
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzodithiophene-based polymer donors for organic photovoltaics
Meng Wei and
Dmytro F. Perepichka*
and
Dmytro F. Perepichka*
Department of Chemistry, McGill University, 845 Sherbrooke St W, Montreal, Quebec H3A 0G4, Canada. E-mail: dmytro.perepichka@mcgill.ca
First published on 17th March 2025
Abstract
The advancement of π-conjugated polymers has contributed to the rapid development of high-performing organic photovoltaic (OPV) devices. The donor (D)–acceptor (A) conjugated polymers dominated the field of OPV materials as these structures offer opportunities to fine-tune material properties such as bandgap, charge transport and solubility. Among all the D building blocks, benzodithiophene (BDT) has received extensive attention owing to its symmetric structure, convenient attachment points for side chains and high stability. This review summarizes the general molecular design strategy for BDT-based D–A copolymers and gives carefully selected examples of such copolymers used in NFA solar cells, aiming to provide a structure–property relationship for rational design of future BDT-based D–A conjugated polymers.
Introduction
Organic photovoltaic (OPV) technology is emerging as one of the promising pathways to transform solar energy into electricity. Compared to conventional solar cells based on silicon and other inorganic semiconductors, OPVs bring several advantages such as flexibility, light weight, low CO2 footprint of fabrication and the possibility of mass production through printing.The first demonstration in OPV diodes based on the planar (bilayer) heterojunction of copper phthalocyanine and a perylenediimide derivative was reported by Tang in 1986, achieving a power conversion efficiency (PCE) of ∼1%.1 The current generation in this device was limited by inefficient exciton dissociation away from the donor/acceptor interface (Fig. 1). The invention of the bulk-heterojunction (BHJ) OPV in 19952,3 addressed this problem by providing an active layer of interpenetrated p-/n-type domains by blending the conjugated poly(2-methoxy-5-(2′-ethylhexyloxy)-1,4-phenylenevinylene) (MEH-PPV, Fig. 2) donor with a derivatized fullerene (PC61BM) acceptor. Polymer-based OPVs have since dominated the field. Other homopolymers such as poly(3-hexylthiophene) (P3HT) were introduced in OPV devices and a PCE of 5% has been achieved with P3HT around 2007.4–6
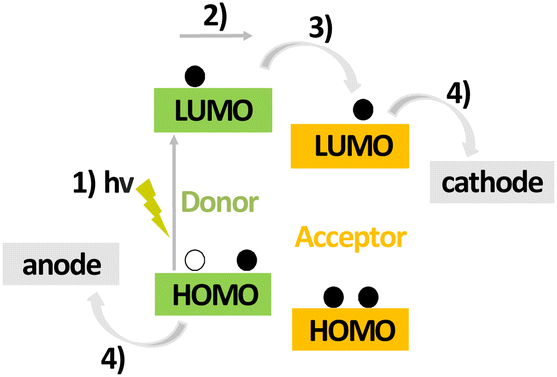 | ||
| Fig. 1 Operation of OPVs: (1) absorption of light and generation of an exciton; (2) diffusion of the exciton to the donor–acceptor interface; (3) exciton dissociation and charge separation at the interface; (4) free charge carrier transport and extraction on electrodes. | ||
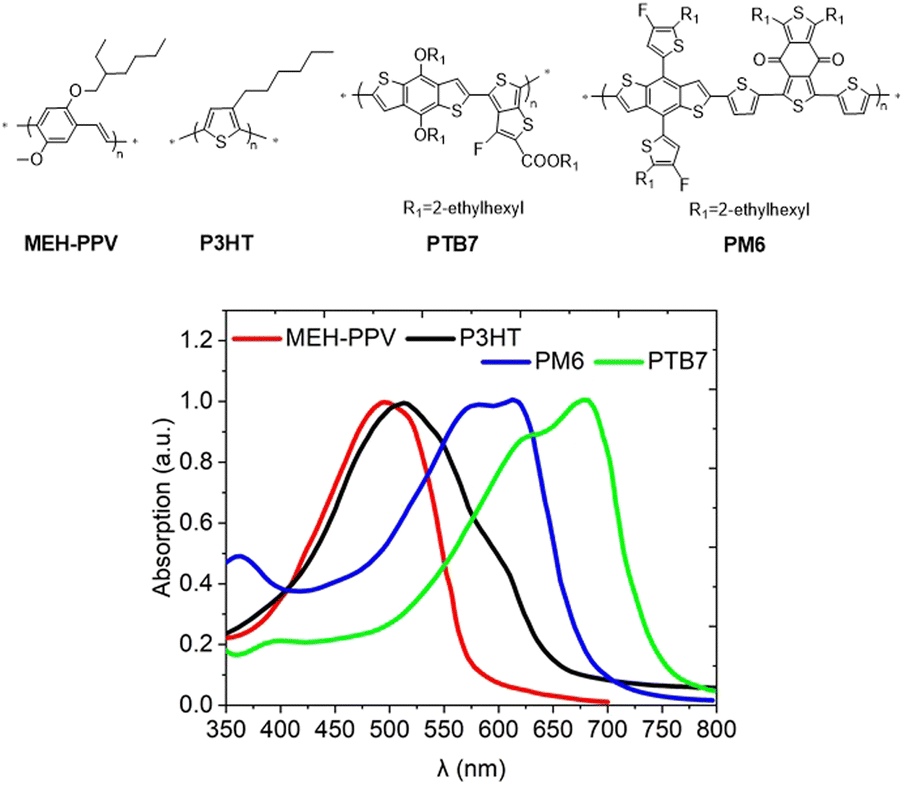 | ||
| Fig. 2 Solid state UV-vis absorption spectra of MEH-PPV, P3HT, PM6 and PTB7 polymers used in OPVs. | ||
The relatively large band-gap (Eg) of homopolymers (∼2.2 eV for MEH-PPV, ∼2.0 eV for regioregular P3HT) leaves unconverted a significant portion of solar radiation which peaks at around 700 nm (1.77 eV). To address this problem, low bandgap donor–acceptor (D–A) copolymers were developed and eventually became dominant in the OPV field. The orbital mixing in a chain of alternating electron-rich D and electron-deficient A moieties results in Eg contraction (Fig. 3) and redshift of the absorption band.7 Also, as established later, quadrupole interactions between the polarized chains of D–A copolymers lead to stronger interchain coupling increasing the charge mobility. The period between 2008 and 2015 witnessed a rapid development of low bandgap D–A copolymers such as PTB7 and OPV devices based on D–A polymer/fullerene blends reached a PCE of ∼12%. With the recent rise of non-fullerene acceptors (NFA), which can strongly absorb in the visible and NIR regions,8 the requirements for donor polymers have slightly changed, and nowadays they are engineered to present a relatively wide Eg with an absorption profile complementary to NFA molecules (e.g., PM6).
 | ||
| Fig. 3 Molecular orbital interactions of the donor and acceptor, resulting in a narrowing of the band gap in D–A copolymers. | ||
Various donor units, such as cyclopentadithiophene and carbazole, have been used as building blocks for conjugated polymers.9–11 Among these, benzo[1,2-b:4,5-b′]dithiophene (BDT) has proved to be particularly versatile and most of the highest efficiency OPVs nowadays employ BDT-containing polymers as donor materials. There are several reasons for this choice. BDT has a symmetric rigid planar structure with a 180° angle between the connection sites, resulting in a linear polymer structure with efficient stacking as well as excellent thermal and chemical stability. Solubilizing chains can be installed on the benzene ring of BDT without affecting the planarity of the conjugated backbone (unlike many other thiophene-based polymers including P3HT12).
BDT monomers were reported for the first time in 1971 (ref. 13) and incorporated in conjugated polymers in the 1990s.14 In 2008, D–A copolymers of BDT with various acceptors were used for the first time in OPVs.15
The development of D–A conjugated polymers for organic solar cells has been extensively reviewed in the past with various focuses, with BDT-containing copolymers also being covered.16–20 The most comprehensive review focused on BDT polymers was published in 2016,21–24 before the recent wave of research in OPVs with NFAs. Several recent reviews have discussed the design and application of BDT polymers in combination with NFAs in BHJ solar cells.25,26 Nevertheless, the continuous and rapid development of the field of OPVs creates a need for an up-to-date review putting the most recent contributions in the perspective of milestone discoveries of the past.
The present paper summarizes the most up-to-date literature to provide the current design principles and structure–property relationship studied in BDT-based D–A conjugated polymers in the context of their applications in OPVs. After a brief introduction to the synthesis of BDT monomers and polymerization approaches, we summarize the main design strategies in engineering their electronic properties, including main-chain engineering and side-chain engineering. We then review key examples of wide bandgap BDT-based polymer donors that are used in combination with NFAs in OPVs. Lastly, we provide an outlook for the remaining challenges and future perspectives in BDT-based polymer materials. Without the intention of comprehensive coverage of all literature on BDT-based polymers, we hope this paper will provide a fair and rounded overview of the recent advances in the field and opportunities for future development.
Synthesis of the BDT monomer and BDT-based polymers
BDT monomers are most commonly synthesized starting from 3-thiophenecarboxamide M1 through lithiation at the 2-position followed by intermolecular cyclization with another molecule of M1 (Scheme 1). The resulting diketone M2 can be reductively aromatized with the addition of solubilizing side groups on the central benzene ring. The alkoxy-substituted BDTs M3 are typically synthesized by first reducing the keto-groups in M2 to the diol intermediate, followed by alkylation with alkyl halide. Alkylthio-substituted BDTs M4 can be accessed by esterifying the diol with N,N-dimethylthiocarbamoyl chloride followed by rearrangement and cleavage of the resulting thiocarbamate, and alkylation of the formed dithiol intermediate.27,28 Reacting M2 with aryllithium reagent yields the corresponding diol intermediate which can be reductively aromatized using SnCl2 to afford aryl-substituted monomer M5. Replacing the aryllithium with alkynyllithium results in the alkynyl-flanked BDT M6. Its hydrogenation on Pd/C produces alkyl-substituted BDT M7,29 which can also be made directly by reacting M2 with alkyllithium followed by SnCl2 reduction.30 BDT flanked with asymmetric side chains M8 can also be synthesized by stepwise monoarylation, reduction and alkylation.31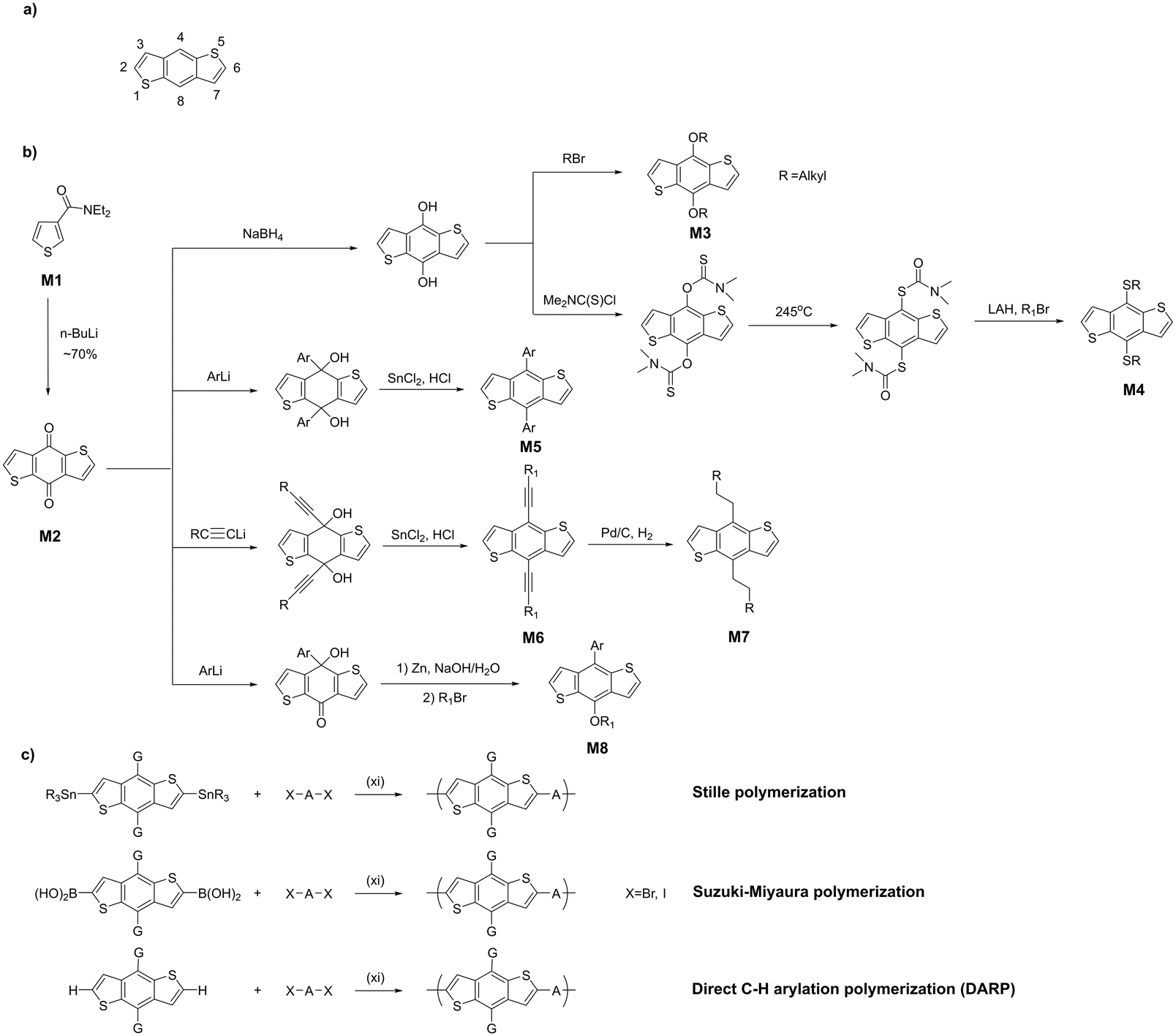 | ||
| Scheme 1 Numbering of BDT (a) and synthesis of (b) BDT monomers and (c) BDT-based polymers. | ||
The most common method for the synthesis of D–A copolymers is Pd-catalysed Stille coupling between distannylated and dibrominated monomers (Scheme 1c). Typically, trimethyl or tributyltin functionalized BDTs (prepared via a lithiation-stannylation sequence) are cross-coupled with dihalogenated acceptor units. In some cases, Stille polymerization of dibromo-BDT derivatives has also been reported.32 The versatility of Stille coupling in the synthesis of thiophene-based polymers makes it a method of choice most of the time. Indeed, it remains the most reliable method used in commercial production of benchmark BDT polymers such as PM6 (polymer 51) and D18 (polymer 75).
However, it also has multiple drawbacks, including the formation of stoichiometric amounts of toxic tin byproduct and the limited shelf-life of tinylated monomers explaining the interest in exploring alternative polymerization methods, especially for large-scale synthesis.
