
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deactivation of copper electrocatalysts during CO2 reduction occurs via dissolution and selective redeposition mechanism†
Blaž
Tomc
ab,
Marjan
Bele
a,
Mohammed
Azeezulla Nazrulla
a,
Primož
Šket
c,
Matjaž
Finšgar
 d,
Angelja Kjara
Surca
a,
Ana Rebeka
Kamšek
ae,
Martin
Šala
f,
Jan
Šiler Hudoklin
g,
Matej
Huš
ghi,
Blaž
Likozar
g and
Nejc
Hodnik
*abj
d,
Angelja Kjara
Surca
a,
Ana Rebeka
Kamšek
ae,
Martin
Šala
f,
Jan
Šiler Hudoklin
g,
Matej
Huš
ghi,
Blaž
Likozar
g and
Nejc
Hodnik
*abj
aLaboratory for Electrocatalysis, Department of Materials Chemistry, National Institute of Chemistry, Ljubljana 1000, Slovenia. E-mail: Nejc.Hodniki@ki.si
bUniversity of Nova Gorica, Nova Gorica 5000, Slovenia
cSlovenian NMR Centre, National Institute of Chemistry, Ljubljana 1000, Slovenia
dFaculty of Chemistry and Chemical Engineering, University of Maribor, Maribor 2000, Slovenia
eFaculty of Chemistry and Chemical Technology, University of Ljubljana, Ljubljana 1000, Slovenia
fDepartment of Analytical Chemistry, National Institute of Chemistry, Ljubljana 1000, Slovenia
gDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Ljubljana 1000, Slovenia
hAssociation for Technical Culture of Slovenia (ZOTKS), Zaloška 65, Ljubljana 1000, Slovenia
iInstitute for the Protection of Cultural Heritage of Slovenia (ZVKDS), Poljanska 40, Ljubljana 1000, Slovenia
jInstitute of Metals and Technology, Ljubljana 1000, Slovenia
First published on 10th December 2024
Abstract
As electrochemical CO2 reduction (ECR) nears industrialisation levels, addressing the uncontrolled stability, restructuring, and deactivation of copper (Cu) catalysts during operation becomes as crucial as achieving high activity and selectivity for a single product. This study used a high-surface area Cu catalyst that exhibited changes in ECR product selectivity over prolonged operation. The detection of dissolved Cu species during electrolysis confirmed an intermediates-mediated Cu dissolution mechanism at ECR potentials (−0.8 to −1.1 V vs. reversible hydrogen electrode). The findings suggest that the electrodeposition of dissolved Cu species is biased towards Cu catalyst sites with lower reaction intermediates coverage, e.g. adsorbed CO (*CO). A dynamic equilibrium between dissolution and subsequent selective redeposition gradually led to morphological restructuring, resulting in a shift in selectivity away from ECR and towards hydrogen production. With the obtained extensive experimental results, theoretical modelling, and literature data, four interconnected parameters governing restructuring and selectivity shifts were recognised: (i) size and (ii) crystallographic orientation of facets of the nanoparticles, (iii) *CO coverage and (iv) CObridgevs. COatop ratio.
Introduction
The increasing concentration of CO2 in Earth's atmosphere1 has emerged as an urgent global concern due to its role in climate change and global warming. ECR on Cu has gained considerable attention as a means not only to reduce CO2 emissions but also to produce valuable chemicals and fuels.2,3 For commercial applications, catalysts must exhibit high faradaic efficiency (FE) and current density for a specific product, and, equally important, long-term operational stability.2,4 In recent years, substantial progress has been made in improving the activity and selectivity,5,6 however, numerous studies have highlighted the substantial morphological restructuring of Cu-based catalysts during operation.7–20 Since morphology is crucial for both the ECR activity and selectivity of Cu catalysts,14,15,21–24 a comprehension of this phenomenon is essential. Two distinct stages of morphological deformation can be identified during the ECR protocol: (i) catalyst immersion in the electrolyte without applied potential, later open circuit potential (OCP), followed by the application of the onset potential at the start of electrochemical measurement (initial stage of the ECR protocol) and (ii) electrochemical measurement, typically chronoamperometry (CA) at a constant ECR potential.In the first stage, the dominant mechanism is dissolution–redeposition, driven by the formation of Cu-oxides on the catalyst surface at OCP.17–20 This degradation process unfolds in two steps: the dissolution of Cu-oxides at OCP, followed by dissolved species electrodeposition onto the catalyst surface when ECR potential is applied.17,18 Dissolved Cu species are in the Cu+ oxidation state,25 and the electrodeposited Cu is low-coordinated.12,14,26,27 Raaijman et al.20 demonstrated that this initial restructuring can be limited if the catalyst gets exposed to the electrolyte under applied potential. Undissolved Cu-oxides undergo reduction to metallic Cu at the initiation of CA,18,28–30 triggering transient dissolution followed by redeposition, leading to some unavoidable restructuring.19
Nevertheless, restructuring during the initial stage of the ECR protocol does not account for all Cu morphology changes, as alterations have been observed even during prolonged CA.7–15 Zhang et al.9 directly observed atomic restructuring of the Cu surface during ECR, noting the formation of a liquid-like amorphous structure at the interface between the Cu catalyst and the electrolyte, composed of Cu, O, H, and C. This process transformed an atomically smooth surface into a rough one. Moreover, their results indicated that Cu+ species exist at this interface. Vavra et al.,25 using density functional theory (DFT) studies, demonstrated that soluble Cu species can exist at ECR potential in the form of [Cu+–ECR intermediates]XH2O complexes. Other studies also suggested that restructuring at ECR potential is facilitated by reaction intermediates, especially *CO.10,15,30,31 Alterations on Cu were observed even in a CO atmosphere.32,33 Therefore, it can be noted that intermediates-mediated dissolution initiates restructuring. Cu instability during the CA is of great importance as it impacts both activity and selectivity, usually leading to deactivation.7,15,34–43 Liu et al.7 suggested that the inactive Cu species formed through this process contain *CO bound to two Cu atoms (CObridge) in contrast to the preferential *CO bound to a single Cu atom (COatop) at active sites. Other studies also proposed that CObridge is inactive,44,45 however, Chou et al.43 elegantly showed that for the successful C–C coupling, a correct ratio of both forms is needed.
Although there have been significant efforts to limit instability6,34–38 and catalyst reactivation using pulsed techniques,7,40–42 deactivation remains an issue. To develop industrially viable catalysts, stability must be prolonged by several orders of magnitude.4 Herein, this study reveals the fundamental mechanisms of Cu catalysts restructuring and deactivation through a dynamic equilibrium between Cu dissolution and selective redeposition, intending to provide the understanding and, consequently, design principles for optimizing operational conditions and/or stable catalysts to effectively address these currently uncontrollable alterations.
