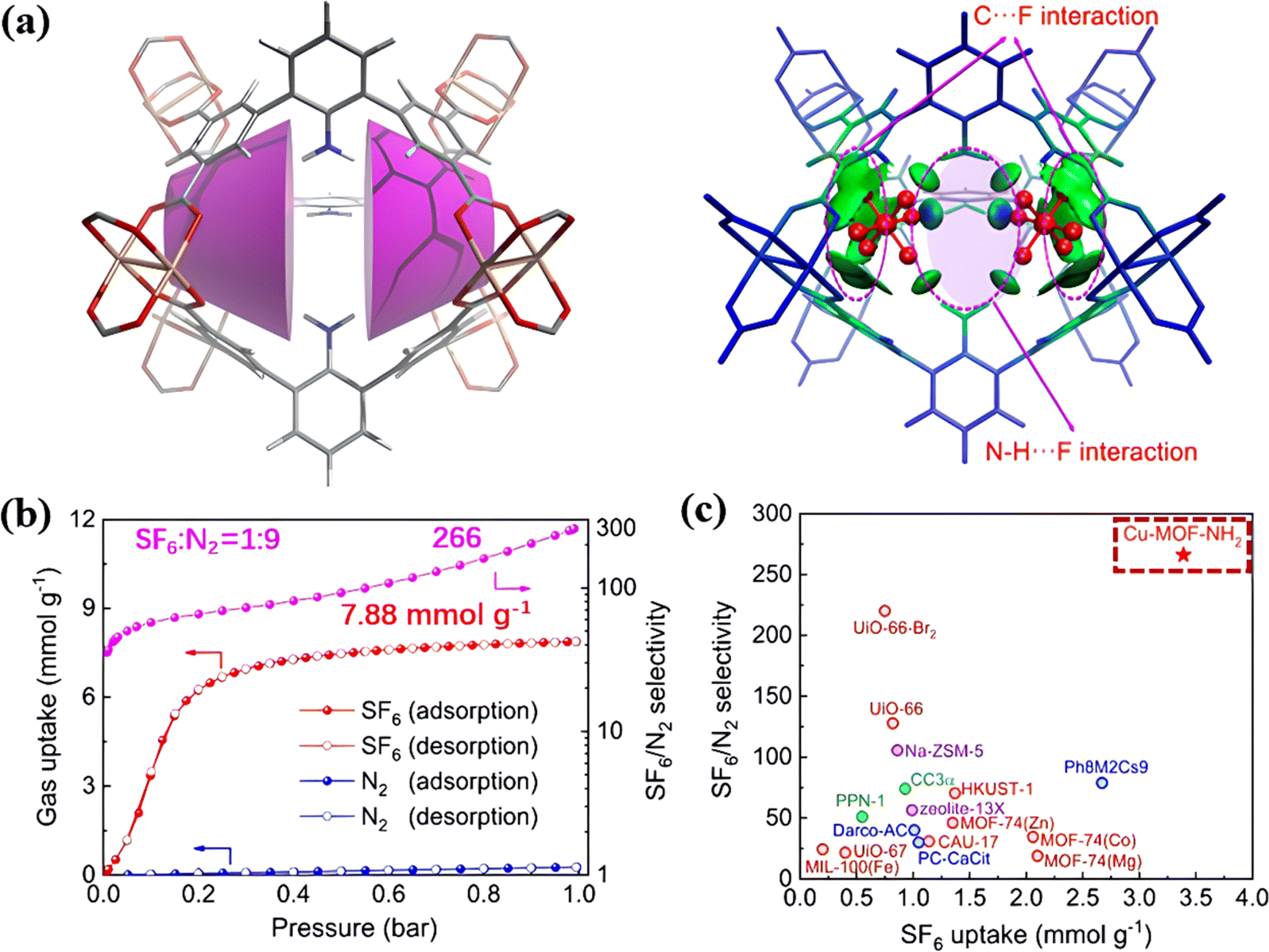 | ||
| Fig. 8 (a) Representation of the [calix3]arene analogous microenvironments present in Cu-MOF-NH2 and IGM-derived isosurfaces of δinterg (isovalue = 0.005 a.u.) for SF6 in cavity A. (b) Experimental N2 (blue) and SF6 (red) single component adsorption isotherms and the IAST-derived SF6/N2 selectivity (pink) in Cu-MOF-NH2 at 298 K. (c) Separation performance of Cu-MOF-NH2 and various benchmark materials. Reproduced with permission from ref. 85. Copyright 2021 American Chemical Society. | ||
The gate-opening phenomenon of flexible MOFs allows gas molecules having larger sizes to enter their pores, thereby achieving higher adsorption capacity and selectivity of SF6 under specific pressure and temperature stimuli.78 Choi et al. investigated the effect of the gate-opening phenomenon via ligand rotation of ZIF-8 under threshold pressure on selective adsorption properties of SF6.98 The ligands of ZIF-8 rotated at a certain angle induced by high pressure, which increased its pore size from 3.4 to 7.6 Å to enhance its adsorption capacity and form a strong interaction with SF6. Experimental results showed that the rotated ZIF-8 presented an increase of SF6 uptake of up to 3.35 mmol g−1 when the pressure was increased to 20 bar. When the pressure was reduced to atmospheric pressure, SF6 molecules were locked in the shrunk pores of ZIF-8. Finally, SF6 can be recovered through variable temperature desorption, achieving efficient separation and capture of SF6 from various gases.
Senkovaska and colleagues developed a type of pillar-layered MOF M2(bdc)2(dabco) (M = Co, Ni, bdc = 1,4-benzenedicarboxylate; dabco = diazabicyclo[2.2.2]octane) with flexible M2 paddle-wheel nodes exhibiting a gate-opening phenomenon (Fig. 9a and b), which showed excellent adsorption performance of SF6.99 As shown in Fig. 9c, MOFs transformed from a narrow pore (np)-phase (5 Å) to a large pore (lp)-phase (7–9 Å) due to the swelling and switching of flexible metal nodes under high pressure stimuli, allowing SF6 molecules to enter the pores and form strong interactions with MOFs. Meanwhile, Co2(bdc)2(dabco) showed a greater phase transition magnitude than Ni2(bdc)2(dabco) since the outer electrons of Co were more tolerable to the changes in the coordination environment. Therefore, Co2(bdc)2(dabco) reached a high SF6 uptake of 4.95 mmol g−1 at 15 bar, which was 7.4 times greater than that of Ni2(bdc)2(dabco).100
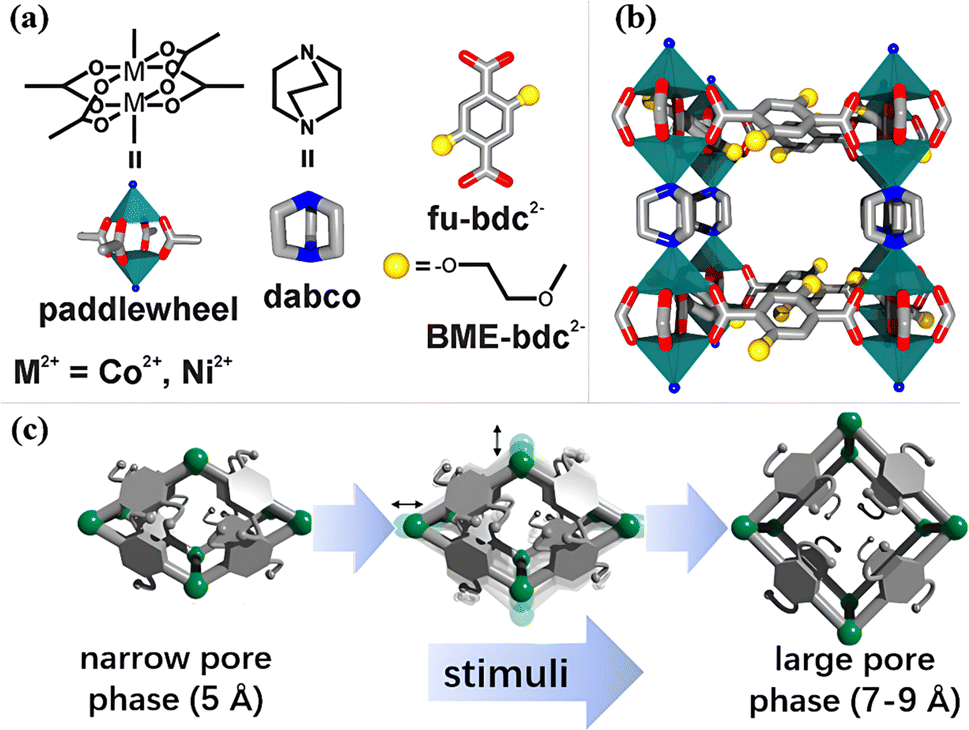 | ||
| Fig. 9 (a) Depiction of the building blocks used for the synthesis of M2(fu-bdc)2(dabco) materials. (b) Side view of a single cavity of the extended 3D network structure. (c) Schematic illustration of the np → lp phase transition of the M2(fu-bdc)2(dabco) materials. Reproduced with permission from ref. 92. Copyright 2018 American Chemical Society. | ||
Other flexible MOFs show thermal expansion features when suffering the temperature variation, and the changing porous structure would affect SF6 adsorption behaviors. Åhlén et al. reported that CAU-33 (1,2,4,5-tetrakis-(4-carboxyphenyl)benzene) exhibited positive thermal expansion properties, which could increase its pore volume to enhance SF6 capacity through temperature increase.101 The covalent bonds between metal centers and ligands were weak due to the low coordination number of Bi molecules and therefore could be elongated under heating conditions. Experiment verified that its pore volume could expand by 9% when the temperature was increased to 60 °C, thereby enhancing the adsorption capacity of SF6 (1.10 mmol g−1) which is twice that of the value observed at 20 °C (0.58 mmol g−1).
