
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual protective layer on lithium metal anodes for improved electrochemical performance – in-depth morphological characterization†
Marlena M.
Bela
 a,
Maximilian
Mense
a,
Sebastian
Greiwe
a,
Marian C.
Stan
b,
Simon
Wiemers-Meyer
a,
Martin
Winter
ab and
Markus
Börner
*a
a,
Maximilian
Mense
a,
Sebastian
Greiwe
a,
Marian C.
Stan
b,
Simon
Wiemers-Meyer
a,
Martin
Winter
ab and
Markus
Börner
*a
aMEET Battery Research Center, University of Münster, Corrensstraße 46, 48149 Münster, Germany. E-mail: markus.boerner@uni-muenster.de
bHelmholtz-Institute Münster, IMD-4, Forschungszentrum Jülich GmbH, Corrensstraße 46, 48149 Münster, Germany
First published on 14th January 2025
Abstract
Lithium metal is a promising electrode material to increase the specific energy and energy density of rechargeable batteries. The high reactivity of lithium in contact with the electrolyte leads to the formation of a solid electrolyte interphase (SEI) that is inhomogeneous in composition and morphology. The SEI is prone to cracking due to volume changes and favors high surface area lithium growth, pit formation and an accelerated Li bulk consumption during electrodeposition/-dissolution due to continuous SEI regeneration. These drawbacks result in low coulombic efficiency and cycle life, and pose a safety risk to rechargeable lithium metal batteries with liquid electrolytes that must be addressed before commercialization. Protective layers by ex situ surface modifications are an attractive strategy to improve the cycle life and performance of lithium metal batteries as they mitigate Li metal degradation reaction and homogenize the Li ion transport from/into electrolyte. Herein, a dual protective layer consisting of an intermetallic LiZn and inorganic Li3N layer deposited by thermal evaporation was investigated. A deeper insight into the durability of the dual protective layer was gained by cross-sections under cryogenic conditions before and after electrodeposition/-dissolution. Galvanostatic cycling experiments in symmetric Li||Li cells revealed an increase in cycle life of 80% and the state of health from LiNi0.6Mn0.2Co0.2O2||Li cells was improved by 50% (SOH 80) for Li electrodes with a dual protective layer compared to pristine Li metal.
Introduction
Li ion batteries (LIBs) are approaching their theoretical and practical limit in specific energy and there are rising concerns that they will meet the increasing demand for various mobile and stationary applications.1–3 Li metal as active material for the negative electrode is a promising candidate for rechargeable next generation battery systems to meet these demands, since Li has the highest capacity (3860 mA h g−1) and lowest electrode potential (−3.04 V vs. standard hydrogen electrode).4,5 The high reactivity of Li metal towards liquid organic electrolytes leads to electrolyte decomposition products that form a solid electrolyte interphase (SEI) on the Li metal surface, which is inhomogeneous in composition and morphology.6,7 The heterogeneous interphase consists of different domains of decomposition products which were firstly described by E. Peled with a mosaic model.8 Between the domains are grain boundaries, which allow faster Li ion diffusion, predisposing to pit formation and various morphologies of high surface area lithium (HSAL, with dendritic Li being the most prominent morphology) growth during electrodeposition/-dissolution.7,9,10 During continuous electrodeposition/-dissolution, the thickness of the HSAL layer increases and inactive “dead” Li is formed, causing low coulombic efficiency, rapid capacity decay and serious safety issues.11,12 In response to these challenges, various strategies were developed to reduce HSAL growth, including the addition of additives to electrolytes,13,14 the creation of artificial 3D structures,15,16 and the application of protective surface layers through in and ex situ surface modifications. These approaches aim to design an artificial protective layer that homogenizes and facilitates Li ion diffusion, prevents further reactions between the Li surface and the electrolyte, and remains stable and effective during electrodeposition/-dissolution.17,18Ex situ coatings have the advantage that their layer thickness and composition are easier to control. As a result, extensive studies have been conducted on organic, inorganic and intermetallic layers on Li metal to evaluate their protection properties.19,20 Organic layers benefit from their mechanical flexibility and possibility to withstand volume changes of the electrode during charge/discharge, but lack in acceptable Li ion conductivity compared to inorganic and intermetallic layers.21 Inorganic layers, such as Li3N, provide a high Li ion conductivity (10−3 S cm−1), low electronic conductivity (>10−12 S cm−1) and (electro-)chemical stability against Li, resulting in an attractive coating material for Li metal batteries (LMBs).22–25 However, nanoscale Li3N layers suffer from poorly interconnected Li3N grains, that lead to coating cracks at grain boundaries during cycling and subsequently facilitated HSAL growth. A pinhole-free Li3N layer was proposed that improved the electrodeposition/-dissolution behavior, but the observed performance improvement was presumably due to a several microns thick Li3N layer.26Intermetallic layers show a noteworthy reduction in overvoltage and in Li metal reactivity towards the electrolyte, and as a result a homogeneously distributed electrodeposition behavior of Li ions.27–29 Nevertheless, HSAL deposition on the surface, attributed to the electronic conductivity of the intermetallic layer was observed, along with coating cracks and accelerated HSAL growth in these regions during cycling. Additionally, the dissolution of the intermetallic layer into HSAL and Li bulk was occurring.29,30
While single protective layers are a viable strategy to overcome specific challenges, they do not offer a comprehensive solution for simultaneously enhancing energy and power performance, cycle life, and safety – key factors needed for the successful application of rechargeable LMBs. Consequently, a combination of artificial layers that meet several requirements for an optimal protective layer was investigated. A combination of an intermetallic layer to provide uniformly distributed nucleation sites for Li ions and an inorganic layer to block the electron transport and ensure the Li ion deposition underneath the protective layer was recently investigated.31–34 Here, liquid-phase reactions were used for surface modification, although the layer thickness is difficult to control.32,33 A dual protective layer based on thermal evaporation was investigated for solid-state batteries with a LiF/LiAg coating showing a considerable enhancement in cycling performance and stability.31 The effective combination of an intermetallic and inorganic layer led to the encouragement of engineering a dual protective layer by thermal evaporation. Hence, a dual protective layer composed of an intermetallic LiZn and an inorganic Li3N layer deposited by thermal evaporation and subsequent gas reaction in the chamber is presented. Thermal evaporation is a physical vapor deposition (PVD) technique that produces high purity coatings, due to solvent- and organic free fabrication of thin films under high vacuum, and is more cost- and energy efficient compared to other PVD techniques.35–37 The thickness control during the coating process is adjustable, as recently demonstrated for an intermetallic LiZn layer.29 Zinc deposition was chosen to create an intermetallic LiZn layer due to its high Li ion diffusion coefficient, homogeneous distributed Li ion nucleation sites, low cost and toxicity.38–40 Li3N has a high Li ion and low electron conductivity, and is therefore a suitable protective layer on top of the intermetallic layer to reduce electronic leakage. Integrating a dual protective layer on Li electrodes is expected to effectively improve the cycle life and performance in Li||Li and NMC622||Li cells compared to single coated or uncoated Li electrodes. To gain a comprehensive insight into the beneficial behavior of the protective layer during cycling, post mortem analysis of cycled electrodes was conducted with a morphology preserving technique.