Results and discussion
Catalyst modification via dissolution–redeposition mechanism at the initial stage of ECR protocol
To address the challenge of dynamic restructuring during CA through dissolution–redeposition, the fundamentally similar dissolution–redeposition mechanism occurring at the initial stage of the ECR protocol was studied. Especially the electrodeposition of dissolved Cu occurred at similar conditions as during the reaction, however, in this first stage of the ECR protocol, its effect was more pronounced. Additionally, promising pulsed electrochemical techniques involve fluctuations between OCP (or near OCP) and ECR potential7,40–42,46–49 at which the Cu dissolution–redeposition mechanism is occurring.A Cu-nanostructured catalyst on carbon support coated on a glassy carbon plate (GCp) was prepared (see Experimental section and Section S1 in ESI†). The catalyst's surface was primarily composed of Cu-oxides (Section S2†) and was therefore appropriate to observe the proposed dissolution–redeposition mechanism. Identical location scanning electron microscopy (IL-SEM) and inductively coupled plasma mass spectrometry (ICP-MS) were employed to track Cu movement (Fig. 1a, b, e, f and S6g†). A rapid dissolution during the first minutes of OCP exposure, followed by a less pronounced dissolution (Fig. S6g† and results from ref. 18 and 25), indicate that Cu dissolution occurs in two stages: (i) an initial rapid dissolution of Cu-oxides up to a few 100 ppb, followed by (ii(1)) a slower process of Cu-oxides dissolution if there were any left after (i), or (ii(2)) an even slower process involving the oxidation of metallic Cu and subsequent dissolution of formed Cu-oxides if all oxides dissolved in the first minutes (Fig. S9†).
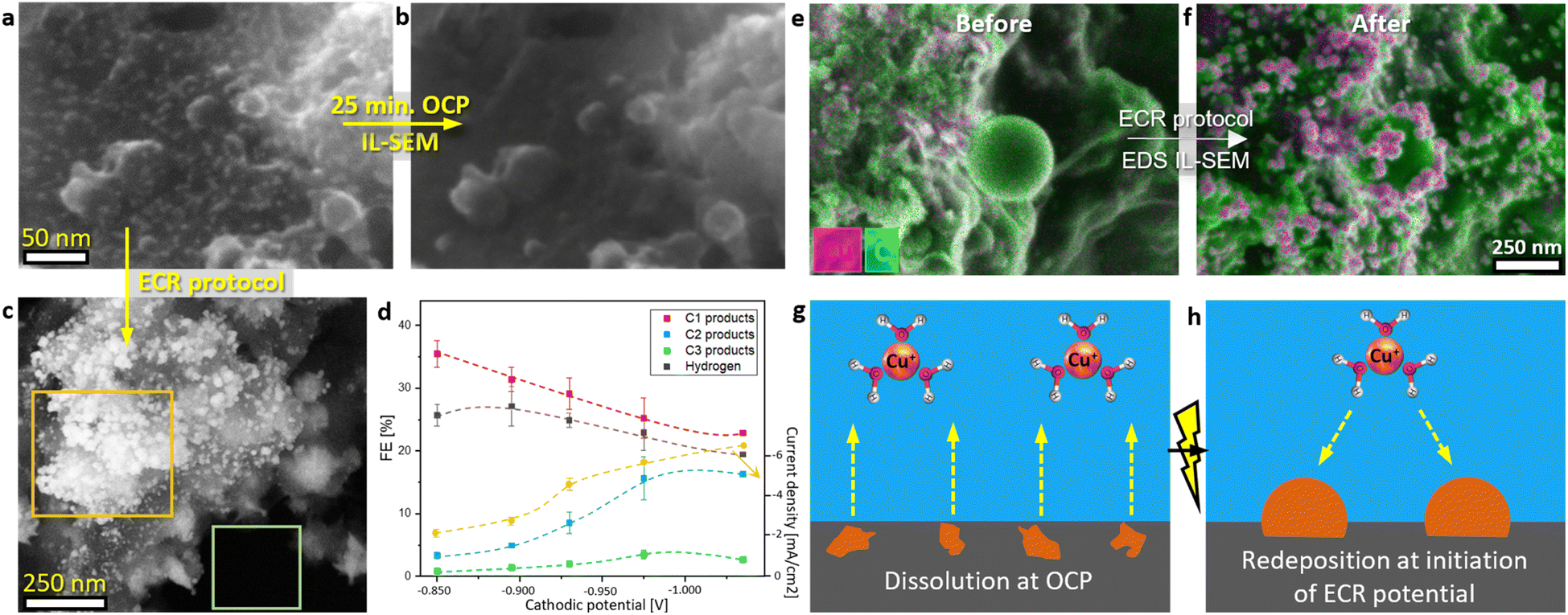 | ||
| Fig. 1 (a and b) IL-SEM of the catalyst before and after OCP exposure in the CO2-saturating 0.1 M KHCO3 solution for 25 minutes. (c) SEM of the catalyst after the ECR protocol: 25 minutes at OCP, followed by the application of a constant −0.895 V vs. reversible hydrogen electrode (RHE) potential for 1 hour in the CO2-saturating 0.1 M KHCO3. The orange rectangle highlights a more exposed area where a greater number of particles formed that were larger in size, while the green rectangle indicates a submerged area where no nanoparticles were observed. (d) Catalyst ECR activity for various types of products and current density as a function of applied potential (detailed product analysis is provided in Section S3†). (e and f) Energy dispersive X-ray spectroscopy (EDS) mapping overlay in association with IL-SEM of the catalyst before and after ECR protocol: 25 minutes at OCP followed by the application of constant −0.975 V vs. RHE potential for 1 hour in the CO2-saturating 0.1 M KHCO3. Schematic representation of (g) Cu nanoparticles dissolution at OCP and (h) subsequent electrodeposition of dissolved Cu species upon applying ECR potential at the start of CA. | ||
Upon applying the potential at the initiation of CA, the electrodeposition of dissolved Cu species resulted in the formation of spherical Cu nanoparticles, ranging from 5 to 80 nm in size, with a higher concentration of particles forming on more exposed surface areas (Fig. 1c, f and S12†). The dissolution–redeposition mechanism is schematically presented in Fig. 1g and h. In detail, the electrodeposition mechanism is presented in the following section (Cu electrodeposition vs. ECR). Through this dissolution–redeposition mechanism, the catalyst morphology was modified before the experiment. Several factors that influence Cu surface transformation were identified: (i) exposure of the catalyst to the electrolyte (Fig. 1c), (ii) amount of dissolved Cu (Fig. 3a and d), (iii) applied potential (Fig. 3a, b and ref. 29), (iv) *CO surface coverage (Fig. 3), (v) type of the catalyst used (comparing the effect of electrodeposition in Fig. 1c with IL-SEM results provided in ref. 17 and 50) and (vi) other experimental conditions.19,20 Since these factors are readily adjusted to optimise ECR, the exact effects of the process are difficult to predict. Therefore, it is important to determine catalyst modification at the initial stage of the ECR protocol for a better understanding of the processes occurring during the reaction. An effective approach to limit the phenomenon was applying the potential before introducing the electrolyte to the catalyst (Fig. S8c and d†), which triggered only the less intense transient dissolution and redeposition.