Senkovaska et al. found that Zn4O(dmcpz)3 (dmcpz = 3,5-dimethyl-4-carboxypyrazolato) exhibited negative thermal expansion properties, which could reduce its pore size to improve the SF6/N2 selectivity by increasing the temperature.65 The effective length of the ligands was shortened with the lateral vibration of benzene rings during the heating process, thereby reducing the pore size of Zn4O(dmcpz)3 and generating stronger van der Waals interactions with SF6. The pore size of Zn4O(dmcpz)3 decreased to 6.5 Å at 60 °C, thereby increasing its SF6/N2 selectivity from 44 (at room temperature) to 80.
3.2. Covalent organic frameworks
Another family of porous organic frameworks is covalent organic frameworks (COFs) constructed by covalent combination of multiple organic linkages.102 They have the similar merits of high porosity and tunable surface properties to MOFs. Differently, their metal-free and fully conjugated structures are beneficial to enhance the chemical stability and surface hydrophobicity and thus expand the SF6 capture application of humid environments.103 Furthermore, the unique covalent structures of COFs are vital to facilitate electron transport efficiency for SF6 selective capture.104Zheng et al. designed a 2-dimensional COF-6 with a graphite-like structure via B–O–C covalent bonds of boronate esters to selectively adsorb SF6 under low pressure (<0.1 bar).100 The large aromatic rings endowed the framework with a highly π-conjugated structure, allowing COF-6 to adsorb SF6 through strong electrostatic interactions. Meanwhile, the graphite-like organic layer partially slipped and generated 6.4 Å grottos along the pore wall of COF-6 and trapped SF6 molecules with suitable pore confinement. As a result, the two-dimensional COF-6 achieved a saturated adsorption of 3.71 mmol g−1, and its SF6/N2 selectivity reached 860, greatly exceeding other previously reported porous materials.
Liao et al. designed RCOF-1 connected by bis-imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) to further enhance the adsorption of SF6 through multiple adsorption sites (Fig. 10a).105 RCOF-1 possessed two adjacent chemical groups of benzene ring and bis-imine and formed a unique intersection angle at the edge of its pore cages. Among them, benzene rings formed F–π interactions at the benzene center and multiple F–H bonds at the edge with the SF6 molecule. Moreover, its imine group could attract electronegative SF6 molecules through lone pairs of N atoms at the same time. These sites formed multisite capture towards the SF6 molecule with a high binding energy of 27.0 kJ mol−1, as illustrated by the green iso-potential surface in Fig. 10b. Its adsorption experiments showed that RCOF-1 had a superior SF6 adsorption capacity of up to 4.13 mmol g−1 and a relatively high SF6/N2 selectivity of up to 125 at 273 K and 100 kPa compared with the state-of-the-art porous materials (Fig. 10c and d).
N) to further enhance the adsorption of SF6 through multiple adsorption sites (Fig. 10a).105 RCOF-1 possessed two adjacent chemical groups of benzene ring and bis-imine and formed a unique intersection angle at the edge of its pore cages. Among them, benzene rings formed F–π interactions at the benzene center and multiple F–H bonds at the edge with the SF6 molecule. Moreover, its imine group could attract electronegative SF6 molecules through lone pairs of N atoms at the same time. These sites formed multisite capture towards the SF6 molecule with a high binding energy of 27.0 kJ mol−1, as illustrated by the green iso-potential surface in Fig. 10b. Its adsorption experiments showed that RCOF-1 had a superior SF6 adsorption capacity of up to 4.13 mmol g−1 and a relatively high SF6/N2 selectivity of up to 125 at 273 K and 100 kPa compared with the state-of-the-art porous materials (Fig. 10c and d).
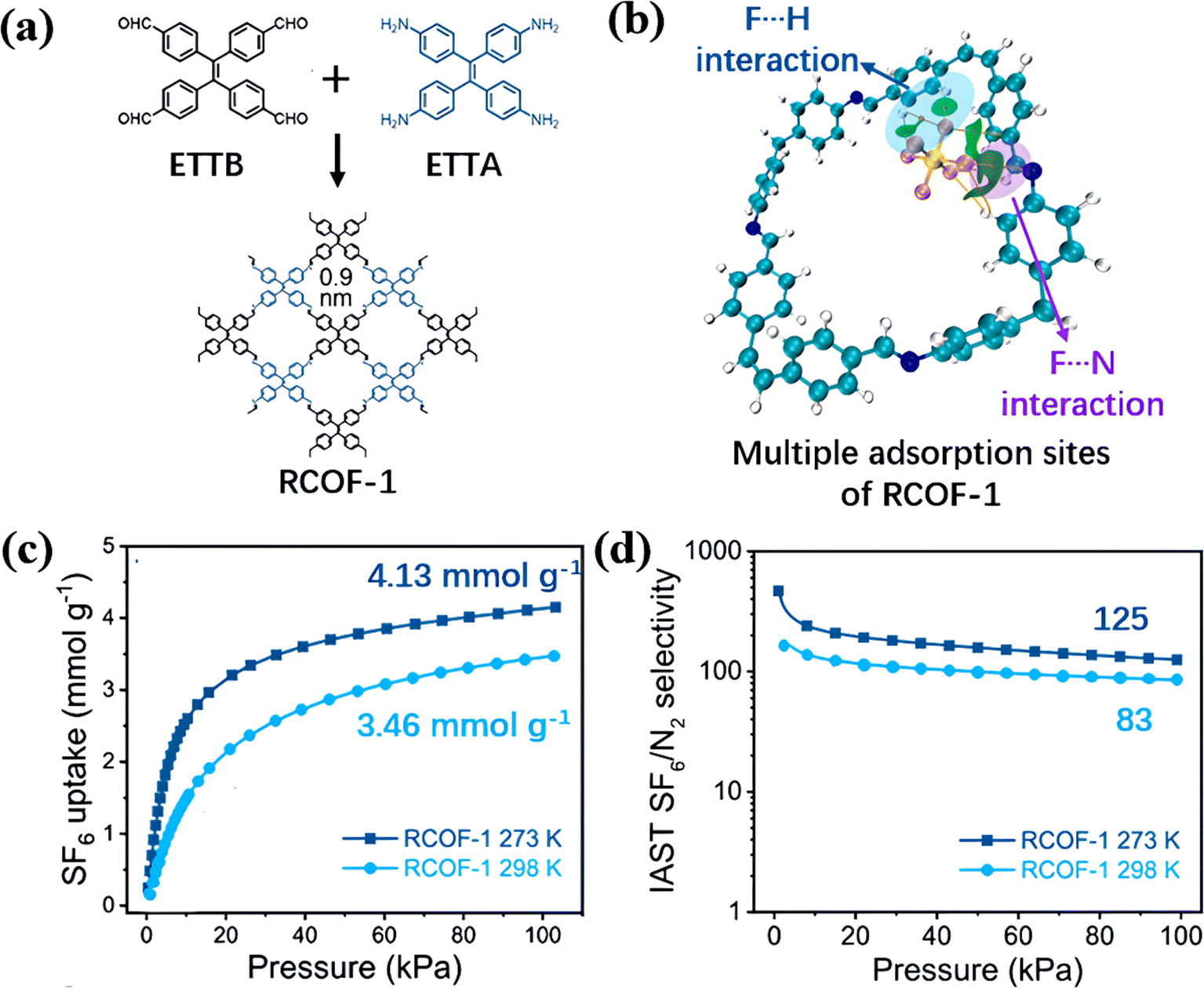 | ||
| Fig. 10 (a) Topological structures of RCOF-1. (b) IGMH map of the cluster model consisting of SF6 and RCOF-1. (c) SF6 adsorption isotherms of RCOF-1. (d) Calculated IAST SF6/N2 (10:90) selectivity of RCOF-1. Reproduced with permission from ref. 98. Copyright 2023 Elsevier. | ||
Polarized modification of the COF pore surface can create high SF6 affinity sites to promote the selectivity for SF6.106 For instance, Xi's group proposed the use of fluorinated monomers to construct FCOF-1 to promote SF6 selective adsorption.105 Benzene rings and F atoms in FCOF-1 generated 3D multiple adsorption sites and formed strong electrostatic attraction towards SF6 through π-conjugation and F–F polar interaction. Therefore, FCOF-1 exhibited a SF6/N2 selectivity of 22.1 and a SF6 uptake of 2.21 mmol g−1 at 273 K and 100 kPa.