Experimental
Materials
Battery grade Li metal was purchased from Honjo Lithium in a thickness of 500 μm and was roll-pressed to a final thickness of 130 μm (Lirp) between two siliconized polyester foils (50 μm, PPI Adhesive Products GmbH) using a roll-press calendar (GK300L, Saueressig) in a dry room (dew point <−60 °C). The roll-pressing method was employed to reduce the surface roughness and dilute the native layer, consisting of Li2CO3, LiOH and Li2O.41–43 Lirp were then transferred to an Ar-filled glovebox (H2O and O2 values <0.1 ppm) for coating or electrode preparation under inert atmosphere. Zinc (Zn) pellets (Sigma Aldrich) with a purity of 99.99% were purchased to form the LiZn-intermetallic layer, and nitrogen gas (N2, Westfalen AG) with a purity of 99.999% was purchased to produce the Li3N layer.Physical vapor deposition by thermal evaporation
The coating process was performed in a ProVap PVD System (MBraun) chamber placed in an Ar-filled glovebox (H2O and O2 values <0.1 ppm). This system is based on a resistive heating approach, where a high electric current and low voltage is applied to melt and vaporize the source material. An SQC-310 thin film deposition controller (Inficon) operating at 6 MHz was used to control the deposition rate during the evaporation process. The substrate was protected by a shutter until the desired deposition rate was reached and a shutter above the deposition material protects the substrate against out-gassing. Lirp metal foil (130 μm) was placed on the substrate holder inside the PVD chamber facing downwards to the evaporation source to deposit different thicknesses at a rate of 0.5 nm s−1. For the deposition process, Zn pellets were placed in a tungsten evaporation boat and Li in a tantalum evaporation boat, both connected to a power supply. To obtain a Li3N layer, 100 nm Li was deposited on the Lirp metal foil, and subsequently the PVD chamber was flooded with N2 gas up to 500 mbar to allow the gas reaction between Li and N2 to form Li3N. The reaction was stopped after three min by evacuating the chamber. For the dual layer, 300 nm of Zn were deposited on the Lirp metal foil to generate a LiZn-intermetallic layer followed by the Li3N-layer fabrication process.Electrochemical investigations
Lirp, the single and dual layer protected electrodes were characterized in a CR2032-type two-electrode coin cell44 for symmetric (Li||Li or LiZn||LiZn) and LiNi0.6Mn0.2Co0.2O2||Li (NMC622||Li, NMC622||LiZn) based cells and cycled at 20 °C using a MACCOR battery cycler (MACCOR series 4000). Symmetric coin cells were assembled in an Ar-filled glovebox (H2O and O2 values <0.1 ppm) using ∅ 12 mm Li electrodes and a ∅13 mm separator (2× Freudenberg (FS2190), 2× Celgard 2500) soaked with 60 μL electrolyte containing 1 M LiPF6 in EC/EMC (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 by weight, EC: ethylene carbonate, EMC: ethyl methyl carbonate). NMC622||Li cells were assembled under equivalent conditions, but a different cell setup was used including a ∅13 mm Li as negative electrode, ∅12 mm NMC622-based positive electrode and ∅14 mm separator soaked with 80 μL electrolyte. For symmetric cells the current density was varied from 0.5 mA cm−2 to 2 mA cm−2 with a constant capacity of 0.5 mA h cm−2 for long-term cycling experiments and cut-off criteria of −1.5 V to +1.5 V. The NMC622||Li cells were cycled after a rest period of 5 h in a voltage range of 3.0 V to 4.3 V at a charge/discharge rate of 0.2C (0.2 mA cm−2, 1C corresponds to 180 mA g−1). As positive electrode 95% NMC622 (BASF) as active material, 2% conductive additive (Super C65, IMERYS) and 3% polyvinylidene difluoride (PVDF) binder (Solef 5130, Solvay) was used to obtain positive electrodes with an areal capacity of 2.3 mA h cm−2 produced in an in-house battery line. The electrode sheets were pre-dried under vacuum (3 × 10−3 mbar) at 100 °C for 12 h and then punched into ∅12 mm electrodes.
:7 by weight, EC: ethylene carbonate, EMC: ethyl methyl carbonate). NMC622||Li cells were assembled under equivalent conditions, but a different cell setup was used including a ∅13 mm Li as negative electrode, ∅12 mm NMC622-based positive electrode and ∅14 mm separator soaked with 80 μL electrolyte. For symmetric cells the current density was varied from 0.5 mA cm−2 to 2 mA cm−2 with a constant capacity of 0.5 mA h cm−2 for long-term cycling experiments and cut-off criteria of −1.5 V to +1.5 V. The NMC622||Li cells were cycled after a rest period of 5 h in a voltage range of 3.0 V to 4.3 V at a charge/discharge rate of 0.2C (0.2 mA cm−2, 1C corresponds to 180 mA g−1). As positive electrode 95% NMC622 (BASF) as active material, 2% conductive additive (Super C65, IMERYS) and 3% polyvinylidene difluoride (PVDF) binder (Solef 5130, Solvay) was used to obtain positive electrodes with an areal capacity of 2.3 mA h cm−2 produced in an in-house battery line. The electrode sheets were pre-dried under vacuum (3 × 10−3 mbar) at 100 °C for 12 h and then punched into ∅12 mm electrodes.
Electrochemical impedance spectroscopy (EIS) was conducted using a VMP3 potentiostat (Bio-Logic) in a frequency range between 0.1 MHz and 0.1 Hz with an amplitude of 10 mV. For the EIS analysis combined with galvanostatic cycling symmetric Li||Li coin cells were used. Initially, EIS was measured three times under open circuit conditions (OCP) at 0 V. Thereafter, Li||Li cells were cycled at 0.5 mA cm−2 and 0.5 mA h cm−2 and EIS was conducted after each cycle for the first ten cycles and then after every fifth cycle up to 50 cycles.
Morphological investigation
The surface morphology of Lirp and coated Lirp before and after charge/discharge cycling was analyzed by scanning electron microscopy (SEM) using a Carl-Zeiss CrossBeam 550 working station with a field emission gun at an acceleration voltage of 3 kV. For SEM characterization, symmetric cells were disassembled after 20 and 50 cycles at 0.5 mA cm−2 to 0.5 mA h cm−2. The samples were integrated inside an Ar-filled glovebox in a custom-built cryo-transfer module (Kammrath&Weiss), which was installed inside the SEM chamber and connected to liquid nitrogen and a temperature control element. The temperature was set at −160 °C and maintained for 10 min before the cross-section was started by focused ion bombardment (FIB). The cryo-conditions resulted in a morphological preservation of the Li metal and Li metal-containing structures, and the acceleration voltage of the FIB was 30 kV with a current ranging from 7 nA to 30 nA.The elemental composition of the electrode surface and cross-section was investigated by energy-dispersive X-ray spectroscopy (EDX) using an Ultim Extreme detector from Oxford Instruments. The spectra were analyzed with the integrated AZtech software (Oxford Instruments) and recorded with an acceleration voltage of 5 kV and a current of 1 nA.