Considering pulsed electrochemical techniques, where the potential switches from OCP to ECR potential, the morphology of the catalyst could be directed in a specific way utilising the dissolution–redeposition mechanism at the initial stage of ECR protocol. It has been demonstrated previously that under ideal conditions, activity, selectivity, and, most importantly, long-term stability can be optimised.7,40–42,46–49
The primary products of the ECR on the catalyst used in this study were formate and ethylene, with hydrogen evolution reaction (HER) accompanying the reaction (Fig. 1d and Section S3†). Aligning with the literature data,18,28–30,51 results in Section S2† also suggested that during operation Cu surface is in the Cu0 oxidation state. After air exposure, the metallic Cu surface oxidised to Cu2O (Fig. S17†).
Restructuring and deactivation of the catalyst via dissolution during operation
During CA, the concentration of dissolved Cu was determined in the electrolyte using ICP-MS (Fig. S6g†). Catholyte was sampled at the 59th minute of CA at −0.975 V vs. RHE, while the ECR potential was still applied. A low concentration of Cu was determined to be 11.8 ppb. Similarly, another sample was taken during a second ECR protocol, again at the 59th minute of CA measurement at −0.975 V vs. RHE. A lower concentration of dissolved Cu was determined (3.7 ppb). This observation aligns with a report by Zhang et al.9 where the Cu surface was observed to be dynamic at ECR conditions, forming an amorphous-like interface containing Cu between the electrolyte and the catalyst. Our results indicate that Cu can leach out of this layer through the proposed formation of [Cu+–ECR intermediates]XH2O complexes at ECR potential.25The catalyst used in this study experienced morphological restructuring during CA (Fig. 2c–e). The well-defined, smooth Cu nanoparticles formed through electrodeposition at the start of CA (Fig. 3a and b) did not change significantly until 2 hours (Fig. 2c). However, anisotropic restructuring of the nanoparticle surface was observed after 6 hours (Fig. 2d). Further nanoparticle restructuring occurred until 16 hours of CA (Fig. 2e). This process followed a mechanism opposite of Ostwald ripening, where smaller nanostructures grew through the dissolution of larger ones. A similar restructuring process was reported by Huang et al.,15 where smaller Cu nanoparticles formed from Cu nanocubes, similar in size to our nanoparticles. Since morphology changed during the reaction, selectivity alterations were expected.
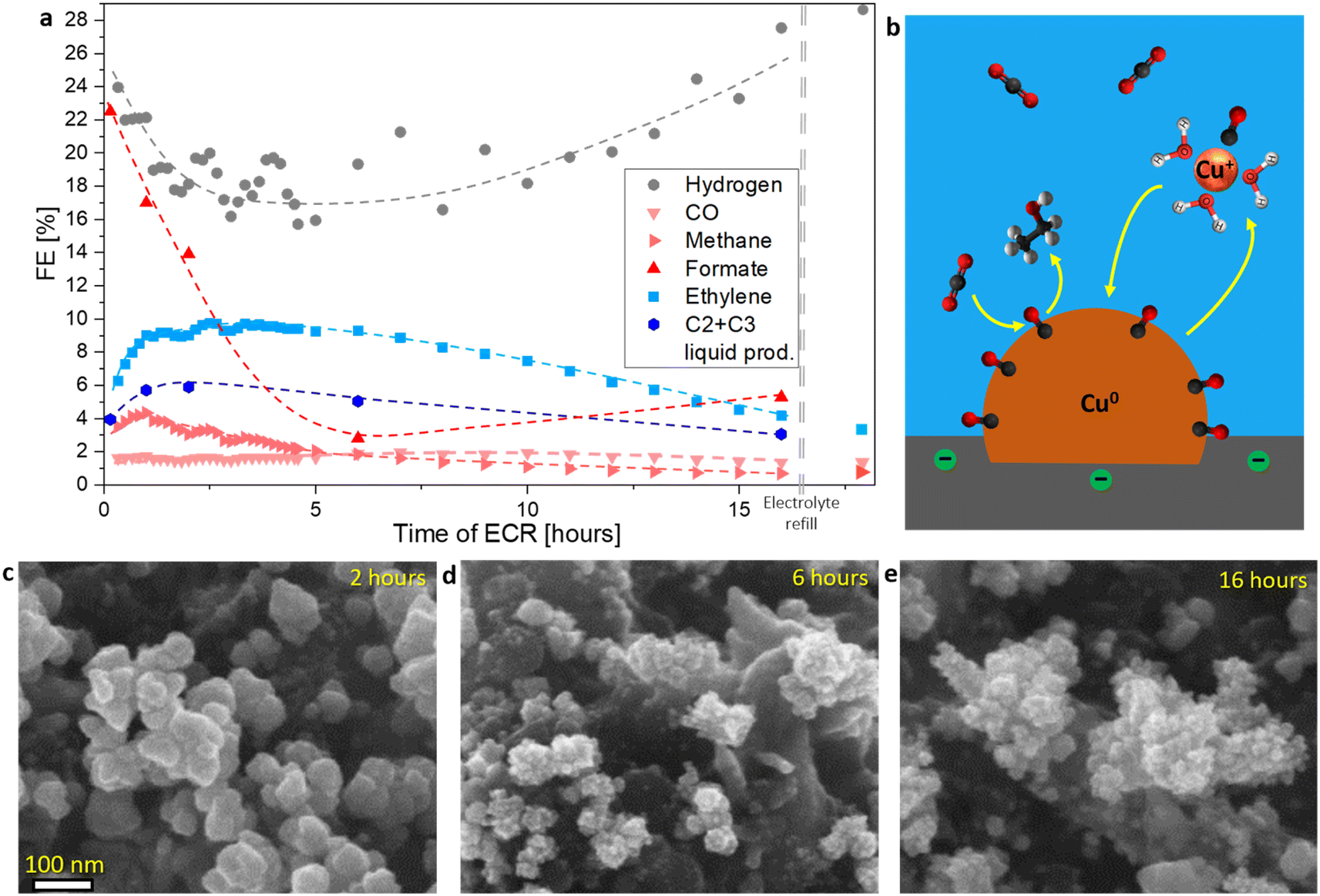 | ||
| Fig. 2 (a) Analysis of ECR product selectivity over an 18-hour CA at a constant potential of −0.975 V vs. RHE in the CO2-saturating 0.1 M KHCO3. The double dashed line indicates the point at which the electrolyte was replaced with a fresh solution without interrupting the applied voltage. Liquid products were measured from five replicate samples after varying durations of electrolysis. Current density can be found in Fig. S28b.† (b) Schematic representation of catalyst restructuring through dissolution and redeposition during operation at ECR potential. (c–e) SEM images of three replicate samples illustrating the catalyst after varying durations of electrolysis. | ||
 | ||
| Fig. 3 SEM images of the catalyst after the ECR protocol: 25 minutes at OCP followed by the application of a constant (a) −0.850 V and (b) −0.975 V vs. RHE potential for 1 hour in the CO2-saturating 0.1 M KHCO3. IL-SEM images showing the catalyst before and after a repeated ECR protocol on catalysts from (a and b): (c and d) at −0.850 V vs. RHE and (e and f) −0.975 V vs. RHE. (g and h) IL-SEM of a repeated ECR protocol at −1.000 V vs. RHE with Ar-saturating electrolyte instead of CO2 on a catalyst prepared at the same conditions as (b). (i and j) Schematic representation corresponding to (c and d), illustrating partial blocking of Cu electrodeposition by ECR. (j–l) Schematic representation corresponding to (e and f), showing complete blocking of Cu electrodeposition by ECR. | ||
Changes in product selectivity were monitored over the 16-hour CA (Fig. 2a). Formate production dropped from approximately 22% FE in the first hour to around 3% FE after 6 hours, before increasing back to around 5% FE. Ethylene formation increased from 6% to 10% FE and reached a plateau after 4 hours. At approximately 6 hours, its production started depleting as well. Other C2+ products experienced a similar trend. Conversely, a decrease in FE for HER occurred from the start, reaching a minimum before 4 hours. After that, HER FE increase was observed from 6 hours onwards. Methane production in the first hour barely increased and then exhibited a depleting trend. On the other hand, CO production remained constant throughout the 16-hour experiment. A repetition of the 16-hour experiment was conducted for gaseous products, yielding a similar trend (Fig. S28b†). By exchanging the electrolyte under potential control, therefore, preventing catalyst oxidation at OCP and subsequent dissolution–redeposition, the selectivity remained unchanged (Fig. 2a). This control experiment ruled out the potential effects of ECR products in the electrolyte, possible pH changes over 16 hours, and other factors on ECR.