Similarly, Xi's group introduced a trifluoromethyl group (–CF3) into BrCOF-2 through post-synthetic modification to improve its SF6/N2 selectivity (Fig. 11a).107 The inclusion of –CF3 altered the charge distribution in BrCOF-2, making it more effective to polarize SF6. Furthermore, the introduction of the bulky –CF3 functional groups reduced the pore size from 22 to 13 Å and further strengthened the binding force towards SF6. As a result, BrCOF-2-CF3 displayed an adsorption energy of 24.8 kJ mol−1 for SF6, increasing by 4.2 kJ mol−1 compared to BrCOF-2. Consequently, BrCOF-2-CF3 exhibited a SF6/N2 selectivity of 50, increasing by 2.8 times that of original BrCOF-2 (Fig. 11c).
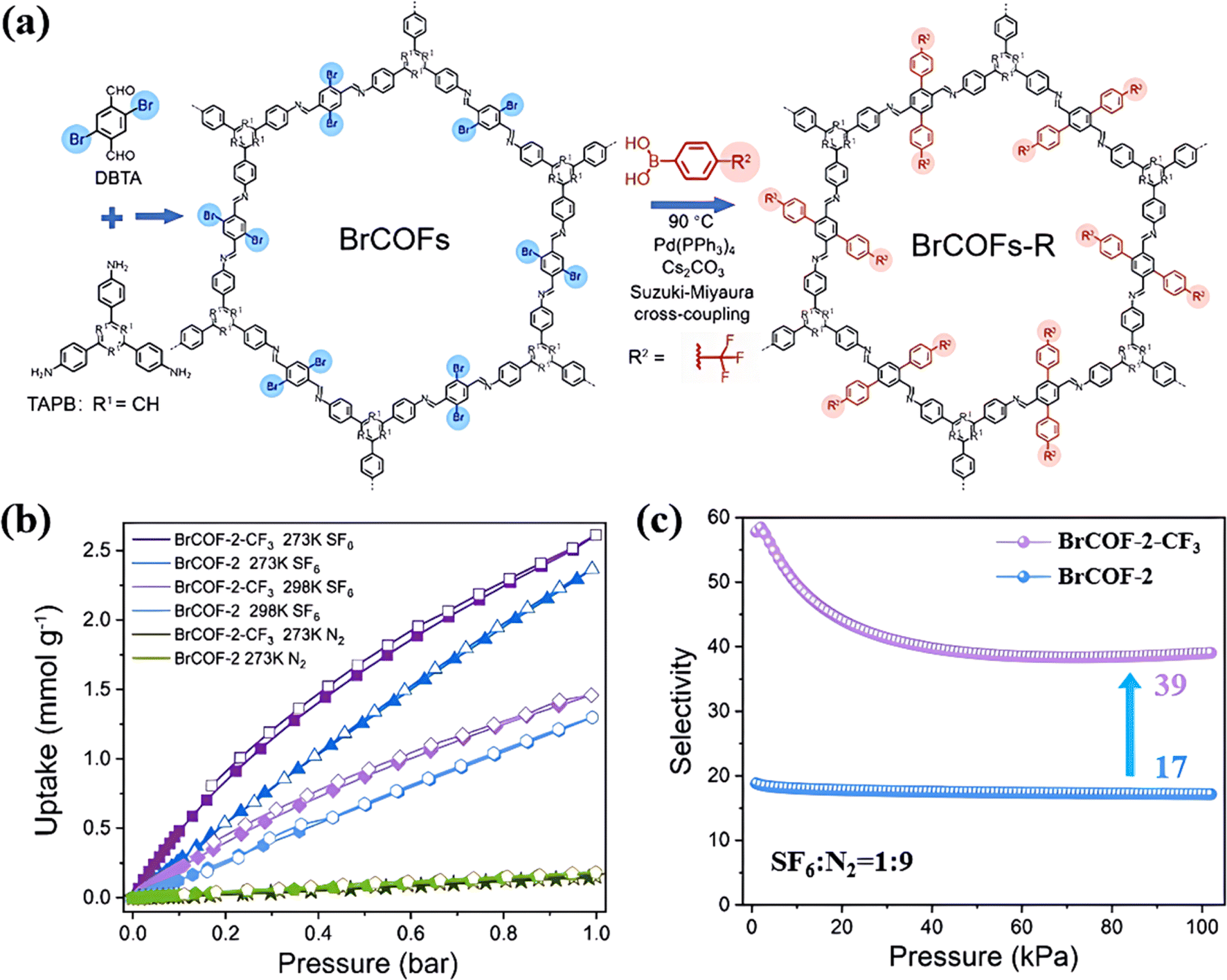 | ||
| Fig. 11 (a) Representation of the synthesis of BrCOFs and the post-synthetic modification via Suzuki–Miyaura cross-coupling. (b) SF6 and N2 sorption isotherms at 273 K and 298 K of BrCOF-2 and BrCOF-2-CF3. (c) Adsorption selectivity of the SF6/N2 (10:90) mixture in BrCOF-2 and BrCOF-2-CF3. Reproduced with permission from ref. 100. Copyright 2021 Wiley-VCH. | ||
Later, Xi's group designed COF-300 consisting of rigid CN double bonds to capture SF6 in humid air.105 The covalent bonds composed of light elements (C and N) were less susceptible to be attacked by water molecules because of their short bond length and strong bond energy. Stability experiment results showed that there were no observable changes in the XRD pattern and specific surface area of COF-300 after being placed in humid air for several weeks, indicating the high water stability of COF-300. Simultaneously, the tetrahedral phenyl linkages in COF-300 created a hydrophobic environment, enabling COF-300 to reach a high SF6 uptake of up to 4.03 mmol g−1.
3.3. Porous aromatic frameworks
Porous aromatic frameworks (PAFs) are a type of porous organic framework composed of exclusively aromatic rigid linkers, which would adsorb SF6 mainly through enriched F–π interactions.108 In contrast to COF materials prepared through reversible polymerizations, PAFs are typically synthesized by irreversible cross-coupling of rigid building blocks connected by C–C bonds and are well known for their top-notch stability.109 These bonds made them structurally stable in the presence of acidic environments and thus showed excellent acid resistance. These properties gained wider applications for SF6 adsorption compared to MOFs and COFs. Table 2 provides a summary of the performance of COFs and PAFs in SF6 adsorption.| Sample name | BET surface area (m2 g−1) | Pore size (Å) | SF6 uptake at 1.0 bar (mmol g−1) | SF6/N2 selectivity (1:9) |
SF6 isosteric heat of adsorption, Qst (kJ mol−1) | Ref. |
|---|---|---|---|---|---|---|
| PAF-4F | 1072 | 9.7 | 2.46 | 145.6 | 30.4 | 68 |
| PAF-8F | 818 | 10.1 | 1.90 | 365.2 | 30 | |
| COF-6 | 750 | 9 | 3.71 | 850 | 93 | |
| RCOF-1 | 1139 | 9 | 4.13 | 125 | 27.0 | 98 |
| RCOF-2 | 763 | 11 | 2.41 | 70 | 25.1 | |
| RCOF-3 | 242 | 13 | 0.91 | 34 | 21.4 | |
| BrCOF-2-CF3 | 1514 | 13 | 2.61 | 50 | 24.8 | 100 |
| New-PAF-1 | 3957 | 12.0 | 2.01 | 28.5 | 22.99 | 103 |
| N–SO3H | 1334 | 9.0 | 4.66 | 74.1 | 29.94 | |
| PAF-XJTU-1 | 2025 | 10.1 | 2.32 | 47.18 | 24.04 | 104 |
| PAF-XJTU-2 | 1924 | 9.2 | 2.43 | 46.64 | 33.64 | |
| PAF-XJTU-3 | 1499 | 8.5 | 2.56 | 36.76 | 31.03 | |
| PAF-XJTU-4 | 1419 | 10.1 | 1.47 | 29.39 | 25.08 | |
| PPN0 | 1480 | 12.0 | 0.48 | 41.6 | 22.3 | 105 |
| PPN1 | 1392 | 12.0 | 0.60 | 51 | 28.1 | |
| PPN2 | 1081 | 12.0 | 0.52 | 45 | 24.2 |
Ma's group constructed an HF resistant PAF (New-PAF-1) with a diamond-like structure to capture SF6 in semiconductor etching exhaust containing the corrosive by-product HF.110 The diamond-like structure of New-PAF-1 allowed sufficient exposure of phenyl rings, enabling the formation of strong electrostatic interaction with SF6 molecules. Simultaneously, the robust tetrahedral C–C linkage of rigid phenyl rings imparted New-PAF-1 with exceptional acidic stability, rendering it resistant to corrosion from acidic HF. Experimental results indicated that there was no significant change in the structure of New-PAF-1 after soaking in HF solution for 24 h, and its SF6 uptake reached 2.01 mmol g−1 at 1 bar.