Time-of-flight secondary ion mass spectrometry (TOF-SIMS)
ToF-SIMS measurements were performed on a TOF.SIMS 5 instrument (Iontof GmbH) equipped with a 30 keV Bi primary ion source and a 2 keV Ar sputter ion source. For the analysis the primary ion source was tuned in the spectrometry mode (bunched mode) for high mass resolution. The analysis was performed in a 100 × 100 μm2 field of view, scanned in random mode with 128 × 128 pixels and a cycle time of 60 μs. Bi3+ ions were used as primary ions with an ion current of 2.3 pA. During the measurement, the surface was sputtered with Ar+ sputter ions at an ion current of 600 nA. Measurements were taken at different but comparable sample positions in both secondary ion polarities. For each sample a set of a minimum of three measurements at different positions was performed. Data evaluation was performed with SurfaceLab7.2 software (Iontof GmbH).45Results and discussion
The fabrication of the single and dual protective layer with a deposited intermetallic LiZn and/or inorganic Li3N layer is schematically illustrated in Fig. 1 and was conducted in a PVD chamber as depicted in Fig. S1.† First, Li metal foil was roll-pressed (Lirp) to dilute the native layer, consisting predominantly of Li2CO3, LiOH and Li2O components, and to decrease the surface roughness, as previously reported by Becking et al.41,43,46 In a second step, an intermetallic LiZn or inorganic Li3N layer was deposited on Lirp by thermal evaporation. For the Li3N layer fabrication, 100 nm Li were deposited on Lirp under vacuum, followed by N2 gas exposure. The freshly deposited, reactive Li quickly formed a Li3N layer in N2 gas atmosphere. After withdrawing the samples from the chamber, a red-brown color was observed on the Li metal surface indicating a successful coating procedure to fabricate a Li3N layer (Fig. S2b;† Lirp|Li3N layer further referred as LN and coated on a Lirp electrode as LiLN).47–49 Intermetallic LiZn layers were thoroughly investigated in a previous report regarding the optimal thickness of the intermetallic layer and the effect of roll-pressing pristine Li foil before coating.29 Especially the preceding roll-pressing showed a beneficial influence regarding the homogeneity of the coating morphology and an enhanced cycling performance by combining these methods. Based on these results, 300 nm Zn were deposited on Lirp and considered as appropriate layer thickness to study the combination of an intermetallic LiZn and inorganic Li3N layer and its behavior during electrodeposition/-dissolution. Fig. S2a† shows the intermetallic LiZn layer on Lirp with a distinctive golden color, indicating the formation of an intermetallic phase.27,28,30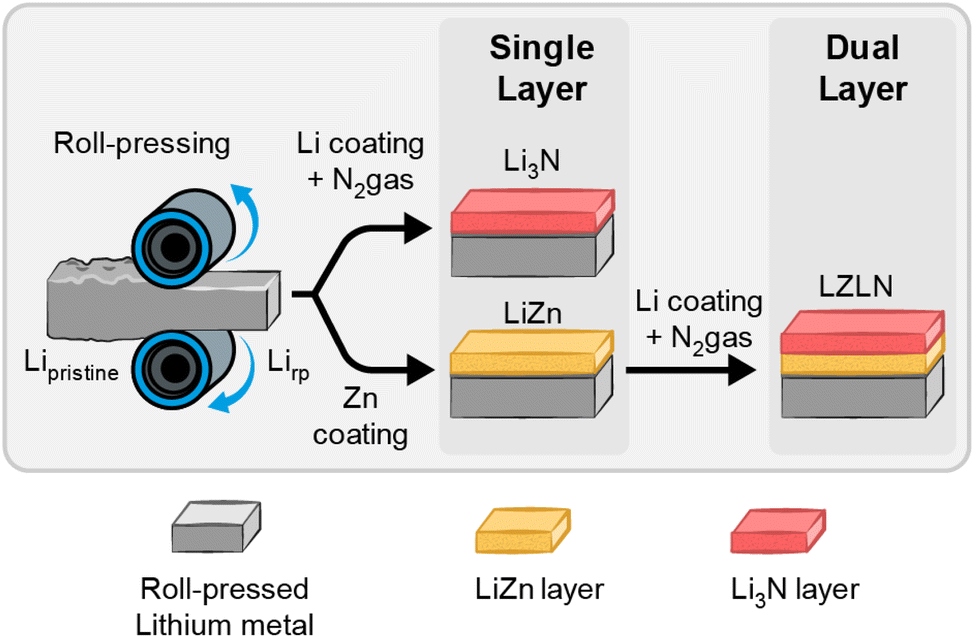 | ||
| Fig. 1 Ex situ fabrication process for the dual protective layer starts with roll-pressing of Li metal foil to obtain Lirp followed by PVD coating with either Zn or Li with subsequent N2 exposure to obtain a single protective layer. An additional Li3N layer is deposited on top of the LiZn-intermetallic layer to form the dual protective layer. | ||
The dual protective layer on Lirp electrodes (Lirp|LiZn|Li3N layer sequence further referred to as LZLN and coated on a Lirp electrode as LiLZLN) was fabricated by applying the two coating methods in sequence by depositing 300 nm Zn on Lirp and subsequently 100 nm Li followed by exposure and reaction with N2 gas. Therein, the deposition of 100 nm Li (prior to N2 exposure) was determined as optimum in preliminary studies including Li thicknesses of up to 400 nm. The optical image of the LZLN layer Lirp foil (Fig. S2c†) shows a homogeneous coating with a mixed golden and red-brown color. The advantage of this method is that the coatings are deposited under ultra-high vacuum, which prevents detrimental side reactions. In addition, thermal evaporation creates homogeneous coatings with a tunable layer thickness that can be adapted to the requirements of the cell chemistry. The Li3N layer was chosen as top layer above the LiZn layer, due to its and electronic insulating properties, which promotes the deposition of Li ions under the protective layer.
The morphology, composition and thickness of the LN and LZLN layer were analyzed by SEM and EDX top-view and cross-section images and compared to the Lirp reference surface (Fig. 2, corresponding EDX images: Fig. S3†). Cross-sections were performed under cryogenic conditions at −160 °C in order to preserve the morphology of the Li containing layers and the Li bulk by avoiding Li melting effects. SEM top-view images of Lirp show linear grooved patterns from the roll-press method (Fig. 2a and b), but the overall surface roughness was reduced and the native layer was diluted compared to the pristine Li metal foil.29,43 The corresponding cross-section image of Lirp (Fig. 2c) presents a flat surface, which is essential for the subsequent coating process, as the surface roughness is forwarded to the applied coating. Fig. 2d–f displays the top-view and cross-section images of LiLN and Fig. S3b and d† the corresponding EDX images. The top-view images show finely dispersed, well-connected particles of the LN layer on the Lirp surface, and the corresponding EDX images reveal a homogeneous distribution of nitrogen. The thickness of the LN layer was determined from cross-section images to be 110 nm (Fig. 2f). Thus, the freshly deposited, highly reactive Li formed primarily with the induced N2 gas and the subjacent Lirp was not involved. The top-view and cross-section images of the LiLZLN are demonstrated in Fig. 2g–i and the corresponding EDX images in Fig. S3f, g, i and j.† The top-view image in Fig. 2g shows a LZLN surface with a homogeneous coating and the formation of protrusions, that were caused by volume expansion during formation of the LiZn-intermetallic phase. After Zn deposition on Li foil (Zn: hcp structure; Li: bcc structure), Li atoms migrate into the Zn layer and form an intermetallic phase (NaTl-type structure) resulting in volume expansion and curvature of the layer.50 The cross-section image (Fig. 2i) confirms the volume expansion, indicating a layer thickness increase to 800 nm of the intermetallic LiZn layer with 300 nm of deposited Zn. Additionally, a distinct and homogeneous Zn signal was detected in the EDX cross-section image (Fig. S3j†). Moreover, it should be noted that beyond the flat Lirp surface the deposition morphology of the LN layer is determined by the lattice structure and chemical properties of the corresponding substrate leading to different appearance in Fig. 2f (Lirp substrate) and Fig. 2i (LiZn substrate) despite having the same chemical composition.
 | ||
| Fig. 2 Top view SEM images before cycling of (a) and (b) Lirp, (d) and (e) LiLN and (g) and (h) LiLZLN electrodes. Cross-section images under cryogenic conditions of (c) Lirp, (f) LiLN at Lirp and (i) LiLZLN. | ||
At higher magnifications, the LZLN layer shows connected grains over the entire surface. The corresponding EDX images confirmed the presence of Li3N as top layer with detection of the nitrogen signal (Fig. 2h with corresponding EDX image in Fig. S3e and f†). However, areas with protrusions exhibited a reduction in the nitrogen signal and an increase in the zinc signal (Fig. S3f and g†). In flat regions, the LN layer is assumed to act as a shield, masking the zinc signal from the LiZn layer. In areas with protrusions, a higher signal intensity of zinc was detected by the EDX detector. The excitation bulb in areas with protrusions likely contains a higher quantity of zinc in the vicinity, which exceeds the detection limit for the nitrogen signal.
Consequently, the contrast between the nitrogen and the zinc signal in the respective regions was leading to the observation of an absence of the nitrogen signal. This is confirmed by the consistent structure of the LZLN layer in the top view images, which demonstrated a homogeneous distribution and well connected Li3N grains (Fig. 2g and h). The cross-section image of the LiLZLN (Fig. 2i) shows a consistent layer thickness of the LN layer with a thickness of 700 nm on top of the LiZn layer compared to 110 nm LN layer on the Lirp surface. The thickness determination was conducted in an additional cross-section with a platinum layer on top of the LZLN layer to exclude the potential co-measurement of and LN edge as total thickness. The observed increase in layer thickness was caused by the reactivity of Li within the intermetallic phase, which reacted with N2 in addition to the vapor deposited Li. Overall, a homogeneously distributed LN and LZLN layer on Lirp metal foil was deposited by thermal evaporation and/or gas treatment. The corresponding layer thicknesses and consistency of the layers were determined by means of cryogenic cross-sections and the EDX analysis revealed the presence of the LiZn and LN layer.
Time-of-flight secondary-ion mass spectrometry (ToF-SIMS) depth profiling was employed to further investigate and confirm the composition of the single LN and dual LZLN layer and to assess the transition of interfaces. Fig. 3 shows the ToF-SIMS sputter depth profiles and the corresponding 3D reconstructions of the LiLN or LiLZLN.