Formate-producing sites deactivated more rapidly than C–C coupling sites (Fig. 2a). The initial increase in C2+ product formation during the first 4 hours could be attributed to the deactivation of formate-producing sites, which made more CO2 available for *CO. We propose that the deactivation of C2+-producing sites began from the start but was initially masked by mass transport effects, leading to the observed activation. Similar could be observed for methane and CO. It could be noted that deactivation of methane-producing sites was faster than at C–C coupling sites but slower than at formate-producing sites. CO-producing sites showed the slowest deactivation, with a subsequent activation trend following the deactivation of C–C coupling sites. A clearer CO site activation was observed at another catalyst, where the deactivation was more rapid (Fig. S28a†). The interconnections of different products were consistent with the established ECR mechanisms,2 where for example by unsuccessful C–C coupling CO will form by desorbing from the Cu-surface, and if the formate reaction pathway is limited, CO2 will reduce to methane, CO and C2+ products. The close interconnection between C–C coupling and HER-producing sites is evident in Fig. 2a, where an inverse relationship was observed: as selectivity for C–C coupling increased, HER selectivity decreased, and vice versa. In contrast, interconnection between formate and HER-producing sites was not observed. C–C coupling requires *CO to remain on the Cu surface for an extended time, and C2+ products involve more than 8 electron transfers.2 This results in a high population of C2+ intermediates on the Cu surface, consequently blocking HER. This is consistent with the report presented by Ooka et al.52
Similar changes in FE for ECR products were reported by Huang et al.,15 though the different deactivation trends observed by other researchers suggest that further investigation is needed to fully understand the deactivation mechanisms for various ECR products. Particularly, the interconnections between different products are of interest and could, with additional data, offer a novel perspective on the ECR mechanism.
Cu electrodeposition vs. ECR
The observed so-called “anti-Ostwald ripening” nanoparticle evolution during CA (Fig. 2c–e and ref. 8, 14, 15) suggested that Cu growth at ECR conditions deviates from the typical ripening process, where curvature (Gibbs–Thomson effect) governs the enlargement of the larger particles at the cost of smaller ones. Consequently, the electrodeposition of dissolved Cu species at the initiation of CA was investigated. The process was, in principle, similar to during the reaction, only on a larger scale. IL-SEM images revealed that the electrodeposition and the ECR are competing processes (Fig. 3). This competition was first evident during the first ECR protocol, where nanoparticles formed at the potential with higher activity and selectivity for C–C coupling (−0.975 V vs. RHE; Fig. 1d) had a rougher surface compared to the surface of nanoparticles formed at a higher potential at which C–C coupling was less pronounced (Fig. 3a and b). Simon et al.26 also reported the formation of rougher Cu surfaces with a higher density of undercoordinated Cu sites at lower potentials.When the ECR protocol was repeated on the modified catalyst (second ECR; Fig. 3c–h), the dissolution during OCP was reduced due to the larger nanoparticles compared to the first ECR. After 40 minutes at OCP, most of the nanoparticles remained intact, resulting in a lower amount of dissolved Cu, compared to the first ECR protocol (Fig. S6e–g†). Consequently, when the onset potential of CA was applied, pre-existing Cu nanoparticles were already present on the catalyst surface, unlike in the first ECR. IL-SEM unveiled an even higher deviation from Ostwald ripening growth after electrodeposition at the onset potential of the second ECR. At −0.850 V vs. RHE, the surface of pre-existing nanoparticles roughened (Fig. 3c and d), similar to evolution during the reaction (Fig. 2c–e). However, when the modified catalyst was subjected to a second ECR protocol at the potential of −0.975 V vs. RHE (optimal for activity and selectivity for C–C coupling), the electrodeposition led to the formation of new nanoparticles alongside pre-existing ones (Fig. 3e and f). A potential of −0.895 V vs. RHE was sufficient to cause full surface obstruction on the catalyst used in this study (Fig. S34†). On the other hand, when the second ECR protocol was repeated with argon purging instead of CO2, Cu nanoparticles marked in green grew evenly at the cost of Cu nanoparticles marked in red (Fig. 3g and h). With CO2, expected ECR activity and selectivity were observed (Section S3†), while no products were detected with argon. Similar anisotropic growth of pre-existing nanoparticles was observed by Popović et al.17,50 using IL-SEM.