Most PAFs were reported to be typical microporous structures due to their short distance linkage of monophenyl ligands, which was not conducive to SF6 diffusion in PAFs. Wu et al. proposed the design of a series of PAFs (PAF-XJTU-1 to PAF-XJTU-4) with hierarchical pore structures using longer polyaromatic blocks (Fig. 12a).111 As expected, these PAF-XJTUs exhibited micropores concentrated at about 0.9 nm and mesopores at around 2.1 nm, maintaining high affinity and fast diffusion for SF6 adsorption. Furthermore, the F–π interaction between SF6 and benzene rings (green area) facilitated the F–F interaction among SF6 molecules (red area) with the increase of SF6 loading (Fig. 12b) and enhanced the affinity of PAF-XJTU for SF6 (Fig. 12c).
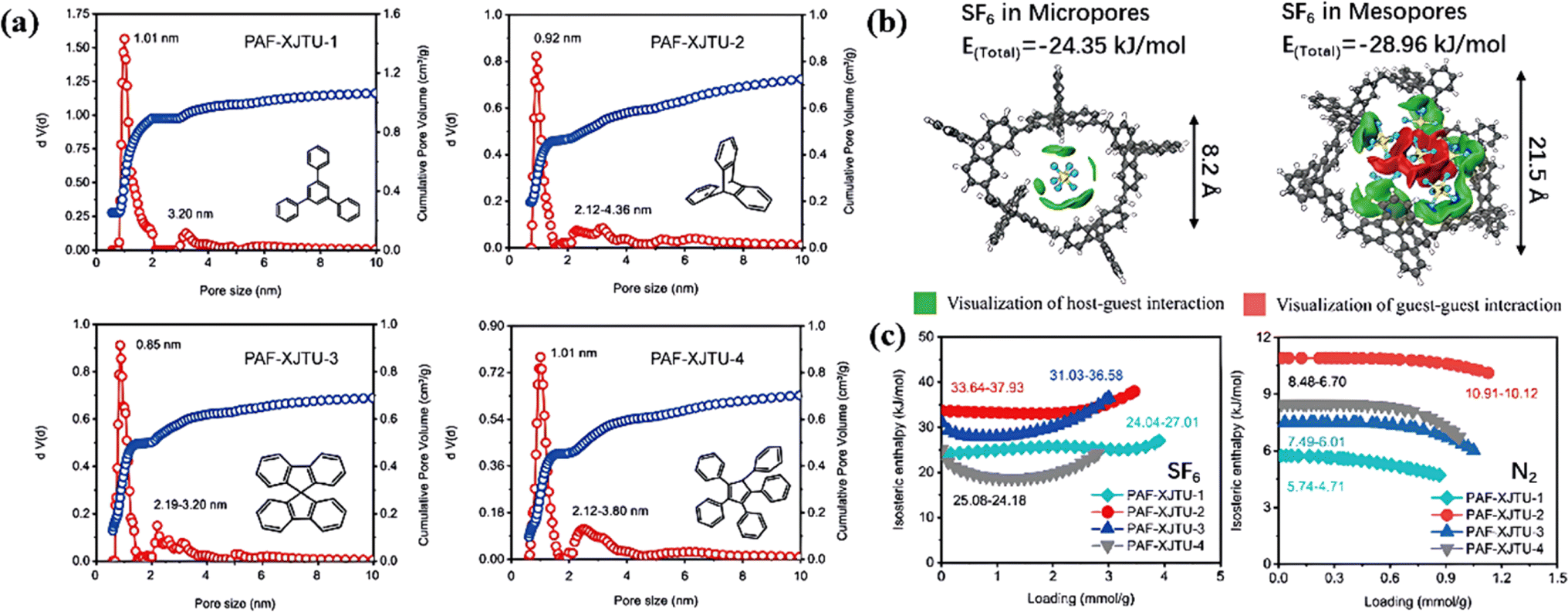 | ||
| Fig. 12 (a) Pore size distribution of PAF-XJTU-1-4. (b) The optimized adsorption structures of different SF6 loadings in the pore models with pore sizes from 0.82 to 2.15 nm, with the visualization of host–guest and guest–guest interaction energies. (c) Isosteric enthalpy of SF6 and N2. Reproduced with permission from ref. 104. Copyright 2023 Elsevier. | ||
Another limitation of using PAFs as SF6 adsorbents is their inferior SF6/N2 selectivity compared to MOFs and COFs. Therefore, researchers adopted a polarized modification strategy to enhance the interaction between SF6 and PAFs, thereby improving their SF6 affinity and SF6/N2 selectivity. For instance, Chuah et al. designed a series of amine-incorporated PAFs (PPNx) using triphenylamine (TPA) struts and investigated the effect of amine content on SF6/N2 selectivity (Fig. 13a).112 The presence of tertiary amines in TPA caused dipole-induced dipole interactions between PPNx and SF6, leading to increases in heat of adsorption of SF6 and SF6/N2 selectivity that were 1.25 times greater for both PPN1 and PPN2 compared to that of original PPN0 (Fig. 13b and c). However, excessive doping of tertiary amines reduced the specific surface area and micropore volume of PAFs (946–717 m2 g−1, 0.442–0.336 cm3 g−1), resulting in a decreased adsorption performance of PPN2 than PPN1.