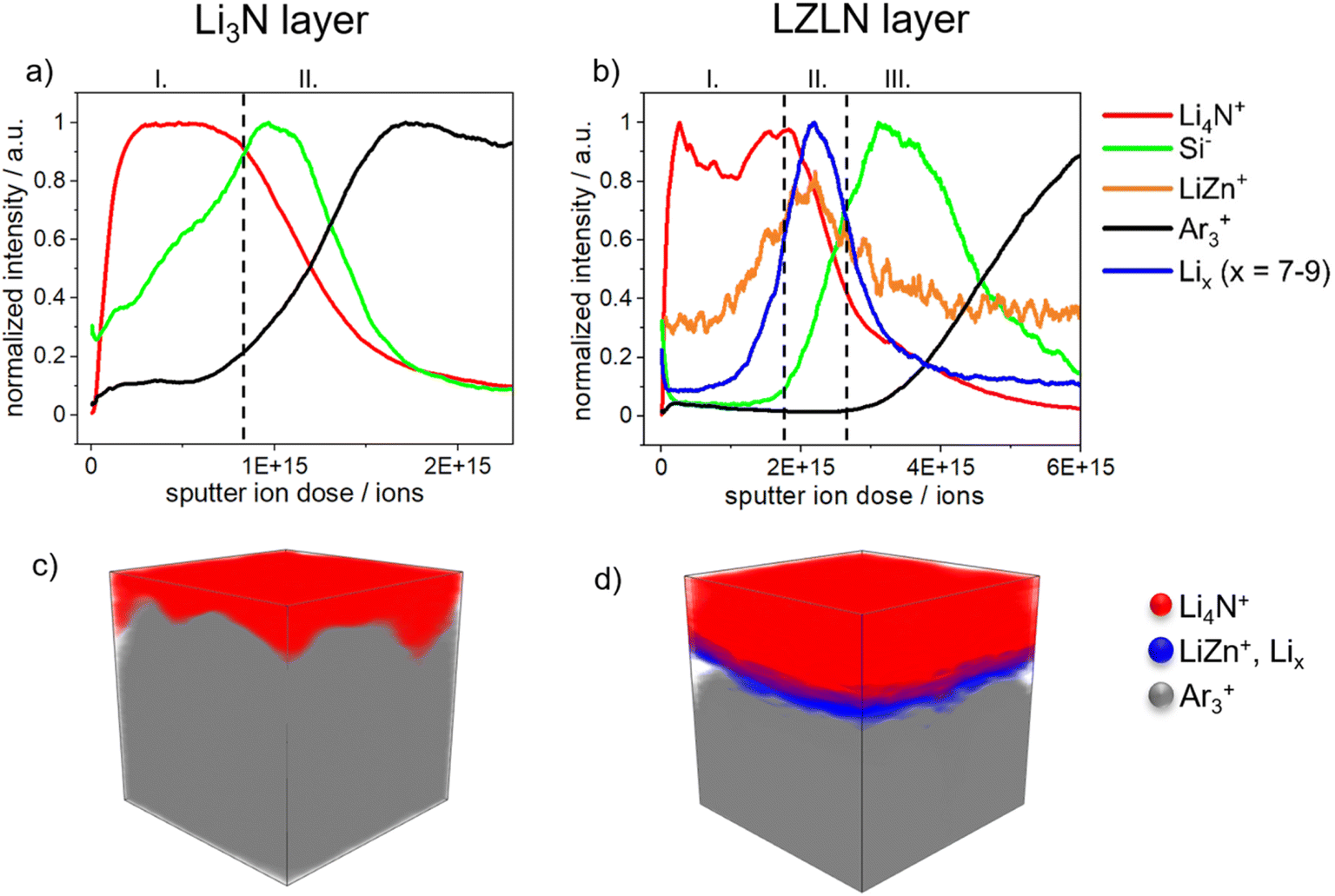 | ||
| Fig. 3 Normalized ToF-SIMS depth profiles reveal the surface composition of (a) LiLN and (b) LiLZLN. 3D reconstruction of the sputtered area (100 × 100 μm2) corresponding to the depth profiles show the composition of (c) LiLN and (d) LiLZLN with selected secondary ions marked in red: Li4N+, blue: LiZn+ and Lix+ (x = 7–9) and grey: Ar3+. | ||
On the surface of the LiLN (Fig. 3a) a Li4N+ fragment (I, red) was detected, assigned to the deposited LN layer. As sputtering continued, the Li4N+ intensity decreased while the Si− intensity increased (II, green). The Si− fragment indicated that the top surface of Lirp was reached and derived from the siliconized Mylar foil, used as release liner during roll-pressing of the Li metal foil. A decrease in the Si− intensity was accompanied by an increase in the Ar3+ intensity (II, black). The Ar3+ fragment was interpreted as an indicator of Li metal foil due to the integration of Ar+ ions into the Li metal surface during sputtering. The described assignment of the Si− and Ar3+ fragments was also reported by Mense et al. in a previous study regarding ToF-SIMS sputter depth profiling of Li metal electrodes.45Fig. 3b illustrates the depth profile of LiLZLN. First, the Li4N+ fragment (I, red) was detected on the outer surface and implied the presence of the LN layer. A higher sputter ion dose was required to sputter through the LZLN layer and indicated a thicker LN layer for the dual layer compared to the single layer. The thicker LN layer was also observed in the cross-section images of LiLZLN as compared to LiLN (LN layer: Fig. 2f, LZLN layer: Fig. 2i). The LN layer was followed by an underlying layer represented by a LiZn+ fragment (II, yellow, Fig. 3b) with an unexpectedly low intensity, but co-located with a more intense Lix+ fragment (x = 7−9, II, blue, Fig. 3b).45 The LiZn+ and Lix+ fragments measured below were associated to the LiZn intermetallic phase as described by Mense et al.45 The observed low intensities for the LiZn+ fragment in the intermetallic phase are likely attributed to matrix-effects during the SIMS ionization process. Since Li has a lower electronegativity compared to Zn it may get preferentially ionized in the collision process resulting in the observed behavior.45 Below the described dual layer consisting of a LN and LiZn layer, the Si− intensity maximum (III, green, Fig. 3b) with subsequent increase of the Ar3+ intensity (III, black, Fig. 3b) indicated the Lirp metal surface. 3D reconstruction of the depth profiles from LiLN and LiLZLN were generated (Fig. 3c and d) and the individual spatial distributions of selected secondary ions are displayed in Fig. S4.† The homogeneous distribution of the LN layer on the outer surface and the thicker LN layer on top of the LiZn intermetallic layer were visualized for both LiLN and LiLZLN in 3D reconstruction (Fig. 3c and d). Underneath the LN layer, detected by the Li4N+ signal (red), the Li bulk (Ar3+ signal, grey, Fig. 3c) or the intermetallic phase (LiZn+ and Lix+ signal, blue, Fig. 3d) is illustrated. Consequently, the individual components of the LN and LZLN layer were confirmed using ToF-SIMS analysis. The sputter ion dose spectra offered a detailed analysis of the single components and the impact of the roll-pressing method, while the 3D reconstructions provided a clear overview of the main fragments (Li4N+, LiZn, Lix+, Ar3+).
Raman spectroscopy was further employed to verify the formation of the LN layer via gas phase reaction, since the EDX and ToF-SIMS analysis solely verified the components of the individual compounds (Zn and Li for LiZn; N for LN). Fig. S6† shows the Raman spectra of the LN layer on Lirp, Li3N powder and Lirp as reference samples. The signal at 1845 cm−1 was assigned to the C–C-triple bond of a carbide species, which was formed as a decomposition product of Li2CO3 from laser irradiation during the measurement and was also observed in the Lirp reference spectra (Fig. S5a and c†).51 Due to the fact that the LN layer on Lirp is relatively thin (100 nm), the underlying native layer was reduced by the laser beam and Li2C2 was detected. A characteristic band at 515 cm−1, 575 cm−1 and 610 cm−1 and a broad band from 930 cm−1 to 1375 cm−1 was observed for the LN layer on Lirp (Fig. S5a†). The characteristic and broad band was assigned to the Li3N species, because both signals were detected in the Li3N powder reference spectra (Fig. S5b†). In summary, the analysis of the LN layer on Lirp by Raman spectroscopy supported the conclusion that Li3N was formed as a top layer through comparison with Li3N powder and Lirp as a reference material.