The electrodeposition of Cu was partially or fully blocked by ECR reaction intermediates, depending on the applied potential. A control experiment with argon purging ruled out any other parameters that could potentially interfere with electrodeposition, e.g. electrolyte, and demonstrated how with classical electrodeposition of Cu, the curvature or size plays an important role, as also predicted by the Ostwald ripening phenomenon. As previously mentioned, *CO is the key ECR intermediate for successful C–C coupling. At the catalyst used in this study, this process was optimal at −0.975 V vs. RHE (Fig. 1d and Section 3†), where the greatest obstruction of Cu electrodeposition was observed (Fig. 3a–f). Gunathunge et al.31,44 found that the total amount of *CO on the Cu surface increases with decreasing potential, but it begins to decline after the optimal C–C coupling potential, a trend also noted by Zhan et al.53 Similarly, Zhang et al.9 observed that the amorphous layer was the thickest at a potential optimal for C–C coupling. Furthermore, as shown in Fig. 1d and 2a, and by Ooka et al.,52 HER was most suppressed when C–C coupling was most favourable. Therefore, it can be concluded that more active C–C coupling sites create a greater obstruction for the electrodeposition of dissolved Cu species compared to less active ones, as schematically illustrated in Fig. 3i–l, consequently making the effect of nanoparticle curvature negligible.
This behaviour is not exclusive to Cu; similar phenomena have been observed in other metals where *CO acts as a capping agent to produce smaller nanoparticles, such as Pt54 and Au.55
Deactivation mechanism
The observations discussed above confirmed that Cu underwent restructuring and deactivation via a dynamic equilibrium between dissolution and redeposition, even at ECR potential. Previous studies have reported that *CO and other ECR intermediates facilitate Cu restructuring.10,15,25,30–33 Zhang et al.9 directly observed that dissolution occurred randomly, leaving behind a rough atomic surface, although the overall nanometer structure remained largely unchanged. The formation of small nanoparticles on top of pre-existing ones during CA on the catalyst used in this study (Fig. 2e and f) and the detection of Cu in the electrolyte (Fig. S6g†) demonstrated that Cu can leach out from the amorphous layer. As shown in Fig. 3, the electrodeposition of the dissolved Cu species was influenced by adsorbed ECR intermediates. These species preferentially electrodeposited onto sites with lower coverage of ECR intermediates, presumably the less ECR-active sites. This is because the highest *CO coverage typically coincides with the potential yielding the highest ratio of C2+ products vs. HER.31,53 Through multiple iterations of this process, these less active sites gradually grew at the expense of more active ones, consequently changing ECR selectivity, leading to deactivation and heightened HER production (Fig. 4a and b). | ||
| Fig. 4 (a) Proposed mechanism depicting dynamic selective dissolution–redeposition of Cu restructuring during CA at a catalyst used in this study, leading to (b) the formation of hydrogen-producing sites. The shapes of Cu sites are only representative. (c) Graph of different products FE vs. nanoparticle size adapted from Reske et al.24 The results from this study are 28.5 nm spherical nanoparticles on GCp (Fig. S12b, c and S13†). (d) Cu (111) and (e) Cu (100) supercells with explicit water layer used in DFT calculations. For the results presented in (f and g) one water molecule was removed at different positions and then CO was placed into the vacancy at different Cu atoms in bridge or atop positions as presented in Fig. S39.† CObinding energies were calculated as demonstrated in Section S6† at an applied potential of −1.0 V vs. RHE. (f) An average of all calculated CObinding energies (Fig. S40†) at (111) vs. (100) Cu-facets. (g) An average of all calculated CObinding energies with *CO bound as bridge vs. top at (111) vs. (100) Cu-facets. The scale of (f) applies to (g) as well. (h) Representation of *CO bound to Cu surface as bridge and atop configurations. | ||
Given that the nanoparticles used in this study were similar in shape and size to those used by Reske et al.,24 some correlations can be drawn. They demonstrated that spherical nanoparticles ranging from 0 to 15 nm on a GCp exhibited lower selectivity for ethylene and methane and higher for CO and HER than Cu foil. In this study, 28.5 nm (on average) spherical nanoparticles on GCp were prepared (Fig. S12b and c†). Product distribution (Fig. S26†) aligned nicely with the trends argued by Reske et al.24 (Fig. 4c). Furthermore, the second ECR process, involving dissolution–redeposition at the initial stage of the ECR protocol, yielded larger nanoparticles than the first ECR (Fig. S16†). These larger nanoparticles had an increased selectivity for C2+ products and reduced selectivity for HER (Section S3†). The most prominent effect was observed at a potential optimal for ECR, −0.975 V vs. RHE, where through anti-Ostwald growth relatively large nanoparticles grew (Fig. 3e and f). The trends therefore suggested that smaller spherical nanoparticles are less active for ECR, indicating that restructuring into smaller particles may have contributed to the observed deactivation (Fig. 2a). Considering the Cu electrodeposition, these sites were less active for ECR and more active for HER, therefore less covered by reaction intermediates and were consequently more pronounced to grow through electrodeposition. Reske et al.24 have nicely demonstrated how with lowering particle size, the coordination number of surface Cu atoms decreases.
However, with Ar instead of CO2 in the second ECR Ostwald nanoparticle growth was observed (Fig. 3g and h), therefore the nanoparticles got bigger, which by this hypothesis would mean a heightened selectivity for ECR. When the selectivity and activity were measured a heightened HER vs. ethylene production was observed compared to the first ECR (Fig. S27†). Similarly to this study, the formation of smaller nanostructures during CA has been reported previously,7,8,14,15 although the impact on ECR selectivity varies across these studies. Liu et al.7 and Huang et al.15 observed a deactivation trend, while Grosse et al.14 reported no significant changes in selectivity. Moreover, Jung et al.8 demonstrated that 20 nm cubic particles gradually fragmented into 2–4 nm particles under negative potential, leading to enhanced C–C coupling at the expense of HER. Therefore, the formation of smaller nanoparticles that cause ECR deactivation is not a general trend, meaning that more parameters govern the alterations.