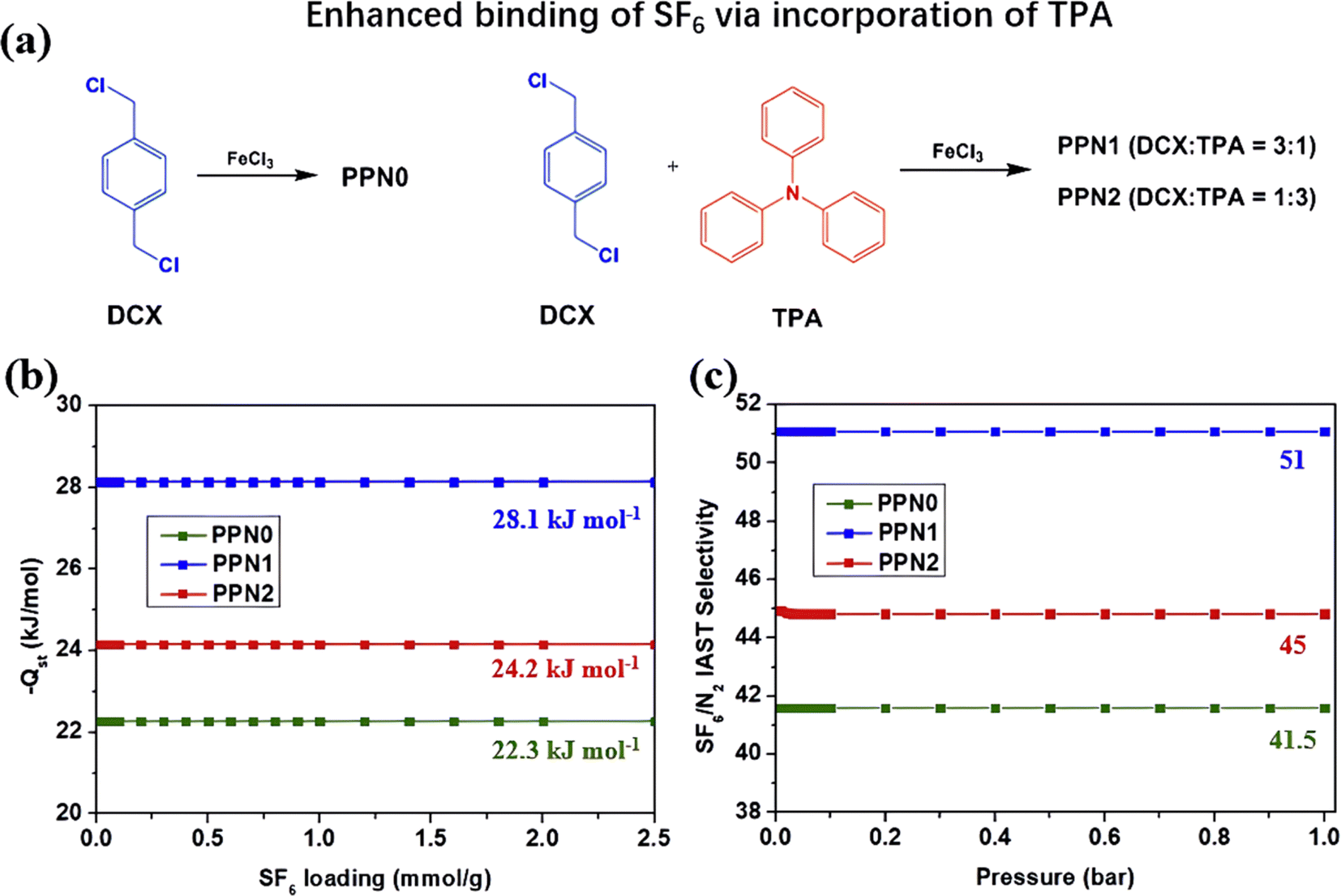 | ||
| Fig. 13 (a) Reaction scheme of PPNx. (b) Isosteric heat of adsorption of PPNx copolymers as a function of SF6 loading. (c) IAST SF6/N2 selectivity of PPNx copolymers as a function of pressure at 25 °C. Reproduced with permission from ref. 105. Copyright 2018 Elsevier. | ||
The introduction of polar functional groups can alter the charge distribution of PAFs to a certain extent to enhance their electrostatic interaction towards SF6. For instance, Zhang et al. proposed fluorinating PAFs (PAF-4F and PAF-8F) by using fluorinated building units and successfully constructed the induced electric field gradients.74 The entry of strongly electronegative F atoms caused localized charge separation on the pore surface of PAFs. This made benzene rings become electropositive sites (red) (Fig. 14a) and formed an interaction angle with adjacent benzene rings, which efficiently intensified the adsorption affinity towards electronegative SF6 (blue) (Fig. 14b). PAF-8F displayed a SF6/N2 selectivity of up to 365.22 and established a new benchmark for PAFs, which was attributed to its strong induced electric field gradient caused by high fluorine content (Fig. 14c).
 | ||
| Fig. 14 (a) Simulated models of PAF-4F@gas and PAF-8F@gas. (b) The electrostatic potential maps of PAF-4F@gas and PAF-8F@gas. (c) The IAST selectivity of PAF-4F and PAF-8F to the SF6/N2 gas mixture at 1 atm. Reproduced with permission from ref. 68. Copyright 2022 American Chemical Society. | ||
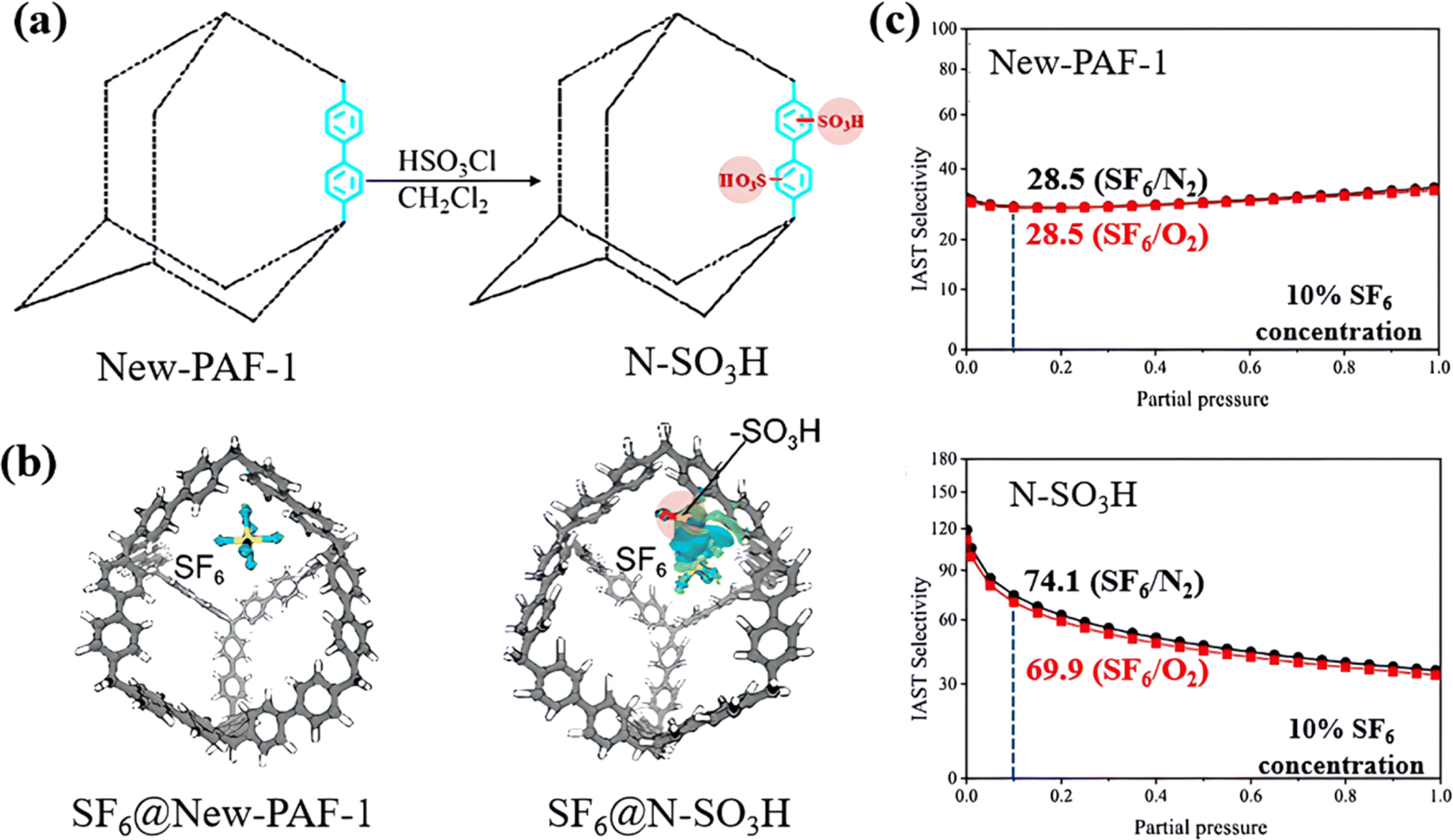
Moreover, Wang et al. also developed a sulfonate-grafted PAF (N–SO3H) to achieve specific capturing of SF6.110 Sulfonate-grafted N–SO3H exhibited significantly increased charge density difference from SF6 compared with the original New-PAF-1, as illustrated in the blue and green areas in Fig. 15a. DFT calculations revealed that the total atomic charge of SF6 changed from neutral to negative after binding in N–SO3H, suggesting that part of the electrons of the framework were transferred to SF6via the –SO3H group and formed a bonding state. By comparison, the original New-PAF-1 adsorbed SF6 only through weak interactions dominated by van der Waals forces, while the modified N–SO3H adsorbed SF6 through charge transfer effects in a synergistic manner, as evidenced by the increase in the heat of adsorption of SF6 from 22.99 kJ mol−1 to 29.94 kJ mol−1. As a result, N–SO3H displayed a remarkable SF6/N2(O2) selectivity of up to 250 in a ternary gas mixture (N2:O2 = 79%:21%), which was 3.4 times greater than that of the original New-PAF-1 (Fig. 15c).
 | ||
| Fig. 15 (a) Schematic representation of New-PAF-1 and N–SO3H. (b) Charge density difference graph of SF6 absorbed on New-PAF-1 and on N–SO3H. (c) IAST selectivity of SF6 in a ternary mixture for New-PAF-1 and N–SO3H at 298 K and 100 kPa. Reproduced with permission from ref. 103. Copyright 2022 American Chemical Society. | ||
4. Conclusion and outlook
Although PFMs have been proven to be promising SF6 adsorbents, they are still facing significant limitations under practical conditions, hindering their widespread application. Addressing these challenges, we propose the following possible research directions to enhance their SF6 adsorption performance in industrial settings:(1) AI-driven creation of high-performance PFMs: currently, the design of PFMs heavily relies on experimental data and computational work, which is not feasible to evaluate the adsorption performance of PFMs using purely experimental methods. Leveraging fast deep learning of artificial intelligence (AI), it is easy to systematically explore the impact of metal species, molecular characteristics of ligand groups and coordination modes on the surface morphology, crystal and pore structures of PFMs, helping to develop materials with a high specific surface area. Furthermore, machine learning algorithms and AI reasoning can be employed to study the design and construction methods of various adsorption sites on PFMs to predict the structure of PFMs with high selectivity and adsorption capacity.