The LN and LZLN protective layers were electrochemically analyzed in a symmetric cell setup with LiLN||LiLN (red) and LiLZLN||LiLZLN (blue) electrode configurations and compared to Lirp||Lirp (grey) as a reference. The beneficial influence of the LN and LZLN layer was observed in the long-term cycling experiment at different current densities (Fig. 4a and b). Notably, the cycling stability was increased with both LN and LZLN layer compared to bare Lirp before reaching the cut-off voltage, due to an enhanced protection of the Li bulk and an improved homogeneity during Li electrodeposition/-dissolution. This trend was observed for low, moderate and high current densities (Fig. 4a for 0.5 mA cm−2, Fig. 4b for 1.0 mA cm−2, Fig. S6† for 2.0 mA cm−2). At moderate current density of 1.0 mA cm−2, an 80% increase in cycle number before reaching the cut-off voltage was reached for symmetric cells built with LiLZLN compared to Lirp (from Table S1:† Lirp: 200 cycles, LiLZLN: 360 cycles), compared to a 42% increase at higher current densities of 2.0 mA cm−2 (Fig. S6 and Table S1†). The LiLN electrodes showed an improved cycling stability compared to Lirp, but a reduced cycling stability compared to the LiLZLN electrodes for the three investigated current densities (Fig. 4a, b, S6 and Table S1†). Fig. 4c–e emphasizes the electrodeposition/-dissolution behavior of symmetric cells with Lirp||Lirp, LiLN||LiLN and LiLZLN||LiLZLN at different cycling stages with a fixed current density of 1.0 mA cm−2. The voltage profile of Lirp||Lirp is magnified in Fig. 4c and shows a small peak at the beginning of the first half cycle and a larger one at the end. The first peak was associated to the nucleation of Li ions onto HSAL at the Li surface.10 Since a large volume of HSAL accumulated after 75 cycles at the Li electrode surface, the energy barrier for nucleation was low (Fig. S7†). Once the initial kinetic barrier for Li ion deposition on existing HSAL was overcome (first peak), there was a continuous deposition of Li ions onto the surface, leading to a decrease in overvoltage (area between the first and second peak). The second peak demonstrated a higher energy barrier for electrodissolution, as Li ions need to dissolve from the Li bulk and diffuse through a porous layer consisting of mossy HSAL and a dead Li layer.10,52,53 After advanced electrodeposition/-dissolution, the second peak becomes more dominant and the overvoltage for the Lirp||Lirp cell continued to increase until the cut-off voltage was reached after 200 cycles. LiLN||LiLN and LiLZLN||LiLZLN showed a similar voltage profile for the initial charge/discharge cycling phase compared to Lirp||Lirp. With increasing cycle number, the voltage profile turned into an arc profile followed by a plateau for each half cycle (Fig. 4c–e). The arcing behavior reflects the state of facilitated Li ion deposition, resulting from a reduced kinetic barrier due to an enhanced Li ion diffusion through the protective layer or the Li ion deposition on HSAL. The plateau profile characterizes a facilitated growth of HSAL at the negative electrode and dissolution from HSAL/pits from the positive electrode. This voltage profile was stable for an extended cycling period, while an overall increase in overvoltage was observed.10,52 The increase in overvoltage for the arc and following plateau profile with advanced electrodeposition/-dissolution was associated to limited Li ion diffusion processes through the protective layer or HSAL/dead Li accumulation at the surface, as the diffusion pathway through the HSAL/dead Li layer was growing. The observed arc and plateau profile for LiLN||LiLN and LiLZLN||LiLZLN was associated to the Li ion diffusion through the single or dual protective layer (Fig. 4c and d). Fig. 4e displays a less dominant plateau and an increase in the second peak overvoltage at higher cycle numbers, which indicated that Li ions need to overcome a larger volume of HSAL and more energy is required for Li ion dissolution from Li bulk at the positive electrode. Overall, the long-term cycling experiment revealed an extended cycle life for symmetric cells with dual layer protected Lirp electrodes compared to Lirp or single layer protection. A detailed analysis of the overvoltage profiles showed a facilitated and more homogeneous Li electrodeposition/-dissolution behavior with single and dual layer protected Lirp, which was accompanied with a decelerated consumption of the Li bulk.
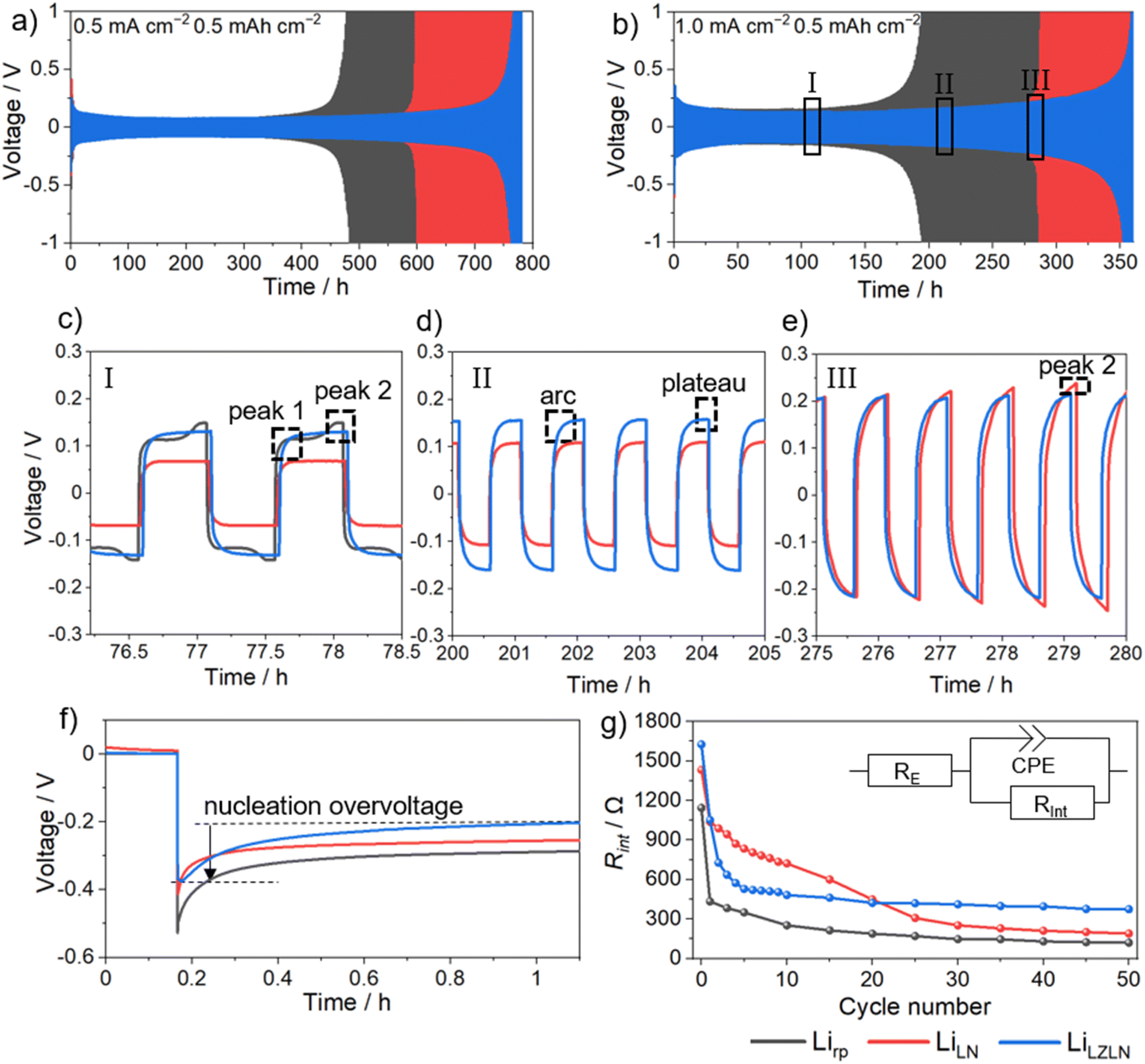 | ||
| Fig. 4 Galvanostatic cycling of symmetric Lirp||Lirp (grey), LiLN||LiLN (red) and LiLZLN||LiLZLN (blue) cells at a fixed current density of (a) 0.5 mA cm−2 and (b) 1.0 mA cm−2 with a capacity of 0.5 mA h cm−2. (c)–(e) Emphasizes the electrodeposition/-dissolution behavior from (b) of Lirp||Lirp, LiLN||LiLN and LiLZLN||LiLZLN after (c) 75, (d) 200 and (e) 275 cycles at a current density of 1.0 mA cm−2. (f) Nucleation overvoltage profiles from the first electrodeposition at 0.5 mA cm−2. (g) Rint evolution determined by EIS within the first 50 cycles at 0.5 mA cm−2. | ||
In order to gain further insight into the Li ion diffusion through the Li3N and LZLN dual layer on Lirp, the nucleation overvoltage of the initial Li electrodeposition and interphase resistance Rint within the first 50 cycles was investigated (Fig. 4f and e). The observed nucleation overvoltage provided information about the kinetic hindrance for Li ions to diffuse through either a protective layer or the SEI layer on Lirp. The nucleation overvoltage is defined as the difference between the sharp voltage tip and the flat voltage plateau, as shown in Fig. 4f for a LiLZLN||LiLZLN symmetric cell.54 Lirp||Lirp has the highest nucleation overvoltage with 0.26 V compared to LN and LZLN protected Lirp electrodes. This trend was observed due to the formation of a SEI which creates an additional barrier for Li ion electrodeposition. For the protected Lirp electrode, a substantial reduction in nucleation overvoltage was observed. Thus, the nucleation overvoltage dropped to 0.20 V for LiLZLN||LiLZLN and to 0.19 V for LiLN||LiLN. The observed nucleation overvoltage decrease was interpreted as either a lowering of the kinetical hindrance for Li ions to diffuse through the protective layer or a deposition of Li ions on top of the protective layer. In order to elucidate the deposition behavior of Li ions by electrochemical analysis, EIS measurements were conducted and analyzed the Rint of symmetric cells before and after electrodeposition/-dissolution (Fig. 4g). The Rint depends on the Li ion and electron conductivity between the interface and electrolyte and allows an assessment of the protective property and the Li electrodeposition/-dissolution behavior. Lirp||Lirp showed a high Rint prior to electrodeposition/-dissolution, due to SEI formation between Li and the electrolyte, followed by a rapid reduction in Rint owing to an increased surface area caused by pit formation and HSAL growth.29,53Rint of LiLN||LiLN and LiLZLN||LiLZLN showed overall a higher value before and during electrodeposition/-dissolution compared to bare Lirp. The increased Rint value at 0 cycles for LiLN and LiLZLN was interpreted as a successful protection of Li surface against degradation processes with the electrolyte. However, after repeated