Liu et al.7 observed that during deactivation, nanostructures preferentially formed with (111) facets from the original (100). In contrast, Kim et al.11 reported the formation of (100) facets during prolonged ECR operation. To understand the discrepancies between these two findings, DFT modelling of *CO bound in bridge and atop configurations (Fig. 4h) on (100) and (111) Cu facets (Fig. 4d and e) was conducted (Section S6†). The calculations were performed using a Solvated Jellium Model56 (SJM) at default parameters and a potential of −1.0 V vs. RHE. Supercells with 36 surface Cu atoms and one water molecule replaced with *CO were used to imitate the real systems where total *CO coverage is around 0.05.57 The CObinding energies at (111) vs. (100) facets showed a slight variation, suggesting that at ECR potential total *CO coverage is different between the crystallographic orientation of facets (Fig. 4f). Since the results in Fig. 3 suggested that different *CO coverages result in different surface obstructions for Cu electrodeposition, and literature data demonstrated different ECR selectivity on different facets,22,23 it can be concluded that different Cu facets are of great importance for the restructuring. The difference in CObinding energies at Cu (111) vs. (100) indicates that through dissolution–redeposition at ECR potential, (111) facets will start predominating over (100), which aligns with the observations by Liu et al.7 In the report a total *CO coverage also changed with time, suggesting the correctness of the explanation. However, different ECR parameters (potential, pH, electrolyte, etc.) could significantly influence the small energy gap between *CO bound at different facets and therefore the resulting formation of specific Cu sites.
Liu et al.7 also observed that during deactivation, CObridge began to prevail over COatop. Similarly, another study reported that deactivation was accompanied by the formation of CObridge and loss of COatop.43 Theoretical data indicated that it is thermodynamically more favourable for CO to adsorb on the surface as a bridge than atop at (111), while on (100) the energy difference between the two *CO forms is not as distinct (Fig. 4g). The observed formation of CObridge during operation could thus be related to the energy released as COatop gradually transitions to CObridge. Especially the report by Liu et al.,7 where (100) sites reformed to (111) and CObridge started prevailing over COatop, could be explained by the results from this study. It has been reported previously that if CObridge is the dominant *CO surface species on Cu, C–C coupling is hindered.43–45 Therefore, the gradual formation of CObridge during CA could be a key factor contributing to the deactivation, as Liu et al.7 have argued.
Data from the literature and this study suggested some trends in alterations during ECR, though with currently limited data a general behaviour remains elusive. Anisotropic morphological restructuring occurred forming smaller nanoparticles, however, it was shown that ECR selectivity changes could be beneficial with this process. The dominance of CObridge over COatop was linked to the deactivation of C–C coupling, though the total amount of *CO, crucial for ECR, also changed. The significant influence of different Cu surfaces on ECR selectivity is evident, yet reports vary as some indicate the formation of (111) facets during operation, while others show a reformation to (100). There are numerous interconnections between these features, and it is possible that additional factors influence ECR selectivity changes, e.g. the applied potential, electrolyte (type and its concentration), cell design (distances, volume), flows (gas and electrolyte), etc. The uncontrolled stability, restructuring, and deactivation of Cu catalysts during operation therefore result from a complex interplay of various parameters. To fully understand and mitigate the effects of the dissolution–redeposition mechanism during CA, further research is needed. This should involve studies on different Cu single crystals and nanoparticles, focusing on the dissolution of Cu, changes in morphology, and alterations in adsorbed CO bands, along with the impact on the catalyst's ability to reduce CO2. Additionally, DFT calculations could be beneficial to comprehend the effects.
Before concluding, one final comprehension considering pulsed techniques can be made based on the results from this study, which may help the researchers in this field. The effect of dissolution–redeposition yielded the highest selectivity for C2+ products at an electrodepositing potential of −0.975 V vs. RHE in a CO2-saturated electrolyte with minimal air exposure during OCP (Fig. S27†), therefore utilizing the dissolution through the slow process of oxidation–dissolution, rather than air exposure, formation of Cu-oxide shells, and rapid dissolution at OCP. Moreover, the dissolution–redeposition in Ar resulted in the formation of sites unfavourable for ECR.
Conclusions
This study delves into the mechanism of the observed dynamic restructuring of Cu-based catalysts at ECR conditions, highlighting a significant challenge in developing stable, industrially viable electrocatalysts. Given that stability must be enhanced by three orders of magnitude, understanding the changes occurring during operation is crucial. Alterations in Cu surface morphology and ECR selectivity were observed during CA, with the detection of dissolved Cu species suggesting the formation of [Cu+–ECR intermediates]XH2O complexes under these conditions. The ECR was found to compete with the electrodeposition of dissolved Cu, leading to atypical Cu nanoparticle growth. The dynamic equilibrium of the dissolution–redeposition mechanism at ECR potential facilitated Cu movement to the less active sites and catalyst deactivation. Alongside literature data, this study identified a complex interplay of four key parameters affecting ECR selectivity: nanoparticle size, the crystallographic orientation of facets, *CO surface coverage, and the CObridge/COatop ratio. Further investigations are needed to elucidate the important steps in FE alterations of different Cu catalysts, which usually experience changes at different rates. While the mechanism is somewhat clear, its impact on different catalysts remains uncertain. The observed restructuring phenomena for other metals indicate broader implications of this knowledge beyond Cu.9,36,58,59We anticipate that our findings and methodologies will inform future endeavours aimed at designing stable Cu catalysts capable of effectively closing humanity's CO2 cycle. In conclusion, we echo the sentiment encapsulated in the following quote summarising this work: “Copper's most powerful tool for fighting global warming is also its greatest weakness.”
Data availability
All other data related to the research content of this paper are provided in the ESI.† The authors have cited additional references within the ESI.†60–82Author contributions
Blaž Tomc: conceptualisation, data curation, investigation, methodology, visualisation, writing – original draft. Marjan Bele: conceptualisation, investigation, methodology, supervision. Mohammed Azeezulla Nazrulla: conceptualisation, investigation, methodology. Primož Šket, Angelja Kjara Surca, Ana Rebeka Kamšek, Martin Šala, Jan Šiler Hudoklin, Matej Huš: data curation, investigation, methodology. Matjaž Finšgar: data curation, investigation, methodology, writing – review & editing, funding acquisition. Blaž Likozar: supervision, funding acquisition. Nejc Hodnik: conceptualisation, funding acquisition, project administration, supervision, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Slovenian Research and Innovation Agency (ARIS) programs P2-0393, P2-0118, P1-0242, P1-0034, P2-0152, I0-0039 and I0-0003; the projects N2-0155, N2-0337, J1-4401, J2-4424, J2-50076 and J7-4638. M. A. N. acknowledges the funding for Marie Skłodowska-Curie Actions, Individual Fellowships, project CO2-CAT-ALOG (grant reference no. 897866) from the European Commission for Horizon 2020 Framework Programme. The project is co-financed by the Republic of Slovenia, the Ministry of Higher Education, Science and Innovation, and the European Union under the European Regional Development Fund. The authors also acknowledge partial support from the Republic of Slovenia, Ministry of Higher Education, Science and Innovation, and from the European Union – NextGenerationEU in the framework of the project HyBReED, that is part of the Slovenian Recovery and Resilience Plan. Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the Republic of Slovenia, Ministry of Higher Education, the European Union or the European Commission. Neither the Republic of Slovenia, Ministry of Higher Education, Science and Innovation, European Union nor the European Commission can be held responsible for them. A. R. K. would like to acknowledge the support of the Milan Lenarčič Foundation and the Janko Jamnik Doctoral Scholarship. Computational resources of the HPC Vega system in Maribor, Slovenia within the HPC RIVR consortium and EuroHPC JU are gratefully acknowledged. The authors acknowledge the efforts of Jaka Birsa in the preparation of the table of contents graphics.References
- Scripps Institution of Oceanography at UC San Diego, The Keeling Curve, https://keelingcurve.ucsd.edu/, accessed 15 April 2024 Search PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- M. W. Schreiber, Curr. Opin. Electrochem., 2024, 44, 101438 CrossRef CAS.
- Y. Yao, T. Shi, W. Chen, J. Wu, Y. Fan, Y. Liu, L. Cao and Z. Chen, Nat. Commun., 2024, 15, 1257 CrossRef CAS PubMed.
- H. Wu, L. Huang, J. Timoshenko, K. Qi, W. Wang, J. Liu, Y. Zhang, S. Yang, E. Petit, V. Flaud, J. Li, C. Salameh, P. Miele, L. Lajaunie, B. Roldán Cuenya, D. Rao and D. Voiry, Nat. Energy, 2024, 9, 422–433 CrossRef CAS.
- Q. Liu, Q. Jiang, L. Li and W. Yang, J. Am. Chem. Soc., 2024, 146, 4242–4251 CrossRef CAS PubMed.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS PubMed.
- Q. Zhang, Z. Song, X. Sun, Y. Liu, J. Wan, S. B. Betzler, Q. Zheng, J. Shangguan, K. C. Bustillo, P. Ercius, P. Narang, Y. Huang and H. Zheng, Nature, 2024, 630, 643–647 CrossRef CAS PubMed.
- W. T. Osowiecki, J. J. Nussbaum, G. A. Kamat, G. Katsoukis, M. Ledendecker, H. Frei, A. T. Bell and A. P. Alivisatos, ACS Appl. Energy Mater., 2019, 2, 7744–7749 CrossRef CAS.
- Y. G. Kim, J. H. Baricuatro, A. Javier, J. M. Gregoire and M. P. Soriaga, Langmuir, 2014, 30, 15053–15056 CrossRef CAS PubMed.
- T. H. Phan, K. Banjac, F. P. Cometto, F. Dattila, R. García-Muelas, S. J. Raaijman, C. Ye, M. T. M. Koper, N. López and M. Lingenfelder, Nano Lett., 2021, 21, 2059–2065 CrossRef CAS PubMed.
- P. Grosse, A. Yoon, C. Rettenmaier, A. Herzog, S. W. Chee and B. Roldan Cuenya, Nat. Commun., 2021, 12, 6736 CrossRef CAS PubMed.
- P. Grosse, D. Gao, F. Scholten, I. Sinev, H. Mistry and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2018, 57, 6192–6197 CrossRef CAS PubMed.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- S. Popović, M. Smiljanić, P. Jovanovič, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef PubMed.
- S. Popović, M. Bele and N. Hodnik, ChemElectroChem, 2021, 8, 2634–2639 CrossRef.
- J. Vavra, T. H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., Int. Ed., 2021, 60, 1347–1354 CrossRef CAS PubMed.
- F. D. Speck and S. Cherevko, Electrochem. Commun., 2020, 115, 106739 CrossRef CAS.
- S. J. Raaijman, N. Arulmozhi and M. T. M. Koper, ACS Appl. Mater. Interfaces, 2021, 13, 48730–48744 CrossRef CAS PubMed.
- C. Hahn, T. Hatsukade, Y. G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS PubMed.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- J. Vavra, G. P. L. Ramona, F. Dattila, A. Kormányos, T. Priamushko, P. P. Albertini, A. Loiudice, S. Cherevko, N. Lopéz and R. Buonsanti, Nat. Catal., 2024, 7, 89–97 CrossRef CAS.
- G. H. Simon, C. S. Kley and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2021, 60, 2561–2568 CrossRef CAS PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P. C. Chen, H. Wang, C. J. Pollock, X. Huang, Y. T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C. T. DInh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- S. H. Lee, J. C. Lin, M. Farmand, A. T. Landers, J. T. Feaster, J. E. Avilés Acosta, J. W. Beeman, Y. Ye, J. Yano, A. Mehta, R. C. Davis, T. F. Jaramillo, C. Hahn and W. S. Drisdell, J. Am. Chem. Soc., 2021, 143, 588–592 CrossRef CAS PubMed.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- B. Eren, D. Zherebetskyy, L. L. Patera, C. H. Wu, H. Bluhm, C. Africh, L.-W. Wang, G. A. Somorjai and M. Salmeron, Science, 2016, 351, 475–478 CrossRef CAS PubMed.
- B. Eren, D. Zherebetskyy, Y. Hao, L. L. Patera, L. W. Wang, G. A. Somorjai and M. Salmeron, Surf. Sci., 2016, 651, 210–214 CrossRef CAS.
- P. P. Albertini, M. A. Newton, M. Wang, O. Segura Lecina, P. B. Green, D. C. Stoian, E. Oveisi, A. Loiudice and R. Buonsanti, Nat. Mater., 2024, 23, 680–687 CrossRef CAS PubMed.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS.
- Z. Q. Liang, T. T. Zhuang, A. Seifitokaldani, J. Li, C. W. Huang, C. S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P. L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S. C. Lo, L. J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
- B. Cheng, J. Du, H. Yuan, Y. Tao, Y. Chen, J. Lei and Z. Han, ACS Appl. Mater. Interfaces, 2022, 14, 27823–27832 CrossRef CAS PubMed.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS PubMed.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- A. Engelbrecht, C. Uhlig, O. Stark, M. Hämmerle, G. Schmid, E. Magori, K. Wiesner-Fleischer, M. Fleischer and R. Moos, J. Electrochem. Soc., 2018, 165, J3059–J3068 CrossRef CAS.
- J. Kok, J. de Ruiter, W. van der Stam and T. Burdyny, J. Am. Chem. Soc., 2024, 146, 19509–19520 CrossRef CAS PubMed.
- T. C. Chou, C. C. Chang, H. L. Yu, W. Y. Yu, C. L. Dong, J. J. Velasco-Vélez, C. H. Chuang, L. C. Chen, J. F. Lee, J. M. Chen and H. L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed.
- C. M. Gunathunge, V. J. Ovalle, Y. Li, M. J. Janik and M. M. Waegele, ACS Catal., 2018, 8, 7507–7516 CrossRef CAS.
- X. Chang, S. Vijay, Y. Zhao, N. J. Oliveira, K. Chan and B. Xu, Nat. Commun., 2022, 13, 2656 CrossRef CAS PubMed.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- A. Herzog, M. Lopez Luna, H. S. Jeon, C. Rettenmaier, P. Grosse, A. Bergmann and B. Roldan Cuenya, Nat. Commun., 2024, 15, 3986 CrossRef CAS PubMed.
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS PubMed.
- S. Popović, M. A. Nazrulla, P. Šket, K. M. Kamal, B. Likozar, L. Suhadolnik, L. Pavko, A. K. Surca, M. Bele and N. Hodnik, Electrochim. Acta, 2022, 436, 141458 CrossRef.
- J. J. Velasco-Velez, R. V. Mom, L. E. Sandoval-Diaz, L. J. Falling, C. H. Chuang, D. Gao, T. E. Jones, Q. Zhu, R. Arrigo, B. Roldan Cuenya, A. Knop-Gericke, T. Lunkenbein and R. Schlögl, ACS Energy Lett., 2020, 5, 2106–2111 CrossRef CAS PubMed.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 33, 9307–9313 CrossRef CAS PubMed.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and B. Roldan Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- M. Gatalo, M. Bele, F. Ruiz-Zepeda, E. Šest, M. Šala, A. R. Kamšek, N. Maselj, T. Galun, P. Jovanovič, N. Hodnik and M. Gaberšček, Angew. Chem., 2019, 131, 13400–13404 CrossRef.
- J. K. Young, N. A. Lewinski, R. J. Langsner, L. C. Kennedy, A. Satyanarayan, V. Nammalvar, A. Y. Lin and R. A. Drezek, Nano Express, 2011, 6, 428 Search PubMed.
- G. Kastlunger, P. Lindgren and A. A. Peterson, J. Phys. Chem. C, 2018, 122, 12771–12781 CrossRef CAS.
- X. Chang, H. Xiong, Q. Lu and B. Xu, JACS Au, 2023, 3, 2948–2963 CrossRef CAS PubMed.
- F. Li, X. V. Medvedeva, J. J. Medvedev, E. Khairullina, H. Engelhardt, S. Chandrasekar, Y. Guo, J. Jin, A. Lee, H. Thérien-Aubin, A. Ahmed, Y. Pang and A. Klinkova, Nat. Catal., 2021, 4, 479–487 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- N. Sreekanth and K. L. Phani, Chem. Commun., 2014, 50, 11143–11146 RSC.
- C. Schneider, W. Rasband and K. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- W. Kohn, A. D. Becke and R. G. Parr, J. Phys. Chem., 1996, 100, 12974–12980 CrossRef CAS.
- J. J. Mortensen, A. H. Larsen, M. Kuisma, A. V. Ivanov, A. Taghizadeh, A. Peterson, A. Haldar, A. O. Dohn, C. Schäfer, E. Ö. Jónsson, E. D. Hermes, F. A. Nilsson, G. Kastlunger, G. Levi, H. Jónsson, H. Häkkinen, J. Fojt, J. Kangsabanik, J. Sødequist, J. Lehtomäki, J. Heske, J. Enkovaara, K. T. Winther, M. Dulak, M. M. Melander, M. Ovesen, M. Louhivuori, M. Walter, M. Gjerding, O. Lopez-Acevedo, P. Erhart, R. Warmbier, R. Würdemann, S. Kaappa, S. Latini, T. M. Boland, T. Bligaard, T. Skovhus, T. Susi, T. Maxson, T. Rossi, X. Chen, Y. L. A. Schmerwitz, J. Schiøtz, T. Olsen, K. W. Jacobsen and K. S. Thygesen, J. Chem. Phys., 2024, 160, 092503 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3858 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Hjorth Larsen, J. JØrgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. SchiØtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.:Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- J. Carrasco, A. Hodgson and A. Michaelides, Nat. Mater., 2012, 11, 667–674 CrossRef CAS PubMed.
- Y. Li, F. Cui, M. B. Ross, D. Kim, Y. Sun and P. Yang, Nano Lett., 2017, 17, 1312–1317 CrossRef CAS PubMed.
- N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061–16064 RSC.
- S. Liu, H. Yang, X. Su, J. Ding, Q. Mao, Y. Huang, T. Zhang and B. Liu, J. Energy Chem., 2019, 36, 95–105 CrossRef.
- R. D. Kent and P. J. Vikesland, Environ. Sci. Technol., 2016, 50, 6772–6781 CrossRef CAS PubMed.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- Y. Li, D. Kim, S. Louisia, C. Xie, Q. Kong, S. Yu, T. Lin, S. Aloni, S. C. Fakra and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9194–9201 CrossRef CAS PubMed.
- N. Hodnik, M. Zorko, M. Bele, S. Hočevar and M. Gaberšček, J. Phys. Chem. C, 2012, 116, 21326–21333 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, NACE International, CEBELCOR, Houston, Brussels, 1966 Search PubMed.
- N. Hodnik, M. Zorko, B. Jozinović, M. Bele, G. Dražič, S. Hočevar and M. Gaberšček, Electrochem. Commun., 2013, 30, 75–78 CrossRef CAS.
- M. Zorko, B. Jozinović, M. Bele, N. Hodnik and M. Gaberšček, Ultramicroscopy, 2014, 140, 44–50 CrossRef CAS PubMed.
- H. F. Goldstein, D.-S. Kim, P. Y. Yu, L. C. Bournet, J.-P. Chaminade and L. Nganga, Phys. Rev. B:Condens. Matter Mater. Phys., 1990, 41, 7192–7194 CrossRef CAS PubMed.
- X. K. Chen, J. C. Irwin and J. P. Franck, Phys. Rev. B:Condens. Matter Mater. Phys., 1995, 52, 130–133 Search PubMed.
- J. Chrzanowski and J. C. Irwin, Solid State Commun., 1989, 70, 11–14 CrossRef CAS.
- L. Debbichi, M. C. Marco De Lucas, J. F. Pierson and P. Krüger, J. Phys. Chem. C, 2012, 116, 10232–10237 CrossRef CAS.
- Y. Deng, A. D. Handoko, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473–2481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06466f |
| This journal is © The Royal Society of Chemistry 2025 |