(2) Construction of PFMs with high water- and acid-resistance: SF6 often coexists with acidic by-products like HF and water vapor in industrial waste gases, which would lead to the collapse and destruction of the framework structure during the adsorption process, resulting in a decline in adsorption performance. Strategies including surface modification of PFMs can be used to enhance the stability of the material in high-humidity environments, such as introducing hydrophobic groups or encapsulating the material in a hydrophobic coating. Utilizing high-valence metal nodes or constructing diamond topological structures can bolster the structural stability of PFMs against an acidic atmosphere.113 Furthermore, researchers should investigate the influence of humidity and acidic gases on SF6 adsorption, optimizing adsorption sites on PFMs through adjusting the hydrophobicity or introducing specific functional groups to enhance the specificity and avoid competitive adsorption.
(3) Shaping of high SF6 adsorption capacity PFMs: traditional shaping methods often lead to pore blockage or structural collapse of PFMs, diminishing their SF6 adsorption performance.114 Shaping by in situ growth such as using a gelation freeze-drying method to obtain a PFM gel or in situ crystallization on a substrate to obtain a PFM film can preserve the specific surface area and avoid the loss of SF6 adsorption capacity of PFMs.115 In addition, monomicelle self-assembly or growth of PFMs on custom-shaped porous scaffolds can create orderly mesoporous and macroporous channels to inhibit pore blockage and further enhance the SF6 mass transfer speed, improving the adsorption/desorption efficiency of PFMs.
(4) Construction of PFMs for SF6 capture via kinetic adsorption: kinetic adsorption driven by the inherent differences between SF6 and N2 molecules in diffusion dynamics is believed to improve the energy efficiency of the separation process, which does not involve strong host–guest interactions and is conducive to the regeneration of adsorbents.116 The diffusion rate of gas molecules mainly depends on the structural characteristics of PFMs and thus could be regulated by adjusting the crystal size and growth orientation. For example, surface-coordinated MOF thin films with preferred growth orientation can be grown by the liquid-phase epitaxial method, enhancing the intragranular diffusion of SF6 to promote kinetic adsorption.117
(5) Green and scale-up synthesis of economically feasible PFMs: in addition to SF6 adsorption performance, the industrial application of PFMs also needs to consider factors such as economic feasibility, scale-up and green synthesis.118,119 However, most of the reported synthesis methods of PFMs involve expensive organic linkers and toxic organic solvents (such as N,N-dimethylformamide) and cannot be synthesized on a large scale, making PFMs unsuitable for large-scale applications.120,121 Choosing mechano-synthesis or using supercritical fluids and ionic liquids as solvents can reduce or eliminate the use of organic solvents, achieving cost reduction and a green synthesis process.122 Besides, the use of continuous flow chemistry can improve the scalability of PFMs, which is attractive in industrial processes considering the efficiency and consistency.123
Porous framework materials (PFMs) with adjustable pores and abundant adsorption sites have been used as ideal materials for capturing SF6, which can efficiently adsorb SF6 through Lewis acid–base interactions, F–π interactions and induced dipole moment effects. This review summarized the latest progress of PFMs for SF6 adsorption based on the adsorption mechanism and discussed various modification strategies to improve their adsorption performance through pore engineering. Finally, possible research directions for the future development of PFMs were proposed, hoping to provide assistance for the development of SF6 adsorbents based on PFMs.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.Author contributions
Xiaoxuan Sun: writing – original draft, investigation. Liqin Zhou: writing – review & editing, investigation, conceptualization. Jianmin Chen: writing – review & editing, supervision, conceptualization. Zhaowei Jia: writing – review & editing, methodology. Zhongxing Zhao: writing – review & editing, supervision, funding acquisition. Zhenxia Zhao: writing – review & editing, conceptualization, funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. U23A20120), Natural Science Foundation of Guangxi Province (No. AD24010026), and National Natural Science of China (No. 22178073, 22268004 and 31401629). We thank Jiabao Wang from Shiyanjia Lab (https://www.shiyanjia.com/) and Jiaqi Li from SCI-GO (https://www.sci-go.com/) for providing the instrument for experimental testing.References
- W. Zhang, Y. Wu, Y. Li, S. Chen, Y. Fu, Z. Zhang, T. Yan, S. Wang and H. Ma, Macromolecules, 2022, 55, 1435–1444 CrossRef CAS.
- L. Ye, Y. Jin, K. Wang, W. Chen, F. Wang and B. Dai, Appl. Energy, 2023, 351, 121866 CrossRef.
- S. D'Auria, A. M. Pourrahimi, A. Favero, P. Neuteboom, X. Xu, S. Haraguchi, M. Bek, R. Kádár, E. Dalcanale, R. Pinalli, C. Müller and J. Vachon, Adv. Funct. Mater., 2023, 33, 2301878 CrossRef.
- Y. Yang, Z. M. Dang, Q. Li and J. He, Adv. Sci., 2020, 7, 202002131 Search PubMed.
- M. Rabie and C. M. Franck, Environ. Sci. Technol., 2018, 52, 369–380 CrossRef CAS.
- E. Guney and O. Ozgonenel, Energies, 2021, 14, 3698 CrossRef CAS.
- T. Magier, M. Tenzer and H. Koch, IEEE Trans. Power Delivery, 2018, 33, 440–446 Search PubMed.
- Q. Han, L. Chen, I. Cotton and J. Khan, IEEE Trans. Transp. Electrif., 2023, 9, 3590–3600 Search PubMed.
- Z. Cui, Y. Li, S. Xiao, S. Tian, J. Tang, Y. Hao and X. Zhang, Sci. Total Environ., 2024, 906, 107347 CrossRef.
- X. Fang, X. Hu, G. Janssens-Maenhout, J. Wu, J. Han, S. Su, J. Zhang and J. Hu, Environ. Sci. Technol., 2013, 47, 3848–3855 CrossRef CAS PubMed.
- D. Arbutina, K. Stanković, L. Perazić and M. Pejović, Int. J. Electr. Power Energy Syst., 2019, 104, 436–442 CrossRef.
- X. Yao, J. Wang, S. Ai, Z. Liu, Y. Geng and Z. Hao, Engineering, 2022, 13, 164–177 CrossRef CAS.
- H. Zhang, Y. Jung Oh, E. Tsegay Tikue, H. Il Lee, S. Kyung Kang and P. Soo Lee, Sep. Purif. Technol., 2022, 303, 122223 CrossRef CAS.
- J. Liu, G. M. Huang, Z. Ma and Y. Geng, IEEE Trans. Smart Grid, 2011, 2, 254–264 Search PubMed.
- A. Tena-Colunga, L. Eduardo Pérez-Rocha, J. Avilés and C. Cordero-Macías, J. Build. Eng., 2015, 4, 21–40 CrossRef.
- P. Wang, W. Chen, J. Wang, J. Tang, Y. Shi and F. Wan, Anal. Chem., 2020, 92, 5969–5977 CrossRef CAS PubMed.
- H. M. Lee, M. B. Chang and K. Y. Wu, J. Air Waste Manage. Assoc., 2012, 54, 960–970 CrossRef PubMed.
- R. S. Pessoa, H. S. Maciel, G. Petraconi, M. Massi and A. S. da Silva Sobrinho, Appl. Surf. Sci., 2008, 255, 749–751 CrossRef CAS.
- M. P. Sulbaek Andersen, M. Kyte, S. T. Andersen, C. J. Nielsen and O. J. Nielsen, Environ. Sci. Technol., 2017, 51, 1321–1329 CrossRef CAS.
- R. Bai, X. Song, W. Yan and J. Yu, Natl. Sci. Rev., 2022, 9, nwac064 CrossRef CAS.
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43–50 CrossRef CAS PubMed.
- T. C. Rolfe and A. L. Rice, Nature, 2020, 583, 171 Search PubMed.
- S. Xiao, X. Zhang, J. Tang and S. Liu, Energy Rep., 2018, 4, 486–496 CrossRef.
- Y. Dai, Q. Li, X. Ruan, Y. Hou, X. Jiang, X. Yan, G. He, F. Meng and Z. Wang, J. Membr. Sci., 2019, 577, 258–265 CrossRef CAS.