charge/discharge cycles, cracking of the LN layer was observed, leading to reduced kinetic hindrance for Li ion deposition, which was observable in the decrease of Rint for LiLN||LiLN to a similar Rint as for Lirp||Lirp. Further cracks in the LN layer, induced by volume changes, lead to accelerated pit formation and HSAL growth in areas of Li electrodeposition/-dissolution. This was indicated by a continuous decrease of Rint for LiLN||LiLN from cycle 30 to 50. The LiLZLN||LiLZLN cells showed a Rint drop within the first 10 cycles, followed by a virtually constant Rint up to 50 cycles. The initial Rint drop implied that the LZLN layer was suffering from coating cracks similarly to the LN layer. Yet, during the subsequent cycling period (cycles 10 to 50), the LZLN layer was found to improve the overall stability of both layers. Therefore, it was concluded that the benefits of the dual layer combination are an electronically insulating top layer, more homogeneously distributed nucleation sites due to the LiZn layer, and an overall higher stability towards volume changes (if LN layer cracks, LiZn layer remains protective character). This improvement resulted in a superior protection of the subjacent Li surface from electrolyte decomposition and an improved behavior during Li electrodeposition and dissolution.
The higher stability and improved protection of LiLZLN was verified by SEM top-view and cross-sections with corresponding EDX images after 25 and 50 cycles and compared to bare Lirp and LiLN (Fig. 5).
 | ||
| Fig. 5 Top-view SEM images of (a) Lirp, (b) LiLN and (c) LiLZLN electrodes after 25 cycles at 0.5 mA cm−2 with red bars that are indicating the cross-section area. The left column belongs to Lirp electrode, the middle column to the LiLN electrode and the right column to the LiLZLN electrode. (d)–(f) Shows the cross-sectional SEM images after 25 cycles and (g)–(i) the corresponding EDX mapping images. (j) and (l) Displays the cross-sectional SEM images after 50 cycles and (m)–(o) the corresponding EDX mapping images. (k) and (l) Display red framed areas that are marking thicker Li deposits. | ||
Fig. 5a–c shows aged Lirp, LiLN and LiLZLN electrode as top-view images after 25 cycles and the area of the cross-section is marked with a red bar. Cross-sections were performed in regions at boundaries between HSAL growth and intact surface to analyze the cracking mechanism of the protective layer on the Lirp surface upon continuous electrodeposition/-dissolution. With repeated electrodeposition/-dissolution, pits are formed and HSAL is deposited on the surface and in the pits. The pits grow deeper with increasing cycle number and were refilled with growing HSAL. The surface analysis of Lirp after 25 cycles proofed the degradation behavior by showing HSAL growth at the surface (Fig. 5a). Cross-section analysis, supported by corresponding EDX images, further demonstrated HSAL penetration in deeper regions (Fig. 5d and g: HSAL in green). HSAL was represented by a mossy/chunk-like morphology and an oxygen signal (green), since Li reacted with the carbonate-based electrolyte to form oxygen-containing species. The LN layer is represented by the associated nitrogen signal (purple) and the intermetallic LiZn layer by the zinc signal (yellow). The top-view image of the LiLN electrode after 25 cycles exhibited HSAL structures on the surface, that cracked the LN layer and penetrated into the Li bulk (Fig. 5b). Cross-section images and corresponding EDX analysis showed pit formation in regions of HSAL growth, while the adjacent LN layer remained intact. The LN layer underneath the HSAL showed a weaker nitrogen signal, indicating the rupture of the rigid layer (Fig. 5h: HSAL in green, Li3N in purple). The HSAL growth was also observed on the surface of the LZLN layer (Fig. 5b). However, in the top-view image, the LZLN layer appeared undamaged in regions of HSAL growth and an undamaged dual layer was also confirmed by the cross-section image and EDX analysis (Fig. 5f and i). HSAL growth (Fig. 5i, green) was observed on top of the LZLN layer (Fig. 5i, Li3N: purple, LiZn: yellow), with an intact LZLN layer and no visible cracks in the region of HSAL growth.
After 50 cycles, deeper pit formation and a higher amount of HSAL on top of the Lirp or protected Lirp electrode surface was observed in both SEM and optical images (Fig. 5j–o and S7†). Cross-section images with corresponding EDX analysis of Lirp showed a distinctive HSAL layer with mossy structure (Lirp: Fig. 5j and m). The HSAL layer of the Lirp electrodes was overall less dense and consisted of a thicker layer compared to the protected Lirp electrodes (Lirp: Fig. 5j, LiLN: Fig. 5k, LiLZLN: Fig. 5l). The LiLN electrode showed a cracked LN layer in areas with advanced HSAL growth as a continuous nitrogen signal was absent (Fig. 5k and n). HSAL was found in a higher depth of penetration into the Li bulk with a dominant mossy structure and a few denser Li deposits in predominantly deeper regions (Fig. 5k, denser HSAL deposits framed red). Denser Li deposits are resulting from a better contact of Li ions with the Li bulk during electrodeposition, while they are effectively protected against electrolyte decomposition and SEI formation during the deposition process. Therefore, the cross-section image derived that the LN layer was first intact, causing thicker Li deposits, and with continued cycling, mossy and less dense HSAL was growing due to a ruptured LN layer with less protective properties (Fig. 5k). Cross-section images and EDX analysis of LiLZLN electrodes showed HSAL growth (green) with a mossy structure on top of the protective layer and a higher amount of denser Li deposits below the protective layer compared to LiLN and Lirp electrodes (Fig. 5l and o, denser HSAL deposits framed red). Presumably, the higher amount of denser Li deposits was found, because the LZLN layer featured a higher degree of intact protective properties shielding the Li deposits from electrolyte decomposition. The LiZn layer (Fig. 5o, yellow) was found after 50 cycles with severe cracks and advanced dissolution into HSAL and the Li bulk. However, the LN layer (purple) on top was found as intact layer in regions of HSAL growth above and below the LZLN layer. Consequently, the improved cycling stability of the LiLZLN electrodes during electrodeposition/-dissolution was explained by the morphological analysis. A single intermetallic LiZn layer dilutes into the HSAL and Li bulk during the cycling process and a rigid, single LN layer cracks in certain areas where HSAL growth and pit formation are subsequently facilitated. The described process occurred delayed with the LZLN layer, since the LN layer protects the LiZn layer from early dissolution into HSAL and the LiZn layer homogenizes the Li ion flux during electrodeposition/-dissolution. Based on the results of the cross-section images and the electrochemical analysis, a combination of protective layers with different properties is proposed. This approach aims to reduce the overall thickness of the protective layer while increasing its effectiveness. Thus, the delayed initiation of coating cracks, higher homogeneity of Li ion electrodeposition/-dissolution and a subsequently delayed exposure of the reactive Li bulk resulted in an enhanced cycle life.
After studying the protective layer on Lirp electrodes in symmetric cells and analyzing their morphology before and after cycling through cryogenic cross sections, the next step was to investigate its practical application in NMC622||Li cells. Therefore, NMC622||Li cells with Lirp, LiLN and LiLZLN electrodes were assembled and their practical application in LMBs was investigated. Fig. 6a shows the progression of the specific discharge capacity and coulombic efficiency of the NMC622||Li cells.
 | ||
| Fig. 6 (a) Electrochemical performance of NMC622||Li cells with Lirp (grey), LiLN (red) or LiLZLN (blue) electrodes as negative electrode. (a) Capacity retention and the coulombic efficiency in a long-term cycling experiment at 0.2C. (b) The corresponding charge/discharge voltage profiles after initial cycle and (c) after 100 cycles. | ||
The initial capacity of NMC622||Li cells with Lirp, LiLN or LiLZLN as negative electrode was ≈160 mA h g−1, based on the discharge capacity of the positive electrode. Fig. 6a shows a rapid capacity decay of NMC622||Lirp followed by NMC622||LiLN and an enhanced capacity retention with NMC622||LiLZLN. The state of health (SOH) values of 80% are visualized in Fig. 6a. For cells with Lirp as negative electrode the SOH (80%) was reached after 70 cycles, for the ones with LiLN after 71 cycles and for the ones with LiLZLN after 105 cycles. This represented a 50% improvement in SOH (80%) for cells with LiLZLN as a negative electrode compared to the ones with Lirp. The comparatively poor cycle life and accelerated capacity fade of NMC622||Li cells in comparison to Li||Li cells was attributed to the 4–5 times higher areal capacity of the NMC622||Li cells associated with a correspondingly enhanced amount of shuttled lithium.
Post mortem analysis of cycled Li electrodes showed that Lirp has already after 25 cycles a higher HSAL growth and pit formation compared to the coated Li electrodes (Fig. 5d–i). It is therefore reasonable to observe a premature capacity fading for Lirp negative electrodes. However, the discharge capacity of NMC622||LiLN cells showed a similar capacity decay as with Lirp electrodes, emphasizing that a single LN layer with 100 nm layer thickness lacks in a comprehensive Li surface protection in combination with NMC622.
A lower surface protection with was visible in the cross-section images of aged LiLN with a cracked LN layer in regions of HSAL growth after 25 cycles (Fig. 5h). These regions accelerate a rollover failure with continues charge/discharge cycles.55 The undamaged LZLN layer after 25 cycles and the intact Li3N layer of the dual layer after 50 cycles (Fig. 5i and o) emphasized the elevated surface protection that was observed in Fig. 6a for NMC622||LiLZLN. The corresponding charge/discharge voltage profile (Fig. 6b) of the initial cycle revealed a similar voltage polarization for LiLN and LiLZLN and a higher polarization for Lirp as negative electrode. The higher polarization of NMC622||Lirp compared to cells with LiLN and LiLZLN electrodes indicated a higher internal resistance and kinetical hindrance for Li ion deposition during charge and for Li ion dissolution during discharge. After 100 cycles, the charge/discharge voltage polarization sharply increased for Lirp and LiLN until reaching 4.2 V, followed by a kink and slower voltage rise. The discharge profile displayed a rapid voltage decline, suggesting low capacity for NMC622 cells equipped with both Lirp and LiLN negative electrodes. In contrast, NMC622||LiLZLN cells retained a classic voltage polarization profile with a lower capacity loss. Overall, the analysis of NMC622||Li cells proved an improved performance of Lirp electrodes with a dual protective LZLN layer compared to single protective LN layer or bare Lirp electrodes.
Conclusion
In this study, a dual protective layer consisting of an intermetallic LiZn bottom layer and an inorganic Li3N (LN) top layer was fabricated via thermal evaporation. The PVD technique offered the possibility of thickness control and high purity of both deposited layers. The selection of the LiZn layer was based on its high Li ion diffusion coefficient, which reduces nucleation overvoltage and promotes a uniformly distributed electrodeposition of Li ions. The Li3N layer was chosen as the top layer for its high Li ion and low electronic conductivity, encouraging Li ions to deposit under the protective layer. SEM, EDX and ToF-SIMS analysis revealed a homogeneously distributed and a well-defined LiZn and LN layer. Intensive analysis of the protected surface before and after cycling by SEM, in particular cross-sections under cryogenic conditions, revealed the reason for the increased protection of the Li metal surface provided by the LZLN layer. Compared to the Lirp surface or LN layer, which showed a cracked surface, pit formation and HSAL growth, the LZLN dual layer presented an intact coating with absence of cracks in areas of HSAL growth. In fact, even after 50 cycles, the LZLN layer was still intact, with the intermetallic layer starting to dissolve into HSAL and Li bulk. Electrochemical analysis revealed an 80% increase in cycle life for symmetric cells and a 50% increase in SOH (80%) for NMC622||Li cells with LiLZLN as negative electrode compared to the ones with Lirp. Overall, an enhanced cycling performance for both symmetric Li||Li cells and NMC622||Li cells was demonstrated with the LZLN protected Lirp electrodes. The performance increase resulted from a mitigation of premature cracking of the brittle Li3N layer due to a more homogeneously distributed Li ion electrodeposition/dissolution attributed the LiZn layer. Additionally, the Li3N layer inhibited an accelerated dissolution of the LiZn layer into HSAL and Li bulk during cycling. In combination, the LZLN layer produced a superior protection of the Li surface and an improved Li electrodeposition/-dissolution behavior.In conclusion, this study highlights the synergistic effects achieved by combining protective layers with diverse properties, leading to a noteworthy improvement in the cycle lifetime of Li||Li and NMC622||Li cells. The study of aged coated/uncoated Li electrodes exhibited the importance in understanding how coating degradation leads to a subsequent capacity loss. Especially the post mortem morphology-preserving analyses of protective layers demonstrated that the failure analysis is essential for a continuous improvement regarding coating development on Li electrodes for symmetric cells, full cells, but also for future investigations involving solid-state batteries. For further thickness optimization of dual protective layers, combined simulation calculations and insights into performance behavior of protective layers from experimental data could be used for predicting the optimal design of next-generation lithium metal anodes.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. Bar for creating the graphical illustration. S. Greiwe co-developed the Li3N layer as a part of a master thesis and performed Raman spectroscopy for characterization; M. Mense: performed ToF-SIMS measurements, analyzed and discussed the results with M. M. Bela; M. C. Stan: discussion and supervision; M. Börner, M. Winter: discussion, supervision, proofreading, contribution to the final draft, and acquisition of funding. All authors were involved in the discussion of the results and approval of the final manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support provided by the EU Horizon Europe – research and innovation action within the research project “PSIONIC” (101069703) and the German Federal Ministry of Education and Research (BMBF) within the research project “ProLiFest” (03XP0253A) is gratefully acknowledged.References
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3(4), 267 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2014, 114(23), 11414 CrossRef CAS PubMed.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135(4), 1167 CrossRef CAS PubMed.
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Electrochem., 2017, 21(7), 1939 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4(3), 180 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- M. Winter, Zeitschrift für Physikalische Chemie, 2009, 223, 1395 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164(7), A1703 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89(2), 206 CrossRef CAS.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K.-H. Chen, J.-G. Zhang, K. Thornton and N. P. Dasgupta, ACS Cent. Sci., 2016, 2(11), 790 CrossRef CAS PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194 CrossRef CAS PubMed.