The application of more benign Suzuki–Miyaura in the thiophene series is often limited by the instability of the corresponding boronic acid monomer under reaction conditions.33 As a result, it is rarely employed in the synthesis of BDT polymers. One notable exception is the work of Xiao et al. who synthesized new BDT monomers and polymerized their dihalides with acceptors equipped with boronic acid groups.34 It is worth noting that in this case, 0.5 h reaction time was sufficient to produce a polymer with a molecular weight of nearly 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, indicating the efficacy of the Suzuki reaction.35
000 g mol−1, indicating the efficacy of the Suzuki reaction.35
Direct C–H arylation polymerization (DArP) is gaining increasing attention in the synthesis of D–A copolymers. This method generates mild byproducts (alkali metal halide) and eliminates the pre-functionalization (stannylation/borylation) step resulting in an overall significant cost reduction compared to the classical cross-coupling reactions.36 The early work on DArP of dibromo-BDT37 with various aromatic comonomers showed high (75–95%) yields of copolymers with similar polydispersities and molecular weights rivalling or exceeding those of reference copolymers synthesized via Stille coupling. Moreover, this study reveals comparable or superior photovoltaic properties versus the same polymers made by Stille coupling, for the first time reaching a PCE of 8% for a DArP-produced material. It should be noted that the presence of different aromatic C–H bonds in comonomers intrinsically limits the regioselectivity of DArP compared to Stille or Suzuki polymerization.37–39 In this context, BDT is well suited for DArP because the steric hindrance of the β-CH makes it significantly less reactive than the α-CH. However, an appropriate choice of arenedibromide comonomer and optimization of polymerization conditions is required to minimize the homocoupling defects in the resulting copolymer.40–42
Regardless of the polymerization methods, batch-to-batch variability43 is a major concern for commercial applications of BDT (and other) conjugated polymers in OPVs. This variability originates from several factors, including molecular weight and polydispersity (chain length distribution), side reactions during synthesis such as homocoupling, remaining traces of catalytic metal, etc.44 A few aspects need to be considered to minimize this problem for existing polymers, including type of catalyst and ligand, reaction setup, etc. For example, replacing the standard Pd(PPh3)4/Pd2(dba)3 with commercial Buchwald catalyst (P(tBu)3Pd G3) allows reducing homocoupling defects in PM6 below the detection level.45 Another promising approach to combat the batch-to-batch variability is flow synthesis which was recently used to produce PM6 with controlled molecular weights and reproducible device performance.45 Last but not least, the monomer purity and post-treatment (e.g. Soxhlet extraction) of the crude polymer play a critical role in reducing batch-to-batch variations.46
Molecular design strategy of BDT-based conjugated polymers for OPVs
One of the major properties of interest in OPV donors is their bandgaps. There are multiple factors that influence the bandgap of a conjugated polymer: bond-length alternation, aromaticity, planarity and the electronic effect of substituents.47 Another important feature is charge mobility, which depends on conjugation within the chain as well as the packing of polymer chains. Last but not least, the solubility and processability of a polymer in green solvents matter when the material is fabricated into the device, especially when it comes to the commercialization stage. All these properties can be fine-tuned via careful engineering of the polymer backbone (main chain) and substituents (side chains).Main-chain engineering
 | ||
| Fig. 4 Conjugated copolymers 1–8 based on BDT and varied acceptor units. | ||
| λmax (nm) | Eg (eV) | HOMO (eV) | LUMO (eV) | |
|---|---|---|---|---|
| 1 | 495 | 2.13 | −5.16 | −2.67 |
| 2 | 511 | 2.06 | −5.05 | −2.69 |
| 3 | 510 | 2.03 | −5.07 | −2.86 |
| 4 | 532 | 1.97 | −4.56 | −2.66 |
| 5 | 591 | 1.7 | −5.1 | −3.19 |
| 6 | 601 | 1.63 | −4.78 | −3.28 |
| 7 | 641 | 1.52 | −4.88 | −3.33 |
| 8 | 780 | 1.05 | −4.65 | −3.46 |
Since then, a number of acceptor units have been copolymerized with BDT to develop donor polymers for OPVs. Early examples include diketopyrrolopyrrole (DPP) (polymer 9)48 and thieno[3,4-b]thiophene (TT) carboxylic ester (polymer 10)49 moieties (Fig. 5). In 2009, a PCE of 5.3% was achieved with a 10:PC71BM blend, demonstrating the potential of D–A copolymers as alternatives to homopolymers for OPV applications (Table 2).
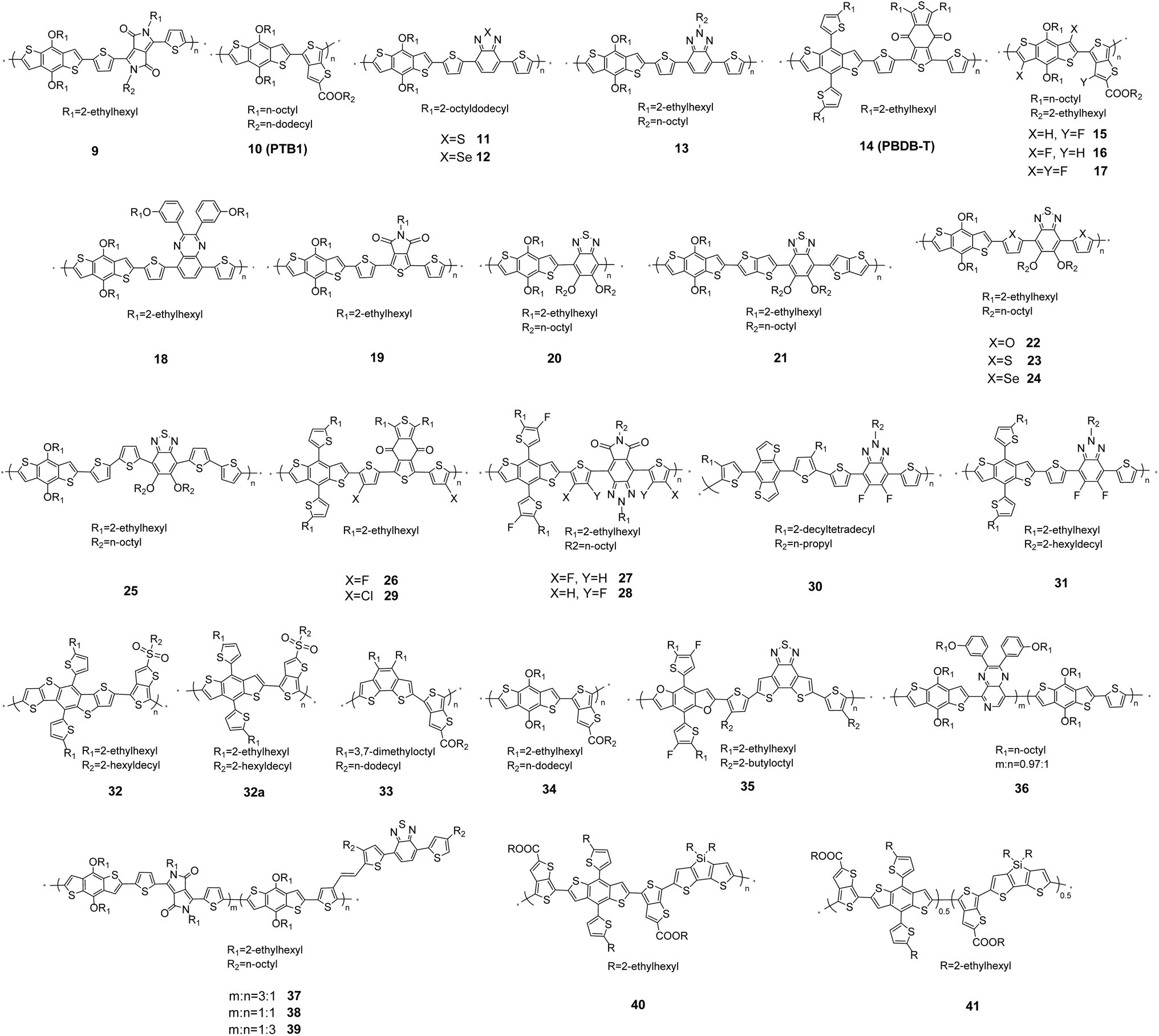 | ||
| Fig. 5 Main chain engineering in D–A copolymers 9–41. | ||
| Polymer properties | OPV device | Ref. | Year of report | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λmaxa (nm) | Eoptg a (eV) | HOMOb (eV) | LUMOb (eV) | wt/wt (acceptor) | VOC (V) | JSC (mA cm−2) | FF (%) | PCE (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a In thin films, bandgap derived from absorption onset.b Measured with cyclic voltammetry. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 750 | 1.31 | −5.16 | −3.51 | 1:1 (PC71BM) |
0.68 | 8.4 | 44.3 | 2.5 | 48 | 2009 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 690 | 1.62 | −4.9 | −3.2 | 1:2 (PC71BM) |
0.56 | 15 | 63.3 | 5.3 | 49 | 2009 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 646 | 1.7 | −5.26 | — | 1:1 (PC61BM) |
0.76 | 8 | 64.4 | 3.9 | 50 | 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 680 | 1.55 | −5.18 | −3.48 | 1:1 (PC71BM) |
0.6 | 13.6 | 64 | 5.2 | 51 | 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 527 | 1.95 | −5.06 | −3.11 | 1:3 (PC61BM) |
0.61 | 4.4 | 55 | 1.5 | 52 | 2010 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 622 | 2.05(EECg) | −5.23 | −3.18 | 1:1 (PC61BM) |
0.86 | 10.7 | 72.3 | 6.7 | 54 | 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 671 | 1.68 | −5.15 | −3.31 | 1:1.5 (PC71BM) |
0.74 | 14.1 | 68.9 | 7.2 | 56 | 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | 670 | 1.75 | −5.41 | −3.6 | 1:1.5 (PC71BM) |
0.68 | 11.1 | 42.2 | 3.2 | 56 | 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | 670 | 1.73 | −5.48 | −3.59 | 1:1.5 (PC71BM) |
0.75 | 9.1 | 39.4 | 2.7 | 56 | 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | 602 | 1.74 | −5.12 | −3.38 | 1:2 (PC71BM) |
0.71 | 7 | 61.5 | 3.1 | 57 | 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19 | 516 | 1.84 | −5.49 | −3.7 | 1:2 (PC61BM) |
0.76 | 2.9 | 43 | 1.0 | 58 | 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20 | 620 | 1.69 | −5.17 | −3.49 | 1:2 (PC71BM) |
0.66 | 1.8 | 41.8 | 0.5 | 60 | 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 21 | 631 | 1.78 | −5.21 | −3.54 | 1:1.5 (PC71BM) |
0.69 | 11.3 | 63 | 4.9 | 59 | 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 22 | 534 | 1.96 | −5.44 | −3.48 | 1:2 (PC71BM) |
0.94 | 6.5 | 46 | 2.8 | 59 | 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 23 | 568 | 1.82 | −5.35 | −3.44 | 1:1.5 (PC71BM) |
0.82 | 9.5 | 48 | 3.7 | 59 | 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 24 | 644 | 1.71 | −5.29 | −3.33 | 1:2 (PC61BM) |
0.65 | 8.6 | 54.5 | 3.1 | 61 | 2012 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 25 | 575 | 1.8 | −5.37 | −3.01 | 1:2 (PC71BM) |
0.66 | 13.3 | 58.1 | 5.1 | 62 | 2015 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 26 | 644 | — | −5.47 | −3.46 | 1:1.25 (C8-ITIC) |
0.94 | 19.6 | 72.0 | 13.2 | 63 | 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 27 | 642 | 1.72 | −5.55 | −3.83 | 1:1 (Y6) |
0.85 | 26.3 | 75.5 | 16.8 | 64 | 2020 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 28 | 505 | 2.09 | −5.67 | −3.58 | 1:1 (Y6) |
0.80 | 4.9 | 36.5 | 1.4 | 64 | 2020 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 29 | 647 | 1.78 | −5.48 | −3.47 | 1.2:1 (IT-4F) |
0.84 | 20.6 | 71.1 | 12.3 | 65 | 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 30 | — | 2.05 | −5.47 | −3.42 | 1:1.5 (o-IDTBR) |
1.08 | 16.3 | 63.6 | 11.6 | 66 | 2017 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 31 | 538 | 1.96 | −5.21 | −2.99 | 1:1 (ITIC) |
0.73 | 13.1 | 57.8 | 5.5 | 67 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 32 | 778 | 1.59 | −5.21 | −1.08 | 1:1.5 (PC71BM) |
0.73 | 16.6 | 64.1 | 7.8 | 68 | 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 32a | 768 | 1.61 | −5.29 | −1.00 | 1:1.5 (PC71BM) |
0.81 | 11.8 | 52.1 | 5.0 | 68 | 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 33 | 662 | 1.55 | −5.35 | −3.80 | 1:2 (PC71BM) |
0.82 | 11.8 | 54.1 | 5.2 | 70 | 2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 34 | — | 1.61 | −5.12 | −3.55 | 1:1.5 (PC71BM) |
0.70 | 14.7 | 64 | 6.6 | 71 | 2009 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 35 | 588 | 1.97 | −5.48 | −3.42 | 1:1.2:0.2 (Y6-1O:PC71BM) |
0.90 | 23.62 | 80.4 | 17.1 | 72 | 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 36 | 634 | 1.6 | −5.2 | −3.28 | 1:4 (PC71BM) |
0.71 | 6.4 | 51.7 | 2.4 | 74 | 2010 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 37 | 746 | 1.43 | −5.29 | −3.7 | 1:2 (PC61BM) |
0.72 | 6.2 | 49.0 | 2.2 | 75 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 38 | 734 | 1.47 | −5.36 | −3.69 | 1:2 (PC61BM) |
0.74 | 12.2 | 46.0 | 4.2 | 75 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 39 | 723 | 1.52 | −5.47 | −3.67 | 1:2 (PC61BM) |
0.78 | 14.1 | 48.0 | 5.3 | 75 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 40 | 691 | 1.45 | −5.19 | −3.62 | 1:1.5 (PC71BM) |
0.64 | 14.4 | 61.8 | 5.6 | 76 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 41 | 649 | 1.28 | −5.08 | −3.62 | 1:1.5 (PC71BM) |
0.50 | 5.6 | 39.2 | 1.1 | 76 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A strong electron-withdrawing benzo[c][1,2,5]thiadiazole (BT) unit connected via a thiophene bridge was incorporated in copolymer 11.50 This combination results in a rather low HOMO of −5.26 eV (cf. −5.16 eV for DPP copolymer 9), contributing to a relatively high open-circuit voltage (VOC) of 0.76 V for the 11:PC61BM solar cell. Replacing benzothiadiazole with a benzoselenadiazole acceptor presents a further reduction of the bandgap from 1.70 for 11 to 1.55 eV for 12 and a PCE of 5.2% was obtained for 12:PC71BM blends.51
Conversely, replacing sulfur in the BT unit with electron-donating nitrogen results in a weaker benzo[d][1,2,3]triazole (BTz) acceptor, which is beneficial for developing wider bandgap polymers. The additional alkyl chains on the nitrogen atom provide an additional handle to tune the solubility and packing of BTz-containing polymers. However, polymer 13 bearing a BTz acceptor displays a wider bandgap (1.95 eV) with blue-shifted absorption by over 100 nm compared to its BT counterpart 11.52 Furthermore, the high-lying HOMO of −5.06 eV of 13 results in a lower VOC of 0.61 V and a PCE of only 1.5% for the BHJ with PC61BM.
Another acceptor that gained significant attention is benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD).20 The first polymer with alternating alkoxy-substituted BDT and BDD was reported in 2012, but the PCE of the corresponding devices was low (0.7% with PC71BM).53 A few months later, a similar polymer 14 (commonly known as PBDB-T) consisting of an alkylthienyl-substituted BDT unit and BDD with thiophene as π-linkers was reported.54 This polymer has a HOMO of −5.23 eV and its device with PC61BM delivered a relatively high VOC of 0.86 V with a PCE of 6.7%. Notably, 14 displays a temperature-dependent aggregation behavior in solution. This behavior enables the control of polymer pre-aggregation before casting the films, leading to a favorable morphology and improving the device performance.55
Fluorination is an extensively studied strategy for tuning the properties of conjugated polymers. Due to the high electronegativity and small size, fluorine effectively lowers the HOMO and LUMO energies of the material without inflicting steric hindrance and twisting the polymer structure. Furthermore, it can planarize/rigidify the polymer backbone through intramolecular F⋯H and F⋯S interactions. The effect of the number and position of fluorine substituents has been explored in the series of polymers 15–17.56 Increasing the number of fluorine atoms results in progressive stabilization of its frontier orbitals, by up to 0.5 eV, as well as an increased planarity of the backbone (due to intramolecular S⋯F interactions). However, the fluorination on BDT decreases its electron-donating ability, resulting in a wider bandgap of polymer 16 (1.75 eV) compared to 15 with a fluorinated acceptor (1.68 eV). The devices with 15 and PC71BM yielded a PCE of up to 7.2%, a significant improvement vs. non-fluorinated analog 10 (5.3%). On the other hand, polymers 16 and 17, which use fluorinated BDT units, showed poor device performance (PCE 3.2% and 2.7%) due to excessive phase separation of the highly fluorinated donor and PC71BM acceptor.