- D.-D. Zhou, X.-W. Zhang, Z.-W. Mo, Y.-Z. Xu, X.-Y. Tian, Y. Li, X.-M. Chen and J.-P. Zhang, EnergyChem, 2019, 1, 100016 CrossRef.
- C. Y. Chuah, Y. Lee and T.-H. Bae, Chem. Eng. J., 2021, 404, 126577 CrossRef CAS.
- C. Y. Chuah, S. Yu, K. Na and T.-H. Bae, J. Ind. Eng. Chem., 2018, 62, 64–71 CrossRef CAS.
- W. Bo, S. Ren, L. You, X. Meng, Y. Ji, X. Peng, M. Zhu, C. Zhang, X. Wang and X. Gu, J. Membr. Sci., 2024, 690, 122200 CrossRef CAS.
- Y. J. Oh, S. K. Kang, A. H. Lee, S. Park, S. Kim, J. Choi and P. S. Lee, Microporous Mesoporous Mater., 2023, 356, 112590 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC.
- K. Tian, S. M. Elbert, X. Y. Hu, T. Kirschbaum, W. S. Zhang, F. Rominger, R. R. Schröder and M. Mastalerz, Adv. Mater., 2022, 34, 2202290 CrossRef CAS PubMed.
- X. Peng, J. M. Vicent-Luna and Q. Jin, ACS Appl. Mater. Interfaces, 2020, 12, 20044–20055 CrossRef CAS.
- X. Huang, F. Chen, H. Sun, W. Xia, Z. Zhang, Q. Yang, Y. Yang, Q. Ren and Z. Bao, Sep. Purif. Technol., 2022, 292, 121059 CrossRef CAS.
- Y. Yin, Y. Zhang, X. Zhou, B. Gui, G. Cai, J. Sun and C. Wang, J. Am. Chem. Soc., 2023, 145, 22329–22334 CrossRef CAS.
- Y. He, X. Cao, Z. Zhang, Z. Jiang, H. Huang, S. Zhang, Q. Han and C. Zhong, Ind. Eng. Chem. Res., 2023, 62, 7642–7649 CrossRef CAS.
- L. Huang and D. Cao, J. Mater. Chem. A, 2013, 1, 9433 RSC.
- P. Bhanja, K. Bhunia, S. K. Das, D. Pradhan, R. Kimura, Y. Hijikata, S. Irle and A. Bhaumik, ChemSusChem, 2017, 10, 921–929 CrossRef CAS.
- S. Chatterjee, P. Bhanja, D. Ghosh, P. Kumar, S. Kanti Das, S. Dalapati and A. Bhaumik, ChemSusChem, 2020, 14, 408–416 CrossRef PubMed.
- S. Ruidas, B. Mohanty, P. Bhanja, E. S. Erakulan, R. Thapa, P. Das, A. Chowdhury, S. K. Mandal, B. K. Jena and A. Bhaumik, ChemSusChem, 2021, 14, 5057–5064 CrossRef CAS.
- T. Wang, M. Chang, T. Yan, Y. Ying, Q. Yang and D. Liu, Ind. Eng. Chem. Res., 2021, 60, 5976–5983 CrossRef CAS.
- M.-B. Kim, T.-U. Yoon, D.-Y. Hong, S.-Y. Kim, S.-J. Lee, S.-I. Kim, S.-K. Lee, J.-S. Chang and Y.-S. Bae, Chem. Eng. J., 2015, 276, 315–321 CrossRef CAS.
- M. I. Nandasiri, S. R. Jambovane, B. P. McGrail, H. T. Schaef and S. K. Nune, Coord. Chem. Rev., 2016, 311, 38–52 CrossRef CAS.
- J. Wang and S. Zhuang, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
- Y. Yuan, Y. Yang and G. Zhu, EnergyChem, 2020, 2, 100037 CrossRef.
- Y. Hu, L. Wang, R. Nan, N. Xu, Y. Jiang, D. Wang, T. Yan, D. Liu, Y. Zhang and B. Chen, Chem. Eng. J., 2023, 471, 145656 CrossRef.
- J.-W. Yan, S.-Q. Gang, Z.-Y. Liu, H.-Y. Xu, R. Wang and J.-L. Du, Sep. Purif. Technol., 2023, 327, 24929 Search PubMed.
- S. Jeoung, S. Kim, M. Kim and H. R. Moon, Coord. Chem. Rev., 2020, 420, 213377 CrossRef CAS.
- Y. Yang, M. Faheem, L. Wang, Q. Meng, H. Sha, N. Yang, Y. Yuan and G. Zhu, ACS Cent. Sci., 2018, 4, 748–754 CrossRef CAS PubMed.
- J. Xie, Y. Liang, Q. Zou, Z. Wang and X. Li, Energy Fuels, 2019, 33, 1910–1921 CrossRef CAS.
- L.-Q. Yang, J. Yu, P. Zhang, Y. Wang, W.-Y. Yuan and Q.-G. Zhai, Sep. Purif. Technol., 2023, 327, 124930 CrossRef CAS.
- Y. Wu, D. Yuan, D. He, J. Xing, S. Zeng, S. Xu, Y. Xu and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 10241–10244 CrossRef CAS PubMed.
- G. Fraux, A. Boutin, A. H. Fuchs and F.-X. Coudert, Adsorption, 2018, 24, 233–241 CrossRef CAS.
- H.-Y. Ren and X.-M. Zhang, Energy Fuels, 2015, 30, 526–530 CrossRef.
- R. Krishna and J. M. van Baten, J. Phys. Chem. C, 2012, 116, 23556–23568 CrossRef CAS.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- Y. Tian and J. Wu, Langmuir, 2017, 33, 996–1003 CrossRef CAS PubMed.
- A. I. Skoulidas and D. S. Sholl, J. Phys. Chem. B, 2002, 106, 5058–5067 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. Wu, X. Zhu, F. Yang, T. Ge and R. Wang, Chem. Eng. J., 2021, 425, 131409 CrossRef CAS.
- Ü. Kökçam-Demir, A. Goldman, L. Esrafili, M. Gharib, A. Morsali, O. Weingart and C. Janiak, Chem. Soc. Rev., 2020, 49, 2751–2798 RSC.
- F. Yi, Y. Chen, Z. Tao, C. Hu, X. Yi, A. Zheng, X. Wen, Y. Yun, Y. Yang and Y. Li, J. Catal., 2019, 380, 204–214 CrossRef CAS.
- F. Zhang, Y. Zhang, L. Ma, J. Yang and J. Li, ACS Sustain. Chem. Eng., 2022, 10, 9359–9368 CrossRef CAS.
- P. Chowdhury, C. Bikkina, D. Meister, F. Dreisbach and S. Gumma, Microporous Mesoporous Mater., 2009, 117, 406–413 CrossRef CAS.
- I. Senkovska, E. Barea, J. A. R. Navarro and S. Kaskel, Microporous Mesoporous Mater., 2012, 156, 115–120 CrossRef CAS.
- S. J. Grabowski, Molecules, 2020, 25(20), 4668 CrossRef CAS.
- M.-B. Kim, K.-M. Kim, T.-H. Kim, T.-U. Yoon, E.-J. Kim, J.-H. Kim and Y.-S. Bae, Chem. Eng. J., 2018, 339, 223–229 CrossRef CAS.
- N. Liu, X. Lv, B. Xiao, D. Kuzuhara, P. Mei, N. Aratani, H. Yamada, F. Qiu, J. Pan and S. Xue, Dalton Trans., 2022, 51, 9606–9610 RSC.
- E. Blum, J. Zhang, J. Zaluski, D. E. Einstein, E. E. Korshin, A. Kubas, A. Gruzman, G. P. Tochtrop, P. D. Kiser and K. Palczewski, J. Med. Chem., 2021, 64, 8287–8302 CrossRef CAS.
- P. Li, J. M. Maier, E. C. Vik, C. J. Yehl, B. E. Dial, A. E. Rickher, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Angew. Chem., Int. Ed., 2017, 56, 7209–7212 CrossRef CAS.
- L. Guo, M. Tian, Z. Li, Q. Wang, Q. Wu, L. Hao and C. Wang, Food Chem., 2023, 427, 136674 CrossRef CAS PubMed.
- Y. Wu, S. Wang, W. Zhang, S. Chen, Z. Zhang, B. Yang, S. Li and H. Ma, Sep. Purif. Technol., 2022, 289, 120739 CrossRef CAS.
- M. Wan, H. Yue, J. Notarangelo, H. Liu and F. Che, JACS Au, 2022, 2, 1338–1349 CrossRef CAS.
- W. Zhang, Y. Li, S. Wang, Y. Wu, S. Chen, Y. Fu, W. Ma, Z. Zhang and H. Ma, ACS Appl. Mater. Interfaces, 2022, 14, 35126–35137 CrossRef CAS PubMed.