- L. Hellweg, T. Beuse, M. Winter and M. Börner, J. Electrochem. Soc., 2023, 170(4), 040530 CrossRef CAS.
- J. Heine, P. Hilbig, X. Qi, P. Niehoff, M. Winter and P. Bieker, J. Electrochem. Soc., 2015, 162(6), A1094 CrossRef CAS.
- H. Zhang, G. Gebresilassie Eshetu, X. Judez, C. Li, L. M. Rodriguez-Martínez and M. Armand, Angew. Chem., Int. Ed., 2018, 57, 5002 Search PubMed.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5(4), 299 CrossRef CAS.
- M.-H. Ryou, Y. M. Lee, Y. Lee, M. Winter and P. Bieker, Adv. Funct. Mater., 2015, 25(6), 834 CrossRef CAS.
- H. Wang, Y. Liu, Y. Li and Y. Cui, Electrochem. Energy Rev., 2019, 2(4), 509 CrossRef CAS.
- N. Delaporte, Y. Wang and K. Zaghib, Front. Mater., 2019, 6, 267 CrossRef.
- D. Kang, M. Xiao and J. P. Lemmon, Batteries Supercaps, 2020, 4(3), 445 CrossRef.
- J. Wellmann, J.-P. Brinkmann, B. Wankmiller, K. Neuhaus, U. Rodehorst, M. R. Hansen, M. Winter and E. Paillard, ACS Appl. Mater. Interfaces, 2021, 13(29), 34227 CrossRef CAS PubMed.
- D. G. Belov, O. V. Yarmolenko, A. Peng and O. N. Efimov, Synth. Met., 2006, 156(9–10), 745 CrossRef CAS.
- M. Wu, Z. Wen, Y. Liu, X. Wang and L. Huang, J. Power Sources, 2011, 196(19), 8091 CrossRef CAS.
- Y. J. Zhang, W. Wang, H. Tang, W. Q. Bai, X. Ge, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2015, 277, 304 CrossRef CAS.
- Z. Li, S. Santhanagopalan and A. Zakutayev, MRS Commun., 2022, 12(3), 352 CrossRef CAS.
- S. Stuckenberg, M. M. Bela, C. T. Lechtenfeld, M. Mense, V. Küpers, T. T. K. Ingber, M. Winter and M. C. Stan, Small, 2023, 2305203, 1 Search PubMed.
- Y. Li, Y. Sun, A. Pei, K. Chen, A. Vailionis, Y. Li, G. Zheng, J. Sun and Y. Cui, ACS Cent. Sci., 2017, 4(1), 97 CrossRef PubMed.
- M. C. Stan, J. Becking, A. Kolesnikov, B. Wankmiller, J. E. Frerichs, M. R. Hansen, P. Bieker, M. Kolek and M. Winter, Mater. Today, 2020, 39, 137 CrossRef CAS.
- J. Deng, Y. Wang, S. Qu, Y. Liu, W. Zou, F. Zhou, A. Zhou and J. Li, Batteries Supercaps, 2020, 4(1), 140 CrossRef.
- M. M. Bela, C. Schmidt, K. Neuhaus, T. Hering, M. C. Stan, M. Winter and M. Börner, Adv. Mater. Interfaces, 2024, 2300836 CrossRef CAS.
- N. Delaporte, A. Perea, S. Collin-Martin, M. Léonard, J. Matton, V. Gariepy, H. Demers, D. Clément, E. Rivard and A. Vijh, Batteries Supercaps, 2022, 5, e202200245 CrossRef CAS.
- S. Lee, K.-s. Lee, S. Kim, K. Yoon, S. Han, M. H. Lee, Y. Ko, J. H. Noh, W. Kim and K. Kang, Sci. Adv., 2022, 8(30), 1 CAS.
- Y. Yang, L. Ai, J. He, C. Zhang, D. Chen and L. Shen, Chem. Commun., 2023, 59, 936–939 RSC.
- S. Wang, J. Chen, H. Lu, Y. Zhang, J. Yang, Y. Nuli and J. Wang, ACS Appl. Energy Mater., 2021, 4(11), 13132 CrossRef CAS.
- J. Wang, J. Yang, Q. Xiao, J. Zhang, T. Li, L. Jia, Z. Wang, S. Cheng, L. Li, M. Liu, H. Liu, H. Lin and Y. Zhang, Adv. Funct. Mater., 2020, 31(7), 2007434 CrossRef.
- S. Lobe, A. Bauer, S. Uhlenbruck and D. Fattakhova-Rohlfing, Adv. Sci., 2021, 8(11), 2002044 CrossRef CAS PubMed.
- M. Nicolaus and M. Schäpers, Modern Surface Technology, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- D. M. Mattox, Handbook of Physical Vapor Deposition (PVD) Processing, Elsevier Science, 2nd edn, 2010 Search PubMed.
- J. Wang, P. King and R. A. Huggins, Solid State Ionics, 1985, 20, 185 CrossRef.
- A. Anani, S. Crouch-Baker and R. A. Huggins, Measurement of lithium diffusion in several binary lithium alloys at ambient temperature, The Electrochemical Society, United States, 1986, http://inis.iaea.org/search/search.aspx?orig_q=RN:18093564 Search PubMed.
- R. A. Huggins, J. Power Sources, 1999, 81–82, 13 CrossRef CAS.
- S.-K. Otto, T. Fuchs, Y. Moryson, C. Lerch, B. Mogwitz, J. Sann, J. Janek and A. Henss, ACS Appl. Energy Mater., 2021, 4(11), 12798 CrossRef CAS.
- C. Zhu, T. Fuchs, S. A. L. Weber, F. H. Richter, G. Glasser, F. Weber, H.-J. Butt, J. Janek and R. Berger, Nat. Commun., 2023, 14, 1300 CrossRef CAS PubMed.
- J. Becking, A. Gröbmeyer, M. Kolek, U. Rodehorst, S. Schulze, M. Winter, P. Bieker and M. C. Stan, Adv. Mater. Interfaces, 2017, 4(16), 1700166 CrossRef.
- R. Nölle, K. Beltrop, F. Holtstiege, J. Kasnatscheew, T. Placke and M. Winter, Mater. Today, 2020, 32, 131 CrossRef.
- M. Mense, M. M. Bela, S. P. Kühn, I. Cekic-Laskovic, M. Börner, S. Wiemers-Meyer, M. Winter and S. Nowak, Commun. Chem., 2025 DOI:10.1038/s42004-025-01426-0.
- S.-K. Otto, Y. Moryson, T. Krauskopf, K. Peppler, J. Sann, J. Janek and A. Henss, Chem. Mater., 2021, 33(3), 859 CrossRef CAS.
- E. Zintl and G. Brauer, Zeitschrift für Elektrochemie und angewandte physikalische Chemie, 1935, 41(2), 102 CrossRef CAS.
- A. Rabenau and H. Schulz, J. Less-Common Met., 1976, 50(1), 155 CrossRef CAS.
- K. Park and J. B. Goodenough, Adv. Energy Mater., 2017, 7(19), 1700732 CrossRef.
- A. D. Pelton, J. Phase Equilib. Diffus., 1991, 12, 42 CrossRef CAS.
- R. Schmitz, R. Müller, S. Krüger, R. W. Schmitz, S. Nowak, S. Passerini, M. Winter and C. Schreiner, J. Power Sources, 2012, 217, 98 CrossRef CAS.
- K.-H. Chen, K. N. Wood, E. Kazyak, W. S. LePage, A. L. Davis, A. J. Sanchez and N. P. Dasgupta, J. Mater. Chem. A, 2017, 5, 11671 RSC.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670 RSC.
- R. Zhang, X.-R. Chen, X. Chen, X.-B. Cheng, X.-Q. Zhang, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56(27), 7764 CrossRef CAS PubMed.
- S. Klein, P. Bärmann, L. Stolz, K. Borzutzki, J.-P. Schmiegel, M. Börner, M. Winter, T. Placke and J. Kasnatscheew, ACS Appl. Mater. Interfaces, 2021, 13(48), 57241 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06366j |
| This journal is © The Royal Society of Chemistry 2025 |