Other prominent examples include BDT copolymers with quinoxaline (18,57) and thiophenedicarboximide (19,58) acceptor units.
Changing the π-linker from furan (22) to thiophene (23) to thienothiophene (21) increases the angle between the two bonds connecting the π-bridge with the other moieties (127°, 152°, and 180°, respectively) and leads to a better packing of the more linear chains of 21.59 21 has redshifted film absorption and reduced bandgap compared to 22 and 23. It is also worth noting that 21–23 all showed better planarity and higher hole mobility than polymer 20 without such linkers. Changing heteroatoms in the π-linker from oxygen to sulfur to selenium results in redshifted absorption (lower bandgap) and increased HOMO energies (22 vs. 23 vs. 24).59,61 The relatively low HOMO level of 22 resulted in a high VOC of 0.94 V in OPV with PC71BM. Fusing the π-linker enhances the backbone planarity which is manifested in a ∼60 nm red shift of thienothiophene polymer 21 compared to bithiophene polymer 25.62
Fluorinating the thiophene π-linker in 26 (ref. 63) stabilizes the HOMO levels of 26 by ∼0.25 eV compared to 14 which leads to a high VOC of 0.94 V and PCE of 13.2% (in the BHJ with NFA C8-ITIC, Fig. 6). It is worth noting that the position of the fluorine on the π-linker can affect the molecular conformation, aggregation morphology, and the optoelectronic properties of the polymer. 27 with fluorine proximal to the BDT unit exhibits an almost flat backbone, while its isomer 28 has a 42° twist between the acceptor and π-bridge.64 This difference leads to a ∼100 nm redshift of absorption and a narrower bandgap of 0.37 eV in 27 which is responsible for its high PCE of 16.8% when used with acceptor Y6 (Fig. 6).
 | ||
| Fig. 6 Non-fullerene acceptors (NFAs) discussed in this paper. | ||
The chlorinated thiophene π-linker in 29 affords similar energy levels and band-gap to those of fluorinated analog 26, indicating the backbone planarity was not disturbed by the larger chlorine atoms.65 It should be noted, though, that the position of chlorine on the π-linker can significantly influence the optoelectronic properties and photovoltaic performance of the polymer.65
Two extra thiophenes are fused on BDT to give dithieno[2,3-d:2′,3′-d′]benzo[1,2-b:4,5-b′]dithiophene. Polymers with this repeat unit feature a more linear structure than their BDT counterparts,68,69 which is beneficial for ordered interchain stacking. As a result, OPV devices with 32 and PC71BM delivered a PCE of 7.8% (cf. 5.0% for BDT counterpart 32a). On the other hand, the strong aggregation of this highly fused polymer requires longer alkyl chains to maintain the solubility69 and the synthesis of the monomer is more complex than BDT, increasing the cost of the material.
Polymer 33 explored the effect of BDT isomerism, using benzo[2,1-b:3,4-b′]dithiophene building block, with two thiophene rings facing the same direction.70 Unlike common BDT with a 180° angle between connection sites, this BDT isomer features a 129° angle connectivity, resulting in a non-linear backbone and reducing the interchain stacking between polymer chains. This difference could be the reason for 33 presenting poorer photovoltaic performance (PCE 5.2%) than the BDT analog 34 (6.6%),71 despite the higher VOC of 0.82 V vs. 0.70 V in the former devices.
A light-element analog of BDT, benzodifuran (BDF), was used to build polymer 35.72 The smaller size of oxygen results in a more planar backbone and closer π–π stacking in 35 (3.57 Å) compared to BDT-bearing analog D18 (3.65 Å, polymer 75 below). Recent comparative studies of ternary devices (with PC71BM and NFA Y6-1O, Fig. 6) showed a higher PCE (>17%) for 35 compared to D18, supporting the potential of the BDF building block in future development of OPV materials.
 | ||
| Fig. 7 Absorption spectra of terpolymers 37–39 as thin films. Copyright 2014 (RSC).75 | ||
One of the drawbacks of the random copolymerization approach to terpolymers is the lack of control over the polymer sequences of the three moieties, which is unfavorable for the crystallinity of the polymer domains and may cause significant batch-to-batch variability. In principle, these problems can be mitigated by developing regioregular terpolymers, albeit at the cost of much higher synthetic complexity. This idea has been probed by Lee and coworkers who compared regioregular 40 with its regiorandom analog 41. The former presents a redshifted absorption with a 0.17 eV smaller bandgap and 40 times higher hole mobility than the latter. As a result, a significantly improved PCE of 5.6% was obtained for 40:PC71BM blends (cf. 1.1% for 41).76
Side-chain engineering
 | ||
| Fig. 8 Side chain engineering in D–A copolymers 42–64. | ||
| Polymer properties | OPV device | Ref. | Year of report | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λmaxa (nm) | Ega (eV) | HOMOb (eV) | LUMOb (eV) | wt/wt (acceptor) | VOC (V) | JSC (mA cm−2) | FF (%) | PCE (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a In thin films, bandgap derived from absorption onset.b measured with cyclic voltammetry. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 42 | 627 | 1.73 | −5.4 | — | 1:1.5 (PC61BM) |
0.85 | 12.5 | 65 | 7.3 | 80 | 2010 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 43 | — | — | — | — | 1:1.5 (PC71BM) |
0.93 | 8.3 | 53 | 3.8 | 80 | 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 44 | 596 | 1.75 | −5.31 | −3.44 | 1:2 (PC71BM) |
0.92 | 10.7 | 57.5 | 5.7 | 84 | 2010 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 45 | — | 1.58 | −5.22 | −3.64 | 1:1.5 (PC71BM) |
0.8 | 15.7 | 74.3 | 9.4 | 85 | 2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 46 | 618 | 1.65 | −5.64 | −3.99 | 1:1.5 (PC71BM) |
0.9 | 4 | 46.6 | 1.7 | 86 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 47 | 720 | 1.55 | −5.19 | −3.64 | 1:1.5 (PC71BM) |
0.69 | 11.8 | 64.8 | 5.3 | 85 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 48 | 699 | 1.58 | −5.29 | −3.71 | 1:1.5 (PC71BM) |
0.81 | 16.6 | 65.6 | 8.8 | 85 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 49 | 619 | 1.82 | −5.43 | −3.48 | 1:1.2 (BTP-eC9) |
0.83 | 26.4 | 68.6 | 15.0 | 88 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 50 | 636 | 1.77 | −5.38 | −3.53 | 1:1.2 (BTP-eC9) |
0.83 | 19.5 | 39.4 | 6.4 | 88 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 51 | 570 | 1.8 | −5.45 | −3.65 | 1:1.2 (Y6) |
0.83 | 25.3 | 74.8 | 15.6 | 89 | 2019 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 52 | — | −5.51 | — | 1:1 (IT-4F) |
0.86 | 21.8 | 77 | 14.4 | 91 | 2018 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 53 | 592 | 1.90 | −5.58 | −3.54 | 1:1.4 (Y6) |
0.82 | 26.6 | 72.5 | 15.8 | 92 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 54 | 675 | 1.68 | −5.10 (UPS) | −3.42 | 1:1.5 (PC71BM) |
0.80 | 17.4 | 67.5 | 9.4 | 31 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 55 | 726 | 1.53 | −5.18 | −3.15 | 1:1.5 (PC71BM) |
0.73 | 15.2 | 64.4 | 7.1 | 93 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 56 | 716 | 1.57 | −5.41 | −3.27 | 1:1.5 (PC71BM) |
0.84 | 15.3 | 65.5 | 8.4 | 93 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 57 | 685 | 1.63 | −5.5 | — | 1:1.5 (PC71BM) |
0.98 | 10.2 | 45 | 4.5 | 94 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 58 | 587 | 1.96 | −5.4 | −3.24 | 1:1 (ITIC) |
0.94 | 17.3 | 70.0 | 11.4 | 95 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 31 | 538 | 1.96 | −5.21 | −2.99 | 1:1 (ITIC) |
0.73 | 13.1 | 57.8 | 5.5 | 67 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 59 | 598 | 1.93 | −5.32 | −3.08 | 1:1 (ITIC) |
0.91 | 16.3 | 60.4 | 9.0 | 96 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 60 | 580 | 1.99 | −5.37 | −2.91 | 1:1.5 (m-ITIC) |
0.92 | 18.1 | 69.8 | 11.6 | 96 | 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 58a | 578 | 1.98 | −5.42 | −3.29 | 1:1.5 (m-ITIC) |
0.96 | 16.4 | 65.0 | 10.2 | 96 | 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 61 | 576 | 1.98 | −5.46 | −2.92 | 1:1.5 (m-ITIC) |
0.97 | 16.5 | 66.9 | 10.7 | 96 | 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 62 | 526 | 1.99 | −5.56 | −3.06 | 1:1.5 (m-ITIC) |
0.99 | 15.9 | 61.2 | 9.6 | 96 | 2018 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 63 | 678 | 1.56 | −5.15 | −3.69 | 1:1.4 (PC61BM) |
0.90 | 14.2 | 61.1 | 7.4 | 97 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 64 | 668 | 1.58 | −5.20 | −3.62 | 1:1.4 (PC61BM) |
0.90 | 10.8 | 60.8 | 5.7 | 97 | 2014 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Fréchet and co-workers found that the size and shape of side chains on BDT can influence the polymer backbone orientation relative to the substrate by comparing 42 bearing branched 2-ethylhexyl chains to 43 with n-tetradecyl chains.80 Grazing-incidence X-ray scattering (GIXS) data shows that the π-stacking (q = 1.76 Å−1; d-spacing ∼3.6 Å) is preferentially aligned in the out-of-plane direction in 42, indicating the preferential “face-on” orientation, but is isotropically distributed in 43 (Fig. 9). Accordingly, 42:PC71BM solar cells displayed an increased JSC and fill factor (FF) leading to almost doubled PCE (7.3%) compared to 43 (3.8%).
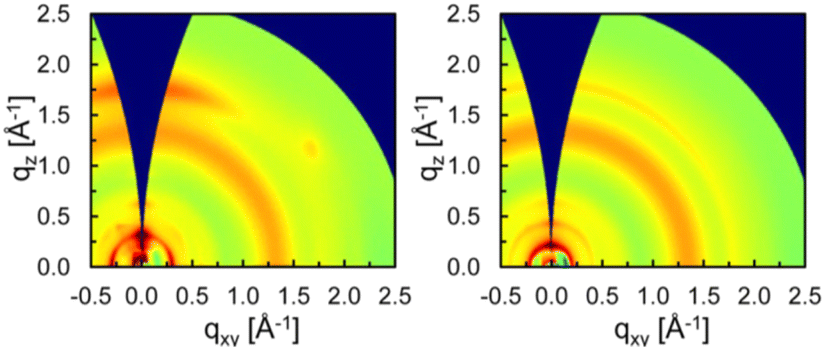 | ||
| Fig. 9 GIXS patterns of (left) 42 and (right) 43 in optimized BHJs with PC71BM. Copyright 2023 (RSC).80 | ||
A more versatile way to extend the side-chain conjugation in BDT polymers is based on aromatic and heteroaromatic substituents. The first D–A alternating copolymer with thienyl-substituted BDT 44 was reported in 2010. 44 has higher absorption in the short-wavelength region (400–500 nm) than its alkoxy counterpart 11 due to conjugated thiophene substituents. A PCE of ∼5.7% was obtained in OPV devices with 44:PC71BM.84 A higher PCE of 7.6% has been achieved with another thienyl-BDT copolymer 45.85 45 has a redshifted absorption by 25 nm and a higher absorption coefficient than its analog 15, which originates from the extended conjugation through alkylthienyl side chains and, possibly, a better π–π stacking. The HOMO level of 45 was 0.08 eV lower than alkoxy-substituted 15, contributing to a higher VOC of 0.79 V and giving a PCE of 7.6% for the 45:PC71BM cell. Installing two more thiophene groups on each thiophene substituent in 45 resulted in a further reduction of the HOMO level to −5.64 eV for 46 affording an enhanced VOC of 0.90 V for the 46:PC71BM device.86 However, the JSC of the corresponding device is only 4 mA cm−2, much lower than that of its mono-thiophene analog 45, which might be due to increased steric hindrance and less ordered intermolecular packing introduced by adjacent thiophene groups.
While thienyl and other aryl rings on BDT are useful for extending the conjugation and broadening the absorption,85 this effect is limited by their twist with respect to the polymer backbone. Zhang et al. found that the smaller furyl substituents in 47 display a lower twist with respect to BDT (calculated dihedral angle of 34°) compared to thienyl (59°) and selenophenyl (61°) groups in the analogous polymers 45 and 48.87 As a result, 47 presents a stronger absorption in the short-wavelength region (at ∼500 nm) than 45 and 48, due to improved side chain conjugation. Perhaps more importantly, 47 packs in the solid state with a closer π–π stacking distance of 3.63 Å, compared to 45 and 48 (3.94 Å), which in principle should be beneficial for the hole transport. However, unfortunately, a relatively low PCE of 5.3% was achieved in the 47:PC71BM BHJ (cf. ∼9% for 45 and 48) which was attributed to the excessive crystallization/phase separation in this highly planar polymer.
Introducing heteroatoms into aryl substituents can lead to a less twisted BDT structure. 1,3-Thiazole connected to BDT at its 5-position inherits the conformation of thiophene, with a dihedral angle of 59° in 49.88 Altering the connecting point to the 2-position results in a significantly smaller dihedral angle of 24° in 50, due to reduced steric repulsion. However, the more planar structure of 50 causes excessive intermolecular interactions and unfavorable phase separation in a blend with NFA BTP-eC9, resulting in a much lower PCE of 6.4% compared to 15.0% for 49.
Halogenation is one of the common strategies employed to lower the energy levels of conjugated polymers. It was explored in BDT polymer 51 by using ring-fluorinated alkylthiophene substituents and resulted in one of the most popular BDT semiconductors, widely known as PM6.89 51 exhibits a HOMO level deeper by 0.22 eV than its non-fluorinated analog 14, similar to that of 26 with a fluorinated thiophene linker on the backbone. Moving fluorine from the 4-position to 3-position on the thienyl substituent generates additional steric clashes between F and β-H on BDT and increases the torsion energy barriers from 5.5 to 12 kcal mol−1 although the minimum energy conformation is not significantly changed (Fig. 10).90 The restricted rotation of the side chains breaks the symmetry of the polymer chains, resulting in improved solubility and processability in a non-halogenated solvent, THF.
 | ||
| Fig. 10 Torsional energy profiles of thienyl-BDT with F on different positions of the thienyl side chain. Copyright 2023 (RSC).86 | ||
52 (also known as PM7) is an analog of 51 where the F atoms are replaced with Cl.91 The chlorinated thiophene building block requires fewer synthetic steps and is easier to purify than the fluorinated thiophene (Scheme 2), lowering the cost of the polymer. Also, 52 exhibits a HOMO of −5.40 eV, lower than −5.33 eV for 51 due to lower π-donation from Cl, but its absorption spectrum, film morphology and hole mobility, are very similar to those of 51.
 | ||
| Scheme 2 Synthesis route of thienyl side chains for 51 and 52. | ||
53 was designed with four side chains on the BDT unit: two alkylthienyl groups attached to 3,7-positions and two alkoxy groups attached to the 4,8-positions.92 It exhibits a λmax of 550 nm in solution, which is blue-shifted compared to that of 14 (581 nm). Such behavior suggests a less efficient conjugation in 53, which can be reasonably attributed to the large dihedral twist of the backbone with such an overcrowded building block.
Most BDT building blocks used in D–A copolymers are substituted symmetrically. Breaking this symmetry was explored in 2016 by the Yang group who made a series of D–A copolymers of BDT bearing one alkoxy and one aryl group, including polymer 54.31 Devices of 54 with PC71BM showed a 0.06 V increase of VOC compared to the symmetric dialkoxy-substituted 15 and also a ∼4 mA cm−2 increase of JSC compared to the symmetric diaryl-substituted polymer, which was attributed to better chain stacking.
Varying the type and number of substituents on the pendant aryl rings on BDT is a widely used strategy to fine-tune the electronic properties of polymers. 55 and 56 have the same backbone structure with alkoxy and alkylthio chains on the thiophene substituents, respectively.93 Due to the stronger electron-donating effect of the alkoxy group, 55 displays a higher HOMO of −5.18 eV than its alkylthio counterpart 56 (−5.41 eV). Accordingly, the VOC of corresponding devices increases from 0.74 V for 55 to 0.84 eV for 56 to as high as 0.98 V for 57 with two alkylthio chains on each thiophene substituent (Table 3).94
The trialkylsilyl group on thienyl substituents can effectively decrease the HOMO level of polymers due to the electron-withdrawing effect of the silyl group through σ*(Si)–π(C) hyperconjugation. As a result, 58 (also known as J71) bearing a bulky trialkylsilyl group presents a deeper HOMO of −5.4 eV, compared to alkylated 31 (−5.21 eV) and alkylthiolated 59 (−5.32 eV).67,95 The modulation of alkyl chains on the trialkylsilyl group was used in fine-tuning the solid state properties of polymers (58, 58a-62).96 The polymers with shorter and linear alkyl chains on the silicone atom displayed closer π–π stacking, higher absorption coefficient and higher JSC and FF values. Of all 5 polymers, devices of 58 with m-ITIC achieved the highest PCE of 12.1%.
Varying the number and the length of alkyl chains on thienyl substituents can be used to fine-tune the interchain stacking of BDT polymers. The importance of such tuning has been demonstrated by Watkins, Muellen and co-workers in polymer 63, which combines both monoalkylthienyl-BDT and dialkylthienyl-BDT units. This polymer packs significantly closer and has a better crystallinity than its analog 64 with all dialkylthienyl-BDT.97 As a result, 63 has higher hole mobility (1.5 × 10−3 cm2 V−1 s−1) and shows a significantly increased PCE of 7.2% compared to 5.7% for 64 (in BHJs with PC61BM).
Optimizing BDT polymer donors for OPV application with non-fullerene acceptors
The rise of low-band-gap NFAs (Fig. 6) in the field of OPV has created a need for polymer donors with new properties, optimized for these acceptors to achieve high PCEs. Currently, the most efficient NFAs have strong absorption in the red-infrared region of the spectrum and are nearly transparent in the blue-green region. Accordingly, they require polymer donors with a wider bandgap and a complementary absorption profile to maximize the harvesting of solar radiation (Fig. 11). Furthermore, there is an increasing demand for polymers that are processable in green solvents and are cost-effective. These properties can be achieved through the abovementioned main-chain engineering and side-chain engineering strategies. In this section, we will summarize the effort on the design of wide bandgap BDT polymers and discuss the key examples of their application in high-performance OPV in conjunction with low band-gap NFAs. | ||
| Fig. 11 Complementary absorption of NFA Y6 and PM6 films showing a complementarity of absorption of wide band-gap polymers with NFAs. | ||
BDT copolymers with benzodithiophenedione
One of the most successful series of BDT polymers is based on its copolymerization with the BDD acceptor, among which 14 (PBDB-T, Fig. 12) is the first example that delivered promising PCE and is still widely used today. The BDD unit is easy to synthesize via Friedel–Crafts acylation, allows the solubility of the polymer to be tuned through additional alkyl chains on the outer thiophene ring, and planarizes the connection with thiophene linkers via non-covalent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯S interactions. In 2016, a device with a 14:ITIC active layer yielded a PCE of 11.2%, for the first time outperforming the control device fabricated with the fullerene acceptor PC71BM (Table 4).98 Since then, 14 has been used in a BHJ with a number of different NFAs.99–103 In 2022, a ternary device with 14 and two NFAs (Y14 and SM16) delivered a PCE of 17.1%, which remains the highest OPV performance of 14 to date.104 In addition to high PCE, 14 can be processed in non-halogenated solvents and showed high stability in OPV devices.105
O⋯S interactions. In 2016, a device with a 14:ITIC active layer yielded a PCE of 11.2%, for the first time outperforming the control device fabricated with the fullerene acceptor PC71BM (Table 4).98 Since then, 14 has been used in a BHJ with a number of different NFAs.99–103 In 2022, a ternary device with 14 and two NFAs (Y14 and SM16) delivered a PCE of 17.1%, which remains the highest OPV performance of 14 to date.104 In addition to high PCE, 14 can be processed in non-halogenated solvents and showed high stability in OPV devices.105
 | ||
| Fig. 12 Representative PBDB-T type polymers with benzodithiophenedione acceptors used in high-performance OPVs (see Table 4 for their properties). | ||
| Polymer properties | OPV device | Ref.a | Year of reporta | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λmax (nm) | Eg (eV) | HOMO (eV) | LUMO (eV) | wt/wt (acceptor) | VOC (V) | JSC (mA cm−2) | FF (%) | PCE (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Refer to the publication of the corresponding OPV device. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 622 | 2.05 (EgEC) | −5.23 | −3.18 | 1:1 (ITIC) |
0.90 | 16.8 | 74.2 | 11.2 | 98 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:1:0.1 (Y14:SM6) |
0.84 | 27.5 | 73.6 | 17.1 | 104 | 2022 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 51 | 570 | 1.8 | −5.45 | −3.65 | 1:1.2 (Y6) |
0.83 | 25.3 | 74.8 | 15.6 | 106 | 2019 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:1.2 (A4T-16) |
0.88 | 21.8 | 79.8 | 15.2 | 107 | 2021 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0.8:0.2:1.2 (51:75:L8-BO) |
0.90 | 26.7 | 81.9 | 19.6 | 108 | 2022 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 52 | — | −5.51 | — | 1:1 (IT-4F) |
0.86 | 21.8 | 77 | 14.4 | 91 | 2018 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 65 | — | 1.84 | −5.55 | −3.7 | 1:1.2 (L8-BO) |
0.90 | 26.1 | 79.3 | 18.7 | 124 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 66 | — | 1.86 | −5.52 | −3.59 | 1:1:0.2 (L8-BO:BTP-2F2Cl) |
0.88 | 27.2 | 80.1 | 19.2 | 129 | 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 67 | 615 | 1.83 | −5.52 | −3.38 | 1:1.2 (L6-BO) |
0.84 | 28.0 | 77.2 | 18.2 | 131 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 68 | — | 1.67 | −5.46 | −3.46 | 1:1.2 (L8-BO) |
0.91 | 26.6 | 78.9 | 19.0 | 132 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 69 | 570 | — | −5.48 | −3.45 | 1:1.2 (L8-BO) |
0.91 | 26.7 | 79 | 19.1 | 133 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 70 | 617 | 1.82 | −5.55 | −3.73 | 1:1.2 (BTP-eC9) |
0.84 | 27.1 | 78.0 | 17.7 | 134 | 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 71 | 619 | 1.84 | −5.56 | −3.72 | (BTP-eC9) | 0.86 | 26.8 | 80.0 | 18.4 | 135 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 72 | — | 1.76 | −5.63 | −3.66 | 1:1.2 (L8-BO) |
0.91 | 25.6 | 78.1 | 18.3 | 136 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 73 | 560 | 1.85 | −5.64 | −3.62 | 1:1.3 (Y6) |
0.82 | 26.9 | 68.2 | 15.0 | 137 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 74 | 564 | 1.84 | −5.61 | −3.57 | 1:1.3 (Y6) |
0.80 | 27.3 | 61.1 | 13.4 | 137 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Polymer properties | OPV device | Ref.a | Year of reporta | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λmax (nm) | Eg (eV) | HOMO (eV) | LUMO (eV) | wt/wt (acceptor) | VOC (V) | JSC (mA cm−2) | FF (%) | PCE (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Refer to the publication of the corresponding OPV device. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 75 | 581 | 1.98 | −5.44 | −3.47 | 1:1.6 (Y6) |
0.86 | 27.7 | 76.6 | 18.2 | 140 | 2020 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:1.2 (L8-BO) |
0.91 | 26.5 | 80.7 | 19.7 | 141 | 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 76 | 576 | 2.00 | −5.49 | −3.49 | 1:1.4:0.2 (Y6:PC71BM) |
0.87 | 26.8 | 77.0 | 18.0 | 147 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| (BTP-4F-P2EH) | 0.92 | 27.9 | 80.8 | 20.8 | 148 | 2024 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 77 | 576 | 1.97 | −5.52 | −3.64 | 1:1.2 (L8-BO) |
0.92 | 25.5 | 80.5 | 18.8 | 149 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 78 | 545 | 1.91 | −5.55 | −3.42 | 1:1 (BTP-eC9-4F) |
0.88 | 27.1 | 76.4 | 18.1 | 128 | 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 79 | 569 | 1.82 | −5.51 | −3.35 | 1:1.2 (L8-BO) |
0.90 | 25.9 | 77.4 | 18.1 | 150 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 80 | 583 | 1.98 | −5.31 | −2.77 | bilayer (Y6) | 0.87 | 27.8 | 78.9 | 19.1 | 151 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 81 | 581 | 1.98 | −5.58 | −3.68 | 1:1.2 (Y6) |
0.87 | 27.0 | 78.7 | 18.4 | 152 | 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A great many analogs of PBDB-T (14) have been developed through the years. Hou et al. introduced the fluorine atom to the alkylthienyl side chains of BDT, making polymer 51, which has become widely known as PM6.89 51 exhibits a deeper HOMO level of 0.22 eV than its non-fluorinated analog 14. In 2019, OPV devices of 51 and Y6 achieved an impressive PCE of 15.6% with VOC of 0.83 V and FF of 74.8%, marking the emergence of the Y6 series as the current state-of-the-art NFAs.106 Later, a similarly high PCE of 15.2% was demonstrated with 51 and a synthetically simpler non-fused NFA A4T-16.107 In a ternary system with 51, 75 and L8-BO, the authors observed the formation of a multi-length scaled double-fibril network which they believe is responsible for the very high device efficiency of 19.6%.108 Recently, a record efficiency of 19.7% was reported for 51-based solar cells.109 51 has now become the benchmark polymer donor, widely used to test new NFAs,99,110–113 unravel photophysical processes in OPVs114,115 and explore new device engineering approaches, e.g. transparent,116 stretchable,117,118 and bilayer (quasi planar heterojunctions)119 devices, interfacial engineering,120 etc.
52 (PM7) is an analog of 51 where the fluorine atoms are replaced with Cl which has a lower HOMO and is synthetically simpler. OPVs with 52 have reached PCE between 14% and 17% for several systems.91,121–123 By incorporating a perfluoroisopropoxyalkyl group as the side chain on the PM7 backbone an enhanced PCE of 18.7% was achieved for polymer 65 with NFA L8-BO.124 The fluoroalkyl chains provide a handle to control the phase separation in the BHJ and a further increase of PCE to 19.1% was realized after fluorous solvent vapor annealing of the active layer.
Many high-efficiency donor polymers have been developed by incorporating a third comonomer and optimizing its ratio in the structure of PM6 51 and PM7 52.125–128 Random terpolymer 66 introduced a moderately electron-withdrawing thiazolothiazole (TTz) with thiophene linkers into 51's backbone.129 The TTz-thiophene connection is almost planar while the BDD-thiophene link has a dihedral angle of 16°, which may explain the improved crystallinity and charge mobility of 66 compared to 51. 66 exhibits excellent batch-to-batch reproducibility, and PCEs of ca. 18% were reported in blends of 66 with various NFAs (Y6, BTP2F2Cl and L8-BO).129,130 The efficiency was further enhanced to 19.2% in a ternary blend of 66:L8-BO:BTP2F2Cl.
A simple 1,4-bis(trifluoromethane)benzene as a third co-monomer in PM6 analog 67 significantly improves the solubility of the polymer, allowing the 67:Y6-BO active layer to be processed from a non-halogenated solvent (xylene), achieving a high PCE of 18.2%.131
Replacing 3% of the BDD unit in PM6 with the zwitterionic borylated dithienopyrazine acceptor in polymer 68 (ref. 132) lowered the HOMO of the polymer allowing it to achieve a high VOC of 0.91 V and a PCE of 19.0% for the 68:L8-BO device. An analogous terpolymer 69 with a structurally related BF2-fused bipyridine acceptor was reported later.133 The out-of-plane dipoles of the BF2 group contribute to the strong interchain interactions and increase the crystallinity of L8-BO in the blend film and OPV devices with the 69:L8-BO active layer delivered a high PCE of 19.1%.
A non-conjugated terpolymer 70 was synthesized by incorporating a flexible penta(ethylene glycol) spacer in the PM6 structure, as a means to enhance the mechanical properties of the film.134 A stretchable device fabricated with BTP-eC9 NFA yielded a PCE of 12.1%, with 80% of the efficiency retained at 22% strain. Meanwhile, regular devices with the same active layer fabricated from 70 in a non-halogenated solvent (toluene) delivered a high PCE of 17.7%.
Other examples of introducing third units in the PM6/PM7 backbone include substituted thienothiophenes in 71 and thienopyrroledione in 72.135,136 71 achieved a PCE of 18.4% and an outstanding FF of 80% with BTP-eC9. Binary devices with 72 and L8-BO yielded a PCE of 18.3% and an even higher PCE of 19.4% was achieved in ternary devices.
Polymer 73 was developed by breaking the symmetry of BDT.137 This polymer with one alkylthio and one alkoxy side chain exhibits a reduced π–π stacking distance compared to its symmetric counterparts (74 and 53), which contributes to an increased hole mobility of 10−3 cm2 V−1 s−1 in its blend with Y6. The BHJ devices 73:Y6 yielded a PCE of 15.0%, higher than the 13.4% for 74 and 14.7% for 53.
BDT copolymers with dithienobenzothiadiazole
Benzo[c][1,2,5]thiadiazole (BT) has been widely used in conjugated polymers and PCEs of up to 12% have been achieved with BDT-BT polymers and ITIC-type NFAs.138 However, the high electron deficiency of BT typically leads to low bandgaps (<1.80 eV) in these polymers, which is not ideal for devices with the NIR-absorbing NFAs as it results in underutilization of the short-wavelength range of the solar spectrum.139 One method to mitigate the strong electron-accepting ability of BT acceptor is by fusing it with electron-donating thiophenes to give dithienobenzothiadiazole (DTBT). In 2020, polymer 75 (also known as D18, Fig. 13) with BDT and DTBT moieties separated by thiophene linkers was reported.140 75 has a wide bandgap of 1.98 eV and a high hole mobility of 1.6 × 10−3 cm2 V−1 s−1. The BHJ device of 75 with Y6 yielded a PCE of 18.2%. To mitigate the low solubility of 75, a high-pressure coating method was developed. This method allowed high molecular weight 75 (85 kDa) to be processed at 100 °C and the fabricated devices with L8-BO showed a very high PCE of 19.7%.141 Other than traditional BHJ architecture, 75 was also utilized in a bilayer p-i-n quasi-planar heterojunction structure with a partial donor/acceptor intermixed phase delivering high PCEs around 19%.141,142 | ||
| Fig. 13 Representative D18-type polymers with dithienobenzothiadiazole for high-performance OPVs (see Table 5 for their properties). | ||
Many modifications of 75 (D18) have been explored to optimize the PCE of NFA-based OPVs.143–146 The chlorination of thienyl substituents in 76 lowers the HOMO level to −5.49 eV (cf. −5.44 eV for 75) and a PCE of 18.0% was achieved in the initial studies in the ternary 76:Y6:PC71BM blend.147 Furthermore, a record-breaking PCE of 20.8% was reported for 76-based bilayer devices with BTP-4F-P2EH acceptor last year.148
The terpolymer strategy has been widely explored for 75. Random terpolymer 77 with a p-trifluoromethylpyridine acceptor presented reduced aggregation tendency compared to 75 and delivered PCEs of >18% in a BHJ with various NFAs.149 Both BDD and DTBT acceptor units were used in polymer 78, which is effectively a mixed copolymer of PM6 (51) and D18 (75).128 The new polymer displayed a much-enhanced solubility and temperature-dependent aggregation behavior in xylene, which is not observed for either of the corresponding parent copolymers. The optimized 78:BTP-eC9-4F device reached a PCE of 18.1%. Increasing the BDD:DTBT ratio from 1:4 in 78 to over 5:1 in 79 resulted in a bandgap reduction by 0.09 eV and a slightly increased HOMO.150 Devices with 79:L8-BO showed high PCEs over 17.5% over the wide range of molecular weights, providing a much better reproducibility of device performance compared to the parent PM6 and D18 polymers.
A series of D18-type terpolymers was developed with a lactone-fused dithiophene unit.151 Increasing the amount of lactone in the polymer backbone gradually changes the polymer film microstructure to edge-on orientation improving the miscibility with the acceptor (Y6). This optimization approach in polymer 80 allowed a PCE of 19.1% to be achieved in bilayer devices.
Another notable example of D18-type terpolymer was developed using an ester-substituted thiazole comonomer.152 With only 5% thiazole vs. DTBT unit, 81 shows a decreased HOMO level (by 0.07 eV) and improved morphology, resulting in a PCE of 18.4% for 81:Y6 devices.
BDT copolymers with benzotriazole
Compared to the widely used benzothiadiazole, benzotriazole (BTz), with its electron-donating nitrogen, acts as a weaker acceptor, leading to wider bandgap polymers. The extra alkyl chain on the nitrogen atom allows for further fine-tuning of the solubility and packing of BTz-containing polymers. To obtain HOMO levels that pair better with NFAs, fluorine atoms are typically placed on 5,6-positions of alkylated BTz units.The first BDT copolymer with difluoro-BTz was reported in 2012 (ref. 153) and later used in combination with NFAs.154,155 A number of thienyl-BDT/difluoro-BTz copolymers with various substituents (e.g. alkyl, alkylthio and alkylsilyl groups) have been developed (Fig. 14 and Table 6).67,95,96 Due to the electron-withdrawing effect of the alkylthio substituents, 59 and 82 display a redshifted absorption (by ca. 10 nm) and decreased HOMO by 0.1 eV compared to its alkyl counterpart 31.67 This HOMO stabilization leads to higher VOC of 0.91 V for 59 and 0.89 V for 82 respectively, compared to 0.73 V for 31 (BHJs with ITIC).
 | ||
| Fig. 14 Representative polymers with BTz, Qx and other acceptors used in OPVs with NFAs (see Table 6 for their properties). | ||
| Polymer properties | OPV device | Ref.a | Year of reporta | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| λmax (nm) | Eg (eV) | HOMO (eV) | LUMO (eV) | wt/wt (acceptor) | VOC (V) | JSC (mA cm−2) | FF (%) | PCE (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Refer to the publication of the corresponding OPV device. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 31 | 538 | 1.96 | −5.21 | −2.99 | 1:1 (ITIC) |
0.73 | 13.1 | 57.8 | 5.5 | 67 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 59 | 550 | 1.93 | −5.32 | −3.08 | 1:1 (ITIC) |
0.91 | 16.3 | 60.4 | 9.0 | 96 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 82 | 552 | 1.93 | −5.32 | −3.08 | 1:1 (ITIC) |
0.89 | 17.4 | 61.5 | 9.5 | 96 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 58 | 587 | 1.96 | −5.4 | −3.24 | 1:1 (ITIC) |
0.94 | 17.3 | 70.0 | 11.4 | 95 | 2016 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 83 | 595 | 1.93 | −5.48 | −3.55 | 1:1.2 (Y18) |
0.85 | 26.8 | 74.9 | 17.1 | 69 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 84 | 544 | 1.97 | −5.30 | −3.50 | 1:1 (ZITI) |
0.94 | 21.3 | 72.5 | 14.4 | 166 | 2019 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 85 | 647 | 1.74 | −5.50 | −3.67 | 1:1.1 (Y6) |
0.86 | 25.4 | 69.5 | 15.2 | 167 | 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 86 | — | 1.87 | −5.45 | −3.14 | 1:1.2 (Y6) |
0.81 | 26.5 | 72.6 | 15.7 | 169 | 2019 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 27 | 642 | 1.72 | −5.55 | −3.83 | 1:1 (Y6) |
0.85 | 26.3 | 75.5 | 16.8 | 64 | 2020 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 87 | 529 | 2.01 | −5.49 | −3.48 | 1:1.2 (Y18) |
0.86 | 21.2 | 68.2 | 12.5 | 170 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 88 | 573 | 1.97 | −5.34 | −3.63 | 1:1.2 (eC9-2F) |
0.81 | 25.3 | 75.7 | 15.5 | 171 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 89 | 636 | 1.64 | −5.05 | — | 1:1 (ITIC) |
0.69 | 16.2 | 59.9 | 6.7 | 177 | 2017 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 90 | 619 | 1.66 | −5.19 | — | — | — | — | — | — | 177 | 2017 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 91 | 600 | 1.68 | −5.19 | — | 1:1 (ITIC) |
0.83 | 17.2 | 62.5 | 8.9 | 177 | 2017 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 92 | 583 | 1.73 | −5.35 | — | 1:1 (ITIC) |
0.95 | 17.9 | 66.8 | 11.3 | 177 | 2017 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 93 | 565 | 1.88 | −5.55 | −2.91 | 1:1.3 (Y6) |
0.84 | 26.0 | 70.8 | 15.6 | 174 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 94 | 634 | 1.71 | −5.64 | −3.18 | 1:1.3 (Y6) |
0.85 | 26.6 | 77.9 | 17.6 | 174 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 95 | — | 2.07 | −5.41 | −2.82 | 1:1:0.2 (eC9-2Cl:F-BTA3) |
0.88 | 26.7 | 80.9 | 19.0 | 175 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1:1.2 (TBT-13) |
0.79 | 25.9 | 77.9 | 16.1 | 179 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 96 | — | 2.05 | −5.39 | −2.86 | 1:1:0.2 (BTP-eC9:BTA3) |
0.84 | 26.9 | 79.6 | 18.0 | 176 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 97 | — | 2.00 | −5.26 | −2.76 | 1:1.2 (PY-IT) |
0.92 | 24.3 | 80.7 | 18.0 | 181 | 2022 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 98 | 530 | 1.80 | −5.47 | −3.72 | 1:1.4 (PY-IT) |
0.90 | 26.5 | 78.6 | 18.8 | 182 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 99 | 518 | 2.06 | −5.43 | −3.37 | 1:1.2 (L8-BO) |
0.90 | 26.7 | 77.6 | 18.6 | 186 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 100 | 531 | 2.02 | −5.66 | −3.47 | 1:1.8 (Y6) |
0.81 | 24.7 | 75.4 | 15.1 | 187 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 101 | 540 | 2.00 | −5.54 | −3.65 | 1:1.2 (PY-IT) |
0.95 | 24.6 | 73.7 | 17.2 | 188 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 102 | — | — | −5.45 | −3.47 | 0.8:0.2:1.2 (51:102:PY-IT) |
0.94 | 24.7 | 76.3 | 17.6 | 189 | 2023 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 103 | — | — | −5.55 | −3.57 | 1:0.15:1.2 (51:103:BTP-eC9) |
0.85 | 28.2 | 78.3 | 18.7 | 190 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 104 | — | — | −5.59 | −3.64 | 1:0.15:1.2 (51:104:BTP-eC9) |
0.85 | 27.5 | 78.2 | 19.0 | 190 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 105 | 557 | 1.98 | −5.47 | −3.49 | 1:1.5 (N3) |
0.86 | 26.6 | 77.3 | 17.6 | 191 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 106 | 562 | 2.05 | −5.39 | −3.49 | 1:1.2 (BTP-eC9-4F) |
0.91 | 26.8 | 67.1 | 16.2 | 192 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 107 | — | 2.12 | −5.64 | −3.52 | 1:1 (IT4F) |
0.96 | 20.5 | 74 | 14.5 | 193 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 108 | 552 | 1.92 | −5.58 | −3.66 | 1:1.35 (Y6) |
0.85 | 27.2 | 74.3 | 17.1 | 194 | 2021 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 109 | 537 | 2.00 | −5.63 | −3.54 | (BTP-eC9-4F) | 0.86 | 26.1 | 66.1 | 15.0 | 195 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 110 | 556 | 1.93 | −5.53 | −3.17 | 1:1.2 (Y6) |
0.87 | 28.2 | 77.3 | 19.0 | 196 | 2024 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Bulky trialkylsilylthienyl substituents are used in a popular BDT-BTz copolymer 58 (also known as J71).95 Apart from improving the solubility of 58, trialkylsilyl groups deepen the HOMO of the polymer compared to its alkyl counterpart 31. The first solar cell based on 58 yielded a PCE of 11.4% and a VOC of 0.94 V with ITIC,95 and further device optimization delivered PCEs of up to 16.5%.156–159
Between 2019 and 2022, more than ten BDT-BTz polymers were reported with PCEs >14%.160–165 In 2023, a record (for BDT-BTz polymers) PCE of 17.1% was reported with polymer 83 and Y18 acceptor.69 This polymer presented a high carrier mobility of ∼10−3 cm2 V−1 s−1 and a high JSC of 26.8 mA cm−2, which is the highest value for all BDT-BTz polymers. Notably, 83 is processable in non-halogenated solvents (xylene), with a respectable PCE of 16.9% for 83:Y18 devices.
The electron-accepting ability of difluoro-BTz can be increased by replacing fluorine with other substituents. 84 with chlorine-bearing BTz presents a slightly lower (by 0.03 V) HOMO and almost identical bandgap to 57.166 It should be noted, however, that the substitution of F with Cl results in a less planar and less rigid polymer backbone (Fig. 15). The BHJ with 84 and NFA ZITI delivered a PCE of 14.4%.
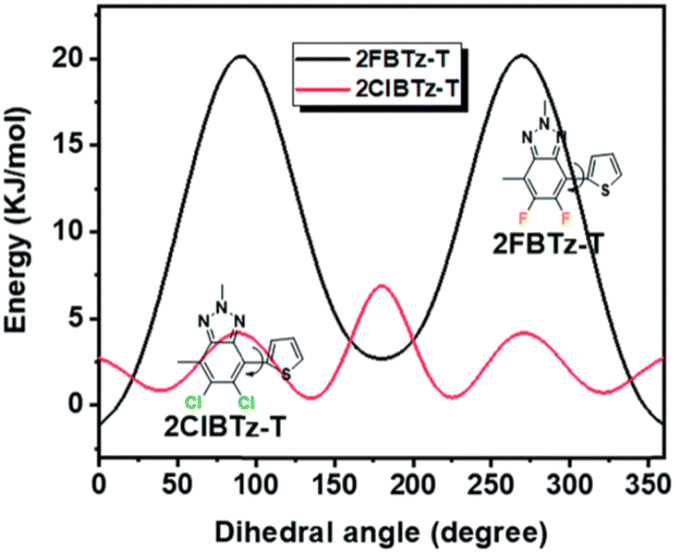 | ||
| Fig. 15 The twisting barriers of chlorine/fluorine substituted BTz with an adjacent thiophene linker calculated by the DFT method. Copyright 2019 (RSC).166 | ||
Electron-withdrawing cyano groups significantly decrease the energy level and 85 exhibits a HOMO of −5.50 eV, 0.29 eV lower than the fluorine analog 31.167 As a result, the 85:Y6 blend yielded a VOC of 0.86 V (0.74 V for 31:Y6) and a PCE of 15.2%. Dicarboxyimide substitution on BTz also results in a lowered HOMO (0.13 eV) and a smaller bandgap (0.15 eV) than 58.168 This strategy was used to develop polymer 86, which has a HOMO of −5.45 eV and delivers a VOC of 0.81 V and a PCE of 16% in the BHJ with Y6.169 The same acceptor was later used in polymer 27 affording a PCE of 16.8% with Y6.64
On the other hand, fusing thiophene on both sides of the BTz unit makes it less electron-deficient than difluoro-BTz and results in a wider bandgap (2.01 eV) for 87 (1.94 eV for difluoro-BTz analog 83).170 Removal of fluorine atoms from the BTz unit eliminates the S⋯F interactions between the acceptor and adjacent π-linkers, resulting in a twisted backbone with a 13° dihedral angle which weakens both intramolecular and intermolecular electronic coupling. The lower absorption of the solar radiation results in the low JSC of 21.2 mA cm−2 of 87:Y18 devices (cf. 26.8 mA cm−2 for 83:Y18 devices). Several other polycyclic BTz acceptors have been explored and a PCE of 15.5% has been reported for 88.171 While the synthetic complexity of these linker/acceptor units will limit its application, this example is notable in using a diphenyl-BDT (rather than dithienyl-BDT) polymer in high-performance OPVs.
BDT copolymers with quinoxaline
The two nitrogen atoms provide quinoxaline (Qx) a moderate electron-withdrawing character for developing wide bandgap copolymers. Multiple positions on the unit allow for installing side chains to fine-tune the optoelectronic properties and aggregation of corresponding materials. Several BDT copolymers with Qx and its derivatives have shown high OPV performance (PCE 11–19%) in combination with NFAs (Fig. 14 and Table 6).172–176 The impact of the number and position of fluorine substituents on the optical properties and energy levels was studied in Qx-based polymers 89–92.177 The absorption coefficient of the polymer is increased when more fluorine atoms are incorporated. HOMO levels of all three fluorine-bearing polymers (90–92) are lower than that of the non-fluorinated 89. When these polymers were paired with ITIC, a VOC of up to 0.95 V and a PCE of up to 11.3% was achieved in 92-based devices.172Y. Li and co-workers studied the role of side chains on the Qx unit in polymers 93 and 94.174 As expected, 94 with the conjugated fluorothienyl substituents on Qx presented a significantly lower bandgap and a 70 nm redshifted absorption compared to 93 with simple alkyl substituents. Furthermore, a reduced π–π stacking distance in 94 results in twice higher hole mobility to 93 and a higher PCE of 17.6% in 94:Y6 devices.
A fused dithienoquinoxaline unit was incorporated in 95.175 Fusion with electron-rich thiophene decreases the electron deficiency of Qx and, as a result, 95 presents a wider bandgap of 2.07 eV, which improves the absorption of the higher energy photons. The device of 95 with eC9-2Cl delivered a PCE of 17.7%, and the device performance can be further enhanced to 19% by introducing a third component, F-BTA3, or other additives.178 In a recent report, a PCE of 16.1% was delivered by the BHJ of 95 and fully non-fused NFA TBT-13.179 The chlorinated polymer 96 has similar optical bandgap and energy levels as its fluorinated analog 95, and similarly high-performing ternary devices with PCEs of 18.0%176 and even 19.0%180 have been reported for this polymer. The processability of 96 in green (non-chlorinated) solvents can be improved by methylating the pyrazine ring. Indeed, the so-designed polymer 97 can be processed from toluene and showed a remarkable PCE of 18.0% in a binary device with a polymer acceptor, PY-IT (Fig. 6), one of the highest values for all-polymer solar cells.181
Enhancing the electron deficiency of Qx with dicarboxyimide substituents lowers the HOMO of the polymer 98 to −5.47 eV, without halogen substitution on the BDT moiety (cf. −5.41 eV for 95)182 The PCE of 18.8% achieved in all-polymer cells with the PY-IT acceptor stands as the record value for non-halogenated polymer donors.
BDT copolymers with other acceptors
A number of other, less common acceptors that have been used for high-performing BDT-based polymers including benzimidazole-fused dithiophene, pentacyclic azepinedione, etc.183–185 Copolymer 99 (Fig. 14 and Table 6) with difluorobenzothiazole has a wide bandgap of 2.06 eV.186 Devices with 99 achieved high performance with several NFAs, among which the 99:L8-BO mixture delivered a PCE of 18.6%. Benzobisthiazole is a stronger acceptor unit than difluorobenzothiazole endowing polymer 100 with a fairly low HOMO of −5.66 eV (cf. −5.43 eV for 99).187 Interestingly, the 100:Y6 BHJ showed a respectable PCE of 15.1% despite the energy offset between 100 and Y6 being only 0.04 V, significantly below the commonly recommended >0.3 eV. A derivative of 100 with different alkyl chains (101) was later combined with the PY-IT acceptor and yielded a PCE of 17.2%.188 Related polymers 102–104 were developed by connecting benzobisthiazole via 2,6-positions and attaching additional alkylthienyl side chains to the 4,8-positions.189,190 This acceptor unit is isoelectronic to the dithienyl-BDT donor unit and the expanded side-chain conjugation contributes to the redshifted absorptions for 102–104. Introducing 102–104 as the third component in a ternary BHJ enhances the absorption of the active layer, resulting in PCEs of 17.9–19.0%.
The large fused polycyclic structure of the thienoacenaphthylenedicarboxyimide acceptor in polymer 105 results in its low solubility in chloroform. Thus, films of 105 cast from chlorobenzene can tolerate the chloroform deposition of the N3 acceptor during bilayer device fabrication.191 The resulting bilayer films achieved PCEs as high as 17.6% and showed very little batch-to-batch variation, even for polymers of different molecular weights, which is critical for commercial applications. 106 with the difluoronaphthodithiophene acceptor presents a higher HOMO and wider bandgap compared to its analog 75 (D18), due to the relatively low electron deficiency of difluoronaphthodithiophene.192 OPV devices with the 106:BTP-eC9 blend yielded a PCE of 16.2%. Although this is no longer a record-breaking efficiency, it is worth noting that the difluoronaphthodithiophene unit can be synthesized in two steps in >70% yield, which compares favorably to the four-step <30% yield synthesis of the DTBT monomer of D18.
In recent years, developing low-cost polymer donors incorporating simple acceptor units has become an important trend. Many of the champion materials discussed above are based on sophisticated acceptor comonomers which require as many as 17 synthetic steps (!) and complex purification in their preparation.54,64,168 In contrast, polymer 107 used a simple acceptor, 1,3,4-thiadiazole.193 107 has a wide bandgap of 2.12 eV and an ultra-deep HOMO level of −5.64 eV. This low HOMO contributes to the high VOC of 0.96 V in a BHJ with IT-4F, and the corresponding device reached a PCE of 14.5%. 108 was built by copolymerizing BDT with cyanoterthiophene, which can be prepared in 3 synthetic steps.194 The OPV devices of 108 with Y6 achieved high PCE in the range of 15.9–17.1%. Notably, the molecular weight of this polymer (Mn ranging from 18 to 74 kDa) showed very little effect on the device efficiency, which is important for batch-to-batch reproducibility. Another simple acceptor, 4-chlorothiazole, was used to synthesize polymer 109.195 109 delivered a PCE of 15.0% in a binary device with BTP-eC9-4F and 19.2% in a ternary system with 75 and L8-BO, outperforming current benchmark polymer donors (PM6, D18, etc.) in terms of ratio of PCE to synthetic complexity.
1,2,3-Benzothiadiazole (iBT), an asymmetric isomer of BT which can be readily made in one step from 2-mercaptoaniline, was used as an acceptor in polymer 110.196 The absorption band of 110 is blue-shifted compared to its BT analog, indicating a lower electron-deficiency of iBT vs. BT unit. On the other hand, single-crystal X-ray analysis of methylthiophene-iBT and methylthiophene-BT shows that the iBT unit provides a more planar connection with the adjacent thiophene linkers, which could explain a two-fold increase of charge carrier mobility of 110 to 2.3 × 10−3 cm2 V−1 s−1. The device with 110 and Y6 achieved a PCE of 19.0% with a high JSC of 28.2 mA cm−2. This is a remarkable efficiency for the binary Y6-only device which shows that there is room for improving the performance of BDT polymers beyond the classical PM6 and D18 structures.
Conclusions and outlook
Over the past decades, great research efforts have been invested in the design of semiconducting polymer donors for OPVs. In particular, several hundreds of BDT-based copolymers were investigated as OPV materials. Molecular engineering of the conjugated backbone, as well as side chain engineering, have been effective in developing p-type polymer semiconductors with controlled bandgap, good charge transport properties and processability. With these strategies, bandgaps between 1.3 and 2.1 eV and hole mobility as high as 8.6 × 10−3 cm2 V−1 s−1 have been realized. As a result, PCEs exceeding 20% have been reliably achieved for small-area devices148,197 and 13.9% for a 55 cm2 sub-module processed in green solvents.198 Nevertheless, the broad deployment of BDT-based polymers in the commercialization of OPV technology still requires solving a number of issues.Morphological control, now well-understood for BHJs based on P3HT and fullerene derivatives,199 remains difficult for high-performing BDT polymers and requires exploring new approaches. For example, matching the solution aggregation of donors and acceptors can favor the formation of an optimal device morphology, crucial for achieving high PCE.200 BDT-based polymers offer opportunities to create a diverse range of polymers with varying aggregation properties, influenced by molecular confirmation and interchain stacking, via modifications to side chains, substituents positions, etc. In this context, quasi-planar heterojunctions prepared by sequential solution coating of the BDT polymer donor and the NFA layers, can now deliver OPVs with very high PCE, and potentially are easier to control than BHJs.201
At the molecular level, a few sub-classes of BDT-based donor–acceptor polymers have emerged as champions of high-performance OPVs. Most of these are based on either benzodithiophenedione (PBDB-T, PM6, PM7 are their variations) or dithienobenzothiadiazole (D18 and variations) acceptor units. While the search for alternative structures with high OPV performance takes place worldwide, it is important to remember that increased chemical complexity comes at a cost and ultimately the simpler structures are more likely to lead to a commercially successful technology.
Given the lengthy synthesis of the BDT-based (and most other high-performing) polymers, more attention should be paid to developing environmentally friendly synthetic routes with fewer steps, such as DArP. The side chains commonly placed at the 4,8-positions of BDT create the steric hindrance of the β-CH, improving the regioselectivity of DArP to the α-CH positions. However, still relatively few papers explore the DArP copolymerization of BDT with other building blocks.37,202,203 Further research establishing the optimal reaction conditions is necessary for the broader implementation of this strategy.
The role of computational chemistry in material development has significantly increased over the past decade, aiding in understanding the structure–property relationships, reducing the workload in screening experiments and design of new, highly complex materials.204 However, most simulations of conjugated materials (including BDT-based polymers) have relied on density-functional theory calculations of individual molecules or single polymer chains (under periodic boundary conditions). While these calculations may accurately represent the molecular properties (confirmation, orbital/band energies, etc.), they give only limited insights into the behavior of such molecules in the condensed phase, especially in heterogeneous semicrystalline bulk heterojunction films. Multiscale modeling of the latter is an infinitely more difficult problem, and most attempts have focused on the simple (and likely no longer technologically relevant) model polymers such as P3HT.205–207 Advancing such modeling of the key parameters of OPV active layers such as phase separation, exciton dissociation/recombination, and charge mobilities is essential for the progress of the field. While machine learning is becoming an increasingly valuable tool in the OPV field,204,208–210 its application in designing BDT-based polymer donors and actively predicting the behaviour of such materials in devices is yet to be fully explored.
Last but not least, while the high (photo)stability of the BDT building block is one of the reasons for its widespread application in OPV materials, the stability of BDT-containing polymers can vary widely with its structure. Although a correlation between the energy levels of the polymer and its resistance to photooxidation has been demonstrated in some cases,211–213 it is also clear that the redox properties are not sufficient to predict the photostability.214 A better understanding of the photodegradation pathways of BDT polymers is required for designing more stable solar cells.
Abbreviations
| A | Acceptor |
| BDD | Benzo[1,2-c:4,5-c′]dithiophene-4,8-dione |
| BDT | Benzo[1,2-b:4,5-b′]dithiophene |
| BHJ | Bulk heterojunction |
| BT | Benzo[c][1,2,5]thiadiazole |
| BTz | Benzo[d][1,2,3]triazole |
| D | Donor |
| DArP | Direct C–H arylation polymerization |
| DPP | Diketopyrrolopyrrole |
| DTBT | Dithienobenzothiadiazole |
| FF | Fill factor |
| GIXS | Grazing-incidence X-ray scattering |
| HOMO | Highest occupied molecular orbital |
| JSC | Short-circuit current density |
| LUMO | Lowest unoccupied molecular orbital |
| NFA | Non-fullerene acceptor |
| OPV | Organic photovoltaics |
| PCE | Power conversion efficiency |
| PC61BM | (6,6)-Phenyl-C61-butyric acid methyl ester |
| PC71BM | (6,6)-Phenyl-C71-butyric acid methyl ester |
| P3HT | Poly(3-hexylthiophene) |
| Qx | Quinoxaline |
| TT | Thieno[3,4-b]thiophene |
| VOC | Open-circuit voltage |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC Discovery and NRC AI4D grants. The authors thank Dr Steven Xiao (1-Material Inc) and Dr Jianping Lu (NRC) for useful discussions.Notes and references
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CAS.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- F. Padinger, R. S. Rittberger and N. S. Sariciftci, Adv. Funct. Mater., 2003, 13, 85–88 CAS.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CAS.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CAS.
- J. Roncali, Chem. Rev., 1997, 97, 173–206 CAS.
- J. Wang, P. Xue, Y. Jiang, Y. Huo and X. Zhan, Nat. Rev. Chem., 2022, 6, 614–634 Search PubMed.
- M. Svensson, F. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersson, Adv. Mater., 2003, 15, 988–991 CAS.
- M. Zhang, H. N. Tsao, W. Pisula, C. Yang, A. K. Mishra and K. Müllen, J. Am. Chem. Soc., 2007, 129, 3472–3473 CAS.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CAS.
- R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–253 CrossRef CAS.
- D. W. H. MacDowell and J. C. Wisowaty, J. Org. Chem., 1971, 36, 4004–4012 CrossRef CAS.
- M. Pomerantz, J. Wang, S. Seong, K. P. Starkey, L. Nguyen and D. S. Marynick, Macromolecules, 1994, 27, 7478–7485 CrossRef CAS.
- J. Hou, M.-H. Park, S. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416–10434 RSC.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CAS.
- C. Cui and Y. Li, Energy Environ. Sci., 2019, 12, 3225–3246 CAS.
- B. Zheng, L. Huo and Y. Li, NPG Asia Mater., 2020, 12, 3 CAS.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CAS.
- C. Lin, R. Peng, J. Shi and Z. Ge, Exploration, 2024, 4, 20230122 CAS.
- Y. Wei, Q. Peng, C. Zhong, S. Ma, T. Wang, Y. Pu, W. Zhang, S. Wang and L. Xie, Dyes Pigm., 2024, 225, 112097 CAS.
- D. Zhou, Y. Wang, S. Yang, J. Quan, J. Deng, J. Wang, Y. Li, Y. Tong, Q. Wang and L. Chen, Small, 2024, 20, 2306854 CAS.
- C. An and J. Hou, Acc. Mater. Res., 2022, 3, 540–551 CAS.
- D. Cevher, S. C. Cevher and A. Cirpan, Mater. Today Commun., 2023, 37, 107524 CAS.
- D. Lee, S. W. Stone and J. P. Ferraris, Chem. Commun., 2011, 47, 10987–10989 CAS.
- R. L. Uy, L. Yan, W. Li and W. You, Macromolecules, 2014, 47, 2289–2295 CAS.
- H. Pan, Y. Li, Y. Wu, P. Liu, B. S. Ong, S. Zhu and G. Xu, Chem. Mater., 2006, 18, 3237–3241 CAS.
- T. M. Pappenfus, D. T. Seidenkranz and E. W. Reinheimer, Heterocycles, 2012, 85, 355–364 CrossRef CAS.
- D. Liu, Q. Zhu, C. Gu, J. Wang, M. Qiu, W. Chen, X. Bao, M. Sun and R. Yang, Adv. Mater., 2016, 28, 8490–8498 CrossRef CAS PubMed.
- S. Sharma, R. Soni, S. Kurungot and S. K. Asha, J. Phys. Chem. C, 2019, 123, 2084–2093 CrossRef CAS.
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed.
- Z. Xiao, J. Subbiah, K. Sun, D. J. Jones, A. B. Holmes and W. W. H. Wong, Polym. Chem., 2014, 5, 6710–6717 RSC.
- D. Zhou, N. Y. Doumon, M. Abdu-Aguye, D. Bartesaghi, M. A. Loi, L. J. Anton Koster, R. C. Chiechi and J. C. Hummelen, RSC Adv., 2017, 7, 27762–27769 RSC.
- R. Matsidik, H. Komber, A. Luzio, M. Caironi and M. Sommer, J. Am. Chem. Soc., 2015, 137, 6705–6711 CrossRef CAS PubMed.
- A. S. Dudnik, T. J. Aldrich, N. D. Eastham, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 15699–15709 CrossRef CAS PubMed.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CAS.
- D. Zimmermann, C. Sprau, J. Schröder, V. G. Gregoriou, A. Avgeropoulos, C. L. Chochos, A. Colsmann, S. Janietz and H. Krüger, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1457–1467 CrossRef CAS.
- F. Lombeck, H. Komber, S. I. Gorelsky and M. Sommer, ACS Macro Lett., 2014, 3, 819–823 CAS.
- A. E. Rudenko and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 135–147 CAS.
- J. Kuwabara, Y. Fujie, K. Maruyama, T. Yasuda and T. Kanbara, Macromolecules, 2016, 49, 9388–9395 CrossRef CAS.
- M. Shi, T. Wang, Y. Wu, R. Sun, W. Wang, J. Guo, Q. Wu, W. Yang and J. Min, Adv. Energy Mater., 2021, 11, 2002709 CrossRef CAS.
- Y. He, L. Huo and B. Zheng, Nano Energy, 2024, 123, 109397 CrossRef CAS.
- S. Smeets, Q. Liu, J. Vanderspikken, T. J. Quill, S. Gielen, L. Lutsen, K. Vandewal and W. Maes, Chem. Mater., 2023, 35, 8158–8169 CrossRef CAS.
- C. K. Lo, R. M. W. Wolfe and J. R. Reynolds, Chem. Mater., 2021, 33, 4842–4852 CrossRef CAS.
- J. A. Schneider and D. F. Perepichka, in Adv. Mater., ed. T. van de Ven and S. Armand, De Gruyter, Berlin, Boston, 2020, pp. 1–50, DOI:10.1515/9783110537734-001.
- L. Huo, J. Hou, H.-Y. Chen, S. Zhang, Y. Jiang, T. L. Chen and Y. Yang, Macromolecules, 2009, 42, 6564–6571 CrossRef CAS.
- Y. Liang, Y. Wu, D. Feng, S.-T. Tsai, H.-J. Son, G. Li and L. Yu, J. Am. Chem. Soc., 2009, 131, 56–57 CrossRef CAS PubMed.
- C.-Y. Mei, L. Liang, F.-G. Zhao, J.-T. Wang, L.-F. Yu, Y.-X. Li and W.-S. Li, Macromolecules, 2013, 46, 7920–7931 CrossRef CAS.
- E. Zhou, J. Cong, K. Hashimoto and K. Tajima, Macromolecules, 2013, 46, 763–768 CrossRef CAS.
- Z. Zhang, B. Peng, B. Liu, C. Pan, Y. Li, Y. He, K. Zhou and Y. Zou, Polym. Chem., 2010, 1, 1441–1447 RSC.
- Y. Ie, J. Huang, Y. Uetani, M. Karakawa and Y. Aso, Macromolecules, 2012, 45, 4564–4571 CrossRef CAS.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. a. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- L. Xu, S. Li, W. Zhao, Y. Xiong, J. Yu, J. Qin, G. Wang, R. Zhang, T. Zhang, Z. Mu, J. Zhao, Y. Zhang, S. Zhang, V. Kuvondikov, E. Zakhidov, Q. Peng, N. Wang, G. Xing, F. Gao, J. Hou, W. Huang and J. Wang, Adv. Mater., 2024, 2403476 CrossRef CAS PubMed.
- H. J. Son, W. Wang, T. Xu, Y. Liang, Y. Wu, G. Li and L. Yu, J. Am. Chem. Soc., 2011, 133, 1885–1894 CrossRef CAS PubMed.
- A. Najari, S. Beaupré, P. Berrouard, Y. Zou, J.-R. Pouliot, C. Lepage-Pérusse and M. Leclerc, Adv. Funct. Mater., 2011, 21, 718–728 CrossRef CAS.
- R. Duan, L. Ye, X. Guo, Y. Huang, P. Wang, S. Zhang, J. Zhang, L. Huo and J. Hou, Macromolecules, 2012, 45, 3032–3038 CrossRef CAS.
- X. Wang, Y. Sun, S. Chen, X. Guo, M. Zhang, X. Li, Y. Li and H. Wang, Macromolecules, 2012, 45, 1208–1216 CrossRef CAS.
- P. Ding, Y. Zou, C.-C. Chu, D. Xiao and C.-S. Hsu, J. Appl. Polym. Sci., 2012, 125, 3936–3945 CrossRef CAS.
- H.-Y. Chen, S.-C. Yeh, C.-T. Chen and C.-T. Chen, J. Mater. Chem., 2012, 22, 21549–21559 RSC.
- L. Mohammad, Q. Chen, A. Mitul, J. Sun, D. Khatiwada, B. Vaagensmith, C. Zhang, J. Li and Q. Qiao, J. Phys. Chem. C, 2015, 119, 18992–19000 CrossRef CAS.
- Z. Fei, F. D. Eisner, X. Jiao, M. Azzouzi, J. A. Röhr, Y. Han, M. Shahid, A. S. R. Chesman, C. D. Easton, C. R. McNeill, T. D. Anthopoulos, J. Nelson and M. Heeney, Adv. Mater., 2018, 30, 1705209 CrossRef PubMed.
- B. Fan, M. Li, D. Zhang, W. Zhong, L. Ying, Z. Zeng, K. An, Z. Huang, L. Shi, G. C. Bazan, F. Huang and Y. Cao, ACS Energy Lett., 2020, 5, 2087–2094 CrossRef CAS.
- Y. Wu, C. An, L. Shi, L. Yang, Y. Qin, N. Liang, C. He, Z. Wang and J. Hou, Angew. Chem., Int. Ed., 2018, 57, 12911–12915 CrossRef CAS PubMed.
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298–6301 CAS.
- H. Bin, Z.-G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS PubMed.
- Y. Wu, Z. Li, W. Ma, Y. Huang, L. Huo, X. Guo, M. Zhang, H. Ade and J. Hou, Adv. Mater., 2013, 25, 3449–3455 CrossRef CAS PubMed.
- M. Du, A. Tang, J. Yu, Y. Geng, Z. Wang, Q. Guo, Y. Zhong, S. Lu and E. Zhou, Adv. Energy Mater., 2023, 13, 2302429 CrossRef CAS.
- L. Huo, X. Guo, Y. Li and J. Hou, Chem. Commun., 2011, 47, 8850–8852 RSC.
- J. Hou, H.-Y. Chen, S. Zhang, R. I. Chen, Y. Yang, Y. Wu and G. Li, J. Am. Chem. Soc., 2009, 131, 15586–15587 CrossRef CAS PubMed.
- J. Yi, M. Pan, L. Chen, Y. Chen, I. C. Angunawela, S. Luo, T. Zhang, A. Zeng, J. Chen, Z. Qi, H. Yu, W. Liu, J. Y. L. Lai, H. K. Kim, X. Zhu, H. Ade, H. Lin and H. Yan, Adv. Energy Mater., 2022, 12, 2201850 CrossRef CAS.
- T. E. Kang, K.-H. Kim and B. J. Kim, J. Mater. Chem. A, 2014, 2, 15252–15267 CAS.
- M.-C. Yuan, M.-Y. Chiu, C.-M. Chiang and K.-H. Wei, Macromolecules, 2010, 43, 6270–6277 CrossRef CAS.
- Y. Huang, M. Zhang, H. Chen, F. Wu, Z. Cao, L. Zhang and S. Tan, J. Mater. Chem. A, 2014, 2, 5218–5223 CAS.
- H. Heo, H. Kim, D. Lee, S. Jang, L. Ban, B. Lim, J. Lee and Y. Lee, Macromolecules, 2016, 49, 3328–3335 CAS.
- L. Huo and J. Hou, Polym. Chem., 2011, 2, 2453–2461 CAS.
- P. Sista, M. C. Biewer and M. C. Stefan, Macromol. Rapid Commun., 2012, 33, 9–20 CAS.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS PubMed.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Fréchet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS PubMed.
- Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS.
- C. Bathula, C. E. Song, S. Badgujar, S.-J. Hong, I.-N. Kang, S.-J. Moon, J. Lee, S. Cho, H.-K. Shim and S. K. Lee, J. Mater. Chem., 2012, 22, 22224–22232 RSC.
- S. Wen, X. Yun, W. Chen, Q. Liu, D. Zhu, C. Gu, M. Sun and R. Yang, RSC Adv., 2014, 4, 58426–58431 RSC.
- L. Huo, J. Hou, S. Zhang, H.-Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500–1503 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed.
- R. Sheng, Q. Liu, W. Chen, M. Sun, H. Zheng, J. Ren and R. Yang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1615–1622 CrossRef CAS.
- S. Zhang, L. Ye, W. Zhao, D. Liu, H. Yao and J. Hou, Macromolecules, 2014, 47, 4653–4659 CAS.
- K. Ma, H. Liang, Y. Wang, X. Jia, W. Shi, Z. Yao, X. Wan, C. Li and Y. Chen, Macromolecules, 2024, 57, 8392–8400 CAS.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- H. W. Cho, S. Y. Jeong, Z. Wu, H. Lim, W.-W. Park, W. Lee, J. V. Suman Krishna, O.-H. Kwon, J. Y. Kim and H. Y. Woo, J. Mater. Chem. A, 2023, 11, 7053–7065 CAS.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 Search PubMed.
- Z. Li, Y. Ma and Q. Zheng, J. Mater. Chem. A, 2023, 11, 5127–5134 CAS.
- C. Cui, W.-Y. Wong and Y. Li, Energy Environ. Sci., 2014, 7, 2276–2284 CAS.
- H. Yao, H. Zhang, L. Ye, W. Zhao, S. Zhang and J. Hou, ACS Appl. Mater. Interfaces, 2016, 8, 3575–3583 CAS.
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CAS.
- H. Bin, Y. Yang, Z. Peng, L. Ye, J. Yao, L. Zhong, C. Sun, L. Gao, H. Huang, X. Li, B. Qiu, L. Xue, Z.-G. Zhang, H. Ade and Y. Li, Adv. Energy Mater., 2018, 8, 1702324 Search PubMed.
- T. Qin, W. Zajaczkowski, W. Pisula, M. Baumgarten, M. Chen, M. Gao, G. Wilson, C. D. Easton, K. Müllen and S. E. Watkins, J. Am. Chem. Soc., 2014, 136, 6049–6055 CAS.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CAS.
- C.-M. Hsieh, M.-R. Chuang, Y. Yamada, C.-J. Su, Y. J. Chang, M. Murata, U. S. Jeng and S.-C. Chuang, ACS Appl. Mater. Interfaces, 2021, 13, 61473–61486 CAS.
- X. Wang, H. Lu, Y. Liu, A. Zhang, N. Yu, H. Wang, S. Li, Y. Zhou, X. Xu, Z. Tang and Z. Bo, Adv. Energy Mater., 2021, 11, 2102591 CrossRef CAS.
- Y. Zhou, M. Li, H. Lu, H. Jin, X. Wang, Y. Zhang, S. Shen, Z. Ma, J. Song and Z. Bo, Adv. Funct. Mater., 2021, 31, 2101742 CrossRef CAS.
- J. Zhou, Z. He, Y. Sun, A. Tang, Q. Guo and E. Zhou, ACS Appl. Mater. Interfaces, 2022, 14, 41296–41303 CrossRef CAS PubMed.
- Y. Che, M. R. Niazi, Q. Chan, P. Ghamari, T. Yu, C. Ruchlin, H. Yu, H. Yan, D. Ma, S. S. Xiao, R. Izquierdo and D. F. Perepichka, Angew. Chem., Int. Ed., 2023, 62, e202309003 CrossRef CAS PubMed.
- H. Lu, W. Liu, H. Jin, H. Huang, Z. Tang and Z. Bo, Adv. Funct. Mater., 2022, 32, 2107756 CrossRef CAS.
- H. Zhao, Z. Yin, P. Gu, Y. Liu, W. Wang, H. Lai, H.-Q. Wang, N. Li and W. Song, ACS Appl. Energy Mater., 2023, 6, 1595–1604 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- L. Ma, S. Zhang, J. Zhu, J. Wang, J. Ren, J. Zhang and J. Hou, Nat. Commun., 2021, 12, 5093 CrossRef CAS PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- X. Yu, P. Ding, D. Yang, P. Yan, H. Wang, S. Yang, J. Wu, Z. Wang, H. Sun, Z. Chen, L. Xie and Z. Ge, Angew. Chem., Int. Ed., 2024, 63, e202401518 CrossRef CAS PubMed.
- S. Li, L. Zhan, Y. Jin, G. Zhou, T.-K. Lau, R. Qin, M. Shi, C.-Z. Li, H. Zhu, X. Lu, F. Zhang and H. Chen, Adv. Mater., 2020, 32, 2001160 CrossRef CAS PubMed.
- T. Duan, W. Feng, Y. Li, Z. Li, Z. Zhang, H. Liang, H. Chen, C. Zhong, S. Jeong, C. Yang, S. Chen, S. Lu, O. A. Rakitin, C. Li, X. Wan, B. Kan and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308832 CrossRef CAS PubMed.
- S. Ono, T. Mikie, K. Nakano, K. Tajima and I. Osaka, ACS Appl. Energy Mater., 2024, 7, 2946–2954 CrossRef CAS.
- R. Zhang, H. Chen, T. Wang, L. Kobera, L. He, Y. Huang, J. Ding, B. Zhang, A. Khasbaatar, S. Nanayakkara, J. Zheng, W. Chen, Y. Diao, S. Abbrent, J. Brus, A. H. Coffey, C. Zhu, H. Liu, X. Lu, Q. Jiang, V. Coropceanu, J.-L. Brédas, Y. Li, Y. Li and F. Gao, Nat. Energy, 2024, 10, 124–134 CrossRef.
- J. Grüne, G. Londi, A. J. Gillett, B. Stähly, S. Lulei, M. Kotova, Y. Olivier, V. Dyakonov and A. Sperlich, Adv. Funct. Mater., 2023, 33, 2212640 CrossRef.
- J. Li, Q. Ji, R. Wang, Z.-G. Zhang, X. Wang, M. Xiao, Y.-q. Lu and C. Zhang, J. Am. Chem. Soc., 2024, 146, 20312–20322 CrossRef CAS PubMed.
- K. Li, Q. Fan, Y. Guo, Z. Wei, X. Yu, J. Zhong and J. Huang, Adv. Opt. Mater., 2023, 11, 2203040 CrossRef CAS.
- C. Guan, C. Xiao, X. Liu, Z. Hu, R. Wang, C. Wang, C. Xie, Z. Cai and W. Li, Angew. Chem., Int. Ed., 2023, 62, e202312357 CrossRef CAS PubMed.
- X. Li, H. Ke, S. Li, M. Gao, S. Li, J. Yu, H. Xie, K. Zhou, K. Zhang and L. Ye, Adv. Funct. Mater., 2024, 34, 2400702 CrossRef CAS.
- R. Sun, Q. Wu, J. Guo, T. Wang, Y. Wu, B. Qiu, Z. Luo, W. Yang, Z. Hu, J. Guo, M. Shi, C. Yang, F. Huang, Y. Li and J. Min, Joule, 2020, 4, 407–419 CrossRef CAS.
- J. Yu, X. Liu, Z. Zhong, C. Yan, H. Liu, P. W. K. Fong, Q. Liang, X. Lu and G. Li, Nano Energy, 2022, 94, 106923 CrossRef CAS.
- M.-A. Pan, T.-K. Lau, Y. Tang, Y.-C. Wu, T. Liu, K. Li, M.-C. Chen, X. Lu, W. Ma and C. Zhan, J. Mater. Chem. A, 2019, 7, 20713–20722 CAS.
- Q. Guo, R. Ma, J. Hu, Z. Wang, H. Sun, X. Dong, Z. Luo, T. Liu, X. Guo, X. Guo, H. Yan, F. Liu and M. Zhang, Adv. Funct. Mater., 2020, 30, 2000383 CrossRef CAS.
- G. Li, D. Li, R. Ma, T. Liu, Z. Luo, G. Cui, L. Tong, M. Zhang, Z. Wang, F. Liu, L. Xu, H. Yan and B. Tang, J. Mater. Chem. A, 2020, 8, 5927–5935 CAS.
- Z. He, S. Li, R. Zeng, Y. Lin, Y. Zhang, Z. Hao, S. Zhang, F. Liu, Z. Tang and H. Zhong, Adv. Mater., 2404824 Search PubMed.
- H. Sun, T. Liu, J. Yu, T.-K. Lau, G. Zhang, Y. Zhang, M. Su, Y. Tang, R. Ma, B. Liu, J. Liang, K. Feng, X. Lu, X. Guo, F. Gao and H. Yan, Energy Environ. Sci., 2019, 12, 3328–3337 CAS.
- J. Liang, M. Pan, G. Chai, Z. Peng, J. Zhang, S. Luo, Q. Han, Y. Chen, A. Shang, F. Bai, Y. Xu, H. Yu, J. Y. L. Lai, Q. Chen, M. Zhang, H. Ade and H. Yan, Adv. Mater., 2020, 32, 2003500 Search PubMed.
- X. Guo, Q. Fan, J. Wu, G. Li, Z. Peng, W. Su, J. Lin, L. Hou, Y. Qin, H. Ade, L. Ye, M. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 2322–2329 CAS.
- H. Lu, H. Wang, G. Ran, S. Li, J. Zhang, Y. Liu, W. Zhang, X. Xu and Z. Bo, Adv. Funct. Mater., 2022, 32, 2203193 CAS.
- J. Wu, G. Li, J. Fang, X. Guo, L. Zhu, B. Guo, Y. Wang, G. Zhang, L. Arunagiri, F. Liu, H. Yan, M. Zhang and Y. Li, Nat. Commun., 2020, 11, 4612 CAS.
- R. Sun, Y. Wu, X. Yang, Y. Gao, Z. Chen, K. Li, J. Qiao, T. Wang, J. Guo, C. Liu, X. Hao, H. Zhu and J. Min, Adv. Mater., 2022, 34, 2110147 CAS.
- Z. U. Rehman, M. Haris, S. U. Ryu, M. Jahankhan, C. E. Song, H. K. Lee, S. K. Lee, W. S. Shin, T. Park and J.-C. Lee, Adv. Sci., 2023, 10, 2302376 CAS.
- B. Pang, C. Liao, X. Xu, S. Peng, J. Xia, Y. Guo, Y. Xie, Y. Chen, C. Duan, H. Wu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2211871 CAS.
- S. Hao, X. Xu, L. Yu, S. Peng, J. Xia, Y. Xie, C. Duan, H. Wu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2301732 CAS.
- J.-W. Lee, C. Lim, S.-W. Lee, Y. Jeon, S. Lee, T.-S. Kim, J.-Y. Lee and B. J. Kim, Adv. Energy Mater., 2022, 12, 2202224 CAS.
- F. Cheng, Y. Cui, F. Ding, Z. Chen, Q. Xie, X. Xia, P. Zhu, X. Lu, H. Zhu, X. Liao and Y. Chen, Adv. Mater., 2023, 35, 2300820 CrossRef CAS PubMed.
- G. Zhang, Q. Wu, Y. Duan, W. Liu, S. Y. Jeong, H. Y. Woo, Q. Zhao and H. Zhou, Adv. Funct. Mater., 2024, 34, 2408678 CrossRef CAS.
- Z. Tao, Y. Li, W. Shen, H. Fu, Y. Ma and Q. Zheng, J. Mater. Chem. C, 2024, 12, 8435–8441 RSC.
- X. Gong, G. Li, S. Feng, L. Wu, Y. Liu, R. Hou, C. Li, X. Chen and Z. Bo, J. Mater. Chem. C, 2017, 5, 937–942 RSC.
- P. Cong, Z. Wang, Y. Geng, Y. Meng, C. Meng, L. Chen, A. Tang and E. Zhou, Nano Energy, 2023, 105, 108017 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- H. Lu, W. Liu, G. Ran, Z. Liang, H. Li, N. Wei, H. Wu, Z. Ma, Y. Liu, W. Zhang, X. Xu and Z. Bo, Angew. Chem., Int. Ed., 2023, 62, e202314420 CrossRef CAS PubMed.
- W. Gao, F. Qi, Z. Peng, F. R. Lin, K. Jiang, C. Zhong, W. Kaminsky, Z. Guan, C.-S. Lee, T. J. Marks, H. Ade and A. K.-Y. Jen, Adv. Mater., 2022, 34, 2202089 CrossRef CAS PubMed.
- T. Jia, J. Zhang, K. Zhang, H. Tang, S. Dong, C.-H. Tan, X. Wang and F. Huang, J. Mater. Chem. A, 2021, 9, 8975–8983 RSC.
- G. Cai, Z. Chen, X. Xia, Y. Li, J. Wang, H. Liu, P. Sun, C. Li, R. Ma, Y. Zhou, W. Chi, J. Zhang, H. Zhu, J. Xu, H. Yan, X. Zhan and X. Lu, Adv. Sci., 2022, 9, 2200578 CrossRef CAS PubMed.
- Z. Ou, J. Qin, K. Jin, J. Zhang, L. Zhang, C. Yi, Z. Jin, Q. Song, K. Sun, J. Yang, Z. Xiao and L. Ding, J. Mater. Chem. A, 2022, 10, 3314–3320 RSC.
- S. Shen, Y. Mi, Y. Ouyang, Y. Lin, J. Deng, W. Zhang, J. Zhang, Z. Ma, C. Zhang, J. Song and Z. Bo, Angew. Chem., Int. Ed., 2023, 62, e202316495 Search PubMed.
- A. Zeng, X. Ma, M. Pan, Y. Chen, R. Ma, H. Zhao, J. Zhang, H. K. Kim, A. Shang, S. Luo, I. C. Angunawela, Y. Chang, Z. Qi, H. Sun, J. Y. L. Lai, H. Ade, W. Ma, F. Zhang and H. Yan, Adv. Funct. Mater., 2021, 31, 2102413 CrossRef CAS.
- L. Zhu, M. Zhang, G. Zhou, Z. Wang, W. Zhong, J. Zhuang, Z. Zhou, X. Gao, L. Kan, B. Hao, F. Han, R. Zeng, X. Xue, S. Xu, H. Jing, B. Xiao, H. Zhu, Y. Zhang and F. Liu, Joule, 2024, 8, 3153–3168 Search PubMed.
- D. Qiu, C. Tian, H. Zhang, J. Zhang, Z. Wei and K. Lu, Adv. Mater., 2313251 Search PubMed.
- X. Chen, C. Liao, M. Deng, X. Xu, L. Yu, R. Li and Q. Peng, Chem. Eng. J., 2023, 451, 139046 CrossRef CAS.
- D. He, J. Zhou, Y. Zhu, Y. Li, K. Wang, J. Li, J. Zhang, B. Li, Y. Lin, Y. He, C. Wang and F. Zhao, Adv. Mater., 2024, 36, 2308909 CrossRef CAS PubMed.
- Z. Liao, D. Hu, H. Tang, P. Huang, R. Singh, S. Chung, K. Cho, M. Kumar, L. Hou, Q. Chen, W. Yu, H. Chen, K. Yang, Z. Kan, F. Liu, Z. Xiao, G. Li and S. Lu, J. Mater. Chem. A, 2022, 10, 7878–7887 RSC.
- J. Min, Z.-G. Zhang, S. Zhang and Y. Li, Chem. Mater., 2012, 24, 3247–3254 CrossRef CAS.
- L. Gao, Z.-G. Zhang, H. Bin, L. Xue, Y. Yang, C. Wang, F. Liu, T. P. Russell and Y. Li, Adv. Mater., 2016, 28, 8288–8295 CrossRef CAS PubMed.
- L. Gao, Z.-G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS PubMed.
- C. Yan, W. Wang, T.-K. Lau, K. Li, J. Wang, K. Liu, X. Lu and X. Zhan, J. Mater. Chem. A, 2018, 6, 16638–16644 RSC.
- J. Gao, R. Ming, Q. An, X. Ma, M. Zhang, J. Miao, J. Wang, C. Yang and F. Zhang, Nano Energy, 2019, 63, 103888 CrossRef CAS.
- M. Hao, T. Liu, Y. Xiao, L.-K. Ma, G. Zhang, C. Zhong, Z. Chen, Z. Luo, X. Lu, H. Yan, L. Wang and C. Yang, Chem. Mater., 2019, 31, 1752–1760 CrossRef CAS.
- G. Xie, Z. Zhang, Z. Su, X. Zhang and J. Zhang, Nano Energy, 2020, 69, 104447 CrossRef CAS.
- A. Tang, Q. Zhang, M. Du, G. Li, Y. Geng, J. Zhang, Z. Wei, X. Sun and E. Zhou, Macromolecules, 2019, 52, 6227–6233 CrossRef CAS.
- X. Xue, B. Zheng, Y. Zhang, M. Zhang, D. Wei, F. Liu, M. Wan, J. Liu, G. Chen and L. Huo, Adv. Energy Mater., 2020, 10, 2002142 CrossRef CAS.
- J. Zhou, P. Cong, L. Chen, B. Zhang, Y. Geng, A. Tang and E. Zhou, J. Energy Chem., 2021, 62, 532–537 CrossRef CAS.
- H. Zhang, H.-E. Wang, T. Zhu, Z. Liu and L. Chen, ACS Appl. Energy Mater., 2022, 5, 5026–5035 CrossRef CAS.
- J. Zhou, P. Lei, Y. Geng, Z. He, X. Li, Q. Zeng, A. Tang and E. Zhou, J. Mater. Chem. A, 2022, 10, 9869–9877 RSC.
- Y. Chen, P. Lei, Y. Geng, T. Meng, X. Li, Q. Zeng, Q. Guo, A. Tang, Y. Zhong and E. Zhou, Sci. China:Chem., 2023, 66, 1190–1200 CrossRef CAS.
- T. Wang, R. Sun, S. Xu, J. Guo, W. Wang, J. Guo, X. Jiao, J. Wang, S. Jia, X. Zhu, Y. Li and J. Min, J. Mater. Chem. A, 2019, 7, 14070–14078 RSC.
- L. Wang, T. Wang, J. Oh, Z. Yuan, C. Yang, Y. Hu, X. Zhao and Y. Chen, Chem. Eng. J., 2022, 442, 136068 CAS.
- L. Lan, Z. Chen, Q. Hu, L. Ying, R. Zhu, F. Liu, T. P. Russell, F. Huang and Y. Cao, Adv. Sci., 2016, 3, 1600032 Search PubMed.
- B. Fan, Z. Zeng, W. Zhong, L. Ying, D. Zhang, M. Li, F. Peng, N. Li, F. Huang and Y. Cao, ACS Energy Lett., 2019, 4, 2466–2472 CAS.
- M. Du, A. Tang, J. Yu, Y. Geng, Z. Wang, Q. Guo, Y. Zhong, S. Lu and E. Zhou, Adv. Energy Mater., 2023, 13, 2302429 CAS.
- N. Sun, M. Du, D. Zhang, J. Du, T. Du, L. Tang, Y. Zhang, Q. Guo and E. Zhou, Chem. Eng. J., 2024, 499, 155970 CAS.
- Z. Zheng, O. M. Awartani, B. Gautam, D. Liu, Y. Qin, W. Li, A. Bataller, K. Gundogdu, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1604241 Search PubMed.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 Search PubMed.
- C. Zhu, L. Meng, J. Zhang, S. Qin, W. Lai, B. Qiu, J. Yuan, Y. Wan, W. Huang and Y. Li, Adv. Mater., 2021, 33, 2100474 CAS.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CAS.
- Y. Xu, Y. Cui, H. Yao, T. Zhang, J. Zhang, L. Ma, J. Wang, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2101090 CAS.
- D. Liu, W. Zhao, S. Zhang, L. Ye, Z. Zheng, Y. Cui, Y. Chen and J. Hou, Macromolecules, 2015, 48, 5172–5178 CAS.
- J. Wang, Y. Wang, P. Bi, Z. Chen, J. Qiao, J. Li, W. Wang, Z. Zheng, S. Zhang, X. Hao and J. Hou, Adv. Mater., 2023, 35, 2301583 CAS.
- N. Yang, Y. Cui, T. Zhang, C. An, Z. Chen, Y. Xiao, Y. Yu, Y. Wang, X.-T. Hao and J. Hou, J. Am. Chem. Soc., 2024, 146, 9205–9215 CAS.
- T. Chen, X. Zheng, D. Wang, Y. Zhu, Y. Ouyang, J. Xue, M. Wang, S. Wang, W. Ma, C. Zhang, Z. Ma, S. Li, L. Zuo and H. Chen, Adv. Mater., 2024, 36, 2308061 CrossRef CAS PubMed.
- J. Wang, Y. Cui, Y. Xu, K. Xian, P. Bi, Z. Chen, K. Zhou, L. Ma, T. Zhang, Y. Yang, Y. Zu, H. Yao, X. Hao, L. Ye and J. Hou, Adv. Mater., 2022, 34, 2205009 CrossRef CAS PubMed.
- Z. Wang, X. Wang, L. Tu, H. Wang, M. Du, T. Dai, Q. Guo, Y. Shi and E. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202319755 CrossRef CAS PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- Q. He, J. Shaw, Y. Firdaus, X. Hu, B. Ding, A. V. Marsh, A. S. Dumon, Y. Han, Z. Fei, T. D. Anthopoulos, C. R. McNeill and M. Heeney, Macromolecules, 2023, 56, 5825–5834 CrossRef CAS PubMed.
- D. Qiu, W. A. Memon, H. Lai, Y. Wang, H. Li, N. Zheng and F. He, J. Phys. Chem. Lett., 2024, 15, 10858–10865 CrossRef CAS PubMed.
- B. Pang, C. Liao, X. Xu, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2300631 CrossRef CAS PubMed.
- S. Wang, Y. Tao, S. Li, X. Xia, Z. Chen, M. Shi, L. Zuo, H. Zhu, X. Lu and H. Chen, Macromolecules, 2021, 54, 7862–7869 CrossRef CAS.
- P. Wu, Y. Duan, Y. Li, X. Xu, R. Li, L. Yu and Q. Peng, Adv. Mater., 2024, 36, 2306990 CrossRef CAS PubMed.
- J. Wang, C. Han, S. Wen, F. Bi, Z. Hu, Y. Li, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2023, 16, 2327–2337 RSC.
- S. Liu, J. Wang, S. Wen, F. Bi, Q. Zhu, C. Yang, C. Yang, J. Chu and X. Bao, Adv. Mater., 2024, 36, 2312959 CrossRef CAS PubMed.
- H. Ning, Q. Jiang, P. Han, M. Lin, G. Zhang, J. Chen, H. Chen, S. Zeng, J. Gao, J. Liu, F. He and Q. Wu, Energy Environ. Sci., 2021, 14, 5919–5928 RSC.
- Z. Su, W. Liu, H. Song, N. Wei, J. Chen, Y. Lin, W. Zhang, X. Xu, Z. Ma, C. Li, A. Zhang, Y. Liu and Z. Bo, ACS Appl. Polym. Mater., 2024, 6, 12644–12653 CrossRef CAS.
- T. Zhang, C. An, P. Bi, Q. Lv, J. Qin, L. Hong, Y. Cui, S. Zhang and J. Hou, Adv. Energy Mater., 2021, 11, 2101705 CrossRef CAS.
- X. Yuan, Y. Zhao, T. Zhan, J. Oh, J. Zhou, J. Li, X. Wang, Z. Wang, S. Pang, P. Cai, C. Yang, Z. He, Z. Xie, C. Duan, F. Huang and Y. Cao, Energy Environ. Sci., 2021, 14, 5530–5540 RSC.
- H. Lu, H. Wang, G. Ran, W. Liu, H. Huang, X. Jiang, Y. Liu, X. Xu and Z. Bo, CCS Chem., 2024, 6, 1757–1766 CrossRef CAS.
- T. Lin, Y. Hai, Y. Luo, L. Feng, T. Jia, J. Wu, R. Ma, T. A. Dela Peña, Y. Li, Z. Xing, M. Li, M. Wang, B. Xiao, K. S. Wong, S. Liu and G. Li, Adv. Mater., 2024, 36, 2312311 CrossRef CAS PubMed.
- S. Guan, Y. Li, C. Xu, N. Yin, C. Xu, C. Wang, M. Wang, Y. Xu, Q. Chen, D. Wang, L. Zuo and H. Chen, Adv. Mater., 2024, 36, 2400342 CAS.
- T. Gokulnath, H. Kim, J. Lee, B. H. Cho, H.-Y. Park, J. Jee, Y. Y. Kim, K. Kranthiraja, J. Yoon and S.-H. Jin, Adv. Energy Mater., 2023, 13, 2302538 CAS.
- S. Agbolaghi and S. Zenoozi, Org. Electron., 2017, 51, 362–403 CAS.
- L. Xu, S. Li, W. Zhao, Y. Xiong, J. Yu, J. Qin, G. Wang, R. Zhang, T. Zhang and Z. Mu, Adv. Mater., 2024, 2403476 CrossRef CAS PubMed.
- Y. Zhu and F. He, ACS Energy Lett., 2025, 10, 935–946 CAS.
- M. Wakioka, N. Torii, M. Saito, I. Osaka and F. Ozawa, ACS Appl. Polym. Mater., 2021, 3, 830–836 CAS.
- L.-J. Yang, N. Chen, X.-M. Huang, Y. Wu, H. Liu, P. Liu, L. Hu, Z.-F. Li and S.-Y. Liu, ACS Appl. Polym. Mater., 2023, 5, 7340–7349 CrossRef CAS.
- W. Li, H. Ma, S. Li and J. Ma, Chem. Sci., 2021, 12, 14987–15006 CAS.
- R. Alessandri, J. J. Uusitalo, A. H. de Vries, R. W. A. Havenith and S. J. Marrink, J. Am. Chem. Soc., 2017, 139, 3697–3705 CAS.
- L. Simine and P. J. Rossky, J. Phys. Chem. Lett., 2017, 8, 1752–1756 CAS.
- T. Koch, J. Bachmann, T. Lettmann and N. L. Doltsinis, Phys. Chem. Chem. Phys., 2021, 23, 12233–12250 CAS.
- D. Padula and A. Troisi, Adv. Energy Mater., 2019, 9, 1902463 CAS.
- H. Sahu, F. Yang, X. Ye, J. Ma, W. Fang and H. Ma, J. Mater. Chem. A, 2019, 7, 17480–17488 CAS.
- N. G. An, J. Y. Kim and D. Vak, Energy Environ. Sci., 2021, 14, 3438–3446 CAS.
- E. T. Hoke, I. T. Sachs-Quintana, M. T. Lloyd, I. Kauvar, W. R. Mateker, A. M. Nardes, C. H. Peters, N. Kopidakis and M. D. McGehee, Adv. Energy Mater., 2012, 2, 1351–1357 CrossRef CAS.
- E. M. Speller, A. J. Clarke, N. Aristidou, M. F. Wyatt, L. Francàs, G. Fish, H. Cha, H. K. H. Lee, J. Luke, A. Wadsworth, A. D. Evans, I. McCulloch, J.-S. Kim, S. A. Haque, J. R. Durrant, S. D. Dimitrov, W. C. Tsoi and Z. Li, ACS Energy Lett., 2019, 4, 846–852 CrossRef CAS PubMed.
- I. V. Martynov, L. N. Inasaridze and P. A. Troshin, ChemSusChem, 2022, 15, e202101336 CrossRef CAS PubMed.
- M. A. Anderson, A. Hamstra, B. W. Larson and E. L. Ratcliff, J. Mater. Chem. A, 2023, 11, 17858–17871 RSC.
| This journal is © The Royal Society of Chemistry 2025 |