- I. Skarmoutsos, M. Eddaoudi and G. Maurin, Microporous Mesoporous Mater., 2019, 281, 44–49 CrossRef CAS.
- C. Y. Chuah, K. Goh and T.-H. Bae, J. Phys. Chem. C, 2017, 121, 6748–6755 CrossRef CAS.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- F. P. Kinik, A. Ortega-Guerrero, D. Ongari, C. P. Ireland and B. Smit, Chem. Soc. Rev., 2021, 50, 3143–3177 RSC.
- G. Wu, J. Ma, S. Li, J. Li, X. Wang, Z. Zhang and L. Chen, J. Mater. Chem. A, 2023, 11, 6747–6771 RSC.
- J. Li, P. M. Bhatt, J. Li, M. Eddaoudi and Y. Liu, Adv. Mater., 2020, 32, 2002563 CrossRef CAS PubMed.
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS.
- M. Åhlén, Y. Zhou, D. Hedbom, H. S. Cho, M. Strømme, O. Terasaki and O. Cheung, J. Mater. Chem. A, 2023, 11, 26435–26441 RSC.
- Y.-P. Li, J.-J. Ni, S. Li, J. Wang, Z.-Y. Sui and X.-F. Xu, J. Solid State Chem., 2024, 329, 124554 Search PubMed.
- M.-B. Kim, S.-J. Lee, C. Y. Lee and Y.-S. Bae, Microporous Mesoporous Mater., 2014, 190, 356–361 CrossRef CAS.
- M. Åhlén, F. M. Amombo Noa, L. Öhrström, D. Hedbom, M. Strømme and O. Cheung, Microporous Mesoporous Mater., 2022, 343, 112161 CrossRef.
- S. M. Wang, X. T. Mu, H. R. Liu, S. T. Zheng and Q. Y. Yang, Angew. Chem., Int. Ed., 2022, 61, e202207066 CrossRef CAS.
- M. Åhlén, A. Jaworski, M. Strømme and O. Cheung, Chem. Eng. J., 2021, 422, 130117 CrossRef.
- X. Liang, P. Wang, C. Li, M. Yuan, Q. Shi and J. Dong, Microporous Mesoporous Mater., 2021, 320, 111109 CrossRef CAS.
- S.-T. Zheng, R.-Y. Jiang, Y. Jiang, S. Ni, G.-W. Guan, S.-Q. Shao, Y.-C. Wang, S.-M. Wang and Q.-Y. Yang, Sep. Purif. Technol., 2023, 318, 123957 CrossRef CAS.
- T. H. Chen, I. Popov, W. Kaveevivitchai, Y. C. Chuang, Y. S. Chen, A. J. Jacobson and O. Š. Miljanić, Angew. Chem., Int. Ed., 2015, 54, 13902–13906 CrossRef CAS PubMed.
- M. Yang, M. Chang, T. Yan and D. Liu, Sep. Purif. Technol., 2022, 295, 121340 CrossRef CAS.
- M. Chang, T. Yan, Y. Wei, J.-X. Wang, D. Liu and J.-F. Chen, Chem. Mater., 2022, 34, 9134–9143 CrossRef CAS.
- J. Ren, M. Chang, W. Zeng, Y. Xia, D. Liu, G. Maurin and Q. Yang, Chem. Mater., 2021, 33, 5108–5114 CrossRef CAS.
- J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CrossRef CAS.
- P. Zhao, S. C. E. Tsang and D. Fairen-Jimenez, Cell Rep. Phys. Sci., 2021, 2, 100544 CrossRef CAS.
- X. Zhang, Y.-L. Zhao, X.-Y. Li, X. Bai, Q. Chen and J.-R. Li, J. Am. Chem. Soc., 2024, 146, 19303–19309 CrossRef CAS.
- J. Choi, J. Im, K. Noh, J. Kim, T. Vogt and Y. Lee, J. Phys. Chem. C, 2019, 123, 25769–25774 CrossRef CAS.
- A. Schneemann, P. Vervoorts, I. Hante, M. Tu, S. Wannapaiboon, C. Sternemann, M. Paulus, D. C. F. Wieland, S. Henke and R. A. Fischer, Chem. Mater., 2018, 30, 1667–1676 CrossRef CAS.
- X. Zheng, Y. Shen, S. Wang, K. Huang and D. Cao, Chin. J. Chem. Eng., 2021, 39, 88–95 CrossRef CAS.
- M. Åhlén, E. Kapaca, D. Hedbom, T. Willhammar, M. Strømme and O. Cheung, Microporous Mesoporous Mater., 2022, 329, 111548 Search PubMed.
- U. Díaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef.
- T. Li, Y. Pan, B. Shao, X. Zhang, T. Wu, Q. He, M. He, L. Ge, L. Zhou, S. Liu, X. Zheng, J. Ye and Z. Liu, Adv. Funct. Mater., 2023, 33, 2304990 CrossRef CAS.
- J. Yang, F. Kang, X. Wang and Q. Zhang, Mater. Horizons, 2022, 9, 121–146 RSC.
- Q. Liao, H. Xu, C. Ke, Y. Zhang, Q. Han, Y. Zhang, Y. Xu, D. Wang and K. Xi, Sep. Purif. Technol., 2023, 306, 122595 Search PubMed.
- I. Ahmed and S. H. Jhung, Coord. Chem. Rev., 2021, 441, 213989 CrossRef CAS.
- Q. Liao, C. Ke, X. Huang, D. Wang, Q. Han, Y. Zhang, Y. Zhang and K. Xi, Angew. Chem., Int. Ed., 2020, 60, 1411–1416 CrossRef.
- Y. Yuan and G. Zhu, ACS Cent. Sci., 2019, 5, 409–418 Search PubMed.
- Y. Yuan, Y. Yang and G. Zhu, ACS Cent. Sci., 2020, 6, 1082–1094 CrossRef CAS.
- S. Wang, Y. Wu, Y. Zhang, Z. Zhang, W. Zhang, X. Li, W. Ma and H. Ma, Langmuir, 2022, 38, 8667–8676 Search PubMed.
- Y. Wu, X. Li, Y. Li, C. Tao, Y. Tang, S. Wang, Y. Fu, W. Ma, W. Zhang and H. Ma, Sep. Purif. Technol., 2023, 315, 123657 Search PubMed.
- C. Y. Chuah, Y. Yang and T.-H. Bae, Microporous Mesoporous Mater., 2018, 272, 232–240 CrossRef CAS.
- R. Liu, Q.-Y. Liu, R. Krishna, W. Wang, C.-T. He and Y.-L. Wang, Inorg. Chem., 2019, 58, 5089–5095 CrossRef CAS PubMed.
- J. Zheng, X. Cui, Q. Yang, Q. Ren, Y. Yang and H. Xing, Chem. Eng. J., 2018, 354, 1075–1082 CrossRef CAS.
- Q. Ma, T. Zhang and B. Wang, Matter, 2022, 5, 1070–1091 CrossRef CAS.
- Q. Ding, Z. Zhang, P. Zhang, J. Wang, X. Cui, C.-H. He, S. Deng and H. Xing, Chem. Eng. J., 2022, 434, 134784 CrossRef CAS.
- Y.-H. Xiao, Y.-B. Tian, Z.-G. Gu and J. Zhang, EnergyChem, 2021, 3, 100065 CrossRef CAS.
- A. M. Wright, M. T. Kapelewski, S. Marx, O. K. Farha and W. Morris, Nat. Mater., 2024 DOI:10.1038/s41563-024-01947-4.
- E. Wu, X.-W. Gu, D. Liu, X. Zhang, H. Wu, W. Zhou, G. Qian and B. Li, Nat. Commun., 2023, 14, 6146 CrossRef CAS PubMed.
- H. M. Wen, C. Yu, M. Liu, C. Lin, B. Zhao, H. Wu, W. Zhou, B. Chen and J. Hu, Angew. Chem., Int. Ed., 2023, 62, e202309108 CrossRef CAS PubMed.
- D. Liu, J. Pei, X. Zhang, X. W. Gu, H. M. Wen, B. Chen, G. Qian and B. Li, Angew. Chem., Int. Ed., 2023, 62, e202218590 CrossRef CAS.
- S. Głowniak, B. Szczęśniak, J. Choma and M. Jaroniec, Mater. Today, 2021, 46, 109–124 CrossRef.
- S. Bose, D. Sengupta, T. M. Rayder, X. Wang, K. O. Kirlikovali, A. K. Sekizkardes, T. Islamoglu and O. K. Farha, Adv. Funct. Mater., 2023, 2307478 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |